

Abstract
Acyltransferase (AT) domains of polyketide synthases (PKSs) select extender units for incorporation into polyketides and dictate large portions of the structures of clinically relevant natural products. Accordingly, there is significant interest in engineering the substrate specificity of PKS ATs in order to site-selectively manipulate polyketide structure. However, previous attempts to engineer ATs have yielded mutant PKSs with relaxed extender unit specificity, rather than an inversion of selectivity from one substrate to another. Here, by directly screening the extender unit selectivity of mutants from active site saturation libraries of an AT from the prototypical PKS, 6-deoxyerythronolide B synthase, a set of single amino acid substitutions was discovered that dramatically impact the selectivity of the PKS with only modest reductions of product yields. One particular substitution (Tyr189Arg) inverted the selectivity of the wild-type PKS from its natural substrate towards a non-natural alkynyl-modified extender unit while maintaining more than twice the activity of the wild-type PKS with its natural substrate. The strategy and mutations described herein form a platform for combinatorial biosynthesis of site-selectively modified polyketide analogues that are modified with non-natural and non-native chemical functionality.
INTRODUCTION
Type I polyketide synthases (PKSs) are huge mega-enzyme assembly lines that catalyze the condensation of acyl-CoA thioester building blocks to form the scaffolds of a large variety of clinically relevant polyketides.1, 2 PKSs are organized into modules of enzyme domains, whereby each discrete module is responsible for the installation and modification of a malonyl-derived extender unit into the growing polyketide chain3 (Figure 1A). The acyltransferase (AT) domain of these modular PKSs controls the specific extender unit selected by each module, which ultimately dictates large portions of polyketide structure as these extender units are assembled into natural product scaffolds. Accordingly, ATs offer powerful potential opportunities for the synthesis of regioselectively-modified analogues for optimization of pharmacological properties4–8 and the development of molecular probes.9
Figure 1.

Assembly of the erythromycin macrolactone core by 6-deoxyerythronolide B synthase (DEBS). (A) Organization of the DEBS PKS from the erythromycin biosynthetic pathway as an example of a type I PKS. (B) Production of erythronolide analogues via AT engineering. The extender unit specificity of a target AT (e.g. from the terminal DEBS module) needs to be manipulated in order to produce selectively-modified macrolactones. In this regard, an engineered PKS module needs to display substrate specificity orthogonal to that of the wild-type assembly line.
Numerous studies have described the ability of AT domains to discriminate between extender units naturally offered to the PKS in the producing organism.10, 11 Consequently, AT-swapping and complementation of inactivated ATs by trans-ATs have been explored in attempts to direct the installation of alternative extender units into polyketides.6, 7, 12, 13 However, chimeric/hybrid PKSs are often completely inactive or display activity reduced by several orders of magnitude, compared to their wild-type counterparts.13–15 Moreover, such approaches have been largely limited to the incorporation of naturally occurring extender units and by the narrow extender unit specificity of wild-type ATs. Thus, the ability to introduce diverse chemical functionality in a regio-selective fashion by these approaches is limited by the inherent extender unit specificity of the AT.
To address these limitations, and in an effort to minimally perturb important structural features of PKSs, attempts have been made to alter the extender unit specificity of individual AT domains by site-directed mutagenesis.14, 16–18 The installation of a non-natural extender unit directed to a single position in a polyketide requires a mutant AT that no longer recognizes its natural extender unit, but instead favors alternative substrates which themselves are not utilized by other ATs in the PKS (Figure 1B). To date, mutant ATs with inverted extender unit specificities have not been reported, largely due to our insufficient understanding of extender unit specificity in PKSs, and partly as a result of the difficulties associated with determining substrate specificity of mutant PKSs in vivo.19
Recently, our group reported that the terminal module from the 6-deoxyerythronolide B synthase (DEBS) displays remarkable in vitro promiscuity towards a variety of non-native and non-natural extender units.20 Our work and that of others16, 17, 21 suggests that extender unit discrimination could be used as a platform to discover AT mutations that shift specificity away from native extender units and toward those poorly incorporated by a given PKS module. In order to discover amino acid mutations that can improve extender unit specificity towards non-natural substrates, we conducted saturation mutagenesis at a small number of amino acid residues in the AT that are hypothesized to be important for extender unit recognition. To probe the capacity of AT mutations to switch extender unit selectivity, engineered Ery6 AT mono-modules from the DEBS3 PKS were assayed in vitro using an acyl-CoA competition assay in conjunction with a chemically synthesized late stage intermediate.22, 23 This strategy successfully led to the identification of a set of mutants with preferences for or against non-natural and non-native extender unit substrates, respectively.
RESULTS
Identification of Ery6 mutants with altered extender unit specificities
Several motifs have been identified that predict extender unit specificity of AT domains from PKSs (Figure 2A),24, 25 yet this information has proven entirely insufficient to switch specificity from one extender unit toward another. Indeed, ATs subjected to site-directed mutagenesis have displayed only relaxed substrate selectivity.14, 17 Rather than attempting to recapitulate specificity determinants found among wild-type PKSs, we hypothesized that saturation mutagenesis at selected AT active site residues could provide novel solutions by reprogramming extender unit specificity in a manner that is distinct from wild-type biosynthetic machinery. Previously, our group and others have shown that the AT and ketosynthase (KS) domain of Ery6, the terminal module from DEBS, are remarkably promiscuous towards non-native and non-natural extender units, although native methylmalonyl-CoA remains the preferred extender unit.17, 20, 26 Here, such promiscuity provided a platform for discovering AT mutations that shifted selectivity towards extender units that were otherwise poor substrates for the wild-type PKS.
Figure 2.

Identification of Ery6 residues for saturation mutagenesis and screening. (A) Multiple amino acid alignment of selected AT domains. Filled circles indicate residues involved in catalysis; open circles indicate residues that likely form the extender unit binding pocket. Two well-known motifs used to predict extender unit specificity are highlighted. Residues chosen as targets for mutagenesis in this study are labeled red, while that from a previous study is labeled green. See Figure S1 for full amino acid sequences and descriptions of sequences. (B) Homology model of the EryAT6 active site constructed using SWISS-Model48 and the DEBS AT5 crystal structure as the template (PDB ID 2HG434). Residues selected for mutagenesis in this study are shown as green sticks. Other potential active site residues involved in catalysis or extender unit binding are shown as teal sticks. (C) Scheme illustrating the extender unit competition assay used to probe the substrate specificity of Ery6 AT mutants. The substrate probe is highlighted in blue and extender unit chains are highlighted in red. CoASH and N-acetylcysteamine (SNAC) products are not shown for brevity.
Inspection of an EryAT6 homology model revealed several residues that likely form the binding pocket for the incoming extender unit (Figure 2B). Three residues were selected for mutagenesis: Leu118, Tyr189, and Ser191. Leu118 does not locate to a previously described specificity-conferring motif, and was chosen because of its close proximity to the extender unit side-chain likely position. Both Tyr189 and Ser191, on the other hand, are located in the well-known ‘YASH motif’ used to predict methylmalonyl-CoA specificity (Figure 2B).27 Partially degenerate mutagenic oligonucleotides (NDT codons)28 were used to introduce on average 12 theoretical different amino acid substitutions at each targeted position. Following saturation mutagenesis, unique variants at each targeted position were identified by DNA sequencing. Subsequently, each Ery6 variant was subjected to in vivo phosphopantetheinylation conditions and each holo-Ery6 variant was purified to homogeneity as a hexa-histidine fusion protein by metal-chelation affinity chromatography. Crucially, in order to discover AT mutations that support installation of non-native and non-natural extender units over the natural substrate, a competition assay was devised that reports activity between competing extender units (Figure 2C).
Each Ery6 AT variant (Figure 2C) was incubated with the diketide-SNAC thioester 3 and a 1:3 mixture of methylmalonyl-CoA (1) and propargylmalonyl-CoA (2) prepared via engineered MatB mutants20, 29, 30 (see Supplemental Information for details). These conditions mimic desired in vivo feeding experiments whereby the non-natural extender unit is usually provided in excess over the natural substrate.16, 18 The alkyne 2 was selected as the competing extender unit given (1) it was previously established to be a substrate for the DEBS system, albeit a poor one, (2) the potential utility of a regioselectively installed alkyne for semi-synthesis or new macrolide derivatives, and (3) the size of 2 might be a good surrogate for other extender units and thus guide the identification of Ery6 AT mutants that could utilize other substrates. Next, the relative amount of the corresponding methyl (4) or propargyl (5) pyrone was determined by HPLC analysis of the product mixture. The DEBS thioesterase (TE) was omitted from these constructs for clarity of analysis given that the resulting triketide spontaneously cyclizes and offloads.31, 32 Notably, variants with activities and product profiles very different from that of the wild-type Ery6 were identified (Figure S2). For example, AT6 Leu118His gave 4 and 5 as a 94:6 mixture, indicating that this variant is more selective toward methylmalonyl-CoA than the wild-type Ery6, which produces 4 and 5 as a 40:60 mixture. In complete contrast, the variant AT6 Tyr189Arg yielded 4 and 5 as an 8:92 mixture, indicating this variant displays extender unit selectivity orthogonal to that of the Leu118His variant.
Mutant Ery6TE-catalyzed generation of 10-deoxymethynolide analogues
Next, we set out to determine whether the newly discovered mutations could be harnessed to direct the synthesis of macrolactones that require TE-catalyzed macrocyclization. The engineered mono-module PikAIII-TE derived from the pikromycin polyketide synthase is able to utilize a chemically synthesized thiophenol-activated pentaketide (6, Figure 3A) that mimics the late intermediate usually handed-off to the final two modules of the pikromycin type I PKS.23, 33 Although 6 is not a native substrate for DEBS, we hypothesized that its structural similarity to the intermediate normally produced by upstream DEBS modules would render it a viable substrate to probe the impact of our AT mutations in Ery6TE. Subsequently, Ery6TE was incubated with 6 and an equimolar mixture of extender units 1 and 2, and the identity and distribution of reaction products was analyzed by LC-HRMS. As expected, wild-type Ery6TE generates the 12-membered macrolactone 10-deoxymethynolide (10-DML, 7) from 6 and the mixture of acyl-CoA’s 1/2 (Figure 3B/C, Figure S3, and Table S1). In addition, a minor product (~8% of the total products by EIC) was detected that had a mass consistent with the corresponding propargyl-modified 10-DML analogue 8 (Figure 3A). This result highlights the ability of the terminal Ery6TE module to incorporate a non-natural extender unit into the polyketide chain and to cyclize the intermediate to provide the corresponding macrolactone, albeit at lower conversion relative to 7. In agreement with the pyrone formation assay (Figure 1C), Ery6TEAT-Leu118His produced 7 and 8 in a ratio of 98:2, indicating that the ability of this variant to incorporate 2 is almost completely abolished (Figure 3B/C, Figure S3, and Table S1). Moreover, the overall yield of this reaction is 1.5-fold greater than that catalyzed by the wild-type Ery6TE, as judged by LC-HRMS analysis. In contrast to the wild-type Ery6TE and the Leu118His mutant, Ery6TEAT-Tyr189Arg provided an 8-fold excess of 8 compared to 7, indicating a preference towards utilization of 2 over the natural substrate 1. Gratifyingly, in addition to the dramatically shifted extender unit selectivity of this variant, Tyr189Arg produced 8 at levels 27-fold higher than that produced by the wild-type Ery6TE, as judged by the extracted ion count by LC-HRMS analysis. Overall, the single mutation Tyr189Arg shifts selectivity 94-fold towards 8 compared to the wild-type Ery6TE, as judged by the ratio of 7:8. As further evidence of the robustness of this mutant, Tyr189Arg supports production of 8 at a yield 2.3-fold higher than the ‘natural’ product 7 produced by wild-type Ery6TE. Additionally, a mutation reported to enhance utilization of 2 in the context of erythromycin A production in Saccharopolyspora erythraea (Val187Ala),17 was introduced into Ery6TE as a single substitution and in combination with Tyr189Arg. Interestingly, while the Val187Ala mutation supported 11-fold improved production of 8 compared to wild-type Ery6TE, the selectivity between 1/2 was merely relaxed under these reaction conditions. Thus, 7 remained the major product in the Ery6TEAT-Val187Ala-catalyzed reaction. This is in complete contrast to Tyr189Arg where 8 was produced preferentially and in greater yield than the wild-type enzyme (Figure 3B/C, Figure S3, and Table S1). Finally, although combination of Tyr189Arg and Val187Ala provided a variant (Ery6TEAT-Val187Ala/Tyr189Arg) with lower overall activity than any other mutant, this double mutant produced a 21-fold excess of 8 compared to 7, respectively, and was therefore almost completely selective towards utilization of 2 from the mixture of 1/2 (Figure 3B/C, Figure S3, and Table S1). The inverted selectivity of Tyr189Arg and relaxed specificity of Val187Ala were also observed when the assays were carried out using a 6-fold excess of 2 to mimic potential in vivo feeding conditions (Table S2, Figure S4, and Figure S5) whereby the concentration of the non-natural extender unit can be manipulated.
Figure 3.
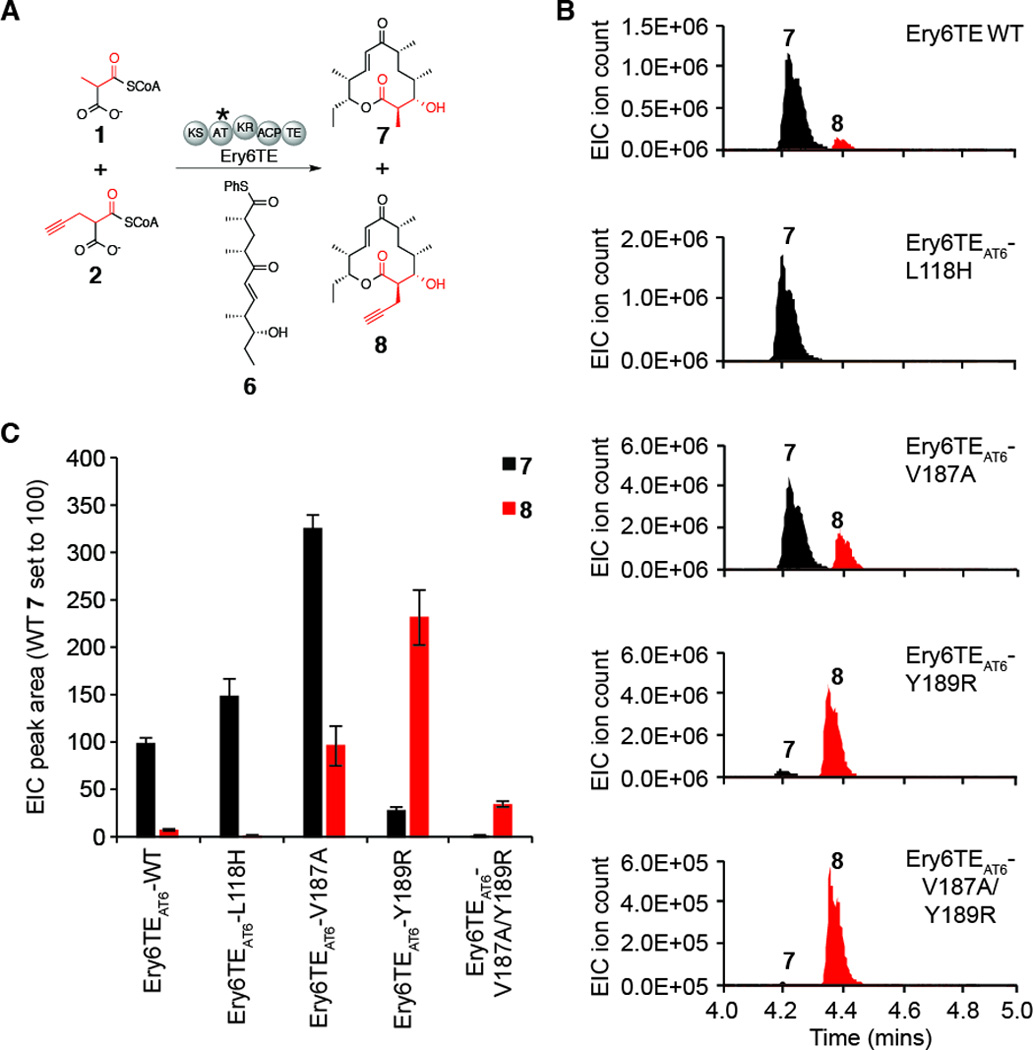
Extender unit selectivity of wild-type and mutant Ery6TEs using equimolar concentrations of natural and non-natural extender units. (A) Scheme illustrating the competition assay to report extender unit selectivity. Asterisk indicates domain location of mutations. (B) Extracted ion chromatograms (EIC) of Ery6TE-catalyzed chain extension reactions (7 [M-H2O+H]+ m/z = 279.1955; 8 [M-H2O+H]+ m/z = 303.1955). (C) Product distribution of wild-type and mutant Ery6TE (values are mean EIC peak area ± SD; n = 3; wild-type production of 7 set to 100).
DEBS3-catalyzed generation of 10-deoxymethynolide analogues
Next, the ability of the wild-type full-length DEBS3 polypeptide to synthesize analogues of the 12- and 14-membered macrolactones 10-DML and narbonolide, respectively, from a mixture of competing extender units was probed by LTQ Orbitrap LC-HRMS analysis (Figure 4A). As expected, upon incubation of wild-type DEBS3 with pentaketide 6 and a 1:6 mixture of 1/2, a product ion (9) corresponding to two extension reactions (route a, Figure 4A) incorporating 1 was detected by LC-HRMS (Figure 4B, Figure S4 and Table S3). Notably, the wild-type DEBS3 failed to provide ions corresponding to production of 10, indicating that utilization of the propargyl extender unit is not detectable under these conditions. However, introduction of Tyr189Arg into AT6 of DEBS3 substantially impacted the distribution of narbonolide products, compared to the wild-type DEBS3. For example, a product ion was detected with a mass consistent with the 14-membered macrolactone 10 derived from propargylmalonyl utilization by Ery6 of DEBS3AT6-Tyr189Arg. Combination of the mutations Val187Ala and Tyr189Arg into AT6 of DEBS3 produced a further dramatic shift in macrolactone product distribution. The proportion of 10 was increased further to 5% of the total products, respectively, albeit at the expense of overall activity.
Figure 4.

Substrate selectivity of wild-type and mutant DEBS3. (A) Scheme illustrating a competition assay to determine extender unit selectivity of DEBS3. Asterisk indicates domain location of mutations. (B) Extracted ion chromatograms of DEBS3-catalyzed chain extension reactions (7 [M-H2O+H]+ m/z = 279.1955; 8 [M-H2O+H]+ m/z = 303.1955; 9 [M-H2O+H]+ m/z = 337.2366; 10 [M-H2O+H]+ m/z = 361.2373). See Experimental Section for a complete description of product identification and quantification. (C) Product distribution of DEBS3-catalyzed chain reactions (values are mean ± SD; n = 3).
Using the wild-type DEBS3 another product ion was detected that was identical in all respects to the Ery6TE-catalyzed synthesis of 7 and thus corresponds to a single extension reaction with 1. This result indicates that the non-native substrate 6 can skip the first extension module of DEBS3 and load to the second module in the polypeptide, Ery6. Notably, wild-type DEBS3 showed little capacity to utilize the non-natural extender unit 2 under these assay conditions; the propargyl-derived macrolactone 8 could be detected at just 1% of the total product mixture, as judged by LC-MS. As expected from the Ery6TE assays described above, introduction of the mutation Leu118His in AT6 of DEBS3 completely eliminated the ability of the synthase to produce 8. In contrast, using the DEBS3 mutant Tyr189Arg, 8 comprised 20% of the total quantified products. Furthermore, DEBS3 Tyr189Arg produces 8 in a higher total yield than the WT DEBS. Combination of the mutations Val187Ala and Tyr189Arg into AT6 of DEBS3 produced a further dramatic shift in 10-DML product distribution; the proportion of 8 was increased further to 35% of the total products.
Extender unit promiscuity of wild-type and engineered DEBS3
To assess the impact of the newly discovered mutations on extender unit specificity beyond the originally targeted propargyl substrate 2, competition assays were used to probe the promiscuity of wild-type DEBS3 and the double mutant Val187Ala/Tyr189Arg in the presence of the natural extender unit 1 and either the allyl (11a), ethyl (11b), propyl (11c), or azidoethylmalonyl-CoA (11d) (Figure 5A) in 6-fold excess compared to 1. Notably, the non-native extender units were used poorly by wild-type DEBS3. For example, the allyl and ethyl substituted 10-DML analogues 12a and 12b, respectively, were produced at 1.8% and 6.1% of the total products, as judged by LC-HRMS analysis of the product mixtures. Moreover, 10-DML analogues were not detected when 11c or 11d were used in the competition assay (Figure 5B, Figure S4, and Table S4). However, the DEBS3 double mutant was able to utilize extender units that were non-detectable substrates with wild-type DEBS, and yielded product distributions that were very different from that of the wild-type enzyme. For example, the allyl, ethyl, propyl, and azidoethyl-substituted 10-DML analogues (12a–d, respectively) were all detected using the DEBS double mutant, at 13%, 8.5%, 35%, and 18% of the total products in each product mixture, respectively, albeit with reductions in overall activity similar to that of wild-type vs. double mutant DEBS3 with 2 (Figure 5B, Figure S4, and Table S4). Interestingly, none of the non-native extender units tested led to the detection of the corresponding non-natural narbonolide analogues (13a–d) with either wild-type or the double mutant DEBS3 (Table S4).
Figure 5.

Extender unit promiscuity of wild-type and mutant DEBS3. (A) Scheme illustrating the competition assay to determine specificity of DEBS3 towards each extender unit. Asterisk indicates location of mutations. (B) Fraction of each 10-DML analogue 12a–d as a percentage of the total products (sum ion counts for 7+9+12+13) of DEBS3-catalyzed chain reactions using each extender unit 11a–d (values are mean ± SD; n = 3). See Experimental Section for complete description of product identification and quantification.
DISCUSSION
Ultimately, the site-selective modification of polyketides via installation of non-natural extender units depends on the ability to manipulate the specificity of a given module to favor the target non-natural extender unit(s) in the presence of the natural extender unit substrate. Despite the intense interest in manipulating the extender unit specificity of type I PKSs, previous approaches have achieved little progress with respect to inverting AT selectivity from one substrate to another. Aside from the difficulties assessing extender unit specificity in vivo,19 this lack of progress can also be attributed to a lack of insight into the molecular basis for extender unit selection by PKSs. For example, although several AT structures are available,34–36 only one example includes an extender unit covalently bound.37 Thus, even though details regarding overall domain movements and protein interactions of type I PKSs are now emerging,38, 39 almost nothing is known with respect to how extender unit loading to the AT active site serine and subsequent transfer to the acyl carrier protein are controlled. Moreover, how to preferentially utilize non-natural extender units (e.g. ethyl, azido, propargyl side-chains) in place of a natural extender unit like methylmalonyl-CoA is an exceedingly subtle enzyme engineering problem to solve. Subsequently, most previous attempts aimed at altering the specificity of type I PKSs have relied on entire AT domain swaps, have been largely limited to naturally occurring extender units, and have generally resulted in greatly reduced product titers. Complementation of inactivated cis-ATs by trans-ATs,6, 40 or approaches that rely on the inherent orthogonality of unusual PKS modules,8, 41, 42 are also hindered by these concerns.
As an alternative to these approaches, and as a strategy that could be applied to any PKS of interest, we have explored the use of active site mutagenesis to shift AT specificity towards non-natural and non-native extender units. Critical to this approach are our previous investigations into extender unit specificity of various type I PKSs, which revealed inherent promiscuity towards various non-native and non-natural extender units.20, 21, 29 Low levels of activity towards poor substrates have proved to be the essential starting point for rational redesign and directed evolution of other enzymes.43, 44 Because high-throughput methods for screening the activity of most PKSs are not yet available, we opted to apply saturation mutagenesis at several AT active site residues and screen the modest panel of variants in vitro. Employing an in vitro assay whereby two extender units compete for loading and extension with a diketide-SNAC provided a simple method to probe the selectivity of the PKS variants by examining the pyrone product profile.
Several AT mutations were discovered that each dramatically shift the extender unit selectivity of the DEBS terminal module Ery6 (lacking a TE domain) towards either extender unit 1 or 2. Gratifyingly, these mutations also shift extender unit selectivity in the context of the Ery6TE when a mimic of a late-stage intermediate from pikromycin biosynthesis was tested, highlighting the tolerance of the TE domain. Introduction of the single mutation Tyr189Arg results in an Ery6TE variant that produces 8, the propargyl analogue of 10-DML (7), as the major product, even in the presence of the competing natural extender unit 1. In contrast, the wild-type enzyme prefers the natural substrate 1 to provide the ‘natural’ macrolactone 7 as the major product. Moreover, the relative yield of the propargyl analogue was 3-fold higher than that of the wild-type enzyme, and was almost 50% that of the wild-type activity with the natural substrate. To the best of our knowledge, this represents the first demonstrable inversion of extender unit selectivity of a PKS AT domain. Additionally, mutations at Tyr189 in DEBS AT6 that shift selectivity towards non-natural extender units have not been identified previously, thus Tyr189 represents a novel AT active site residue for further exploration in other PKSs. Notably, this result represents a significant improvement compared to a previously reported Ery6 mutation, Val187Ala, which shown herein serves to relax specificity but not invert it. Although Val187Ala was previously shown to support production of the desired propargyl analogue in vivo, the yield was low and could not be quantified.17 Described herein, the Tyr189Arg mutation in combination with Val187Ala in DEBS3 was able to utilize extender units not originally screened for (e.g. 11a–d) and provided access to products not detectable via the wild-type enzyme (e.g. 12c–d). At the same time, the fidelity of DEBS3 with the non-natural extender units was not as high as that of the Ery6TE variants. Given that the pentaketide 6 is not the native substrate for DEBS module 5, this likely explains the discrepancy between the Ery6TE and DEBS3 substrate selectivity. This feature also likely contributes to the significant module skipping observed with the DEBS3 reactions. Perhaps saturation mutagenesis of DEBS3 (versus the standalone module Ery6) and subsequent selectivity screening using 6 as the acceptor substrate might address this in the future. Thus, these mutants are likely to provide a platform for further enzyme engineering efforts, which are ongoing in our laboratories.
Cumulatively, these results suggest that assaying PKS variants using easily accessible acyl-SNAC acceptor substrates can lead to the discovery of AT mutations that shift extender unit specificity towards non-native and non-natural substrates. Moreover, these mutations can also impact selectivity with acceptor substrates that better mimic ACP-bound intermediates provided by PKSs in vivo, as demonstrated using the advanced pikromycin biosynthesis intermediate 6. Thus, while the pikromycin inspired pentaketide 6 is likely a much poorer substrate for Ery6TE and DEBS3 than for PikAIII, these mutations are likely to provide access to the corresponding erythronolide analogues in vivo whereby Ery6 receives the natural ACP-bound hexaketide intermediate from Ery5. Moreover, the in vitro extender unit competition assay described here provides an indication of selectivity that is likely relevant in vivo, given that these conditions approximate what is usually achieved by feeding extender units as malonyl-SNAC derivatives to engineered or producing organisms.
In summary, the strategy presented here enables production of site-selectively modified macrolactones and could be applied to a broad range of PKSs and alternative extender units. For example, a similar approach could be applied to other extension modules within DEBS, other macrolide PKSs, trans-ATs, or PKS modules from other systems that display extender unit promiscuity. In future work, the anticipated ability to site-selectively introduce alkynyl-, azido-, and allyl-modified extender units in place of native substrates in vivo could be leveraged by various chemistries to rapidly diversify the structure of polyketides.
MATERIALS AND METHODS
General
Unless otherwise stated, all materials and reagents were of the highest grade possible and purchased from Sigma (St. Louis, MO). Isopropyl β-D-thiogalactoside (IPTG) was from Calbiochem (Gibbstown, NJ). Primers were ordered from Integrated DNA Technologies (Coralville, IA). Plasmid pBP13045 was a gift from Prof. Pfeifer, University of Buffalo. pET24b-DEBS3 was as previously described. Plasmid pET28a-MatB was as previously described.29, 30 E. coli NovaBlue was used as a bacterial host for manipulation of plasmid DNA. E. coli BL21(DE3) pLysS competent cells were from Promega. E. coli K207-3 strain was a gift from Prof. Keatinge-Clay, University of Texas at Austin. The substrates 3 and 6 were prepared as previously described.20, 33 DNA sequence analysis of all clones generated in this study was performed by GeneWiz. Orbitrap LC-HRMS analysis was carried out by the Mass Spectrometry Facility at UNC-Greensborough.
Construction of Ery6 (TE-null)
The gene for the DEBS TE was PCR amplified from pBP130 using primers TE_HindIII_F and TE_XhoI_R. The PCR mixture contained: 5x HF Phusion DNA polymerase buffer (10 µL), DNAse free water (33.5 µL), forward/reverse primer mix (2 µL, 10 µM), template DNA (1 µL, 45 ng/µL), dNTP (1 µL, 2.5 mM each dNTP), DMSO (1.5 µl), and Phusion High Fidelity DNA Polymerase (1 µL). PCR cycling parameters: step 1) 98 °C, 30 s; step 2) 29×[a) 98 °C, 10 s; b) 60 °C 20 s; c) 72 °C, 1 min]; step 3) 72 °C, 10 min. The PCR product was purified by agarose gel electrophoresis, digested with HindIII and XhoI, and ligated with similarly treated pET28a vector. The gene for Ery6 was PCR amplified from pBP130 using primers Ery6_NdeI_F and Ery6_HindIII_R. The PCR mixture contained: 5x HF Phusion DNA polymerase buffer (10 µL), DNAse free water (33.5 µL), forward/reverse primer mix (2 µL, 10 µM), template DNA (1 µL, 60 ng/µL), dNTP (1 µL, 2.5 mM each dNTP), DMSO (1.5 µl), and Phusion High Fidelity DNA Polymerase (1 µL). PCR cycling parameters: step 1) 98 °C, 30 s; step 2) 29x [a) 98 °C, 10 s; b) 60 °C 20 s; c) 72 °C, 5 min]; step 3) 72 °C, 10 min. PCR product was purified by agarose gel electrophoresis, digested with NdeI and HindIII, and ligated with similarly treated TE-pET28a plasmid. This procedure generated a plasmid which housed Ery6-TE with a stop codon at the 3’-terminus of the Ery6 gene, thus producing a TE-null module.
Saturation mutagenesis of Ery6
Ery6 L118X, Y189X, and S191X libraries were prepared using the ‘round the horn‘ site-directed mutagenesis method46, using pET28a-Ery620 as template and the oligonucleotide sequences described in the Supplemental Information. Each PCR contained: 5x HF Phusion DNA polymerase buffer (10 µL), DNAse free water (33.5 µL), forward/reverse primer mix (2 µL, 10 µM), template DNA (1 µL, 20 ng/mL), dNTP (1 µL, 2 mM each dNTP), DMSO (1.5 µl), and Phusion High Fidelity DNA Polymerase (1 µL). PCR cycling parameters: step 1) 98 °C, 30 s; step 2) 29x [a) 98 °C, 10 s; b) 60 °C 20 s; c) 72 °C, 5 min]; step 3) 72 °C, 10 min. Next, PCR mixture was subjected to restriction enzyme digest with DpnI to remove any remaining template DNA. The DpnI reaction mixture contained: 10x CutSmart Buffer (5 µL), the PCR mixture (44 µL), and DpnI (1 µL). The mixture was vortexed, centrifuged, and incubated for 4 h at 37 °C. The digested reaction mixture was then purified by agarose gel electrophoresis, with total 8 µl of DNA eluted at the final step. The PCR product was then ligated using T4 DNA ligase in a reaction containing: 1 µl of the 10x T4 DNA ligase buffer, 8 µl of the DpnI-treated and purified PCR product, and 1 µl of the T4 DNA ligase (overnight incubation at 16 °C). Each subsequent ligation reaction was transformed directly into E. cloni 10G electrocompetent cells (Lucigen). Individual transformants from each library were sequenced to identify incorporation of mutant codons. Plasmids for unique mutants were used to transform chemically competent E. coli K207-3 strain for protein expression.
Construction of Ery6TE
The gene for the Ery6TE was PCR amplified from pET24b-DEBS3 using primers Ery6TE_F/ Ery6TE_R and cloned into pET24a via NdeI and EcoRI restriction sites. PikA4 docking domain was PCR amplified from pET24b-pikA433 using the primers Pik4dd_F and Pik4dd_R, and subsequently digested with the restriction enzyme XbaI. pET24a-Ery6TE was double digested with XbaI and EcoRV, and ligated with the digested PikA4 docking domain.
Construction of Ery6TE and DEBS3 AT mutants by site-directed mutagenesis
Ery6TE and DEBS3 mutants were prepared using KOD Hot Start DNA Polymerase Kit (EMD Millipore, Billerica, MA), using pET24a-Ery6TE and pET24b-DEBS3 DNA as template and the primers listed in the Supplemental Information. Each PCR contained: 2x Xtreme buffer (25 µL), DNAse free water (9 µL), forward/reverse primer mix (2 µL, 1 µM), template DNA (1 µL, 115 ng/mL), dNTP (10 µL, 2 mM each dNTP, Hotstart Xtreme kit), DMSO (2 µl), and Hotstart Xtreme KOD DNA Polymerase (1 µL). PCR cycling parameters: step 1) 94 °C, 2 min; step 2) 14x [a) 95 °C, 10 s; b) 60 °C 30 s; c) 68 °C, 11 min (ery6) or 14 min (DEBS3)]; step 3) 68 °C, 30 s. The PCR mixture was buffer exchanged using a Zymo DNA Clean & Concentrator-5 kit. The eluted DNA was used for restriction enzyme digest with DpnI to remove remaining template DNA. The DpnI reaction mixture contained: 10x CutSmart Buffer (2 µL), DNA (17 µL), and DpnI (1 µL). The mixture was vortexed, centrifuged, and incubated for 4 h at 37 °C. The digested reaction mixture was used to transform E. coli DH5α competent cells. Successful incorporation of mutations was confirmed by sequencing. Each mutant plasmid was used to transform the E. coli BAP1 expression strain47 that also contained the plasmid pRARE (Novagen).
Expression and purification of wild-type and mutant Ery6TE and DEBS3
Wild-type and mutant Ery6TE and DEBS3 enzymes were over-expressed in E. coli as C-terminally His6-tagged fusion proteins and purified as previously described.20, 33 A single colony was transferred to LB (3 mL) supplemented with kanamycin (30 µg/mL) and grown at 37 °C and 250 rpm overnight. The culture was used to inoculate TB media (1L) supplemented with kanamycin (30 µg/mL). One liter culture was incubated at 37 °C and 250 rpm to an OD600 of 1, at which time protein synthesis was induced by the addition of IPTG to a final concentration of 0.4 mM. After incubation at 18 °C and 200 rpm for 18 h, cells were collected by centrifugation at 4,000 g for 30 min, and resuspended in 100 mM Tris-HCl pH 8.0 (15 mL) containing NaCl (300 mM) and then lysed by sonication. Following centrifugation at 10,000 g, the soluble extract was loaded onto a 1 mL HisTrap HP column (GE Healthcare, Piscataway, NJ) and purified by fast protein liquid chromatography using the following buffers: wash buffer [20 mM phosphate (pH 7.4) containing 0.5 M NaCl and 20 mM imidazole] and elution buffer [20 mM phosphate (pH 7.4) containing 0.5 M NaCl and 250 mM imidazole]. The purified protein-containing fractions were pooled and exchanged into storage buffer (100 mM Tris-HCl pH 8.0 containing NaCl (300 mM) and 20% glycerol) using PD-10 desalting columns (GE Healthcare). Protein purity was verified by SDS-PAGE. Protein quantification was carried out using the Bradford Protein Assay Kit from Bio-Rad.
Supplementary Material
Acknowledgments
This study was supported in part by an NSF CAREER award (CHE-1151299, G.J.W.), NIH grant GM104258 (G.J.W.) and GM076477 (D.H.S.), and the Hans W. Vahlteich Professorship (D.H.S.). The Authors thank Tashina Robinson and Jarrett S. Johnson for technical assistance with plasmid preparation and Edward Kalkreuter for useful discussions.
Footnotes
SUPPORTING INFORMATION
Includes supplementary tables, supplemental figures, and detailed methods that describe expression and purification of MatB, synthesis of acyl-CoAs, expression and purification of wild-type and mutant Ery6, the Ery6, Ery6TE, and DEBS3 competition assays, and LC-HRMS analysis.
REFERENCES
- 1.Williams GJ. Engineering polyketide synthases and nonribosomal peptide synthetases. Curr Opin Struct Biol. 2013;23:603–612. doi: 10.1016/j.sbi.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ladner CC, Williams GJ. Harnessing natural product assembly lines: structure, promiscuity, and engineering. J Ind Microbiol Biotechnol. 2015;43:371–387. doi: 10.1007/s10295-015-1704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Yan YJ, An J, Huang SX, Wang XJ, Xiang WS. Designed biosynthesis of 25-methyl and 25-ethyl ivermectin with enhanced insecticidal activity by domain swap of avermectin polyketide synthase. Microb Cell Fact. 2015;14:152. doi: 10.1186/s12934-015-0337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nigam A, Almabruk KH, Saxena A, Yang J, Mukherjee U, Kaur H, Kohli P, Kumari R, Singh P, Zakharov LN, Singh Y, Mahmud T, Lal R. Modification of rifamycin polyketide backbone leads to improved drug activity against rifampicin-resistant Mycobacterium tuberculosis. J Biol Chem. 2014;289:21142–21152. doi: 10.1074/jbc.M114.572636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MC. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341:1089–1094. doi: 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel K, Piagentini M, Rascher A, Tian ZQ, Buchanan GO, Regentin R, Hu Z, Hutchinson CR, McDaniel R. Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem Biol. 2004;11:1625–1633. doi: 10.1016/j.chembiol.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Mo S, Kim DH, Lee JH, Park JW, Basnet DB, Ban YH, Yoo YJ, Chen SW, Park SR, Choi EA, Kim E, Jin YY, Lee SK, Park JY, Liu Y, Lee MO, Lee KS, Kim SJ, Kim D, Park BC, Lee SG, Kwon HJ, Suh JW, Moore BS, Lim SK, Yoon YJ. Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues. J Am Chem Soc. 2011;133:976–985. doi: 10.1021/ja108399b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X, Zhang W. Tagging polyketides/non-ribosomal peptides with a clickable functionality and applications. Frontiers in chemistry. 2015;3:11. doi: 10.3389/fchem.2015.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Tolerance and specificity of polyketide synthases. Annu Rev Biochem. 1999;68:219–253. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]
- 11.Liou GF, Lau J, Cane DE, Khosla C. Quantitative analysis of loading and extender acyltransferases of modular polyketide synthases. Biochemistry. 2003;42:200–207. doi: 10.1021/bi0268100. [DOI] [PubMed] [Google Scholar]
- 12.Stassi DL, Kakavas SJ, Reynolds KA, Gunawardana G, Swanson S, Zeidner D, Jackson M, Liu H, Buko A, Katz L. Ethyl-substituted erythromycin derivatives produced by directed metabolic engineering. Proc Natl Acad Sci U S A. 1998;95:7305–7309. doi: 10.1073/pnas.95.13.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruan X, Pereda A, Stassi DL, Zeidner D, Summers RG, Jackson M, Shivakumar A, Kakavas S, Staver MJ, Donadio S, Katz L. Acyltransferase domain substitutions in erythromycin polyketide synthase yield novel erythromycin derivatives. J Bacteriol. 1997;179:6416–6425. doi: 10.1128/jb.179.20.6416-6425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reeves CD, Murli S, Ashley GW, Piagentini M, Hutchinson CR, McDaniel R. Alteration of the substrate specificity of a modular polyketide synthase acyltransferase domain through site-specific mutations. Biochemistry. 2001;40:15464–15470. doi: 10.1021/bi015864r. [DOI] [PubMed] [Google Scholar]
- 15.Lau J, Fu H, Cane DE, Khosla C. Dissecting the role of acyltransferase domains of modular polyketide synthases in the choice and stereochemical fate of extender units. Biochemistry. 1999;38:1643–1651. doi: 10.1021/bi9820311. [DOI] [PubMed] [Google Scholar]
- 16.Bravo-Rodriguez K, Ismail-Ali AF, Klopries S, Kushnir S, Ismail S, Fansa EK, Wittinghofer A, Schulz F, Sanchez-Garcia E. Predicted incorporation of non-native substrates by a polyketide synthase yields bioactive natural product derivatives. ChemBioChem. 2014;15:1991–1997. doi: 10.1002/cbic.201402206. [DOI] [PubMed] [Google Scholar]
- 17.Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, Schulz F. Enzyme-directed mutasynthesis: a combined experimental and theoretical approach to substrate recognition of a polyketide synthase. ACS Chem Biol. 2013;8:443–450. doi: 10.1021/cb300505w. [DOI] [PubMed] [Google Scholar]
- 18.Bravo-Rodriguez K, Klopries S, Koopmans KR, Sundermann U, Yahiaoui S, Arens J, Kushnir S, Schulz F, Sanchez-Garcia E. Substrate Flexibility of a Mutated Acyltransferase Domain and Implications for Polyketide Biosynthesis. Chem Biol. 2015;22:1425–1430. doi: 10.1016/j.chembiol.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Dunn BJ, Khosla C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J R Soc Interface. 2013;10:20130297. doi: 10.1098/rsif.2013.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koryakina I, McArthur JB, Draelos MM, Williams GJ. Promiscuity of a modular polyketide synthase towards natural and non-natural extender units. Org Biomol Chem. 2013;11:4449–4458. doi: 10.1039/c3ob40633d. [DOI] [PubMed] [Google Scholar]
- 21.Bonnett SA, Rath CM, Shareef AR, Joels JR, Chemler JA, Hakansson K, Reynolds K, Sherman DH. Acyl-CoA subunit selectivity in the pikromycin polyketide synthase PikAIV: steady-state kinetics and active-site occupancy analysis by FTICR-MS. Chem Biol. 2011;18:1075–1081. doi: 10.1016/j.chembiol.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mortison JD, Kittendorf JD, Sherman DH. Synthesis and biochemical analysis of complex chain-elongation intermediates for interrogation of molecular specificity in the erythromycin and pikromycin polyketide synthases. J Am Chem Soc. 2009;131:15784–15793. doi: 10.1021/ja9060596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aldrich CC, Beck BJ, Fecik RA, Sherman DH. Biochemical investigation of pikromycin biosynthesis employing native penta- and hexaketide chain elongation intermediates. J Am Chem Soc. 2005;127:8441–8452. doi: 10.1021/ja042592h. [DOI] [PubMed] [Google Scholar]
- 24.Yadav G, Gokhale RS, Mohanty D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J Mol Biol. 2003;328:335–363. doi: 10.1016/s0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 25.Haydock SF, Aparicio JF, Molnar I, Schwecke T, Khaw LE, Konig A, Marsden AF, Galloway IS, Staunton J, Leadlay PF. Divergent sequence motifs correlated with the substrate specificity of (methyl)malonyl-CoA:acyl carrier protein transacylase domains in modular polyketide synthases. FEBS Lett. 1995;374:246–248. doi: 10.1016/0014-5793(95)01119-y. [DOI] [PubMed] [Google Scholar]
- 26.Pohl NL, Hans M, Lee HY, Kim YS, Cane DE, Khosla C. Remarkably broad substrate tolerance of malonyl-CoA synthetase, an enzyme capable of intracellular synthesis of polyketide precursors. J Am Chem Soc. 2001;123:5822–5823. doi: 10.1021/ja0028368. [DOI] [PubMed] [Google Scholar]
- 27.Del Vecchio F, Petkovic H, Kendrew SG, Low L, Wilkinson B, Lill R, Cortes J, Rudd BAM, Staunton J, Leadlay PF. Active-site residue, domain and module swaps in modular polyketide synthases. J Ind Microbiol Biotechnol. 2003;30:489–494. doi: 10.1007/s10295-003-0062-0. [DOI] [PubMed] [Google Scholar]
- 28.Reetz MT, Kahakeaw D, Lohmer R. Addressing the numbers problem in directed evolution. ChemBioChem. 2008;9:1797–1804. doi: 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- 29.Koryakina I, McArthur J, Randall S, Draelos MM, Musiol EM, Muddiman DC, Weber T, Williams GJ. Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem Biol. 2013;8:200–208. doi: 10.1021/cb3003489. [DOI] [PubMed] [Google Scholar]
- 30.Koryakina I, Williams GJ. Mutant malonyl-CoA synthetases with altered specificity for polyketide synthase extender unit generation. ChemBioChem. 2011;12:2289–2293. doi: 10.1002/cbic.201100383. [DOI] [PubMed] [Google Scholar]
- 31.Vagstad AL, Bumpus SB, Belecki K, Kelleher NL, Townsend CA. Interrogation of global active site occupancy of a fungal iterative polyketide synthase reveals strategies for maintaining biosynthetic fidelity. J Am Chem Soc. 2012;134:6865–6877. doi: 10.1021/ja3016389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Complete reconstitution of a highly reducing iterative polyketide synthase. Science. 2009;326:589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen DA, Rath CM, Eisman EB, Narayan AR, Kittendorf JD, Mortison JD, Yoon YJ, Sherman DH. Biocatalytic synthesis of pikromycin, methymycin, neomethymycin, novamethymycin, and ketomethymycin. J Am Chem Soc. 2013;135:11232–11238. doi: 10.1021/ja404134f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. The 2.7-Angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci U S A. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong FT, Jin X, Mathews II, Cane DE, Khosla C. Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase. Biochemistry. 2011;50:6539–6548. doi: 10.1021/bi200632j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keatinge-Clay AT, Shelat AA, Savage DF, Tsai SC, Miercke LJ, O'Connell JD, 3rd, Khosla C, Stroud RM. Catalysis, specificity, and ACP docking site of Streptomyces coelicolor malonyl-CoA:ACP transacylase. Structure. 2003;11:147–154. doi: 10.1016/s0969-2126(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 37.Oefner C, Schulz H, D'Arcy A, Dale GE. Mapping the active site of Escherichia coli malonyl-CoA-acyl carrier protein transacylase (FabD) by protein crystallography. Acta Crystallogr D Biol Crystallogr. 2006;62:613–618. doi: 10.1107/S0907444906009474. [DOI] [PubMed] [Google Scholar]
- 38.Whicher JR, Dutta S, Hansen DA, Hale WA, Chemler JA, Dosey AM, Narayan AR, Hakansson K, Sherman DH, Smith JL, Skiniotis G. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature. 2014;510:560–564. doi: 10.1038/nature13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, Narayan AR, Hakansson K, Sherman DH, Smith JL, Skiniotis G. Structure of a modular polyketide synthase. Nature. 2014;510:512–517. doi: 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar P, Koppisch AT, Cane DE, Khosla C. Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase. J Am Chem Soc. 2003;125:14307–14312. doi: 10.1021/ja037429l. [DOI] [PubMed] [Google Scholar]
- 41.Zhu X, Liu J, Zhang W. De novo biosynthesis of terminal alkyne-labeled natural products. Nat Chem Biol. 2015;11:115–120. doi: 10.1038/nchembio.1718. [DOI] [PubMed] [Google Scholar]
- 42.Lechner A, Wilson MC, Ban YH, Hwang J-y, Yoon YJ, Moore BS. Designed biosynthesis of 36-methyl-FK506 by polyketide precursor pathway engineering. ACS Synthetic Biology. 2012;2:379–383. doi: 10.1021/sb3001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams GJ, Zhang C, Thorson JS. Expanding the promiscuity of a natural-product glycosyltransferase by directed evolution. Nat Chem Biol. 2007;3:657–662. doi: 10.1038/nchembio.2007.28. [DOI] [PubMed] [Google Scholar]
- 44.Carr R, Alexeeva M, Enright A, Eve TS, Dawson MJ, Turner NJ. Directed evolution of an amine oxidase possessing both broad substrate specificity and high enantioselectivity. Angew Chem Int Ed Engl. 2003;42:4807–4810. doi: 10.1002/anie.200352100. [DOI] [PubMed] [Google Scholar]
- 45.Boghigian BA, Zhang H, Pfeifer BA. Multi-factorial engineering of heterologous polyketide production in Escherichia coli reveals complex pathway interactions. Biotechnol Bioeng. 2011;108:1360–1371. doi: 10.1002/bit.23069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.OpenWetWare-contributors. Round-the-horn site-directed mutagenesis, OpenWetWare. 2010 26 February 2015, http://openwetware.org/index.php?title=%27Round-the-horn_site-directed_mutagenesis&oldid=458415.
- 47.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E-coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 48.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.