

Abstract
A single nucleotide variant (SNV) of the cadherin 23 gene (Cdh23c.753A), common to many inbred mouse strains, accelerates age-related hearing loss (AHL) and can worsen auditory phenotypes of other mutations. We used homologous recombination in C57BL/6 NJ (B6N) and 129S1/SvImJ (129S1) embryonic stem cells to engineer mouse strains with reciprocal single base pair substitutions (B6-Cdh23c.753A>G and 129S1-Cdh23c.753G>A). We compared ABR thresholds and cochlear pathologies of these SNV mice with those of congenic (B6.129S1-Cdh23Ahl+ and 129S1.B6-Cdh23ahl) and parental (B6N and 129S1) strain mice. Results verified the protective effect of the Cdh23c.753G allele, which prevented high frequency hearing loss in B6 mice to at least 18 months of age, and the AHL-inducing effect of the Cdh23c.753A allele, which worsened hearing loss in 129S1 mice. ABR thresholds differed between 129S-Cdh23c.753A SNV and 129S1.B6-Cdh23ahl congenic mice, and a linkage backcross involving these strains localized a Chr 10 QTL contributing to the difference. These results illustrate the large effects that strain background and congenic regions have on the hearing loss associated with Cdh23c.753alleles. Importantly, the B6-Cdh23c.753Gstrain can be used to eliminate the confounding influence of the Cdh23c.753Avariant in hearing studies of B6 mice and mutant mice on the B6 background.
Age-related hearing loss (AHL) is the most prevalent sensory impairment in elderly individuals and is becoming a major health concern with world population aging1. AHL has been shown to have a significant genetic component2,3; however because of its complex nature and late onset, causative gene variants influencing AHL susceptibility have been difficult to identify in human populations4. Mice serve as valuable model systems for studying human hearing loss because their auditory system is similar to that of humans and because genetic and environmental variation can be manipulated and controlled in mice5,6,7. Genetically homogeneous inbred strains of mice vary widely in onset times and progression of hearing loss8 making them highly amenable to genetic studies of AHL. The C57BL/6 (B6) strain, widely used in many research fields, has long been studied as a model for AHL9,10,11.
A major genetic factor contributing to AHL in B6 mice was mapped to a locus (named ahl) on Chr 1012 and later shown to be a common variant in many other inbred strains13. The coincident map position of ahl with cadherin 23 (Cdh23) and the correspondence of specific Cdh23 alleles with hearing loss severity in multiple inbred strains provided evidence that a single nucleotide variant (SNV) of Cdh23 underlies the ahl phenotype14. This SNV corresponds to SNP rs257098870 and is at the 753rd nucleotide position from the ATG translation start site of Cdh23 cDNA (c.753), which corresponds to the last nucleotide of exon 7 in GenBank sequence AF308939 and to the last nucleotide of exon 9 in Ref Seqs NM_023370 and NM_001252635. A G nucleotide at this site (Cdh23c.753G) results in normal exon splicing, whereas an A nucleotide (Cdh23c.753A) disrupts the canonical donor splice site sequence and causes in-frame exon skipping14. The Cdh23c.753G allele is associated with resistance to AHL and is dominant to the recessive Cdh23c.753A allele, which is associated with AHL susceptibility.
The analyses of congenic strains, including B6.CAST-Cdh23Ahl+/Kjn12 and B6.CBACa-Cdh23CBA/CaJ/Kjn15, provided evidence that CAST/EiJ- and CBA/CaJ-derived allelic substitutions at the ahl locus could alleviate the progression of hearing loss in B6 mice, but the protective effect could not be definitively attributed to the Cdh23 gene because of possible contributions from linked genes in the congenic regions of these strains. Recently, Cdh23c.753A>G single base pair substitutions created by CRISPR-Cas9 genome editing were shown to prevent AHL in B6 strain mice16 and to prevent hearing loss in Ush1gjs heterozygotes on the B6 strain background17, verifying the causative nature of the Cdh23c.753A variant.
Here, we describe our analyses of C57BL/6NJ mice with a Cdh23c.753A>G single nucleotide substitution and 129S1/SvImJ mice with the reciprocal Cdh23c.753G>A substitution produced by means of the traditional targeting approach using homologous recombination in embryonic stem (ES) cells. These strains were chosen because of their widespread use and the availability of strain-matched ES cells and BAC clones. We compared, over an 18-month time course, the auditory phenotypes (hearing loss and cochlear pathology) of these single nucleotide variant mice with those of mice from their parental strains and with mice from corresponding congenic strains. Our results provide insight into the genetic and pathological mechanisms underlying progressive hearing loss in these two commonly used inbred strains of mice with potential implications for human genetic studies of age-related hearing loss.
Results
Full strain nomenclature and Jackson Laboratory stock numbers are given in Table 1 for the abbreviated strain designations used henceforth. 129S1 and B6 ES cells were used in a recombineering approach to create the 129S-Cdh23c.753A and B6-Cdh23c.753G SNV strains (Fig. 1), and the targeted single nucleotide substitutions in these strains were verified by DNA sequence analysis (Fig. 2A). A simple PCR method was developed to identify mice with the genetically engineered Cdh23c.753A/G substitutions and distinguish their genotypes (Fig. 2B).
Table 1. Abbreviated inbred strain designations used throughout the paper, followed by the type of strain, the full strain name and the Jackson Laboratory stock number.
| Abbreviation | Type | Full strain name | Stock # |
|---|---|---|---|
| B6Ja | parental | C57BL/6 J | 664 |
| B6Na | parental | C57BL/6NJ | 5304 |
| 129S1 | parental | 129S1/SvImJ | 2448 |
| B6.129S1-Cdh23Ahl+ | congenic | B6.129S1-(rs13480546-rs13480629)/Kjn | 21479 |
| 129S1.B6-Cdh23ahl | congenic | 129S1.B6-(rs3696307-rs257098870)/Kjn | 18926 |
| B6-Cdh23c.753G | SNVb | B6N(Cg)-Cdh23tm2.1Kjn/Kjn | 18399 |
| 129S-Cdh23c.753A | SNVb | 129S/Sv-Cdh23tm1.1Kjn/Kjn | 16208 |
aThe designation C57BL/6 (or B6) is used when referring to either C57BL/6 J or C57BL/6 NJ.
bGenetically engineered strain with a single nucleotide variant (SNV).
Figure 1. Recombineering strategy used to produce 129S-Cdh23c.753A and B6-Cdh23c.753G SNV mice.
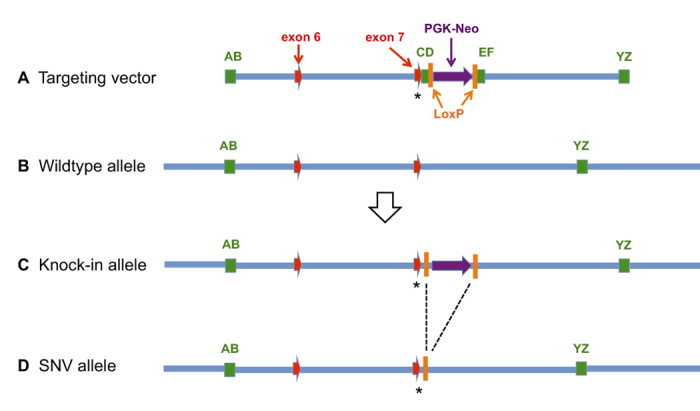
To produce B6N strain mice with the Cdh23c.753G SNV, a targeting vector with DNA from a 129S1/Sv BAC clone was constructed for recombination with C57BL/6 N ES cells (JM8.A3). To produce 129S strain mice with the Cdh23c.753A SNV, a targeting vector with DNA from a C57BL/6 J BAC clone was constructed for recombination with 129S1/SvImJ ES cells. (A) Targeting vector: The Cdh23 targeted region (9,729 bp) was cloned into a linearized pBlight plasmid (4695 bp) by retrieval from a BAC clone with AB (300 bp) and YZ (320 bp) homology arms. PGK-Neo and LoxP sites then were inserted into the plasmid by retrieval from a synthesized DNA cassette (2320 bp) with CD (200 bp) and EF (203 bp) homology arms. The targeted Cdh23c.753 SNV (marked by asterisk) is separated from the cassette insertion site by 142 bp and from PGK-Neo by 178 bp. In this figure, the targeted Cdh23c.753 SNV is shown as the last nucleotide of exon 7 according to Genbank transcript AF308939, which is equivalent to exon 9 in other transcripts such as NM_023370. (B) Wildtype allele: The targeted Cdh23 region of wildtype mouse genomic DNA showing positions of AB and YZ homology arms relative to exons 6 and 7. (C) Knock-in allele: After homologous recombination between the targeting vector and the wildtype Cdh23 allele, ES cells were selected for the presence of the PGK-Neo cassette and the closely linked targeted SNV (asterisk). Restriction enzyme cleavage sites and genomic probes (5′ and 3′) were used to distinguish wildtype and knock-in alleles. (D) The PGK-Neo cassette was subsequently removed by Cre-Lox recombination to produce the final SNV allele.
Figure 2. DNA sequence validation, PCR identification of targeted SNVs and assessment of exon skpping.
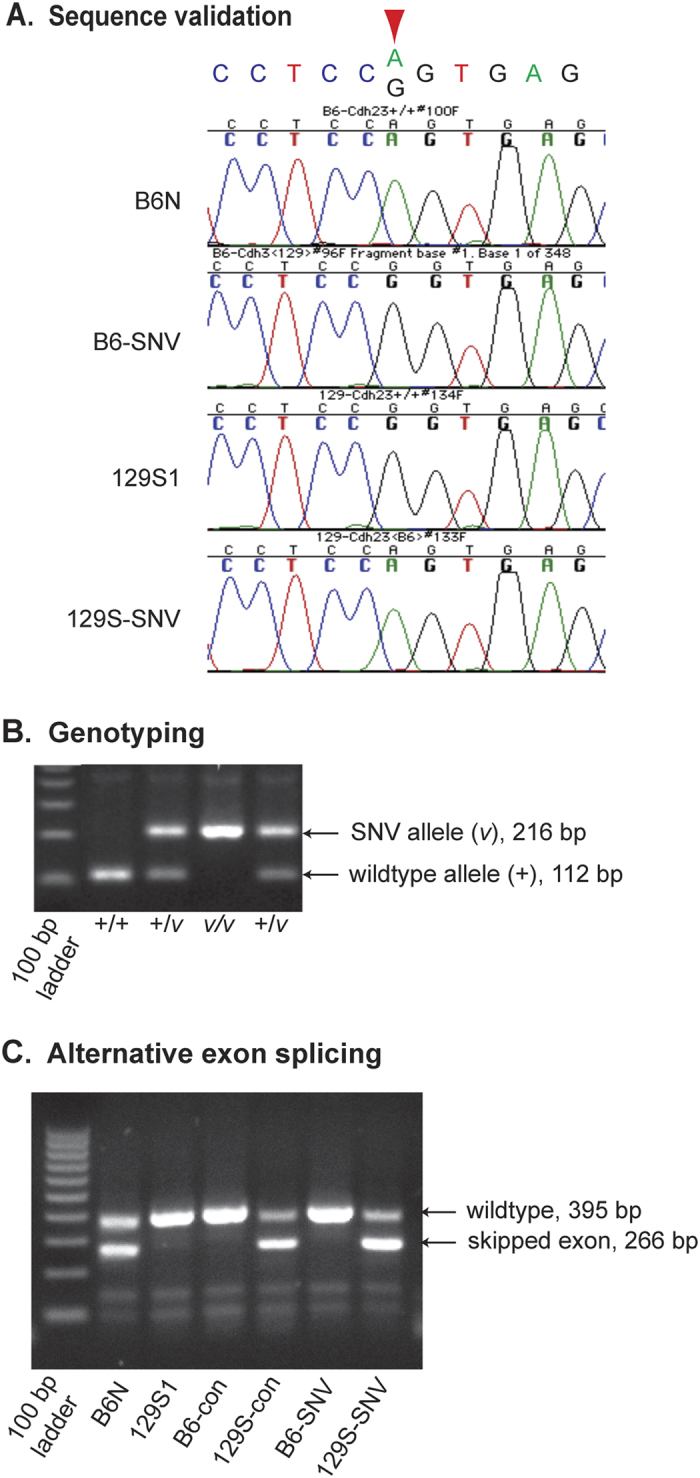
(A) Sequence chromatograms of PCR amplified DNA surrounding the targeted Cdh23c.753 nucleotide (indicated by the red downward-pointing arrow) confirm that C57BL/6 NJ (B6N) and 129S-Cdh23c.753A (129S-SNV) mice are homozygous for the Cdh23 c.753 A nucleotide, while 129S1/SvImJ (129S1) and B6N-Cdh23c.753G (B6N-SNV) mice are homozygous for the Cdh23 c.753 G nucleotide. (B) Identification of Cdh23 alleles with targeted SNVs by PCR amplification of the closely linked PGK-Neo insertion remnant. Primers flanking the PGK-Neo cassette insertion site were used to amplify PCR products that differ in size between the wildtype allele and the targeted SNV allele, which retains an intronic 104 bp remnant of the PGK-Neo cassette after Cre deletion. Because of its close proximity (178 bp) to the targeted SNV, the presence or absence of the PGK-Neo remnant can be used to distinguish the wildtype allele (+, 112 bp) from the targeted SNV allele (v, 216 bp). Lane 1, 100-bp DNA size ladder; lanes 2–5, genotypes of individual mice. (C) RT-PCR to evaluate the extent of exon skipping related to the Cdh23c.753A variant. cDNA primers flanking the alternatively spliced exon of Cdh23 (containing the c.753 nucleotide) were used to amplify alternatively spliced products. The PCR product size from the wild-type Cdh23 transcript (395 bp) is larger than the PCR product from the alternatively spliced, in-frame transcript (266 bp), which lacks the 129 bp skipped exon. Lane 1, 100-bp DNA size ladder; lane 2, B6N; lane 3, 129S1; lane 4, B6.129S1-Cdh23Ahl+ congenic (B6-con); lane 5, 129S1.B6-Cdh23ahl congenic (129S-con); lane 6, B6-Cdh23c.753G SNV (B6-SNV); and lane 7, 129S-Cdh23c.753ASNV (129S-SNV). Lanes 2, 5, and 7 show alternatively spliced transcripts caused by the Cdh23c.753A variant.
For comparisons with the SNV strain mice, we generated two corresponding congenic strains, B6.129S1-Cdh23Ahl+ and 129S1.B6-Cdh23ahl, by recurrent backcrossing and selection. The 44–48 Mb 129S1-derived Chr 10 congenic region of B6.129S1-Cdh23Ahl+ extends from a breakpoint between rs3665690 (23.19 Mb position) and rs13480546 (24.28 Mb position) to a breakpoint between rs13480629 (67.62 Mb position) and rs13480646 (71.25 Mb position). The 7–18 Mb B6J-derived Chr 10 congenic region of 129S1.B6-Cdh23ahl extends from a breakpoint between rs3661754 (44.97 Mb position) and rs3696307 (53.74 Mb position) to a breakpoint between rs257098870 (60.53 Mb position) and rs29330969 (63.15 Mb position).
Exon skipping is associated with the Cdh23 c.753A variant
RNA extracted from eight cochleae (four mice) from each of the SNV and congenic strains and from the B6 and 129S1 parental strains were evaluated for Cdh23 alternative exon splicing by RT-PCR (Fig. 2C). As shown previously by others14,17, exon skipping was specifically associated with the Cdh23c.753A variant. Our results (Fig. 2) showed exon skipping in cochlear transcripts from B6N parental (lane 2), 129S1.B6-Cdh23ahlcongenic (lane 5), and 129S-Cdh23c.753A SNV (lane 7) strains, but not in 129S1 parental (lane 3), B6.129S1-Cdh23Ahl+ congenic (lane 4), and B6-Cdh23c.753G SNV (lane 6) strains. For each of the three strains exhibiting exon skipping, we compared the image density of the gel band corresponding to the wildtype allele (larger, upper band) to the density of the band corresponding to the exon-skipped allele (smaller, lower band) using the ImageJ software program. From these gel band density ratios, we estimated that the wildtype allele comprises about 45% of the Cdh23 transcripts in cochleae of B6 mice, about 24% in 129S1.B6-Cdh23ahl congenic mice, and only about 18% in 129S-Cdh23c.753A SNV mice.
Hearing loss progression differs in B6N, B6.129S1-Cdh23 Ahl+, and B6-Cdh23 c.753G mice
Hearing was assessed by ABR threshold analysis. The high frequency 16 kHz and 32 kHz thresholds of B6-Cdh23c.753G SNV mice remained at normal levels throughout the 18 month duration of the study (Fig. 3G,K) and were much lower than those of B6N controls (Fig. 3H,L), indicating a strong influence of the Cdh23c.753G allele in preventing the progression of high frequency hearing loss. The 32 kHz threshold means of B6N mice were statistically significantly higher than those of SNV mice by 3 months of age, and the 16 kHz threshold means were significantly higher by 9 months of age (Table 2). The Cdh23c.753G variant, however, had little effect on slowing the progression of lower frequency hearing loss, as shown by the 8 kHz thresholds of B6N and B6-Cdh23c.753G SNV mice (Fig. 3D), which were not statistically significantly different at any test age (Table 2).
Figure 3. Progression of hearing loss in B6-Cdh23c.753G SNV mice compared with congenic and inbred strain mice.

Progression of hearing loss was assessed in C57BL/6 NJ (B6N) inbred strain mice (3 females and 7 males; A,E,I), B6.129S1-Cdh23Ahl+ (Congenic) strain mice (3 females and 7 males; B,F,J), and B6-Cdh23c.753G (SNV) strain mice (9 females and 9 males; C,G,K). ABR thresholds of individual mice tested at 3, 6, 9, 12, 15, and 18 months of age are shown for 8 kHz (A–C), 16 kHz (E–G), and 32 kHz (I–K) pure-tone stimuli, with sexes (F, M) of mice in each strain indicated by slightly different colors. Average ABR thresholds for each strain are shown for 8 kHz (D), 16 kHz (H), and 32 kHz (L) stimuli; vertical bars indicate standard errors of the means. Two congenic mice with exceptionally high thresholds (B,F,J) died before completion of the study and were excluded from the analysis; therefore, threshold averages for the congenic strain mice at the 15- and 18-month test ages are not presented (D,H,L).
Table 2. Statistical comparisons of ABR threshold means at different ages: C57BL/6NJ (B6N, N = 10), B6.129S1-Cdh23 Ahl+ (Congenic, N = 10), and B6-Cdh23 c.753A (SNV, N = 18).
| ABR thresholds | Tukey-Kramer HSD test P values | |||||||
|---|---|---|---|---|---|---|---|---|
| Pairwise Comparisons | 3 months | 6 months | 9 months | 12 months | 15 months | 18 months | ||
| 8 kHz | B6N | Congenic | 0.11 | 0.95 | 0.99 | 0.87 | — | — |
| 8 kHz | B6N | SNV | 0.06 | 0.71 | 0.44 | 0.39 | 0.13 | 0.32 |
| 8 kHz | Congenic | SNV | 0.99 | 0.90 | 0.44 | 0.73 | — | — |
| 16 kHz | B6N | Congenic | 0.86 | 0.004 | 0.08 | <0.0001 | — | — |
| 16 kHz | B6N | SNV | 0.86 | 0.04 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 16 kHz | Congenic | SNV | 0.99 | 0.35 | 0.008 | 0.03 | — | — |
| 32 kHz | B6N | Congenic | <0.0001 | <0.0001 | <0.0001 | <0.0001 | — | — |
| 32 kHz | B6N | SNV | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 32 kHz | Congenic | SNV | 0.46 | 0.33 | 0.06 | 0.10 | — | — |
Although a reduction in high frequency hearing loss also was observed in the B6.129S1-Cdh23Ahl+ congenic strain mice (which also have the Cdh23c.753G allele) compared with parental B6N mice (Fig. 3H,L), two of the 10 congenic mice tested exhibited rapid threshold increases between 6 and 12 months of age and died before the final 18 month test age (Fig. 3B,F,J), phenotypes not seen in B6-Cdh23c.753G SNV mice.
Hearing loss progression differs in 129S1, 129S1.B6-Cdh23 ahl , and 129S-Cdh23 c.753A mice
Hearing loss progression was more rapid in both 129S1.B6-Cdh23ahl congenic and 129S-Cdh23c.753A SNV mice than in parental 129S1 mice (Fig. 4), indicating a deleterious effect of the Cdh23c.753A variant. Although both strains share the same Cdh23c.753A variant and the same 129S1 strain background, the progression of hearing loss in the 129S-Cdh23c.753A SNV strain mice was more severe than that in 129S1.B6-Cdh23ahl congenic mice. Already at one month of age, ABR thresholds of 129S-Cdh23c.753A SNV mice were statistically significantly higher than those of 129S1.B6-Cdh23ahl congenic mice for all test frequencies (Table 3).The differences in hearing loss progression between these strains suggest the influence of another locus (or loci) in the B6-derived congenic region of 129S1.B6-Cdh23ahl that modifies the hearing loss effect of the Cdh23c.753A variant in these mice.
Figure 4. Progression of hearing loss in 129S-Cdh23c.753A SNV mice compared with congenic and inbred strain mice.

Progression of hearing loss was assessed in 129 S1/SvImJ (129S1) strain mice (6 females and 4 males; A,E,I), 129S1.B6-Cdh23ahl (Congenic) strain mice (5 females and 6 males; B,F,J), and 129S-Cdh23c.753A (SNV) strain mice (7 females and 13 males; C,G,K). ABR thresholds of individual mice tested at 2, 3, 4, 6, and 9 months of age are shown for 8 kHz (A–C), 16 kHz (E–G), and 32 kHz (I–K) pure-tone stimuli, with sexes (F,M) of mice in each strain indicated by slightly different colors. Average ABR thresholds for each strain are shown for 8 kHz (D), 16 kHz (H), and 32 kHz (L) stimuli; vertical bars indicate standard errors of the means. Average ABR threshold values for the 1-month test age were obtained from additional mice not included in the longitudinal study: 6 males of the 129S1 strain, 4 females and 2 males of the Congenic strain, and 6 females and 4 males of the SNV strain.
Table 3. Statistical comparisons of ABR threshold means at different ages: 129S1/SvImJ (129S1, N = 10), 129S1.B6-Cdh23 ahl (Congenic, N = 11), and 129S-Cdh23 c.753A (SNV, N = 20).
| ABR thresholds | Tukey-Kramer HSD test P values | |||||||
|---|---|---|---|---|---|---|---|---|
| Pairwise Comparisons | 1 month | 2 months | 3 months | 4 months | 6 months | 9 months | ||
| 8 kHz | 129S1 | Congenic | 0.12 | 0.99 | 0.6 | 0.23 | 0.02 | 0.15 |
| 8 kHz | 129S1 | SNV | 0.04 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 8 kHz | Congenic | SNV | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0016 |
| 16 kHz | 129S1 | Congenic | 0.99 | 0.59 | 0.42 | <0.0001 | <0.0001 | <0.0001 |
| 16 kHz | 129S1 | SNV | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 16 kHz | Congenic | SNV | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 32 kHz | 129S1 | Congenic | 0.0007 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 32 kHz | 129S1 | SNV | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 32 kHz | Congenic | SNV | <0.0001 | <0.0001 | <0.0001 | 0.0014 | 0.71 | 1.0 |
Cochlear hair cell loss corresponds with hearing loss
Consistent with the ABR results, there was minimal OHC loss and negligible IHC loss in B6-Cdh23c.753G SNV mice and B6.129S1-Cdh23Ahl+ congenic mice at 18 months of age (Fig. 5C,D, n = 4/group; mean +/−SEM). In contrast, there were large OHC and IHC lesions in age-matched C57BL/6NJ mice (Fig. 5C,D, n = 4; mean +/−SEM; Supplementary Fig. S1). These results suggest a strong influence of the Cdh23c.753G allele in preventing the age-related progression of OHC and IHC losses in the basal high-frequency region of the cochlea. In C57BL/6NJ mice, OHC losses were greater than the IHC loss and the lesions were greater in the base than the apex of the cochlea (Fig. 5C,D).
Figure 5. Cochlear hair cell loss associated with Cdh23c.753 variants.

Cochleograms show the mean (+/−SEM, n = 4 cochlea per group) percent outer hair cell loss (OHC; A,C) and inner hair cell loss (IHC; B,D) in 20% intervals plotted as a function of percent distance from the apex of the cochlea. Cochlear locations related to frequency36 are shown on the x-axis. (A,B) Increased hair cell loss in 6-month-old 129S-Cdh23c.753A (SNV) and 9-month-old 129S1.B6-Cdh23ahl(Congenic) mice compared with 9-month-old 129S1/SvImJ (129S1) parental strain mice indicates a strong influence of the Cdh23c.753A allele in accelerating age-related hair cell degeneration. (C,D) Minimal hair cell loss in 18-month-old B6-Cdh23c.753G (SNV) and B6.129S1-Cdh23Ahl+ (Congenic) mice compared with age-matched C75BL/6 NJ (B6N) parental strain indicates a strong influence of the Cdh23c.753G allele in preventing age-related hair cell degeneration.
Consistent with ABR thresholds, mean (+/−SEM, n = 4/group) OHC and IHC losses in 9-month-old 129S1.B6-Cdh23ahl congenic mice and 6-month-old 129S-Cdh23c.753A SNV mice were much greater than in 9-month-old mice of the parental 129S1/SvlmJ strain (Fig. 5A,B; Supplementary Fig. S1) indicating the deleterious effect of the Cdh23c.753A variant. Even though both strains share the same Cdh23c.753A variant and the same 129S1 strain background, the degree of OHC and IHC loss in the 6-month-old 129S-Cdh23c.753A SNV mice was more severe than the hair cell lesions seen in the 9-month-old 129S1.B6-Cdh23ahl congenic mice. Hair cell lesions in the 129S1.B6-Cdh23ahl congenic mice and 129S-Cdh23c.753A SNV mice were more severe in the high-frequency basal region than the low-frequency apex, and OHC lesions were greater than IHC lesions at all cochlear locations. In contrast, OHC and IHC lesions in mice of the parental 129S1/SvlmJ strain were negligible.
A linked locus on Chr 10 (Mahl) modifies Cdh23 c.753A (ahl)-related hearing loss
To genetically investigate the different rates of hearing loss progression in 129S1.B6-Cdh23ahl congenic and 129S-Cdh23c.753A mice, we produced F1 hybrids between these two strains and measured their ABR thresholds at three months of age, when ABR thresholds differ significantly between the parental strains (Fig. 6A). The F1 hybrid mice are homozygous for the Cdh23c.753Aallele, heterozygous for 129S1 and B6 alleles at other loci within the congenic region, and homozygous for 129S1 alleles everywhere else in the genome. The thresholds of F1 hybrid mice were similar to those of 129S1.B6-Cdh23ahl congenic mice and much lower than those of 129S-Cdh23c.753A SNV mice (Fig. 6A), indicating a dominant effect of a postulated B6-derived, AHL-protective allele at a locus within the congenic region. To reveal the phenotype of the corresponding recessive 129S1-derived, AHL-promoting allele and reduce genetic complexity for mapping, the F1 hybrids were backcrossed to 129S-Cdh23c.753A SNV mice rather than intercrossed. The ABR thresholds of the resulting N2 backcross progeny showed a strongly bimodal frequency distribution (Fig. 6B), indicating the strong influence of a single locus. If a single locus is primarily responsible for a trait, then only two genotypes affecting the trait will segregate in a backcross, resulting in a bimodal distribution. If multiple loci are responsible for a trait, there will be additional contributing genotypes, resulting in a more bell-shaped phenotypic distribution.
Figure 6. Localization of a hearing loss modifier in the 129S1.B6-Cdh23ahl congenic region.

(A) Average 16 kHz ABR thresholds (±standard deviations) of 3-month-old 129 S1 inbred strain mice (N = 10), 129S1.B6-Cdh23ahlcongenic mice (Congenic; N = 10), 129S-Cdh23c.753A mice (SNV; N = 19), and (Congenic × SNV) F1 hybrids (N = 6). (B) Frequency distribution of 16 kHz ABR thresholds among 3-month-old N2 mice (N = 182) from a backcross of (129S-Cdh23c.753A SNV × 129S1.B6-Cdh23ahl congenic) F1 hybrids with 129S-Cdh23c.753A SNV mice. The threshold ranges and averages of age-matched parental strain mice (F1 hybrids, SNV mice) are indicated at the top of the figure as horizontal gray lines. N2 mice with homozygous and heterozygous Mahl genotypes are distinguished by red and blue colors. (C) Chr 10 haplotype analysis of N2 mice. SNP markers and their Mb positions along Chr 10 are shown above the corresponding genotypes of parental strain and N2 backcross mice. Haplotypes C and D contain crossovers within the congenic region, and the correlations of ABR thresholds with SNP genotypes further refine the candidate region of the modifier locus (designated Mahl). The 129S1.B6-Cdh23ahlcongenic region and the Mahl candidate gene region are indicated by dotted lines. The non-recombinant rs13480621 and Cdh23c.753A markers were used to assign Mahl genotypes in panel B above. Allele designations: B, C57BL/6-derived; S, 129S1-derived; S*, 129S1-derived with targeted Cdh23c.753G>A substitution.
Allelic segregation analysis identified a locus within the Chr 10 congenic region of 129S1.B6-Cdh23ahl that strongly associated with the hearing loss variation observed in the N2 progeny. We designated this locus Mahl for its modifying effect on the hearing loss of 129S1 mice homozygous for the Cdh23c.753A(ahl) variant. A total of 182 N2 generation progeny were produced from the backcross, and each mouse was assessed for ABR thresholds at three months of age and genotyped for 13 SNPs that differ between B6- and 129S1-strain DNA and that span the 20–70 Mb region of Chr 10. SNP marker genotypes were used to assign haplotypes to individual N2 mice to better define the 129S1.B6-Cdh23ahl congenic region and to identify informative crossover events for narrowing the Mahl candidate interval. The strain of origin for the Cdh23c.753A allele was distinguished by the presence or absence of the 104 bp PGK-Neo insertion remnant (Fig. 2B), which was inherited from the 129S-Cdh23c.753A SNV strain. The 87 non-recombinant N2 mice that inherited B6-derived alleles from the F1 hybrid parent (haplotype A, Fig. 6C) exhibited low ABR thresholds (average 51 dB SPL), and the 91 non-recombinant N2 mice that inherited 129S1-derived alleles (haplotype B, Fig. 6C) exhibited high thresholds (average 97 dB SPL). Four N2 mice had recombinant chromosomes with informative crossovers in the Chr 10 congenic region. Analysis of these recombinant mice (haplotypes C and D, Fig. 6C) and their associated ABR thresholds reduced the candidate interval for Mahl to an approximately 5 Mb region (Fig. 6C).
Genotypes at the Mahl locus could account for about 80% of the total 16 kHz ABR threshold variation that was observed in the N2 backcross mice (Fig. 6B). Because total variation also includes environmental and experimental variation, the Mahl locus by itself may account for all of the underlying genetic variation. The amount of the total ABR threshold variance that could be explained by the Mahl locus was calculated as the difference between the total variance and the residual variance, expressed as a percent of the total variance. The total variance was calculated as the sum of squares for ABR thresholds of all 182 backcross mice, and the residual variance was calculated as the sum of squares for ABR thresholds of the 90 mice with the Mahl BS genotype plus the sum of squares for ABR thresholds of the 92 mice with the Mahl SS genotype.
Discussion
Our results using homologous recombination in ES cells to produce single base pair substitutions in Cdh23 add to the evidence from recent in vivo CRISPR-Cas9 gene editing studies16,17 showing that Cdh23c.753A is the causative variant responsible for the age-related hearing loss effects ascribed to the Chr 10 ahl locus. C57BL/6 N mice with a CRISPR/Cas9-generated Cdh23c.753A>G substitution were shown to retain normal ABR thresholds up to the final test age of 36 week16. The B6-Cdh23c.753G SNV mice we generated also retain normal ABR thresholds at old ages, indicating that the loxP site retained after PGK-Neo excision (Fig. 1D) does not affect the phenotype of these mice. Our ABR (Fig. 3H,L) and cochleogram (Fig. 5C,D) results extend the time frame for high frequency (16 and 32 kHz) hearing loss protection conferred by the Cdh23c.753A>G substitution in B6 mice to 78 weeks (18 months). The Cdh23c.753G allele, however, did not fully prevent progression of lower frequency (8 kHz) hearing loss (Fig. 3D) and apical OHC loss (Fig. 5C) in B6 mice. A similar attenuation of high frequency but not low frequency hearing loss was reported in threshold comparisons between B6.CAST-Cdh23Ahl+ congenic mice and B6 mice6 and in comparisons between B6.CBA-Cdh23Ahl+ congenic mice and B6 mice15. Histological evidence for a frequency specific protective effect of the Cdh23c.753G allele was also provided by the Kane et al.15 study, which showed that the degree of OHC loss in 18-month-old B6.CBA-Cdh23Ahl+ congenic mice is similar to that of age matched C57BL/6 J mice in cochlear regions corresponding to low frequencies (8 kHz and lower) but is much reduced in more basal regions corresponding to higher frequencies. Taken together, the results of these studies suggest that genetic factors other than the Cdh23c.753A variant are responsible for the less commonly recognized low frequency hearing loss exhibited by B6 mice.
Cdh23 knockout mice exhibit defects in hair bundle development and are congenitally deaf, whereas mice with a missense mutation of Cdh23 exhibit normal hair bundle development but have defective tip links and exhibit a progressive hearing loss18, supporting the concept that non-severe Cdh23 mutations may cause age-related hearing loss. The Cdh23c.753A variant disrupts a splice donor site and is predicted to cause in-frame skipping of an exon encoding part of the 2nd and 3rd ectodomains of CDH2314. Because CDH23 is an integral component of stereocilia tip links19, the defect caused by exon skipping likely increases tip link breakage and slows repair mechanisms, which eventually could lead to cellular stress and hair cell apoptosis20. Our results are consistent with results from other studies14,17 showing that the Cdh23c.753A splice site variant causes exon skipping (Fig. 2C). In our study, the estimated degree of exon skipping in mice with the Cdh23c.753A variant appeared to correspond with the extent of high frequency hearing loss. 129S-Cdh23c.753A SNV mice, which exhibit the earliest onset of 16 kHz hearing loss (<2 months), had the smallest estimated proportion of wild-type Cdh23 transcripts (18%) followed by 129S1.B6-Cdh23ahl congenic mice (onset ~ 4 months, 24% wild-type transcripts) and B6N mice (onset ~ 9 months, 45% wild-type transcripts). These results suggest that genetic factors that modify the fidelity of Cdh23 exon splicing may contribute to some of the hearing loss variability associated with the Cdh23c.753A variant in different inbred mouse strains.
The Cdh23c.753A variant (ahl) not only influences the progression of hearing loss in many inbred mouse strains5 but also has been shown to affect noise-induced hearing loss21 and to modify the hearing phenotypes of other gene mutations, including Atp2b2dfw 22, Adgrv1frings 23, Sod1tm1Leb 24, Myo7ash1-8J 25, Actbtm1.1Erv 26 and Ush1gjs 17. Thus an important advance of this study is the development of the B6-Cdh23c.753G strain, which can be used to eliminate the potentially confounding effects of the Cdh23c.753A variant when assessing other auditory phenotypes of mutations produced on the commonly used C57BL/6 J and C57BL/6 N sub-strains. To accomplish this, B6 mice carrying the mutation to be studied would first be crossed with B6-Cdh23c.753G mice. The resulting F1 hybrid progeny (Cdh23c.753A/G) that have inherited the mutation would then be selected and backcrossed to mice of the B6-Cdh23c.753Gstrain. Finally, N2 progeny that have inherited the mutation and are homozygous for the Cdh23c.753G allele would be interbred to establish a new mutant line for phenotype evaluation. An advantage of using the B6-Cdh23c.753G strain described here is the ability to easily identify the targeted Cdh23c.753G allele by PCR genotyping of size differences due to the closely linked 104 bp remnant of the PGK-Neo selection cassette (Figs 1D and 2B).
Although they have the same Cdh23c.753A/Gsubstitutions and are on the same inbred strain backgrounds, congenic and SNV strain mice showed differences in their auditory phenotypes (Figs 3, 4, 5) that must be due to the effects of genetic factors located in the introgressed regions of the congenic strains. Three of the ten B6.129S1-Cdh23Ahl+ congenic strain mice examined exhibited decreased longevity and elevated ABR thresholds compared with B6-Cdh23c.753G SNV mice (Fig. 3), suggesting that genetic incompatibilities may exist between the 129S1-derived congenic region and the B6 host genome in the B6.129S1-Cdh23Ahl+ mice. Because of the possible complications of co-transferred linked genes in congenic strains, genetically engineered mice with specific base pair changes are preferred for evaluating gene-specific phenotypic effects.
129S1.B6-Cdh23ahl congenic strain mice and 12S1-Cdh23c.753A SNV mice exhibited large differences in ABR thresholds (Fig. 4) and hair cell loss (Fig. 5). We used the differences in ABR thresholds of backcross mice derived from these two strains to genetically map a locus that modifies the hearing loss effect of the Cdh23c.753A variant. We refined the map position of this locus, which we designate Mahl, to a 5 Mb interval within the 129S1.B6-Cdh23ahl congenic region (Fig. 6C) and hypothesize that the 129S1 allele at this locus worsens Cdh23c.753A-related hearing loss relative to the B6 allele’s effect. The combinations of Cdh23 and Mahl alleles can help explain the hearing loss differences observed among the 129S1 parental strain, 129S1.B6-Cdh23ahl congenic strain, and 129S-Cdh23c.753A SNV strain mice examined in this study (Supplementary Fig. S2).
On the basis of map position and inbred strain ancestry, we suspect that the Mahl locus may be equivalent to the previously described Phl2 locus, which contributes to the progressive hearing loss of 101/H strain mice27. Phl2 was mapped to the 51–78 Mb region of Chr 10, which overlaps with the 45–63 Mb congenic region of the 129S1.B6-Cdh23ahl strain. The strains involved in the Phl2 mapping crosses (101/H, MAI/Pas, and MBT/Pas) all have the Cdh23c.753A allele, thereby excluding this variant as the underlying cause of the Phl2-related hearing loss. The 101/H strain and the 129S1 strain were derived from a recent ancestor28 and therefore may share the same deleterious allele at the Phl2 locus. If so, the deleterious 129S1 allele at this locus (Phl2/Mahl) would be present in 129S-Cdh23c.753A SNV strain mice (which exhibit high ABR thresholds), but replaced by the non-deleterious B6J allele in 129S1.B6-Cdh23ahl congenic strain mice (which exhibit lower ABR thresholds).
We narrowed the candidate region for Mahl to the 58–63 Mb position on Chr 10, which includes the Cdh23 gene. A comparative search of whole genome sequences of mouse inbred strains (http://www.sanger.ac.uk/science/data/mouse-genomes-project) did not reveal any sequence differences between C57BL/6 J (or C57BL/6NJ) and 129S1/SvImJ DNA that would affect splicing or cause amino acid changes in any of the 65 protein-coding genes located in the ~5 Mb Mahl candidate region. These results suggest that DNA differences in non-coding sequences or regulatory elements may be responsible for the modifier effect of the Mahl locus on hearing loss caused by the Cdh23c.753A variant. Non-coding DNA differences within or surrounding the Cdh23 gene itself cannot be ruled out, including those that may influence Cdh23 expression levels or exon splicing fidelity.
The Cdh23c.753A variant in the context of the 129S strain background (129S-Cdh23c.753A SNV or 129S1.B6-Cdh23ahl congenic mice) accelerates the progression of hearing loss (Fig. 4). In contrast, the presence of the same Cdh23c.753A allele in CBA/CaJ mice (CBA.B6-Cdh23ahl congenic mice) has no effect on hearing thresholds15. The effect that the Cdh23c.753A variant has on hearing loss thus can vary greatly depending on strain background, having little or no effect on the CBA background15, a moderate effect on the B6 background (Fig. 3A,E,F), and a large effect on the 129S background (Fig. 4C,G,K). The strong effect of strain background on the severity of Cdh23c.753A-related hearing loss has potential implications for genetic studies of human age-related hearing loss. Specific screens of CDH23 mutations in human subjects have identified variants that are weakly associated with both age-related29 and noise-induced30 hearing loss, but genome-wide association studies (GWAS) have failed to find any associations. Like the case for the Cdh23c.753A variant in mice, the phenotypic expression of hypomorphic CDH23 mutations in human populations may depend on the diverse genetic backgrounds of individuals, and the resulting decreased phenotypic penetrance would make associations with age-related hearing loss difficult to detect by GWAS.
Methods
Mice
All mice examined in this study originated from The Jackson Laboratory (http://www.jax.org/), and all procedures involving their use were performed at The Jackson Laboratory and approved by the Institutional Animal Care and Use Committee. All methods used in the study were performed in accordance with the guidelines and regulations of the U.S. National Institutes of Health (NIH) Office of Laboratory Animal Welfare (OLAW) and the Public Health Service (PHS) Policy on the Humane Care and Use of Laboratory Animals. The Jackson Laboratory is accredited by the American Association for the Accreditation of Laboratory Animal Care.
Congenic strain development
The B6.129S1-Cdh23Ahl+ and 129S1.B6-Cdh23ahl reciprocal congenic strains were developed by genetic introgression of B6J- and 129S1-derived segments of Chromosome 10 (containing the Cdh23 gene) into the genomes of 129S1 and B6J, respectively. To accomplish this introgression, mice from the B6J and 129S1 parental strains were first intercrossed to generate F1 hybrids. The F1 hybrids were then backcrossed to B6J mice to produce N2 generation mice for development of the B6.129S1-Cdh23Ahl+ strain and to 129S1 mice to produce N2 mice for development of the 129S1.B6-Cdh23ahl strain. Repeated backcrossing of hybrid progeny (selected on the basis of Chromosome 10 marker genotypes) to the parental (B6J or 129S1) strain continued until 10 backcross generations (N10) were completed. Finally, the N10 generation mice of each line were sibling inbred to produce the two homozygous congenic strains. To define the extent of the congenic regions in each strain, single nucleotide polymorphisms (SNPs) along the length of Chromosome 10 were genotyped.
Genetic engineering of B6-Cdh23 c.753G and 129S-Cdh23 c.753A mice
Single base pair substitutions in the Cdh23 gene of C57BL/6 (B6) and 129S1/SvImJ (129S1) strain mice were produced using a combination of recombineering31,32 and standard molecular cloning techniques. Briefly, a 9,728 bp fragment (GRCm38 assembly, Chr10: 60,527,162–60,536,889) of the Cdh23 gene containing the targeted nucleotide (c.753) was retrieved from bacterial artificial chromosome (BAC) clones and integrated into embryonic stem (ES) cells by homologous recombination (Fig. 1). To produce B6-Cdh23c.753Gknock-in mice, the Cdh23 fragment from a 129S1 BAC clone was incorporated into the genome of B6N ES cells (JM8.A3), and to produce 129S-Cdh23c.753A knock-in mice, the Cdh23 fragment from a B6 BAC clone was incorporated into the genome of 129S1-derived ES cells (ES-CJ7). The Cdh23c.753A/G single nucleotide polymorphism (SNP) is the only DNA sequence difference between B6 and 129S1 in the entire targeted region (9,728 bp), and the natural strain variants precluded the need to create mutations in the targeting vectors.
To create the targeting vector, a synthetic gene (Genscript, Piscataway, NJ) consisting of a 300 bp homology arm (Chr10: 60,536,591–60,536,889) and a 320 bp homology arm (Chr10: 60,527,162–60,527,481) and including restriction enzyme sites (HindIII and Xho1, spaced by Kpn1) for retrieval and Not1 for construct release was cloned into the pBlight-TK plasmid33. This plasmid then was used to retrieve the targeted Cdh23 fragments from BAC DNA by gap repair in competent bacteria, and DNA was sequenced to confirm the presence of the targeted nucleotide. A PGK-Neo cassette with flanking LoxP sites and homology arms was synthesized and inserted by recombineering into the Cdh23 fragment of the targeting vector, 178 bp downstream of the targeted c.753 nucleotide. The final plasmid vector was confirmed by DNA sequence analysis, linearized with NotI and electroporated into ES cells. ES cells under G418 positive selection were screened by a loss of allele assay against the PGK-Neo insertion site, and positive ES cell clones were confirmed by Southern blot and DNA sequence analysis.
Verified B6N JM8A3 ES cell clones were microinjected into 3.5 dpc blastocyst-stage albino C57BL/6 J host embryos. After surgical transfer of the microinjected embryos and development to term, chimeras were identified by coat color and high chimera males were mated with C57BL/6NJ females. Progeny were screened for the presence of PGK-Neo and the targeted Cdh23c.753G substitution. The floxed PGK-Neo cassette then was deleted by mating with B6N.Cg-Tg(Sox2-cre) 1Amc/J mice (Stock #14094). Mice heterozygous for the Cdh23c.753G substitution, with the PGK-Neo cassette excised and lacking the Cre transgene, were mated to C57BL/6NJ mice, and progeny heterozygous for the Cdh23c.753G substitution then were intercrossed to establish a homozygous B6-Cdh23c.753G line, confirmed by DNA sequence analysis (Fig. 2A). The formal name for this strain is B6N(Cg)-Cdh23tm2.1Kjn/Kjn (Jackson Laboratory Stock #18399).
Verified 129S1/SvImJ ES cell clones were microinjected into 3.5 dpc blastocyst-stage non-agouti B6 host embryos. After surgical transfer of the microinjected embryos and development to term, chimeras were identified by coat color, and high chimera males were mated with 129S1/SvImJ females. Progeny were screened for the presence of PGK-Neo and the closely linked targeted Cdh23c.753A substitution. The floxed PGK-Neo cassette then was deleted by mating with 129S/Sv-Tg(Prm-cre)58Og/J mice (Stock #3328). Mice heterozygous for the Cdh23c.753A substitution and lacking the PGK-Neo cassette and Cre transgene were mated to 129S1/SvImJ mice, and progeny heterozygous for the Cdh23c.753A allele then were intercrossed to establish the homozygous 129S-Cdh23c.753Astrain, confirmed by DNA sequence analysis (Fig. 2A). The formal name for this strain is 129S/Sv-Cdh23tm1.1Kjn/Kjn (Stock #16208).
PCR genotyping method to identify genetically engineered Cdh23 c.753A/G substitutions
A simple PCR-based genotyping method was developed to distinguish wild type alleles from those with genetically engineered single base pair substitutions (Fig. 2B). The forward primer CTTAAGCTCGGCCAAACATC and the reverse primer CACAACAGGAAGAAAGCAAGC, which flank the site of the excised PGK-Neo selection cassette, were used to amplify PCR products that distinguish alleles based on the presence or absence of the post-excision 104 bp cassette remnant, which serves as a convenient marker for the targeted Cdh23c.753 SNVs.
cDNA synthesis and RT- PCR analysis
For each of the six strains analyzed in this study, total RNA from eight cochleae (pooled from four 6-week-old male mice) was extracted and purified with the RNeasy Mini Kit (Cat. No. 74104, Qiagen, Valencia, CA) using the manufacturer’s procedures. Total RNA quantity and quality were determined using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). First strand synthesis of cDNA was performed using the High Capacity cDNA Reverse Transcription Kit (Cat. No. 4368814, Thermo Fisher Scientific, Waltham, MA) in accordance with manufacturer’s instructions, but using a Cdh23 gene-specific primer (RT-Cdh23R, AGACAATAGTGTAGCCAATCCCACGG) rather than random primers.
To evaluate alternative splicing, RT-PCR primers were designed from DNA sequences located in the Cdh23 exons that flank the variably skipped exon: forward primer (RT-Cdh23F, ACACCAGTGGGGACACCCATCTTCATC) and reverse primer RT-Cdh23R (the same primer shown above for reverse transcription). The same RT-PCR primers were used in a previously reported assay of Cdh23 exon skipping17. RT-PCR amplifications for each of the cDNA samples were performed using the MasterTaq Kit (Cat. No. 2200210, 5 PRIME, Inc., Gathersburg, MD) with HotStart-IT Taq DNA polymerase (Cat. No. 71195, Affymetrix, Santa Clara, CA), and run in a Bio-Rad Peltier Thermal Cycler. Amplification consisted of one cycle of denaturation at 97 °C for 30 seconds followed by 40 cycles, each consisting of 94 °C for 30 seconds, 58 °C for 30 seconds, and 72 °C for 30 plus 1 second per cycle. After the 40 cycles, the final product was extended for 10 minutes at 72 °C. PCR products were visualized on 2% agarose gels stained with ethidium bromide. The ImageJ software program (https://imagej.nih.gov/ij/) was used to process digital images of the gels and quantify the densities of gel bands corresponding to alternatively spliced transcripts.
Auditory brainstem response (ABR) measurements to assess hearing loss
ABR thresholds were measured at 8, 16 and 32 kHz in a sound attenuating chamber using the SmartEP auditory evoked potential diagnostic system from Intelligent Hearing Systems (IHS, Miami, FL) as described previously8. Briefly, mice were anesthetized with tribromoethanol (0.2 ml of 20 mg/ml stock per 10 g of body weight, i.p.) and placed on a temperature controlled heating pad to maintain body temperature at 37 °C. Three subdermal electrodes, placed at the vertex and behind each ear, were used to record brain stem responses to defined tone-bursts (3 ms duration. 1.5 ms cosine-gated rise/fall time). The responses were then amplified, filtered (100–3000 Hz) and averaged (25 kHz sampling rate, 10 ms analysis window). Stimulus intensity was initially decreased in 10 dB steps until the response began to disappear and then lowered in 5 dB steps; ABR threshold was defined as the lowest intensity at which an ABR response could be reliably obtained. With our testing system, average ABR thresholds for normal hearing mice are about 40, 20, and 45 dB SPL for 8, 16 and 32 kHz stimuli, respectively. The same individual performed all of the ABR threshold determinations throughout the study and was uninformed of the ages, strains, and genotypes of mice at the time of the tests.
Cochleograms to assess hair cell loss
Our procedures for preparing cochleograms have been described in detail in earlier publications34,35. Briefly, after completing the ABR measurements at The Jackson Laboratory, the mice were euthanized with CO2, decapitated, and their bullae quickly removed and opened to expose the inner ear. A small hole was carefully made in the round window and 10% formalin fixative was gently perfused through the opening. Afterward, the cochlea was immersed in fixative and shipped to University of Buffalo for analysis. Cochleae were stained with Ehrlich’s hematoxylin solution. A person, blind to the ABR results, dissected out the organ of Corti into two to three half turns. The samples were then mounted as a flat surface preparation in glycerin on glass slides, coverslipped and examined with a light microscope (Zeiss Standard, 400X). Inner hair cells (IHC) and outer hair cells (OHC) were counted along successive 0.12–0.24 mm intervals from the apex to the base of the cochlea by a second person blind to the experimental conditions. A hair cell was counted as present if both the cuticular plate and hair cell nucleus were clearly visible and considered missing if either were absent. Using lab norms and custom software, the percentages of missing IHC and OHC were determined for each animal and a cochleogram constructed showing the percentage of missing OHC and IHC as a function of percent distance from the apex of the cochlea. Position in the cochlea was related to frequency using a mouse tonotopic map36. Mean percent OHC loss and mean percent IHC loss were computed over 20% intervals of the cochlea for four cochleae in each of the six experimental groups, at 6 months of age for 129S-Cdh23c.753A mice, at 9 months of age for 129S1/SvlmJ and 129S1.B6-Cdh23ahl mice, and at 18 months of age for C57BL/6NJ, B6.129S1-Cdh23Ahl+ and B6-Cdh23c.753G mice. Cochlear surface preparations were photographed with a digital camera (SPOT Insight, Diagnostic Instruments Inc.) attached to a Zeiss Axioskop microscope, processed with imaging software (SPOT Software, version 4.6), Adobe Photoshop 5.5.
Genotyping markers for genetic mapping
For genetic mapping analysis of the (129S-Cdh23c.753A × 129.B6-Cdh23ahl) × 129S-Cdh23c.753A backcross, tail-tip samples from 182 individual N2 progeny mice were provided to the Jackson Laboratory’s Genotyping Service, which outsources SNP genotyping to LGC Genomics (Beverly MA). SNP genotyping was performed for 13 selected SNPs on Chr 10 that differed between the B6 and 129S1 strains. For genotyping the Cdh23c.753 locus, 129S1- and B6-derived alleles were distinguished by the presence or absence of the of the 104 bp PGK-Neo insertion remnant (Fig. 2B).
Statistical analysis
ABR data analysis was performed using the JMP 7.0 interactive statistics and graphics software program (www.JMP.com). Statistical significance of the differences among means was determined by one-way ANOVA with post-hoc Tukey HSD test for multiple pair-wise comparisons.
Additional Information
How to cite this article: Johnson, K. R. et al. Effects of Cdh23 single nucleotide substitutions on age-related hearing loss in C57BL/6 and 129S1/Sv mice and comparisons with congenic strains. Sci. Rep. 7, 44450; doi: 10.1038/srep44450 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors would like to thank Sandra Gray for mouse colony management and production of linkage cross mice, Kelly Kane for congenic strain development, and Chantal Longo-Guess for the initial ABR testing of mutant mice. We also thank the Genetic Engineering Technologies Service at The Jackson Laboratory for producing the B6-Cdh23c.753G and 129S-Cdh23c.753A mice. This research was supported by US National Institutes of Health (NIH) Grant DC005827 (KRJ).
Footnotes
The authors declare no competing financial interests.
Author Contributions K.R.J. conceived and designed the experiments. K.R.J. and R.S. wrote the manuscript text and prepared the figures. L.H.G. performed RT-PCR and DNA genotyping. C.T. measured ABR thresholds. H.J. and D.D. generated the cochleograms. All authors contributed to data analyses and manuscript review.
References
- Bainbridge K. E. & Wallhagen M. I. Hearing loss in an aging American population: extent, impact, and management. Annual Review of Public Health 35, 139–52 (2014). [DOI] [PubMed] [Google Scholar]
- Newman D. L. et al. GRM7 variants associated with age-related hearing loss based on auditory perception. Hear Res 294, 125–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyken E., Van Camp G. & Van Laer L. The complexity of age-related hearing impairment: contributing environmental and genetic factors. Audiol Neurootol 12, 345–58 (2007). [DOI] [PubMed] [Google Scholar]
- Fransen E. et al. Genome-wide association analysis demonstrates the highly polygenic character of age-related hearing impairment. European Journal of Human Genetics 23, 110–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K. & Johnson K. R. Inheritance patterns of progressive hearing loss in laboratory strains of mice. Brain Res 1277, 42–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller K. K. Contributions of mouse models to understanding of age- and noise-related hearing loss. Brain Res 1091, 89–102 (2006). [DOI] [PubMed] [Google Scholar]
- Bowl M. R. & Dawson S. J. The mouse as a model for age-related hearing loss - a mini-review. Gerontology 61, 149–57 (2015). [DOI] [PubMed] [Google Scholar]
- Zheng Q. Y., Johnson K. R. & Erway L. C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res 130, 94–107 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry K. R. & Lepkowski C. M. Evoked potential correlates of genetic progressive hearing loss. Age-related changes from the ear to the inferior colliculus of C57BL/6 and CBA/J mice. Acta Otolaryngol 86, 366–74 (1978). [DOI] [PubMed] [Google Scholar]
- Spongr V. P., Flood D. G., Frisina R. D. & Salvi R. J. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J Acoust Soc Am 101, 3546–53 (1997). [DOI] [PubMed] [Google Scholar]
- Willott J. F., Kulig J. & Satterfield T. The acoustic startle response in DBA/2 and C57BL/6 mice: relationship to auditory neuronal response properties and hearing impairment. Hear. Res. 16, 161–167 (1984). [DOI] [PubMed] [Google Scholar]
- Johnson K. R., Erway L. C., Cook S. A., Willott J. F. & Zheng Q. Y. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear Res 114, 83–92 (1997). [DOI] [PubMed] [Google Scholar]
- Johnson K. R., Zheng Q. Y. & Erway L. C. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics 70, 171–180 (2000). [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K., Zheng Q. Y. & Johnson K. R. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet 35, 21–23 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane K. L. et al. Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hear Res 283, 80–88 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mianne J. et al. Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology directed repair. Genome Med 8, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka Y. et al. Heterozygous mutation of Ush1g/Sans in mice causes early-onset progressive hearing loss, which is recovered by reconstituting the strain-specific mutation in Cdh23. Hum Mol Genet (2016). [DOI] [PubMed] [Google Scholar]
- Schwander M. et al. A mouse model for nonsyndromic deafness (DFNB12) links hearing loss to defects in tip links of mechanosensory hair cells. Proc Natl Acad Sci USA 106, 5252–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak P. et al. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449, 87–91 (2007). [DOI] [PubMed] [Google Scholar]
- Someya S. et al. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc Natl Acad Sci USA 106, 19432–19437 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. R. et al. Genetic basis for susceptibility to noise-induced hearing loss in mice. Hear Res 155, 82–90 (2001). [DOI] [PubMed] [Google Scholar]
- Zheng Q. Y. & Johnson K. R. Hearing loss associated with the modifier of deaf waddler (mdfw) locus corresponds with age-related hearing loss in 12 inbred strains of mice. Hear Res 154, 45–53 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. R., Zheng Q. Y., Weston M. D., Ptacek L. J. & Noben-Trauth K. The Mass1(frings) mutation underlies early onset hearing impairment in BUB/BnJ mice, a model for the auditory pathology of Usher syndrome IIC. Genomics 85, 582–90 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. R. et al. Separate and combined effects of Sod1 and Cdh23 mutations on age-related hearing loss and cochlear pathology in C57BL/6J mice. Hear Res 268, 85–92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q. Y. et al. Digenic inheritance of deafness caused by 8J allele of myosin-VIIA and mutations in other Usher I genes. Hum Mol Genet 21, 2588–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin B. J. et al. beta-Actin and fascin-2 cooperate to maintain stereocilia length. J Neurosci 33, 8114–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T., Erven A. E., Spiden S. L., Guenet J. L. & Steel K. P. Two quantitative trait loci affecting progressive hearing loss in 101/H mice. Mamm Genome 17, 841–50 (2006). [DOI] [PubMed] [Google Scholar]
- Witmer P. D. et al. The development of a highly informative mouse Simple Sequence Length Polymorphism (SSLP) marker set and construction of a mouse family tree using parsimony analysis. Genome Res 13, 485–91 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa M., Nishio S. Y. & Usami S. Prevalence and clinical features of hearing loss patients with CDH23 mutations: a large cohort study. PLoS One 7, e40366 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski T. J., Pawelczyk M., Rajkowska E., Dudarewicz A. & Sliwinska-Kowalska M. Genetic variants of CDH23 associated with noise-induced hearing loss. Otol Neurotol 35, 358–65 (2014). [DOI] [PubMed] [Google Scholar]
- Liu P., Jenkins N. A. & Copeland N. G. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13, 476–84 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S., Costantino N., Court D. L., Jenkins N. A. & Copeland N. G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33, e36 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S., Rachel R. A., Jenkins N. A. & Copeland N. G. Zfp423 is required for normal cerebellar development. Molecular and Cellular Biology 26, 6913–22 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D., McFadden S. & Salvi R. J. Cochlear hair cell densities and inner ear staining techniques. In The Auditory Psychobiology of the Mouse (ed. Willott J. F.) 189–204 (CRC Press, Boca Raton, FL, 2001). [Google Scholar]
- Ding D. et al. N-acetyl-cysteine prevents age-related hearing loss and the progressive loss of inner hair cells in γ-glutamyl transferase 1 deficient mice. Aging (Albany NY) 8, 730–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M., von Hunerbein K., Hoidis S. & Smolders J. W. A physiological place-frequency map of the cochlea in the CBA/J mouse. Hear Res 202, 63–73 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.