

Dear Editor
The mammalian Ras proteins, H-, N-, and K-Ras, belong to the small GTPase superfamily. They cycle between a GTP-bound active form and a GDP-bound inactive form (1, 2). The conversion from the GTP-bound form to the GDP-bound form is mediated by the intrinsic GTPase activity of Ras proteins and is greatly accelerated by Ras-associated GAPs (GTPase-activating proteins). Canonical, cancer-associated Ras mutations at codons G12, G13, or Q61 severely compromise the hydrolysis of GTP to GDP, leading to the accumulation of Ras-GTP and hyperacivation of Ras downstream signaling. Mutations in the KRAS and NRAS genes (not in HRAS) are frequently identified in myeloid disorders (15-60%), including acute myeloid leukemia (AML), atypical chronic myeloid leukemia, chronic myelomonocytic leukemia (CMML), and juvenile myelomonocytic leukemia (JMML) (3). In these diseases, the frequency of NRAS mutations is significantly higher than that of KRAS mutations.
Although mutations at codons G12, G13, and Q61 of NRAS are all perceived oncogenic equivalents, they display distinct biochemical and functional properties. NRAS G12 and G13 mutations affect the intrinsic GTPase activity of Nras by preventing proper position of the Ras-GAP arginine finger within the catalytic site, while Q61 mutations lead to more severe impairment of Nras intrinsic GTPase function (1). Therefore, G12 and G13 mutations are less activating than Q61 mutations. In human cancers with NRAS mutations, G12 mutations are prevalent in myeloid leukemia, while Q61 mutations are predominant in plasma cell myeloma and melanoma (Fig. S1A). In hematopoietic malignancies, ~70% of NRAS G12 mutations are identified in patients with myeloid diseases but these mutations are rare in those with plasma cell myeloma (Fig. S1B). In contrast, the incidence of Q61 mutations is equal in patients with myeloid leukemias or plasma cell myeloma (~40% each, Fig. S1B). Of note, the acquisition of two copies of oncogenic NRAS G12D or G13D alleles is associated with JMML/CMML progression in humans and mice (COSMIC database and (4)), indicating that incremental activation of Ras signaling is a pathological mechanism contributing to JMML/CMML development. In a recent study of mutation-specific Nras oncogenicity, only an endogenous Nras Q61R mutation could promote spontaneous melanoma formation in mice (5). However, the ability of endogenous Nras G12D and Q61R mutants to drive leukemogenesis has never been evaluated in a side-by-side comparison.
We and others previously showed that expression of one or two copies of oncogenic Nras G12D (NrasG12D/+ and NrasG12D/G12D) in the hematopoietic compartment induces a chronic or an acute myeloproliferative neoplasm (MPN) respectively, closely resembling human JMML/CMML (6-9). Here, we used the same interferon-inducible Mx1-Cre to generate NrasQ61R/+ and NrasQ61R/Q61R mice (see details in Supplementary Methods). To our surprise, NrasQ61R/Q61R mice were not found at the time of weaning, likely due to the leaky expression of NrasQ61R on the paternal parent side, which was more detrimental than that of the equivalent NrasG12D allele (Table S1). Therefore, our entire study focused on the comparison among NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D animals.
After the induction of oncogenic Nras expression using polyinosinic-polycytidylic acid (pI-pC) injections (Fig. S2A), we evaluated the levels of GTP-bound Ras isoforms in total bone marrow cells (Fig. 1A). Consistent with our previous results (6), Nras-GTP level moderately but significantly increased in NrasG12D/+ cells compared to that in control cells (Fig. 1A). This increase was slightly higher in NrasQ61R/+ cells and maximal in NrasG12D/G12D cells. In contrast to what is observed in KrasG12D/+ cells, wherein levels of GTP bound wild-type (WT) H- and N-Ras are significantly elevated (10), no increase in WT Hras and Kras activation was observed in NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D cells (Fig. 1A). Consistent with this incremental increase of Nras-GTP levels, NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D mice displayed incrementally more severe MPN phenotypes in general, as measured in spleen weight and morphology (Fig. 1B and S2B), white blood cell count (Fig. S3), and the abundance of monocytes (Mac1+ Gr1-) and neutrophils (Mac1+ Gr1+) within various hematopoietic tissues (Fig. S2C). We closely monitored NrasQ61R/+ mice and found that they exhibited an intermediate survival rate between NrasG12D/+ and NrasG12D/G12D mice (Fig. 1C). Detailed evaluation of moribund mice revealed that 100% of them died with an advanced MPN (Fig. S4) and ~10% simultaneously developed acute T-cell lymphoblastic leukemia/lymphoma (T-ALL) (Fig. 1D). In contrast, 100% of NrasG12D/+ mice developed MPN and/or histiocytic sarcoma without T-ALL, while 100% of NrasG12D/G12D mice developed MPN and ~30% simultaneously developed T-ALL (Fig. 1D). These results indicate that NrasQ61R/+ has an intermediate leukemogenic activity between NrasG12D/+ and NrasG12D/G12D.
Figure 1. Somatic activation of NrasQ61R/+ leads to intermediate leukemia phenotypes between NrasG12D/+ and NrasG12D/G12D.
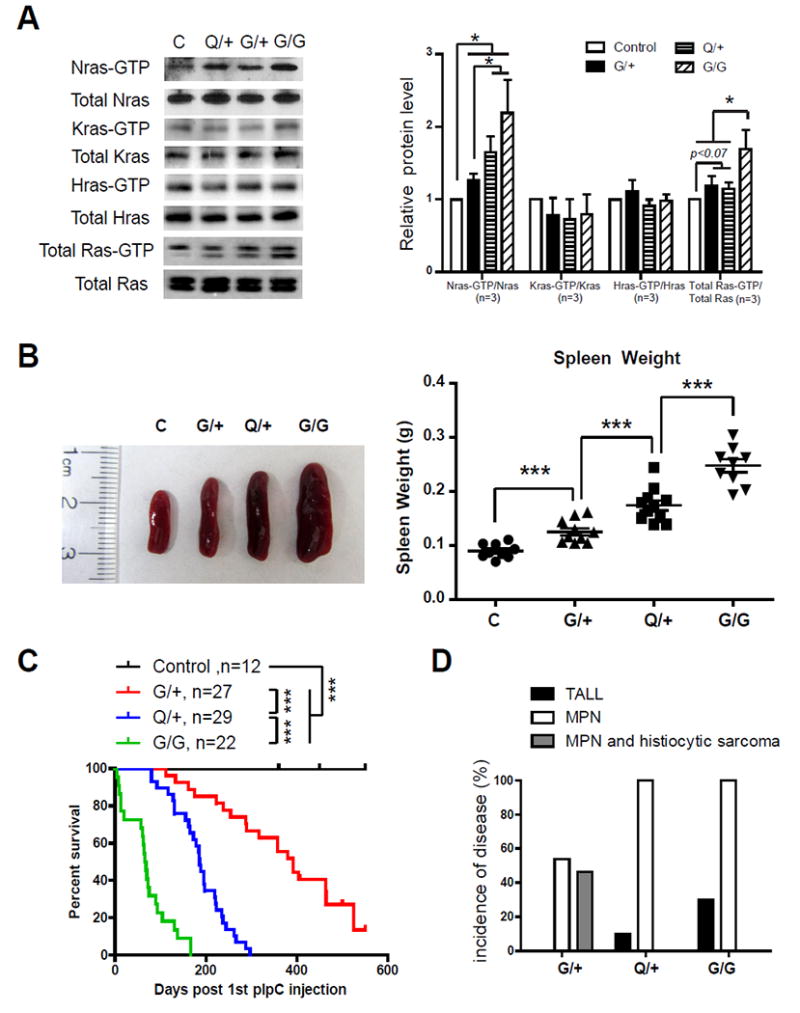
(A, B) Six-seven weeks old control (C), NrasG12D/+ (G/+), NrasQ61R/+ (Q/+), and NrasG12D/G12D (G/G) mice were injected with pI-pC and sacrificed on Day 5 for analysis as described in Methods. (A) Whole cell lysates were extracted from total bone marrow cells and assayed for expression levels of different Ras isoforms. Ras-GTP was affinity purified from WCL using a GST fusion with the Ras binding domain of Raf (Raf RBD) immobilized on agarose beads. Different Ras isoforms and total Ras bound with GTP were analyzed using Western blot analyses. The results were quantified using ImageJ software. The ratios of Ras-GTP/Ras in control cells are arbitrarily set at 1. (B) Quantification of spleen weight. (C) Kaplan-Meier survival curves were plotted against days after 1st pI-pC injection. P values were determined using the Log-rank test. (D) Disease incidences of moribund mice. The results are presented as mean ± SD. * P<0.05; *** P<0.001.
We further investigated the underlying cellular mechanisms contributing to the phenotypes observed in NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D mice. Compared to control mice, the numbers of Lin- c-Kit+ Sca1+ (LSK) cells and myeloid progenitors (MPs) were generally increased in the bone marrows of all three groups of oncogenic Nras mice and incrementally increased in their spleens (Fig. S5A and 2A), correlating with their respective spleen size. Consistent with our previous results (4, 11), the common lymphoid progenitor (CLP) compartment was only expanded in NrasQ61R/+ and NrasG12D/G12D mice but not in NrasG12D/+ mice (Fig. S5B). This observation may explain why TALL was developed in NrasQ61R/+ and NrasG12D/G12D mice, but not in NrasG12D/+ animals. Cell cycle analysis revealed a significant increase in the proliferation of NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D LSK cells (Fig. S5C), but increased proliferation was only significant in NrasQ61R/+ and NrasG12D/G12D MPs but not in NrasG12D/+ MPs (Fig. 2B). Colony forming assays demonstrated that in the absence of cytokines or in the presence of IL-3/mGM-CSF, NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D bone marrow cells formed incrementally increased number and size of colonies (Fig. 2C and S6). Together, the MP phenotypes in NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D mice are highly consistent with the increasing severity of MPN phenotypes found in these mice.
Figure 2. NrasQ61R/+ displays intermediate capability to promote proliferation, colony formation, and cytokine signaling in myeloid progenitors.
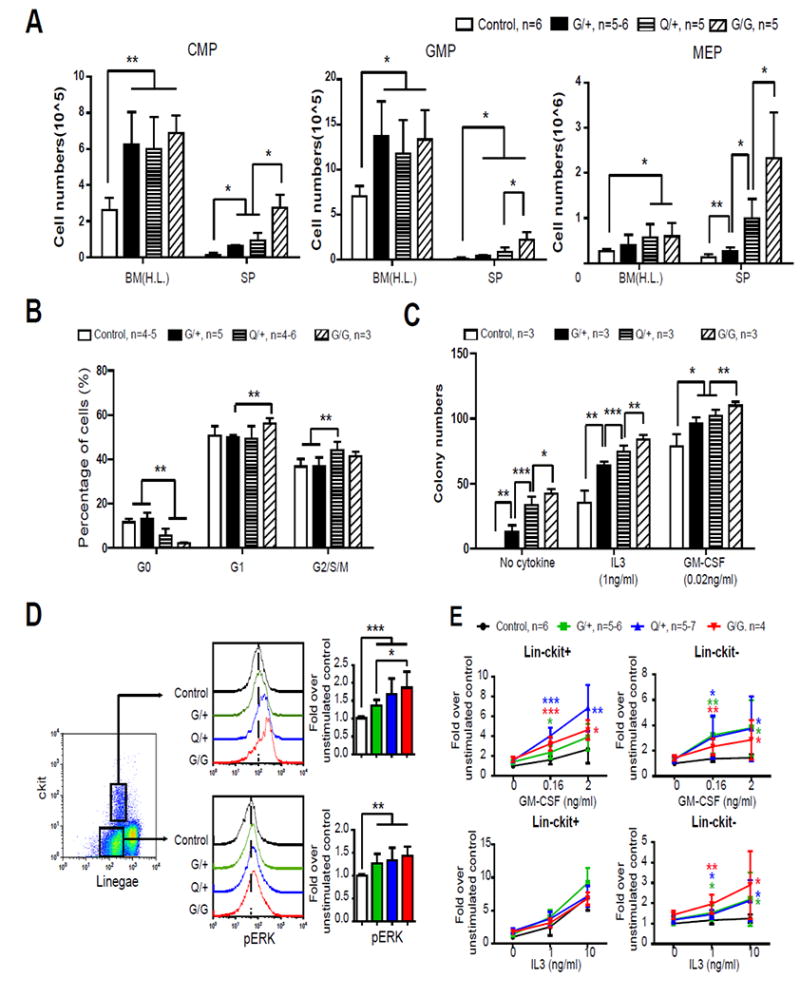
Six-seven weeks old control (C), NrasG12D/+ (G/+), NrasQ61R/+ (Q/+), and NrasG12D/G12D (G/G) mice were injected with pI-pC and sacrificed on Day 5 for analysis as described in Methods. (A) Quantitative analysis of common myeloid progenitors (CMPs), granulocyte-monocyte progenitors (GMPs), and megakaryocyte-erythrocyte progenitors (MEPs) in hind limb bone marrow [BM(H.L.)] and spleen (SP). (B) Cell cycle analysis of myeloid progenitor cells using Ki67 and DAPI staining. (C) Bone marrow cells were cultured in methylcellulose-based medium M3234 supplemented with purified mGM-CSF or mIL-3 as described in Methods. The colonies were counted after 7 days in culture. (D, E) Whole bone marrow cells were serum- and cytokine-starved for 2 hours at 37°C. Levels of p-ERK1/2 were measured using phospho-flow cytometry (D). Cells were then stimulated with different concentration of mGM-CSF or mIL-3 for 10 minutes at 37°C. Levels of p-ERK1/2 were measured using phospho-flow cytometry (E). Non-neutrophil bone marrow cells were gated for data analysis. Myeloid progenitors are enriched in Lin-/low c-Kit+ cells, whereas myeloid precursors are enriched in Lin-/low c-Kit- cells. To quantify the activation of ERK1/2, median intensities of p-ERK1/2 at different GM-CSF or IL-3 concentrations are compared to their respective control cells at 0 ng/ml, which is arbitrarily set at 1. Representative plots from one experiment are shown. The results are presented as mean ± SD. Asterisks of different colors represent P values when comparing corresponding group with control. * P<0.05, ** P<0.01 and *** P<0.001.
To determine whether the expansion of the MP compartment is associated with the hyperactivation of cytokine signaling, we performed phos-flow analysis in Lin- c-Kit+ cells (enriched for MPs) and Lin- c-Kit- cells (enriched for myeloid precursors) from NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D mice. Total bone marrow cells were first serum- and cytokine-deprived for 2 hours before fixation and permeabilization to measure phospho-ERK1/2 (p-ERK1/2) and p-STAT5 levels (Fig. 2D and S7A). As expected, the ERK pathway was hyperactivated in both populations of cells from NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D mice, while pSTAT5 levels in these cells were comparable to those in control cells. Upon cytokine stimulation, both Lin- c-Kit+ and Lin- c-Kit- cells from NrasG12D/+, NrasQ61R/+, and NrasG12D/G12D mice showed significant hyperactivation of ERK1/2 in response to GM-CSF but no or marginal hyperactivation in response to IL-3 (Fig. 2E). Interestingly, p-STAT5 level was moderately increased in Lin- c-Kit- cells from all 3 genotypes but not in Lin- c-Kit+ cells following GM-CSF or IL-3 stimulation (Fig. S7B).
Interestingly, despite the more potent leukemogenic activity of the NrasQ61R allele, NRAS Q61 mutations are less frequent than G12 mutations in hematopoietic malignancies (Fig. S1). One possible explanation of this observation is that NRAS G12 mutations could be initiating events in leukemogenesis or acquired during leukemia progression, while Q61 mutations are more likely to serve as secondary events during leukemia development. This possibility is supported by the observation that NRAS G12 mutations are identified in both chronic and acute stages of hematopoietic malignancies, while Q61 mutations are enriched in acute or more advanced stage of these diseases (3).
In summary, our data indicate that endogenous NrasQ61R/+ induces an increase of Nras-GTP and cytokine-evoked signaling, which is intermediate between NrasG12D/+ and NrasG12D/G12D. Therefore, the leukemogenic activity of NrasQ61R/+ is stronger than NrasG12D/+ but weaker than NrasG12D/G12D. The simplicity of breeding and the intermediate leukemogenic activity of the NrasQ61R/+ model make it ideal to test whether additional genetic mutations that might accelerate or alleviate its leukemia phenotypes. In addition, given the prevalence of NRAS Q61 mutations in certain hematopoietic malignancies (e.g. plasma cell myeloma), the conditional NrasQ61R allele, when combined with appropriate transgenic Cre lines, could be used to physiologically mimic these specific diseases in vivo.
Sincerely,
Supplementary Material
Acknowledgments
We would like to thank the University of Wisconsin Carbone Comprehensive Cancer Center (UWCCC) for use of its Shared Services (Flow Cytometry Laboratory and Experimental Pathology Laboratory) to complete this research. This work was supported by R01 grants R01CA152108 and R01HL113066, and a Scholar Award from the Leukemia & Lymphoma Society to J.Z. This work was also supported in part by NIH/NCI P30 CA014520--UW Comprehensive Cancer Center Support.
Footnotes
Author Contributions
The contributions of individual authors are listed below: GK for experimental design & execution as well as writing manuscript; YIC, XY, and YZ for experimental execution; EAR for histopathology analysis and writing manuscript; CEB for generating NrasLSL Q61R/+ allele and writing manuscript; JZ for experimental design and writing manuscript.
Conflict of Interest Disclosures
We declare no competing financial interests.
Methods are described in Supplementary Information.
References
- 1.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003 Jun;3(6):459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 2.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011 Nov;11(11):761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012 Aug 16; doi: 10.1182/blood-2012-05-378596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong G, Wunderlich M, Yang D, Ranheim EA, Young KH, Wang J, et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. J Clin Invest. 2014 Jun 2;124(6):2762–2773. doi: 10.1172/JCI74182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer discovery. 2014 Dec;4(12):1418–1429. doi: 10.1158/2159-8290.CD-14-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang JY, Liu YG, Li ZY, Du J, Ryu MJ, Taylor PR, et al. Endogenous oncogenic Nras mutation leads to aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116(26):5991–6002. doi: 10.1182/blood-2010-04-281527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang JY, Liu YG, Li ZY, Wang ZD, Tan LX, Ryu MJ, et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood. 2011;118(2):368–379. doi: 10.1182/blood-2010-12-326058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011 Feb 10;117(6):2022–2032. doi: 10.1182/blood-2010-04-280750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Haigis KM, Firestone AJ, McNerney ME, Li Q, Davis E, et al. Dominant role of oncogene dosage and absence of tumor suppressor activity in Nras-driven hematopoietic transformation. Cancer discovery. 2013 Sep;3(9):993–1001. doi: 10.1158/2159-8290.CD-13-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong G, Chang Y-I, Damnernsawad A, You X, Du J, Ranheim RA, et al. Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia. 2016 doi: 10.1038/leu.2016.40. in print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Kong G, Liu Y, Du J, Chang Y-I, Zhang X, et al. Nras G12D/+ promotes leukemogenesis by aberrantly regulating haematopoietic stem cell functions. Blood. 2013;121(26):5203–5207. doi: 10.1182/blood-2012-12-475863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.