

Abstract
Large-scale analysis of the GC-content distribution at the gene level reveals both common features and basic differences in genomes of different groups of species. Sharp changes in GC content are detected at the transcription boundaries for all species analyzed, including human, mouse, rat, chicken, fruit fly, and worm. However, two substantially distinct groups of GC-content profiles can be recognized: warm-blooded vertebrates including human, mouse, rat, and chicken, and invertebrates including fruit fly and worm. In vertebrates, sharp positive and negative spikes of GC content are observed at the transcription start and stop sites, respectively, and there is also a progressive decrease in GC content from the 5′ untranslated region to the 3′ untranslated region along the gene. In invertebrates, the positive and negative GC-content spikes at the transcription start and stop sites are preceded by spikes of opposite value, and the highest GC content is found in the coding regions of the genes. Cross-correlation analysis indicates high frequencies of GC-content spikes at transcription start and stop sites. The strong conservation of this genomic feature seen in comparisons of the human/mouse and human/rat orthologs, and the clustering of genes with GC-content spikes on chromosomes imply a biological function. The GC-content spikes at transcription boundaries may reflect a general principle of genomic punctuation. Our analysis also provides means for identifying these GC-content spikes in individual genomic sequences.
Keywords: gene clustering, gene ontology, transcription start site, transcription stop site
The rapid accumulation of sequencing data covering a vast variety of genomes underscores the need to identify functional elements in the genomes and to reveal their roles in nucleosomal structure, gene regulation, gene duplication, and other biological processes. The functional importance of these genomic elements has been explored many times (1–9). For instance, several studies have been performed with a direct goal of revealing possible correlations between the stability of duplex DNA and its functional potential (1–5, 9). These results were quite variable, ranging from almost perfect correlation to no correlation at all. Nevertheless, an algorithm for gene identification based on DNA stability has been developed (6, 10).
The availability of genomic sequences of different species and a large number of gene annotations provide us with an unprecedented opportunity to study genome organization at different scales. Here we present the results of a large-scale analysis of genomic GC-content distribution at the gene level for different species. We found both common features and basic differences in compositional distributions in different groups of species. Most interestingly, we observed sharp transitions of GC-content (GC-content spikes) around transcriptional boundaries in all species studied. These results suggest a general feature of genomic punctuation, likely important for gene transcription, that has not previously been reported.
Methods
Data Set. Our study is based on the genomic sequences of six species, including human (Homo sapiens, ≈18,000 genes), mouse (Mus musculus, ≈17,000 genes), rat (Rattus norvegicus, ≈6,000 genes), fruit fly (Drosophila melanogaster, ≈18,000 genes), chicken (Gallus gallus, ≈1,500 genes), and worm (Caenorhabditis elegans, ≈20,000 genes).
For each species, the genomic sequence of each refseq (11) gene (including 5 kb upstream and downstream intergenic regions, 5′ and 3′ UTRs, introns, and exons) was extracted from its genome assembly by using the refgene annotations available at the University of California, Santa Cruz, Genome Browser database (12). In our analysis, we used the builds hg17, mm5, rn3, galGal2, ce2, and dm1 as the genome assemblies and reggene annotations of human, mouse, rat, chicken, worm, and fruit fly, respectively.
In addition, 5,213 pairs of human and mouse ortholog genes and 1,050 pairs of human and rat ortholog genes were obtained based on the homology maps of the National Center for Biotechnology Information's locuslink (11).
GC Content at Different Genomic Regions. For each species, the GC-content distributions in different genomic regions, including 5′ UTR, coding sequences (CDS), and 3′ UTR, were measured. Very short UTRs (<20 nt) or CDSs (<50 nt) were not included in our study.
GC-Content Transitions at Transcription Boundaries. The GC-content profiles along the gene at the transcription start/stop sites were characterized by using an overlapping sliding window of 71 bp. For each gene, the window is shifted from 5 kb upstream to 5 kb downstream of the transcription start/stop site at a step of 10 bp, and the GC content of the DNA sequence within the window is recorded. It is noteworthy that similar results have been obtained by using different window sizes (50–100), nonoverlapping sliding windows, or overlapping sliding windows of different step lengths.
We used the sliding window to generate the GC-content profile of the 10-kb genomic sequence centered at the transcription start/stop site of each gene. For each species, the consensus GC-content profile at the transcription start/stop site was generated by averaging the corresponding GC-content profiles of all genes at each position along the sequence.
Cross-Correlation Analysis. Cross correlation analysis, widely used in radar, sonar, ultrasound imaging, and other applications, is an efficient tool to detect known signals buried in noise (13). Consider two sequences x(i) and y(i), where i = 1, 2,..., N, the cross correlation coefficient of these two sequences, rxy, is defined as
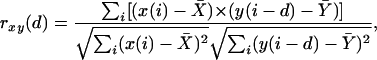 |
[1] |
where d is a variable that designates a shift of one sequence with respect to the other, and X̄ and Ȳ are the means of the corresponding sequence. The correlation rxy(0) gives an indication of pattern similarity of the two series of data without any shifting. The denominator in the expression above serves to normalize the correlation coefficients such that –1 ≤ rxy(d) ≤ 1. The bounds indicate maximum correlation and 0 indicates no correlation. A high negative correlation indicates a high correlation but of the inverse of one of the series.
We used cross correlation analysis to assess the frequency of GC-content spikes around the transcription boundaries. First, for each species, the characteristic region of the consensus GC-content profile around the transcription start/stop site was selected and used as the consensus GC-content spikes. For different species, these characteristic regions are of different lengths, and we took the –1.2- to 1.2-kb region for human, mouse, rat, and chicken transcription start sites, the –900- to 900-bp region for fruit fly transcription start sites, the –300- to 300-kb region for worm transcription start sites, the –700- to 700-bp region for human, mouse, rat, and fruit fly transcription stop sites, the –400- to 400-bp region for chicken transcription stop sites, and the –400- to 400-bp region for worm transcription stop sites.
We used these defined transcription start/stop sites to define positions >250 bp but <5 kb away from them as nonsites. The consensus GC-content spike was compared with the GC-content profile at each site and nonsite of each individual gene, and the similarity was evaluated by using the cross-correlation coefficient. For instance, given the consensus GC-content spike of the human transcription start site, s(i), where i =–M, –M + 1,..., 0, 1,..., M – 1, M, and position j (either a site or a nonsite) in the GC-content profile [p(i), where i = 1, 2,..., N and M < N] of a human gene around the transcription start site, the similarity between the consensus GC-content spike and the GC-content profile at position j is given by the no-shift cross-correlation coefficient of series s(i) and q(i), where q(i) is the subsequence of p(i) from j – M to j + M.
Note that the cross-correlation coefficient evaluates how correlated or how similar two series are and does not account for their absolute amplitude. Therefore, a very small spike in the GC-content profile may result in a strong correlation coefficient as long as its waveform is similar to that of the consensus spike. To use the cross-correlation coefficient to detect GC-content spikes in individual genes, we require that at each position j of the individual GC-content profile, the range(q(i)) = max(q(i)) – min(q(i)) must be larger than 0.6 × range(s(i)) before doing the cross correlation. However, in our study, >99.9% of the nonsites and all sites satisfy this criterion. Therefore, we believe that the cross-correlation coefficient provides a reasonable evaluation of the strength of GC-content spikes.
Mann–Whitney Test. The Mann–Whitney test is a nonparametric test used to test for the differences between two groups of sampled data (14). We compared the distributions of cross-correlation coefficients at the sites and the nonsites by using the Mann–Whitney test with a confidence level of 0.05.
Spearman's Rank Correlation. By using Spearman's rank correlation (15), we studied first the relationship between the GC-content spikes at the transcription start and stop sites of individual genes. Then, we assessed the correlation of the GC-content spikes at the transcription start/stop sites and the length of 5′ UTRs, CDS, and 3′ UTRs.
We investigated the evolutionary conservation of GC-spikes at the transcription start/stop sites across related species. A total of 5,213 pairs of human and mouse ortholog genes and 1,050 pairs of human and rat were obtained based on locuslink. Then, the correlation between the GC-spikes at the transcription start/stop sites on human/mouse (or human/rat) orthologs was evaluated by using Spearman's rank correlation.
Evaluation of Overrepresentation of Gene Ontology (GO) Terms Within Genes With GC-Content Spikes. We empirically selected 0.7 as the threshold of the cross-correlation coefficient to decide whether there is a GC-content spike at the transcription start/stop site of individual gene. For each species, we calculated which GO terms were statistically overrepresented within the set of genes with GC-content spikes with respect to the expected frequencies of the terms based on all of the genes analyzed (16). The statistical significance of the overrepresentation was evaluated by using the online expression analysis systematic explorer (ease) (17).
Clustering of Genes with GC-Content Spikes Along the Chromosomes. We studied the chromosomal distribution of genes with GC-content spikes (cross-correlation coefficient >0.7) in each species by using their chromosomal positions that are available in the refgene annotations in University of California, Santa Cruz, Genome Browser database (12). Adjacent genes on the chromosomes were clustered together, and the number of genes found in clusters and the size distribution of clusters were calculated. Then, we investigated whether the observed size distribution of clusters differed from a random distribution that was generated by using a stochastic model (18). For instance, there are 1,786 genes on human chromosome 1 (nonpredicted genes in refgene), and 404 genes among them have GC-content spikes in transcription start sites. We generated 404 random, nonrepetitive numbers in the range of 1 to 1,786 by using a random number generator. With the assumption that each of these numbers from 1 to 1,786 comprises the order of genes on the chromosome, each iteration assigns random genomic positions to the 404 genes with GC-content spikes. The same procedure was repeated for each chromosome. The proportion of genes found in clusters and the size distribution of clusters in the whole genome were calculated, and the values were averaged for 1,000 reiterations.
Results
GC-Content Distribution Across Genes. Fig. 1 shows the GC-content distribution in CDS and 5′ and 3′ UTRs of the human genome. A clear separation of distributions is visible, with the highest GC-content in the 5′ UTR, lowest GC-content in the 3′ UTR, and intermediate GC-content in the CDS. Similar results were obtained for mouse, rat, and chicken genomes (Table 1). However, no such dependence has been found for the fruit fly and worm genomes, where the GC content is highest in CDSs, intermediate in 5′ UTRs, and lowest in 3′ UTRs (Fig. 1 and Table 1).
Fig. 1.

Distributions of GC content in 5′ UTRs, CDS, and 3′ UTRs of human and fruit fly genes.
Table 1. GC content in different genomic regions of several species.
| Species | Class | N | %GC ± SD |
|---|---|---|---|
| H. sapiens | 5′ UTR | 21,364 | 60.6 ± 12 |
| CDS | 22,606 | 52.3 ± 8.5 | |
| 3′ UTR | 22,420 | 42.4 ± 11 | |
| M. musculus | 5′ UTR | 16,737 | 59 ± 11 |
| CDS | 17,980 | 51.8 ± 6.5 | |
| 3′ UTR | 17,783 | 43 ± 8.5 | |
| R. norvegicus | 5′ UTR | 5,943 | 58.9 ± 11 |
| CDS | 6,726 | 51.8 ± 6.0 | |
| 3′ UTR | 6,552 | 44.2 ± 9 | |
| G. gallus | 5′ UTR | 718 | 59.7 ± 14.1 |
| CDS | 849 | 50.1 ± 6.9 | |
| 3′ UTR | 826 | 41.4 ± 9.4 | |
| D. melanogaster | 5′ UTR | 8,941 | 41.5 ± 8 |
| CDS | 9,228 | 54 ± 5 | |
| 3′ UTR | 9,157 | 34 ± 7 | |
| C. elegans | 5′ UTR | 568 | 40.1 ± 9.7 |
| CDS | 981 | 44.3 ± 4.3 | |
| 3′ UTR | 975 | 29.7 ± 5.9 |
N, the number of sequences analyzed.
GC-Content Profiles at Transcription Boundaries. GC-content profiles at the transcription start/stop site were studied by using an overlapping window scanning 5 kb upstream and downstream of the corresponding transcription site. The consensus GC-content profiles were generated by averaging the GC-content profiles for each individual gene. Sharp changes in GC content have been observed at both transcription boundaries of all species analyzed. Interestingly, two substantially distinct groups of GC-content profiles were observed: warm-blooded vertebrates including human (Fig. 2), mouse, rat, and chicken, and invertebrates including fruit fly (Fig. 2) and worm. In vertebrates, sharp positive and negative spikes of GC content are observed at the transcription start and stop sites, respectively. In invertebrates, the profiles are more complex, and the positive and negative spikes at the transcription start and stop sites are preceded by the spikes of opposite value. Very similar GC-content profiles (data not shown) have been observed based on those human genes with validated refseq annotations or with full-length annotation, which confirmed that the consensus GC-content profiles for these species are generally accurate, regardless of the fact that many refseq annotations of the transcription boundaries are predicted but not confirmed.
Fig. 2.

Consensus GC-content profiles in 10-kb regions centered at the transcription start (blue) and stop (red) sites of human and fruit fly genes. The consensus GC-content profile at the transcription start/stop site was generated by averaging the GC-content profiles of all genes, which were calculated by using an overlapping sliding window of 71 bp at a step of 10 bp.
Frequency of Characteristic GC-Content Spikes at Transcription Boundaries or Other Genomic Regions in Individual Genes. We analyzed the probabilities of GC-content spikes at the transcription start/stop sites and compared them to the probability of spikes in other genomic locations (nonsites). Cross correlation analysis is an efficient technique to detect GC-content spikes in individual gene. The radically different distributions of the cross-correlation coefficient at sites and nonsites for human and fruit fly genes, as shown in Fig. 3, implies that the chance of finding a characteristic GC-content spike in sites is significantly greater than in nonsites. For instance, with an empirical cross-correlation coefficient threshold of 0.7, 31% of the human transcription start sites have GC-content spikes, but only 1.1% of the corresponding nonsites have GC-content spikes. Similarly, the chance of finding GC-content spikes is 4.0% at human transcription stop sites, 0.3% at corresponding nonsites; 14% at fruit fly transcription start sites, 2.4% at corresponding nonsites; 18% at fruit fly transcription stop sites, and 3% at corresponding nonsites. For all six species studied, it is clear that the mean of the cross-correlation coefficient at sites is significantly larger than at nonsites, where it is practically zero and implies no correlation. The difference in the distributions of cross-correlation coefficients at sites and nonsites was also confirmed by using the Mann–Whitney rank order test. For all cases, the probability of the null hypothesis that the two samples come from identical populations is 0 with a level of significance of 0.05.
Fig. 3.

Distributions of the cross-correlation coefficients of the consensus GC-content spikes and the GC-content profiles at sites and corresponding nonsites in human and fruit fly genomes. Distributions for human transcription start sites, human transcription stop sites, fruit fly transcription start sites, and fruit fly transcription stop sites are shown.
No Correlations Between GC-Content Spikes at Transcription Start and Stop Sites and Between GC-Content Spikes and the Length of the 5′ UTRs, CDS, and 3′ UTRs. We computed the Spearman's rank correlation (Rs) between the GC-content spikes (cross-correlation coefficient) at the transcription start and stop sites. As shown in Table 2, no correlation is observed in any of the six species. This finding implies that the GC-content spikes at the transcription start and stop sites are generally two independent genomic features.
Table 2. Relationship between the GC-content spikes at the transcription start site and stop site, measured by the Spearman rank correlation.
| Species | Rs | P |
|---|---|---|
| H. sapiens | 0.04 | 0.00 |
| M. musculus | 0.03 | 0.00 |
| R. norvegicus | -0.02 | 0.05 |
| D. melanogaster | 0.13 | 0.00 |
| G. gallus | -0.01 | 0.79 |
| C. elegans | 0.08 | 0.00 |
Rs, Spearman rank correlation; P, probability of mistakenly rejecting the null hypothesis of Rs = 0.
The relationships between the GC-content spikes (cross-correlation coefficient) and the length of the 5′ UTR, CDS, and 3′ UTR have also been assessed by using the Spearman's rank correlation. As shown in Table 3, there is generally no strong link between the GC-content spikes and the length of the UTRs and CDS.
Table 3. Relationship between the GC-content spikes at the transcription start/stop site and the length of CDS, 5′ UTR, and 3′ UTR, measured by the Spearman rank correlation.
| Transcription start site
|
Transcription stop site
|
||||
|---|---|---|---|---|---|
| Species | Class | Rs | P | Rs | P |
| H. sapiens | 5′ UTR | -0.03 | 0.00 | -0.00 | 0.79 |
| CDS | 0.06 | 0.00 | 0.01 | 0.34 | |
| 3′ UTR | 0.12 | 0.00 | 0.02 | 0.11 | |
| M. musculus | 5′ UTR | -0.05 | 0.00 | -0.00 | 0.84 |
| CDS | 0.07 | 0.00 | 0.02 | 0.09 | |
| 3′ UTR | 0.14 | 0.00 | -0.08 | 0.00 | |
| R. norvegicus | 5′ UTR | -0.00 | 0.78 | 0.01 | 0.71 |
| CDS | 0.02 | 0.27 | 0.03 | 0.05 | |
| 3′ UTR | 0.12 | 0.00 | -0.13 | 0.00 | |
| D. melanogaster | 5′ UTR | -0.24 | 0.00 | -0.16 | 0.00 |
| CDS | -0.02 | 0.05 | -0.04 | 0.00 | |
| 3′ UTR | -0.15 | 0.00 | -0.36 | 0.00 | |
| G. gallus | 5′ UTR | -0.09 | 0.01 | -0.04 | 0.28 |
| CDS | -0.04 | 0.25 | -0.04 | 0.28 | |
| 3′ UTR | 0.01 | 0.77 | -0.25 | 0.00 | |
| C. elegans | 5′ UTR | -0.21 | 0.00 | -0.01 | 0.84 |
| CDS | -0.06 | 0.10 | -0.18 | 0.00 | |
| 3′ UTR | -0.01 | 0.85 | -0.16 | 0.00 | |
Rs, Spearman rank correlation; P, probability of mistakenly rejecting the null hypothesis of Rs = 0.
Evolutionary Conservation of GC Spikes. Biologically functional genomic elements tend to be conserved through evolution. We studied the conservation of the GC-content spikes by using human/mouse and human/rat orthologs. Strong correlation of the GC-content spikes (cross-correlation coefficient) between human and mouse (or rat) has been observed, as shown in Table 4.
Table 4. Spearman rank correlation of the cross-correlation coefficient between human/mouse orthologs and human/rat orthologs.
| Transcription start site
|
Transcription stop site
|
|||
|---|---|---|---|---|
| Cross species | Rs | P | Rs | P |
| H. sapiens/M. musculus | 0.54 | 0.00 | 0.31 | 0.00 |
| H. sapiens/R. norvegicus | 0.53 | 0.00 | 0.14 | 0.00 |
Rs, Spearman rank correlation; P, probability of mistakenly rejecting the null hypothesis of Rs = 0.
Overrepresentation of GO Terms Within Genes With GC-Content Spikes. The GO project is developing three structured, controlled vocabularies (ontologies) that describe gene products in terms of their associated biological processes, cellular components and molecular functions in a species-independent manner (16). We evaluated the overrepresentation of the subset of genes with GC-content spikes (empirically, correlation coefficient >= 0.7). We used the online expression analysis systematic explorer (ease) (17) to calculate the overrepresentation of GO terms in this subset relative to the expected frequencies of these terms in all genes analyzed. The ease score evaluates the statistical significance of the overrepresentation. In Table 5, we list the GO terms with the most statistical significance in each structured category (i.e., biological processes, cellular components, and molecular functions). As shown in Table 5, overrepresentations of genes with GC-content spikes at the transcription start or stop sites are visible in a wide variety of functional categories. For instance, human genes with GC-content spikes at the transcription start sites are significantly overrepresented in intracellular transport with an ease score of 1.1E-13. A detailed analysis of the correlation of GC-content spikes and functional overrepresentation is required.
Table 5. Overrepresentation analysis of genes with strong-spikes at the transcription start or stop site.
| Species | Class | System | Category | LH/LT | PH/PT | ease score |
|---|---|---|---|---|---|---|
| H. sapiens | Transcription start site (>0.7) | Biological process | Intracellular transport | 165/2,106 | 414/8,886 | 1.1E-13 |
| Cellular component | Cytoplasm | 723/1,860 | 2,366/8,026 | 9.43E-24 | ||
| Molecular function | Catalytic activity | 924/2,214 | 3,271/9,277 | 3.1E-13 | ||
| Transcription stop site (>0.7) | Biological process | Homophilic cell adhesion | 22/183 | 112/8,886 | 5.7E-15 | |
| Cellular component | Integral to membrane | 68/170 | 2,464/8,026 | 0.00777 | ||
| Molecular function | Calcium ion binding | 30/200 | 456/9,277 | 1.2E-07 | ||
| D. melanogaster | Transcription start site (>0.7) | Biological process | Physiological process | 430/475 | 2,888/3,419 | 2.7E-05 |
| Cellular component | Intracellular | 366/463 | 2,269/3,302 | 7.9E-08 | ||
| Molecular function | Aspartic-type endopeptidase activity | 9/751 | 14/5,665 | 0.00013 | ||
| Transcription stop site (>0.7) | Biological process | Response to temperature | 15/550 | 31/3,419 | 0.00012 | |
| Cellular component | Proteaqsome complex | 20/549 | 46/3,302 | 5.5E-05 | ||
| Molecular function | Catalytic activity | 518/953 | 2,703/5,665 | 4.7E-06 |
The human and fruit fly genes with high cross-correlation coefficient (>0.7) at either transcription boundaries are compared with respective background genes (all the genes used in our study) by using the online expression analysis systematic explorer (ease). In each system, only the most significantly overrepresented category is shown. LH, list hits; LT, list total; PH, population hits; PT, population total.
Clustering of Genes with GC-Content Spikes Along the Chromosomes. Clustering of genes along the chromosomes is demonstrated by using the relationship of the cluster size and the number of clusters in the genome. As shown in Fig. 4, the proportion of genes found in each cluster of the genes with GC-content spikes significantly differs from the stochastic distribution. Especially for clusters of three or more genes, the number of such clusters observed within the genes with GC-content spikes was significantly higher than chances predicted by our stochastic modeling.
Fig. 4.

Human or fruit fly genes with GC-content spikes at the transcriptional start or stop sites tend to cluster along chromosomes. Cluster size is the number of genes per cluster. The height of the bars represents the number of clusters of genes, with GC-content spikes of corresponding size (black) or estimated for the stochastic distribution (gray).
Discussion
The large-scale analysis of GC-content distribution in genomes of different species revealed two major features. First, in all species analyzed, we observed sharp GC-content spikes at transcription start and stop sites. The GC-content spikes at the transcription boundaries of these six genomes are clearly visible in the large sets of genomic sequences. Although the features are noisier on the level of individual genes, which may partly explain why they have been overlooked until now, our analysis shows that the presence of GC-content spikes at the transcription boundaries is statistically significant.
Attempts to find correlations between physical properties of DNA and its functional units have been undertaken in many studies for many years (1–9). In several publications, correlations between the GC-content and physical borders of functional elements (genes or complexes of genes) have been detected. For example, pathogenicity islands and gene transfer events were detected in bacteria by using wavelet transformations (19). The same method allowed also detection of isochore boundaries (20). Translation initiation sites were also characterized by sharp GC-content changes (21).
Thus, several lines of evidence indicated the possibility of correlation between GC-content profiles and genome functional units. However, only the large-scale analysis of thousands of genes reveals GC-spikes as a major characteristic of genomic organization. We suggest that the GC-content spikes may represent important compositional factors that define different functional genomic units, particularly transcription boundaries. Regions with sharp GC-content changes should be structurally different from the rest of the genome, where the GC content is constant or gradually changing and thus should be recognizable from the rest of genome (9, 22, 23). Therefore, GC-content spikes may play a general role in delineating different functional regions of the genome. Evolutionary conservation of these features supports a functional role. Although the biological function of these GC-content spikes is still unknown, an ease analysis shows that GC-spikes are enriched in genes with certain specific biological functions. In addition, it has been reported that, in eukaryotes, coexpressed genes are often found in clusters, and clustering of these genes may suggest coregulation (18, 24, 25). In our study, the clustering of genes with GC-content spikes further supports the assumption that they play a biological function, especially in transcriptional regulation.
The second finding from our large-scale analysis is that the distribution of GC content from 5′ UTR to 3′ UTR along genes is a characteristic feature, and different in vertebrates and invertebrates. In vertebrates, there is a gradual decrease of GC content from 5′ UTR to the 3′ UTR along the gene (Table 1), whereas in invertebrates, the highest GC-content is found in coding regions. It will be of great interest when additional species can be added to the analysis to clarify the evolutionary border between the two general types of patterns we observe. Our results also corroborate data from Xia et al. (26), who detected decreasing GC content along the coding regions in genes of various animals, from humans to Xenopus laevis. Compositional gradients were also found in genes of monocot, but not dicot, plants (27).
What might be the biological and evolutionary meaning of the differences in GC-content distribution between these two groups of genomes? It has been known for a long time that the warm-blooded vertebrates and monocot plants differ from the rest of animal and plant kingdoms in having the most compositionally heterogeneous genomes. They contain large genomic regions (>300 kb long) with different and fairly homogeneous GC-content called isochors (28, 29). Although GC-rich isochores are known to contain high concentrations of genes, their role at a functional and evolutional level is still not clear (9). The GC-content features revealed in our study are detected at the level of genes, a much smaller scale compared to isochores. However, on this level, the GC-content distribution shows substantial differences between the two groups of species. In line with the major biological trend that more complex organisms have more complex genomes, we speculate that changes in GC content along genes in warm-blooded vertebrates may present additional kinetic or thermodynamic guides for transcription that have appeared only in the most complex genomes.
Further study of the biological function of these compositional features and how their impact differs from gene to gene may provide invaluable understanding of genomic punctuation, gene transcription, and gene regulation. Independent of the underlying mechanisms, the GC spike is already a unique feature for in silico gene identification (10).
Acknowledgments
We are thankful to Temple Smith, Konstantin Khrapko, Chris Passaglia, Vadim Demidov, and Edward Trifonov for valuable suggestions and discussions. L.Z. was supported by Sequenom, Inc.
Author contributions: L.Z. and N.E.B. designed research; L.Z. performed research; L.Z., S.K., C.R.C., and N.E.B. analyzed data; and L.Z., S.K., C.R.C., and N.E.B. wrote the paper.
Abbreviations: CDS, coding sequence; GO, gene ontology.
References
- 1.Suyama, A. & Wada, A. (1983) J. Theor. Biol. 105, 133–145. [DOI] [PubMed] [Google Scholar]
- 2.Ikemura, T. & Aota, S. (1988) J. Mol. Biol. 203, 1–13. [DOI] [PubMed] [Google Scholar]
- 3.Hanai, R., Suyama, A. & Wada, A. (1988) J. Biomol. Struct. Dyn. 6, 51–62. [DOI] [PubMed] [Google Scholar]
- 4.Wada, A. & Suyama, A. (1984) J. Biomol. Struct. Dyn. 2, 573–591. [DOI] [PubMed] [Google Scholar]
- 5.Yeramian, E. (2000) Gene 255, 139–150. [DOI] [PubMed] [Google Scholar]
- 6.Yeramian, E. (2000) Gene 255, 151–168. [DOI] [PubMed] [Google Scholar]
- 7.Bina, M. & Crowely, E. (2001) Biopolymers 59, 347–355. [DOI] [PubMed] [Google Scholar]
- 8.Zhang, C. T. & Zhang, R. (2003) Gene 317, 127–135. [DOI] [PubMed] [Google Scholar]
- 9.Vinogradov, A. E. (2003) Nucleic Acids Res. 31, 1838–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeramian, E. & Jones, L. (2003) Nucleic Acids Res. 31, 3843–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pruitt, K. D. & Maglott, D. R. (2001) Nucleic Acids Res. 29, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karolchik, D., Baertsch, R., Diekhans, M., Furey, T. S., Hinrichs, A., Lu, Y. T., Roskin, K. M., Schwartz, M., Sugnet, C. W., Thomas, D. J., et al. (2003) Nucleic Acids Res. 31, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice, J. A. (1995) in Mathematical Statistics and Data Analysis (Duxbury, Belmont, CA), pp. 129–134.
- 14.Ewens, W. J. & Grant, G. R. (2002) in Statistical Methods in Bioinformatics: An Introduction (Springer, New York), p. 120.
- 15.Altman, D. G. (1991) in Practical Statistics for Medical Research (Chapman and Hall, London), pp. 285–288.
- 16.Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C., et al. (2004) Nucleic Acids Res. 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosack, D. A., Dennis, G., Jr., Sherman, B. T., Lane, H. C. & Lempicki, R. A. (2003) Genome Biol. 4, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boutanaev, A. M., Kalmykova, A. I., Shevelyov, Y. Y. & Nurminsky, D. I. (2002) Nature 420, 666–669. [DOI] [PubMed] [Google Scholar]
- 19.Lio, P. & Vannucci, M. (2000) Bioinformatics 16, 932–940. [DOI] [PubMed] [Google Scholar]
- 20.Wen, S. Y. & Zhang, C. T. (2003) Biochem. Biophys. Res. Commun. 311, 215–222. [DOI] [PubMed] [Google Scholar]
- 21.Mizuno, M. & Kanehisa, M. (1994) FEBS Lett. 352, 7–10. [DOI] [PubMed] [Google Scholar]
- 22.Vinogradov, A. E. (2001) Mol. Biol. Evol. 18, 2195–2200. [DOI] [PubMed] [Google Scholar]
- 23.Tenzen, T., Yamagata, T., Fukagawa, T., Sugaya, K., Ando, A., Inoko, H., Gojobori, T., Fujiyama, A., Okumura, K. & Ikemura, T. (1997) Mol. Cell. Biol. 17, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lercher, M. J., Urrutia, A. O. & Hurst, L. D. (2002) Nat. Genet. 31, 180–183. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, X. & Smith, T. F. (1998) Microb. Comp. Genomics 3, 133–140. [DOI] [PubMed] [Google Scholar]
- 26.Xia, X., Xie, Z. & Li, W. H. (2003) J. Mol. Evol. 56, 362–370. [DOI] [PubMed] [Google Scholar]
- 27.Wong, G. K., Wang, J., Tao, L., Tan, J., Zhang, J., Passey, D. A. & Yu, J. (2002) Genome Res. 12, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang, C. T. & Zhang, R. (2004) Genomics 83, 384–394. [DOI] [PubMed] [Google Scholar]
- 29.Bernardi, G., Olofsson, B., Filipski, J., Zerial, M., Salinas, J., Cuny, G., Meunier-Rotival, M. & Rodier, F. (1985) Science 228, 953–958. [DOI] [PubMed] [Google Scholar]