

Significance
Iron is an essential element in biology but has limited bioavailability. Ferritins are 24-mer iron-storage nanocage proteins that concentrate iron in their inner compartment as a bioavailable iron oxide biomineral. In L-type subunits, abundant in ferritins from organs involved in long-term iron storage, the biomineralization has been proposed to proceed through nucleation events involving iron(II) oxidation at the inner cage surface. Here, we demonstrate the nature and structural features of these nucleation sites. Structures captured during iron uptake show that the formation of the iron biomineral proceeds via the assembly of a tri-nuclear iron cluster, anchored to the protein through glutamic acid side chains, and involving oxo and peroxo ligands that are produced during the iron(II) oxidation by dioxygen.
Keywords: L-ferritin, metallocluster, nucleation site, biomineralization, X-ray
Abstract
X-ray structures of homopolymeric L-ferritin obtained by freezing protein crystals at increasing exposure times to a ferrous solution showed the progressive formation of a triiron cluster on the inner cage surface of each subunit. After 60 min exposure, a fully assembled (μ3-oxo)Tris[(μ2-peroxo)(μ2-glutamato-κO:κO′)](glutamato-κO)(diaquo)triiron(III) anionic cluster appears in human L-ferritin. Glu60, Glu61, and Glu64 provide the anchoring of the cluster to the protein cage. Glu57 shuttles incoming iron ions toward the cluster. We observed a similar metallocluster in horse spleen L-ferritin, indicating that it represents a common feature of mammalian L-ferritins. The structures suggest a mechanism for iron mineral formation at the protein interface. The functional significance of the observed patch of carboxylate side chains and resulting metallocluster for biomineralization emerges from the lower iron oxidation rate measured in the E60AE61AE64A variant of human L-ferritin, leading to the proposal that the observed metallocluster corresponds to the suggested, but yet unobserved, nucleation site of L-ferritin.
Twenty-four–mer ferritins are ubiquitous iron-biomineralizing nanocage proteins. In mammals, they are generally heteropolymers composed by two different types of subunits, the heavy H and the light L (183 and 175 amino acids, respectively, in the human chains). The subunits self-assemble to form a hollow structure with a central cavity capable of accommodating thousands of iron atoms in the form of an oxoferric biomineral (1, 2) (Fig. 1A). The relative ratios of the two types of subunits in the 24-mers vary in different tissues; ferritins in iron storage organs, such as liver and spleen, are richer in L-subunits whereas those with fast iron metabolism, such as brain and heart, are richer in H-subunits (3). The H-subunit contains a ferroxidase center, composed by the so-called Fe1 and Fe2 sites, capable of rapidly oxidizing Fe2+ to Fe3+ (4, 5); a relatively low number of amino acid ligands characterize the two sites. In the human H-subunit, Glu62 acts as a bridging ligand between the two metals; the metal at Fe1 is also bound to His65 and monodentate Glu27 whereas the metal at Fe2 binds bidentate Glu107 (4). Accessory transient metal sites (Fe3, Fe4, and, in some cases, Fe5) have been identified by X-ray crystallography in the proximity of the ferroxidase site, and they have been demonstrated to play a key role in the reaction turnover (4, 6). L-subunits lack the ferroxidase site whereas a number of other residues are conserved with respect to the H-subunit (homology 53% between human L- and H-chains (Fig. 1B) (7). Consequently, iron incorporation in nanocages rich in L-subunits is much slower. Nevertheless, even homopolymeric L-ferritins are able to biomineralize iron. The proposed mechanism involves the presence of a putative nucleation site on the inner cage surface of L-subunits. In several studies, the role of residues Glu57, Glu60, Glu61, and Glu64 (human H-chain aligned numbering) (Fig. 1B) for an efficient biomineralization was inferred by site-directed mutagenesis or chemical modifications (8, 9). Nonphysiological metal ions like Cd2+ have been observed bound to the corresponding glutamates of the above-mentioned human Glu residues in several mammalian L-ferritins (10–14) whereas the structure with iron of these homopolymeric L-ferritins has not yet been reported.
Fig. 1.

Ferritin main structural features and sequence alignment. (A Left) Schematic representation of the four-helix bundle subunit: Helices III and I are solvent-exposed and linked by a long loop (magenta); helices II and IV locate on the inner cage surface; a fifth short helix at the C terminus is tilted 60° with respect to the bundle axis. (Right) Section of the human L-ferritin (HuLf) cage. The trinuclear iron clusters sprout into the 8-nm cavity, surrounded by the protein shell of 12 nm external diameter. (B) Alignment of amino acid sequences of human H-ferritin (HuHf), HuLf, and horse spleen L-ferritin (HoLf) (FRIH_HUMAN, FRIL_HUMAN, and FRIL_HORSE, respectively), performed by Clustal Omega. All sequences are depleted of the initiator methionine. Shown in cyan are the residues binding the metal cluster in L-ferritin; in orange, the iron-binding amino acids in the ferroxidase site of the catalytically active H-subunit and the corresponding amino acids in the L-chains of HuLf and HoLf (both lacking the ferroxidase site); and, in magenta, the amino acids in the conserved pore responsible for iron entry.
Here, we describe a structural study of iron binding to human and horse L-ferritins (HuLf and HoLf, respectively) where we observe the ferritin-mediated assembling of a trinuclear iron cluster on the inner surface of L-ferritin cages, with a scaffold provided by Glu60, Glu61, and Glu64 ligands. The presence of these residues facilitates the biomineralization process, as demonstrated by different kinetics of biomineral formation between recombinant WT homopolymeric HuLf and its triple variant E60AE61AE64A. All together, these results provide direct evidence of the glutamate-bound triiron cluster as the mineral nucleation site in L-ferritins. The cluster structure is unprecedented in biological systems although it shows striking structural similarities to a synthetic hexanuclear iron cluster proposed about 20 years ago as a possible model of a ferritin biomineral (15).
Results
Structural Studies on Iron-Free HuLf and HoLf: Overall Structures.
Crystals of iron-free HuLf were exposed to a concentrated ferrous ammonium sulfate (Mohr’s salt) solution and frozen after 15, 30, and 60 min of diffusion of Fe2+ ions into the crystals. The same experimental approach was used to characterize the iron-loaded state of HoLf ferritin. The structures of HuLf and HoLf before iron exposure were determined as reference structures. They show the typical quaternary structure of maxi-ferritins, consisting of the 24-subunit assembly to form a hollow cage. The asymmetric unit in both structures contains a single protein chain, representing the spatial and time average of all of the molecular subunits found in the crystals. Each ferritin subunit shows the characteristic four-helix bundle tertiary structure completed by a fifth helix (Fig. 1A) (13, 16). The protein models are almost complete, with the exception of the two initial and the three final residues of HoLf and the two starting residues in the HuLf sequence. The structure of the purified horse spleen ferritin is that of an L-chain homopolymer.
Several cadmium ions have been observed bound to both iron-free HuLf and HoLf, in the threefold axis channel, on the twofold axis, and on the internal surface of the protein shell, but none on the fourfold axis channel (SI Channels and Metal Ions Binding Sites) (17, 18). In the iron-free structures of both L-type ferritins, two Cd2+ ions are coordinated by Glu57 and Glu60, and by Glu61 and Glu64, respectively.
L-Ferritin Subunits Provide the Scaffold for a Trinuclear Peroxo-Bridged Cluster.
Despite several structures of HuLf and HoLf, ferritins have been determined in complex with different metal ions (13, 16, 19), and no structural observation of iron binding to the mineralization site has been reported in the literature, the only iron(III) ion reported in HoLf being bound to an exogenous ligand (20). To the goal of obtaining insights into the ferritin biomineralization process, we have obtained the crystal structures of the iron-loaded HuLf and HoLf cages. Besides observing the expected iron ions in the access threefold channels (SI Channels and Metal Ions Binding Sites), we could monitor iron binding inside the cavity. Iron binding to the inner cage surface was achieved with the involvement of the carboxylic side chains of three glutamic acids: namely Glu60, Glu61, and Glu64 in HuLf and the corresponding residues in HoLf (Figs. 2 and 3). Three iron ions are coordinated to the above carboxylate side chains, generating an oxo-centered trinuclear cluster (Fig. 2). The anomalous difference maps show that the triiron cluster is formed at 30 min of iron exposure (the 15-min structure does not show any iron atom bound), but the best defined ligand set of the iron ions appears in the structures obtained after 60 min (Fig. 3). Fourier difference maps of both HuLf and HoLf show the presence of an atom in the middle of the iron cluster that has been interpreted as an oxide anion forming a μ3-oxo bridge (Fig. 3 B and D and SI Structural Features of the Metal Cluster). In the 60-min structure of HuLf (1.98 Å resolution), the oxide anion is located almost centrally in the plane formed by the three iron ions, with coordination distances in the 1.8- to 2.0-Å range, all identical at 2σ significance level (Figs. 2 and 3B and Table S1). The coordination displayed by the iron ions in the cluster is a distorted octahedral geometry. The carboxylate groups of Glu60, Glu61, and Glu64 symmetrically bridge the three iron couples (Fig. 2) on the same side of the cluster. On the opposite side, each pair of iron ions is connected by an elongated electron density compatible with a diatomic molecule. The observed densities have been interpreted as being due to three peroxide anions generating a second series of bridges (Fig. 3B). The arrangement of the three carboxylates, the three iron ions, and the three peroxides follows an almost regular threefold symmetry centered on the oxide anion. A sixth ligand is present in the basal plane of each iron ion: a water molecule for Fe1 and Fe2, and an oxygen from the carboxylate group of Glu57 (monodentate) for Fe3. Glu57 bridges a nearby fourth iron aqua-ion present at lower occupancy (about 50%). A likely explanation of the described arrangement of the iron ions in this site is that we have detected a (μ3-oxo)Tris[(μ2-peroxo)(μ2-glutamato-κO:κO′)](glutamato-κO)(diaquo)triiron(III) anion. Remarkably, the L-ferritin cluster shows the same structure of the analogous portion present in a synthetic hexanuclear iron(III) cluster reported as a model of the ferritin mineral core (a detailed structural comparison is reported in SI Structural Features of the Metal Cluster and Table S1) (15). The striking similarity to that model compound supports the presence of iron ions in the ferric state resulting in an overall trinegative charge of the assembly in HuLf, which can provide the driving force to attract new iron(II) ions to promote oxidation and biomineralization, as exemplified by the observed fourth iron ion approaching the cluster via Glu57.
Fig. 2.

Different stereoviews of the trinuclear iron binding site in HuLf. (A) Electrostatic surface representation of the trinuclear cluster binding site. Iron ions and water molecules are represented as cyan and red spheres, respectively. Peroxide molecules are shown as red ball-and-stick models. (B) Structure of the oxo-centered trinuclear iron cluster. Coordinating glutamates are highlighted as sticks. Iron ions, water, and peroxide molecules are represented as in A. Coordination bonds to iron ions are shown as dashed red lines; coordination distances and angles are reported in Table S1. Two different orientations are proposed in A and B to better appreciate the structural features of the protein site and metal cluster.
Fig. 3.

Stereoviews of the iron cluster in HuLf and HoLf. (A) Iron ions (cyan spheres) in HuLf are surrounded by the anomalous difference map (dark-red mesh) contoured at 4.0 σ. (B) Omit map (green mesh) in HuLf contoured at 3.0 σ of peroxide and oxide anions involved in iron coordination. In both HuLf pictures: light-blue schematic, helix II; light-blue sticks, residues involved in iron coordination. (C) Iron ions (cyan spheres) in HoLf are surrounded by the anomalous difference map (dark-red mesh) contoured at 5.0 σ. (D) Omit map (green mesh) in HoLf contoured at 3.0 σ of peroxide and oxide anions and the water molecule involved in iron coordination. In both HoLf pictures: yellow schematic, helix II; yellow sticks, residues involved in iron coordination.
Table S1.
Coordination distances and angles in the (μ3-oxo)-Tris[(μ2-peroxo)]triiron(III) cluster observed in the mineralization site of HuLf and HoLf and in the inorganic complex Tris(µ4-peroxo) bis(µ3-oxo) nonakis(µ2-acetato) hexa-iron(III). Empty lines were added to facilitate the comparison with the corresponding measurement in each cluster. In HoLf-Fe complex, some ligands are missing due to the incomplete coordination of Fe(III) ions determined in this structure
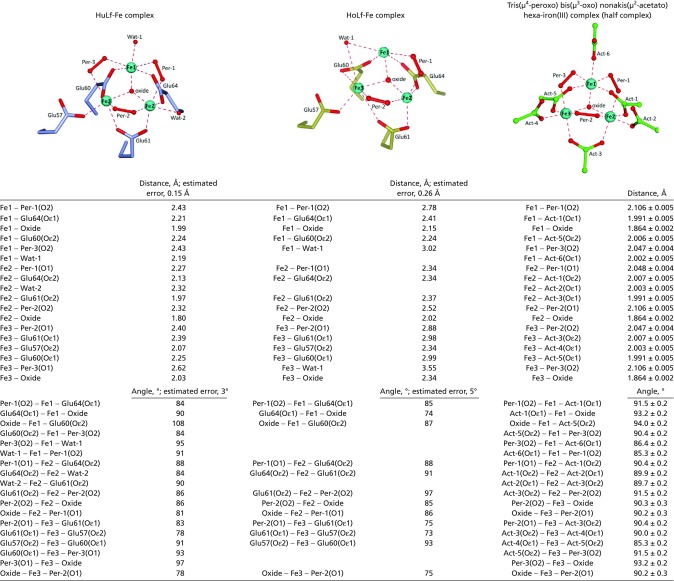 |
Wat1 in HoLF interprets electron density present in that position, but it refines outside the Fe coordination sphere. It should also be noticed that the Fe coordination spheres in HoLf are not complete.
The similarity between the trinuclear iron cluster in L-ferritin and the above model extends also to spectroscopic properties. The UV-vis spectrum of the model compound is characterized by the absence of any spectral feature between 500 and 800 nm and the presence of poorly defined shoulders (at 408, 456, and 534 nm) superimposed on a broad underlying tail (15). In HuLf, a broad shoulder at 490 nm is present that forms in the crystals after soaking times corresponding to those of the formation of the trinuclear cluster observed by us in crystal structures of HuLf and HoLf (Fig. S1). Such a feature is absent in the spectra of HuHf crystals exposed to Fe2+ for the same time.
Fig. S1.

UV-vis spectra of HuLf and HuHf crystals recorded after 30-min (A) and 60-min (B) exposure to Mohr’s salt. The characteristic absorption band at about 490 nm, indicative of Fe-bound peroxide, is visible only for HuLf.
The comparison with the iron-free HuLf structure reveals that the formation of the oxo-centered trinuclear iron cluster occurs without modification of the protein fold, but with a rearrangement of the glutamate side chains involved in iron coordination. The Cd2+ ions present in the same site in the iron-free structure are displaced by iron. Upon formation of the iron cluster, the carboxylate of Glu57 shifts by ∼2.5 Å (and rotates on the β-carbon by 60°) whereas the carboxylate of Glu60 moves by about 1.3 Å (and rotates on the γ-carbon by 50°). The formation of the cluster induces also the rearrangement of the side chains of several surrounding residues. In particular, the hydrophilic side chains of Arg68 and Glu140 contribute to stabilize the cluster through the formation of a network of water-mediated interactions involving Glu61 and the water molecule indicated as WatL in Fig. S2. The structures of HoLf exposed to an iron salt solution for the same time intervals confirmed the formation of the oxo-centered trinuclear iron cluster in the mineralization site of L-type ferritins (Fig. 3 C and D and Table S1). Only two peroxide anions can be modeled bridging the Fe1-Fe2 and Fe2-Fe3 couples (Fig. 3D). No density is present in the omit map in correspondence to the third peroxide observed in HuLf. Only an elongated maximum is found close to Fe1 and incoming Fe4 that has been interpreted as a water molecule. The lower resolution of this structure (2.22 Å) does not provide a satisfying modeling of all aqua-ligands that should complete the iron coordination in the cluster. However, the arrangement of the three iron ions in the cluster and of the incoming fourth iron ion, at lower occupancy, is almost superimposable with that present in HuLf (Fig. 3 A and C), which confirms that the formation of such clusters is a general feature of L-subunits and suggests its functional relevance.
Fig. S2.

Least-squares superimposition of iron-loaded HuLf (light blue) with iron-free HuLf (green). The three Cd2+ ions bound in the mineralization site in iron-free HuLf are replaced by Fe3+ ions and oxide after 60 min of exposure to Fe2+. Upon iron binding, the side chains of Glu57, Glu60, Glu61, Arg68, and Glu140 change conformation. The two latter residues are involved in an H-bond network with three water molecules, one of which (WatL) ligates Fe2.
Assessing the Functional Relevance of the Trinuclear Iron Cluster.
The crystal structure indicates the key role of Glu60, Glu61, and Glu64 to act as template for the formation of the cluster. Our functional working hypothesis was that substituting these negatively charged glutamate residues with nonpolar alanine could alter the biomineralization process. To prove the functional role of the residues linking the triiron cluster to the protein matrix, we designed and produced the triple mutant E60AE61AE64A. The protein yield was 30 mg/L, meaning that these multiple mutations did not affect the expression efficiency. Moreover, the typical quaternary structure was maintained as demonstrated in size exclusion chromatography experiments (Fig. S3). A comparative kinetic analysis of the formation of ferric biomineral between WT HuLf and its variant E60AE61AE64A was performed spectrophotometrically. Spectral changes of A350nm (Materials and Methods) were monitored over time, following the addition to protein solutions of different numbers of Fe2+ ions (3 to 18 Fe2+ per subunit) in 200 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 200 mM NaCl, pH 7 (Fig. 4). The same measurements were repeated also in the absence of the protein to observe the contribution of spontaneous ferrous ion oxidation. In the absence of ferritin, precipitation of iron-oxo species was clearly observed through a decrease of the A350nm; the effect was stronger at higher concentrations of Fe2+ (Fig. 4 C and D, blue curves). Differently, in the presence of HuLf, either WT or E60AE61AE64A variant, the iron was maintained in a soluble form, and the A350nm reached a plateau at incubation times that were dependent upon the number of added Fe2+ equivalents (Fig. 4, red and green curves). The initial velocity of Fe2+ oxidation was more rapid in the presence of WT HuLf with respect to the E60AE61AE64A variant, thus demonstrating a direct role of the mutated residues in the iron oxidation kinetics and therefore attributing a functional significance to the trinuclear iron cluster that forms at their site. From the expansion of the initial reaction rate (center graphs in Fig. 4 A–D), the differences between the WT and the E60AE61AE64A variant cages are clearly visible, accounting for reductions of the initial reaction rates by 33%, 55%, 68%, and 70% at 3, 6, 9, and 18 Fe per subunit ratios, respectively. The behavior of the E60AE61AE64A variant was always closer to that of the iron oxidation in the absence of ferritin rather than to that of the WT L-ferritin.
Fig. S3.

Purification of WT and E60AE61AE64A HuLf proteins. (A) Anionic exchange chromatograms obtained by a Q-Sepharose Fast Flow column with a 0 to 1 M NaCl gradient (dashed green line). In red and cyan, the elution profiles of WT and E60AE61AE64A HuLf, respectively, followed by UV detector (FPLC; Akta system). (B) SDS/PAGE gel of fractions obtained by anionic exchange chromatography. Lane M, marker; lanes 1 to 6, main peak of WT HuLf profile; lanes 8 to 13, main peak of E60AE61AE64A profile (bands around 20 kDa corresponding to the protein monomers). (C) Size exclusion elution chromatograms (UV measure) of WT and E60AE61AE64A HuLf superimposed with one of an already-characterized ferritin (violet curve; HuHf sample) to demonstrate their 24-meric conformation. (D) SDS/PAGE analysis of fractions eluted with HiLoad 16/60 Superdex 200 column. Lanes M, marker; lanes 1 to 6 main peak of WT HuLf profile; lanes 7 to 12, main peak of E60AE61AE64A profile (bands around 20 kDa corresponding to the protein monomers).
Fig. 4.

Kinetic measurements in WT and E60AE61AE64A HuLf variant. Kinetics of oxo-ferric species formation followed spectrophotometrically as the change in absorbance at 350 nm over 16 h (960 min). Different concentrations of Fe2+ were incubated, at room temperature, in 200 mM MOPS, 200 mM NaCl, pH 7 buffer with 25 µM protein subunits of WT HuLf (red curves) and triple mutant E60AE61AE64A (green curves) or without protein (blank, blue curves). Panels reflect addition of (A) 3 Fe2+ ions per subunit, (B) 6 Fe2+ ions per subunit, (C) 9 Fe2+ ions per subunit, (D) 18 Fe2+ ions per subunit. In the left graphs, the curves were recorded over 960 min, and the expansion of the curves during the first 100 min are provided in the central graphs, to highlight the differences between WT and triple mutant E60AE61AE64A. The bar plots of the right graphs provide the percent reduction in ferric-oxo species formation rate in the triple mutant with respect to the 100% rate assumed for the WT; rates were calculated from the linear fitting of the first 30 min (3 to 9 Fe2+ per subunit) and 15 min (18 Fe2+ per subunit).
SI Structural Features of the Metal Cluster
Structural Features of the Trinuclear Iron Cluster and Similarities to Model Systems.
Fig. 3 B and D shows the residual electron density present around the iron ions forming the cluster in HuLf and HoLf, respectively. The assignment of the residual electron density present in the center of the three iron ions to an oxide anion is based on the comparison of the coordination distances and on the chemical environment, as well as on the similarity to several model compounds. Examples of Fe3(μ3-O) units are abundant in crystal structures of iron(III) complexes synthesized to mimic intermediates in the catalytic cycle of nonheme iron proteins (15, 35–38). Fe3+-(μ3-O) distances in carboxylate-bridged ferric ions in model compounds are reported between 1.865(2) Å and 2.01(1) Å (35) whereas, in R2 ribonucleotide reductase, example (23) range between 1.7 Å and 2.2 Å. Fe3+-peroxide distances in multiple peroxo-bridged models range between 1.997(7) Å and 2.106(5) Å (15, 35).
In our HuLf 60-min structure, we observed Fe-oxo distances between 1.80(15) Å and 2.03(15) Å and Fe-peroxo distances between 2.27(15) Å and 2.62(15) Å. The peroxo O–O bond lengths determined in our structure were all identical between experimental error to 1.46(15) Å, which compares well with the canonical 1.48(2) Å found in μ2-peroxo models (39). The Fe–Fe distances in HuLf range between 3.21(15) Å and 3.45(15) Å and are comparable with those found in Fe3+-(μ3-O) models [e.g., 3.278(3) to 3.196(2) Å) (15).
The Fe-peroxide coordination distances observed in HuLf are all longer than those observed in model compounds. However, the lowest estimate on coordinate error (from maximum likelihood refinement) is 0.12 Å, which means that the most optimistic error on Fe-O distances is 0.15 Å. So, the experimental distances found in our structure are within 2–3 σ from those observed in the model compounds. The 2.62-Å distance falls just a bit above 3σ, but it should be observed that this peroxide is the only one that appears asymmetrically bound to the two Fe(III) of the cluster due to an interaction with the fourth Fe ion outside the cluster. Furthermore, at variance with the model compound of Shweky et al. (15), the cluster in L-ferritin is “open,” with the peroxides not sandwiched between two layers of Fe ions, which reasonably lengthens the Fe-O distances (see the comparison in Table S1) and which also applies to the cluster observed in the HoLf structure where only two peroxide anions could be modeled. Here, the average coordinate error amounts to 0.26 Å (Table S1).
Finally, the Protein Data Bank (PDB) and the literature report other Fe-bound peroxide-containing structures where Fe-O distances are the same as those obtained here (or longer), as follows: (i) In the μ-η1,η1 bridged Fe2-peroxo cluster reported in three independent rubrerythrin molecules present in the crystal asymmetric unit (PDB ID code 3QVD, 2 Å resolution) (40), the Fe-O peroxide distances are in the range 2.1 to 2.5 Å; (ii) Fe-O peroxide distances of 2.2 Å are reported for the μ-η1,η1 bridged Fe2-peroxo cluster present in human deoxyhypusine hydroxylase (PDB ID code 4D50, 1.7 Å resolution) (41); (iii) in 2,3-dihydroxy biphenyl dioxygenase from Rhodococcus sp. (PDB ID code 3LM4, 1.8 Å resolution) for an Fe side-on η2 peroxide, the Fe-O reported distances are 2.5 to 2.8 Å; (iv) the Fe side-on η2 bound peroxide reported distances are 2.2 to 2.4 Å for the iron oxidase UndA (PDB ID code 4WX0, 1.7 Å resolution) (42).
Long distances are also reported for Fe-Cu–bound peroxides: i.e.: Cu(II)-O, 2.6 to 2.7 Å; Fe(heme)-O, 2.3 to 3.2 Å for a μ-η2,η2-bound peroxide in a cytochrome ba3 oxidase mutant (PDB ID code 4GP4, 2.8 Å resolution) (43).
The structure of the ferric cluster in the HoLf is somewhat less regular than that of HuLf because the Fe triangle shows one Fe-Fe distance of 3.3(2) Å and two longer Fe-Fe distances [3.8(2) Å and 4.0(2) Å]. Only two μ2-bridged peroxide anions could be modeled into the residual electron density (Fig. 3D).
The above structural analysis led to our interpretation of the cluster as a stable (μ3-oxo)Tris[(μ2-peroxo)(μ2-glutamato-κO:κO′)](glutamato-κO)(diaquo)triiron(III) anion that constitutes a likely candidate for the nucleation site of the ferric mineral in L-ferritins.
SI Channels and Metal Ions Binding Sites
Twenty-four–mer ferritin cages have a 432 (O, octahedral)-point symmetry. In correspondence to the threefold and fourfold symmetry axes, there are eight and six channels, respectively, connecting the protein exterior to the inner cavity. The fourfold channels form at the encounter point of the C termini of four subunits, and the four symmetry-related helices V define the inner wall of the channel. The threefold channels occur where three subunits come into contact through the N-terminal ends of helices IV and the C-terminal ends of helices III. The two types of channels differ in their size and hydrophilicity (17).
Fourfold Symmetry Channel.
The channel located on the fourfold symmetry axis is not occupied by any metal ion. This finding can be ascribed to the prevalent hydrophobic nature of the residues present in HuLf and HoLf channels whereas Fe-bound His residues are present in both human H-ferritin and Rana catesbeiana H′-ferritin (4, 6).
Threefold Symmetry Channel.
It has been largely accepted that the channels present on the threefold symmetry in the ferritin protein shell constitute the preferential entry path of iron ions proceeding to the catalytic center. Very recently the transit of iron ions within these channels has been demonstrated through X-ray crystallography in homopolymeric human H-type ferritin and H′-type ferritin from R. catesbeiana (4, 6). The hydrophilic nature of these channels is strongly conserved among the different ferritin types.
A path of Cd2+ ions (discriminated according to the anomalous signal, still strong in the data collected below the Fe K-edge) is present inside this funnel-shaped channel. Three symmetry-related Cd2+ ions are visible on the external portion of the channel (largest part) at a distance of ∼4.8 Å. Cd2+ ions in these sites are coordinated by two protein residues, His118 and Cys130, whereas the other four ligands completing the octahedral coordination geometry have been assigned to water molecules. The partial occupancy of Cd2+ ions in this site is also suggested by the two alternative conformations adopted by Cys130. In the first conformation, showing the side chain exposed on the threefold channel, it coordinates a Cd2+ ion (and has been refined with the same occupancy) whereas, in the second, the side chain points to the adjacent helix III (and adopts the same conformation observed in HuHf showing magnesium ions inside the channel) (6). Going from the outer to the internal part of the channel, three further Cd2+ ions are clearly visible. One of them can be modeled in three symmetry-related alternative positions coordinated by the carboxylates of Glu134. A further Cd2+ is placed at the inner end of the channel, located exactly on the threefold symmetry axis where it is coordinated by three symmetry-related Asp131 and water molecules. A similar cluster of six Cu2+ ions has been previously observed in the threefold symmetry channel of R. catesbeiana H′-ferritin (PDB ID code 3RE7) (32). In our HoLf structure, only one Cd2+ ion is bound to Glu130 in the threefold axis channels.
Other Cd2+ Binding Sites.
In HuLf, four Cd2+ ions are bound at partial occupancy to the cage surface by Asp15, Glu90, His136, Glu49, and Asp178, with their coordination spheres completed by water molecules. In HoLf, two Cd2+ ions are bound to the protein surface, one to Asp84 and the other to Glu15 side chains.
Discussion
It is generally accepted that, in ferritin cages lacking ferroxidase-active subunits, the core biomineral formation proceeds via a few key steps. The iron entry into the cage cavity proceeds through the iron channels at the threefold symmetry axes (Fig. 5) and is followed by oxidation at specific, yet unobserved, nucleation sites that facilitate the subsequent growth of the mineral (2, 4, 21, 22). Using X-ray crystallography, we detected iron bound at inner cage sites of L-subunits of recombinant homopolymeric human ferritin in the form of (μ3-oxo)Tris[(μ2-peroxo)(μ2-glutamato-κO:κO′)](glutamato-κO)(diaquo)triiron(III) anion; a very similar arrangement was here observed in HoLf. The protein ligands to iron are provided by Glu60, Glu61 and Glu64 whereas Glu57 seems to be involved in shuttling additional iron ions toward the cluster. The peroxo cluster was fully formed in HuLf after 60 min of Fe2+ diffusion through the crystals whereas, in the lower resolution structure of HoLf at the same diffusion time, only two fully formed peroxide bridges could be modeled. The functional role to the observed trinuclear iron cluster emerged from the comparative kinetic analysis of the ferritin-mediated iron oxidation reaction of WT and E60AE61AE64A variant HuLf cages. The three mutated residues had a clear role in accelerating the oxidation reaction at low iron contents. These results allowed us to propose the three iron-carboxylate cluster as the site for the iron mineral nucleation site in L-ferritins. Indeed, the observed metallocluster seems to be a general characteristic of animal L-type ferritins because it is found also in HoLf crystals. The chemical features of the observed triiron cluster partially confirm previous proposals about the nature of biomineral precursors. The observed L-ferritin trinuclear cluster can be considered as a portion of the hexanuclear synthetic cluster discussed in Results, which was proposed to represent a ferritin biomineral model (15). Other authors have suggested that an oxo-centered trinuclear iron cluster, which forms spontaneously on the surface of class Ib ribonucleotide reductase R2 protein from Corynebacterium ammoniagenes in crystals subjected to iron soaking, could mimic an early stage in the mineralization of iron in ferritins (23). In this case, however, the bridging ligands were provided by six carboxylates whereas no peroxide anions were detected (23).
Fig. 5.

The properties of the threefold ion channel in ferritin. Electrostatic surface representation of a trimer of L-ferritin subunits where three monomers (gray schematic) come in contact generating a threefold channel responsible for iron entry. The external (Left) and the internal (Right) views of the pore circled with black lines show the negatively charged environment, due to the presence of conserved Asp131, Glu134, and Thr135 side chain residues. In HoLf, in position 135, there is a Ser, which is highly similar to Thr.
In analogy with the ribonucleotide reductase case, in L-ferritins, the presence of three adjacent Glu residues at positions 60, 61, and 64 is perfectly suited to provide cooperative binding of iron in a trinuclear center. In H-type subunits, like human H-ferritin (HuHf), this ideal arrangement of carboxylate side chains is disrupted by replacement of Glu60 with a His, with the concomitant increase in the conformational freedom of the Glu61 side chain (6). Also, Glu57 is replaced by His in HuHf (Fig. 1B). Consistently, the biomineralization reaction in H-ferritin cages does not require a nucleation site (24, 25), and His57 and Glu61 are involved in controlling the access of ferrous ions to the ferroxidase site (6); the reaction at the ferroxidase site is much faster than in L-subunits, with time scales for the iron oxidation in homopolymeric H-cages of <1 s. Even with a low number of H-subunits (1 to 2 per cage), the reaction rate is dominated by the catalytic oxidation of iron occurring at the ferroxidase sites. The formation of ferric clusters of high nuclearity in ferroxidase-active (H or H′) subunits has been inferred from Mössbauer data (26–28) and NMR data (29, 30), but these clusters have never been detected by time-lapse anomalous crystallography approaches. This observation supports the occurrence of a different iron mineralization process in ferroxidase-active subunits, leading to spatially disordered clusters that escape detection by X-ray crystallography. The observed peroxide ligands in the L-ferritin cluster might result from the initial oxidation of ferrous iron bound to the nucleation site and by the excess reductant (Fe2+) present in solution (31). The formation of a peroxide-complex in L-ferritins supports a mechanism for the mineral growth in the absence of the ferroxidase catalytic site, where the reaction between glutamate-bound Fe2+ and dioxygen provides the Fe-peroxide anionic catalyst able to attract and oxidize incoming Fe2+ aqua-ions. This mechanism is consistent with the previous proposal (31) that, at iron load up to 500 Fe per cage, hydrogen peroxide is involved in iron oxidation whereas, at higher Fe/protein ratios, the core biomineralization occurs via iron deposition directly on the biomineral surface (crystal growth model). The peroxide-driven reaction could be relevant in the context of the proposed ferritin-mediated detoxification reaction involving both Fe(II) and H2O2 (31). In vivo, this reaction is important to attenuate harmful Fenton chemistry under conditions of iron overload. In vitro, it becomes the dominant one due to the lack of the protective role exerted by catalase, which could disproportionate H2O2 produced upon iron oxidation. Time-lapse crystallography shows here once more (4, 6, 32) its merit in capturing reaction intermediates within ferritin cages, thus leading to the identification of functional iron-binding sites and to a dynamic picture of their population by iron ions.
Materials and Methods
Protein Preparation.
The plasmid encoding WT HuLf (33) was transformed into Escherichia coli BL21(DE3)-pLysS–competent cells. The protein was expressed and purified following a homemade protocol developed for HuHf (34) with some modifications [17 °C with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 48 h in rich medium Luria Bertani (LB)]. The subunit mass was analyzed by MALDI mass spectrometry. The retention volume obtained in the size exclusion chromatography demonstrated the 24-mer formation (Fig. S3). The E60AE61AE64A HuLf variant was expressed and purified as for WT; mutations were verified by MALDI peptide fingerprint analysis.
HoLf samples were obtained by purification of the commercially available protein.
Details on proteins’ preparation, purification, initial characterization, and mineralization are provided in SI Materials and Methods.
UV-Vis Measurements.
The rate of the biomineralization reaction in solution was determined spectrophotometrically upon addition of variable numbers of Fe2+ (3 to 18 Fe2+ per subunit). The characteristic absorbance at 490 nm was measured in HuLf crystals. Experimental details are provided in SI Materials and Methods.
Crystallography.
A series of L-ferritin structures were determined by soaking HuLF and HoLf ferritin crystals with a freshly prepared [(NH4)2Fe(SO4)2]∙6H2O solution for exposure times of 15 to 60 min, followed by flash freezing in liquid nitrogen. The procedure, detailed in SI Materials and Methods, follows with some modifications an established protocol that already allowed us to characterize iron oxidation in the ferroxidase site of human H- and Rana catesbeiana H′-type ferritins (4, 6). Data collection, structure solution, and refinement procedures are given in SI Materials and Methods and in Tables S2 and S3. The final coordinates and structure factors for 60-min iron-loaded HuLf and HoLf have been deposited in the Protein Data Bank under the accession codes 5LG8 and 5LG2, respectively.
Table S2.
Data collection and processing
| HuLf + Fe(II) 60′ | HoLf + Fe(II) 60′ | |||
| Structure | High energy remote | Above iron K-edge | Below iron K-edge | Above iron K-edge |
| PDB ID codes | 5LG8 | 5LG8 | 5LG8 | 5LG2 |
| Diffraction source | I03 (DLS) | XRD-1 (Elettra) | XRD-1 (Elettra) | XRD-1 (Elettra) |
| Wavelength, Å | 0.9184 | 1.7220 | 1.7587 | 1.7220 |
| Temperature, K | 100 | 100 | 100 | 100 |
| Detector | Pilatus 6M-F | Pilatus 2M | Pilatus 2M | Pilatus 2M |
| Crystal-detector distance, mm | 342 | 100 | 100 | 90 |
| Rotation range per image, ° | 0.15 | 0.5 | 0.5 | 0.5 |
| Total rotation range, ° | 75 | 60 | 60 | 60 |
| Exposure time per image, s | 0.25 | 10 | 10 | 15 |
| Space group | I432 | I432 | I432 | F432 |
| a, b, c, Å | 151.42 | 151.47 | 151.64 | 181.35 |
| Mosaicity, ° | 0.73 | 0.68 | 0.68 | 0.54 |
| Resolution range, Å | 29.70–1.98 (2.09–1.98) | 25.98–2.25 (2.37–2.25) | 26.00–2.28 (2.40–2.28) | 23.61–2.22 (2.34–2.22) |
| Total no. of reflections | 241,127 (34,962) | 128,079 (19,118) | 149,132 (22,337) | 138,083 (20,118) |
| No. of unique reflections | 20,846 (2,990) | 14,378 (2,052) | 13,874 (1,978) | 13,146 (1,862) |
| Completeness, % | 99.7 (100.0) | 99.9 (100.0) | 99.8 (100.0) | 99.9 (100.0) |
| Anomalous completeness, % | 99.7 (100.0) | 99.8 (100.0) | 99.9 (100.0) | 99.9 (100.0) |
| Redundancy | 11.6 (11.7) | 8.9 (9.3) | 10.7 (11.3) | 10.5 (10.8) |
| Anomalous redundancy | 5.9 (5.8) | 4.6 (4.8) | 5.7 (5.8) | 5.6 (5.6) |
| 〈I/σ(I)〉 | 16.8 (4.9) | 12.6 (4.1) | 14.0 (4.9) | 12.6 (4.9) |
| Rr.i.m. | 0.096 (0.481) | 0.113 (0.456) | 0.113 (0.449) | 0.126 (0.401) |
| Overall B factor from Wilson plot, Å2 | 15.4 | 15.9 | 16.6 | 14.2 |
Values for the outer resolution shell are given in parentheses. Rr.i.m., redundancy-independent merging R factor.
Table S3.
Structure solution and refinement
| Structure | HuLf + Fe(II) 60’ | HoLf + Fe(II) 60’ |
| PDB ID codes | 5LG8 | 5LG2 |
| Resolution range, Å | 29.70–1.98 (2.03–1.98) | 23.61–2.22 (2.28–2.22) |
| Completeness, % | 99.6 (100.0) | 99.8 (100.0) |
| No. of reflections, working set | 19,824 (1,437) | 12,489 (905) |
| No. of reflections, test set | 1,021 (67) | 656 (49) |
| Final Rcryst | 14.2 (21.0) | 16.5 (19.0) |
| Final Rfree | 17.7 (23.8) | 22.4 (28.0) |
| Cruickshank DPI | 0.103 | 0.185 |
| No. of non-H atoms | ||
| Protein | 1,437 | 1,364 |
| Metal Ions (Fe, Cd) | 12 (4, 8) | 7 (4, 3) |
| Ions (oxide, peroxide, acetate, chloride, sulfate) | 16 (1, 6, 4, 0, 5) | 6 (1, 4, 0, 1, 0) |
| Other | 0 | 6 |
| Water | 208 | 154 |
| Total | 1,674 | 1,537 |
| rms deviations | ||
| Bonds, Å | 0.026 | 0.015 |
| Angles, ° | 2.214 | 1.625 |
| Average B factors, Å2 | 25.88 | 28.35 |
| Ramachandran plot | ||
| Most favored, % | 98.0 | 98.0 |
| Allowed, % | 2.0 | 2.0 |
Values for the outer resolution shell are given in parentheses.
SI Materials and Methods
Protein Preparation.
The WT HuLf homopolymer was expressed and purified according to an in-house–developed procedure, which presents some modifications with respect to that developed for the human H protein (34). The pET-21c plasmid encoding native HuLf was a kind gift of S. Torti, Department of Molecular Biology and Biophysics, University of Connecticut Health Center, Farmington, CT (33). This vector was transformed into Escherichia coli BL21(DE3)-pLysS–competent cells (already used in our laboratory for ferritin expression). Several expression trials were performed to optimize the final protein yield. We tested different incubation temperatures (17 °C, 25 °C, and 37 °C), incubation times (4, 8, 16, 24, and 48 h), and IPTG concentrations (0.2, 0.5, and 1 mM). Our best condition turned out to be 17 °C, 0.2 mM IPTG for 48 h in LB, avoiding the protein to be found in inclusion bodies as occurred at higher temperatures or higher IPTG concentrations. In detail, bacteria were grown in LB-rich medium containing 100 mg/L ampicillin and 34 mg/L chloramphenicol at 37 °C. Once the A600nm reached 0.6 to 0.8, the overexpression of HuLf was induced with 0.2 mM IPTG, shifting the incubation temperature to 17 °C for 48 h. The cells were harvested (7,500 rpm for 15 min; JA-10 Beckman Coulter rotor) and resuspended in 20 mM Tris⋅HCl, pH 7.5. After sonication, the crude lysate was clarified by ultracentrifugation (40,000 rpm for 40 min; 70Ti Beckman Coulter rotor), and the soluble fraction was heated at 65 °C for 15 min to precipitate undesired proteins (40,000 rpm for 15 min; 70Ti Beckman Coulter rotor). Subsequently, the supernatant was dialyzed against 20 mM Tris⋅HCl, pH 7.5 buffer. Before chromatographic steps, the protein was precipitated with ammonium sulfate at 70% of saturation. Once the pellet containing protein was resolubilized, it was extensively dialyzed in 20 mM Tris⋅HCl, pH 7.5. Then the protein-containing fractions were recovered through a Q-Sepharose anionic exchange column eluting the sample with a linear NaCl gradient (0 to 1 M) in 20 mM Tris⋅HCl, pH 7.5. Next, fractions with ferritin (identified with SDS/PAGE analysis) were pooled and further purified by size exclusion chromatography (HiLoad 16/60 Superdex 200 column; GE Healthcare) in 20 mM Tris⋅HCl, 200 mM NaCl, pH 7.5 (Fig. S3).
Maldi-mass spectrometry (MALDI-MS; Toscana Life Sciences Foundation) confirmed the calculated mass; the experimental value of 19,888 Da corresponds to the mass of the WT HuLf subunit (20,019.6 Da from ProtParam tool; web.expasy.org/protparam/) without the first methionine (131 Da). Trypsin digestion of the protein and the analysis of the fragmentation pattern obtained by MALDI-MS demonstrated the identity of the HuLf subunit.
Apoferritin from equine spleen was purchased from Sigma Aldrich (product code A3641) and further purified by size exclusion chromatography (HiLoad 16/600 Superdex 200 pg; GE Healthcare). Mass spectra on the purified protein and on the dissolved crystal show the homogeneous presence of HoLf, as later confirmed by the crystal structure (as described in Data Collection, Structure Solution, and Refinement).
Design and Production of the Triple Mutant E60AE61AE64A.
The proper oligonucleotide primers containing the three point mutations were designed (QuikChange Primer Design tool) and purchased accordingly (Eurofins Genomics). We used pET-21c vector harboring the native gene as the DNA template, and we set up the site-directed mutagenesis reaction following the QuikChange Site-Directed Mutagenesis kit protocol (Agilent Technologies). The correct introduction of mutations was checked through Next-generation sequencing (Eurofins Genomics). The triple mutant E60AE61AE64A was expressed and purified in the same way as the WT protein. An aliquot of purified E60AE61AE64A was digested with trypsin and subjected to Maldi Peptide Mass Fingerprint analysis, confirming the insertion of the desired mutations.
Demineralization and Iron-Loading Procedures.
Iron and other metal ions were removed from purified ferritins by four dialysis steps using 4 L each of 20 mM Tris⋅HCl, 200 mM NaCl, 3 mM EDTA, ammonium thioglycolate (1:400), pH 7.5 buffer to reduce and chelate the iron, followed by four dialysis steps against 4 L each of 20 mM Tris⋅HCl, 200 mM NaCl, pH 7.5. After removing metal ions from ferritins, for kinetic studies, known iron equivalents were added to ferritin variants as previously described (18), preparing fresh ferrous sulfate stock solution in 1 mM HCl to avoid instantaneous oxidation of iron(II) to iron(III).
Kinetic Measurements.
Kinetics of ferric-oxide species formation in the absence and in the presence of WT HuLf and E60AE61AE64A HuLf were monitored as the change in A350nm over time. Then, 25-µM protein subunits in 200 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 200 mM NaCl, pH 7, were incubated at room temperature after addition of 3, 6, 9, and 18 Fe2+ per subunit. Spectra were acquired at a Varian Cary 50 Bio UV-vis spectrophotometer, collecting one point every 30 s in the first 5 h and every 15 min until 16 h. The rates of ferric-oxo species formation were calculated from the linear fitting of the first 30 min (3 to 9 Fe2+per subunit) and 15 min (18 Fe2+ per subunit).
Crystallization.
Demineralized HuLf in 20 mM Tris, pH 7.5, was concentrated to 9 mg/mL. Crystals of HuLf were grown using the hanging-drop vapor-diffusion technique at 281 K (44). Drops were prepared by mixing equal volumes of demineralized HuLf and a precipitant solution composed of 80 to 100 mM Cd(NO3)2, 80 to 100 mM ammonium sulfate, and 200 mM sodium acetate, pH 5. Drops were equilibrated over 800 µL of reservoir solution. Octahedral crystals appeared in 3 to 6 days and grew to final dimensions of about 200 to 400 µm in several days.
HoLf crystals were grown by the vapor diffusion hanging drop technique at room temperature. Drops were prepared by mixing 2 µL of HoLf solution (12 to 24 mg/mL in 0.15 M NaCl) with 2 µL of precipitant constituted by 80 mM Cd(NO3)2, 800 mM ammonium sulfate, and 200 mM sodium acetate, pH 5. Octahedral crystals, similar to those observed for HuLf, grew in a few days in drops equilibrated over 800 µL of reservoir solution.
Iron loading was performed by soaking both L-type ferritin crystals with a freshly prepared [(NH4)2Fe(SO4)2]∙6H2O solution (∼40 mM) for the monitored exposure times of 15, 30, 60, 90, and 120 min. Before flash freezing in liquid nitrogen, crystals were washed in cryoprotectant solutions constituted by 40% (vol/vol) glycerol added to the precipitant solutions.
UV-Vis Spectra on Crystals.
Spectra on HuLf and HuHf crystals (Fig. S1), obtained using the above crystallization solutions and treated with Mohr’s salt for 30 and 60 min, were recorded between 220 and 750 nm using a NanoDrop 1000 spectrophotometer (Thermo Scientific).
Data Collection, Structure Solution, and Refinement.
Complete datasets for iron-free HuLf and HoLf and for iron-loaded HuLf and HoLf were collected on the Elettra beamline XRD1 (Trieste, Italy) equipped with a Pilatus 2M detector, and on the DLS beamline I03 (Didcot, UK) equipped with a Pilatus 6M for crystals soaked for 15, 30, and 60 min. Crystals frozen after 90 and 120 min of exposure to iron seemed to have lost diffraction. The iron-free HuLf and HoLf data were collected for comparison. Data were integrated using either XDS (45) or Mosflm (46) and scaled with Scala (47) from the CCP4 suite (48). The HuLf crystals belonged to the body-centered cubic space group I432, with unit-cell parameter a of ∼152 Å, varying slightly among the different crystals. On the other hand, the HoLf crystals belonged to the face-centered cubic space group F432, having unit-cell parameter a of ∼182 Å. Data collection and processing statistics are reported in Table S2. Initial models were obtained by molecular replacement using MOLREP (49) from the CCP4 suite. A subunit of either HuLf or HoLf was used as a search model, excluding nonprotein atoms and water molecules.
To unambiguously prove that the observed cluster was formed by iron ions, full three-wavelength anomalous data (remote high energy 13,500 eV, peak of the Fe K-edge at 7,200 eV and immediately below the iron edge at 7,050 eV) were collected for HuLf whereas, for HoLf, we collected only the 7,200 and 7,050 eV data. The positions of metal ions were determined from the anomalous Fourier difference maps calculated at 7,200 eV and at 7,050 eV, using FFT from the CCP4 suite. Iron can be easily distinguished from cadmium due to the disappearance of its anomalous signal in the 7,050-eV map, where the anomalous signal of Cd2+ ions is still evident. Anomalous signals indicative of iron placement ranged between 15 σ and 4 σ in the anomalous difference map calculated with data collected at 7,200 eV.
The structures were refined with REFMAC5 (50) by using the high energy remote data in the case of HuLf or the 7,200-eV data in the case of HoLf, which were those at the highest resolution. The refinement protocol involved sequential iterative manual rebuilding of the model and maximum-likelihood refinement. Manual rebuilding and modeling of the missing atoms into the electron density and water molecules addition were performed with the molecular graphic software Coot (51). Partial occupancies for the iron and cadmium ions, for other ligands, and for several amino acid residues were adjusted to keep atomic displacement parameters close to those of the neighboring protein atoms in fully occupied sites. Metal ions were refined with anisotropic temperature factors. The occupancies of the water molecules or other ligands bound to the metal ions were kept the same as the parent metal ion.
The interpretation of the electron density surmounting the iron atoms in the cluster as peroxide anions relies on the following steps. We first modeled water molecules, coordinating the three Fe ions in the cluster, but additional, significant electron density surrounding each water molecule was observed, which suggested the presence of diatomic species. We then tried to model dioxygen molecules as Fe-bridging species, but, after the refinement, the interatomic distances between the oxygens were too long (above 1.4 Å), suggesting the presence of molecules having longer interatomic distances. Finally, we modeled peroxide ion as bridging species, obtaining O-O distances comparable with canonical peroxide anion distances. No constrained distances for the peroxide molecules were used, but, like any other residue in protein crystallography at medium resolution, restraints were used for modeling the peroxide anions. In summary, the interpretation of the residual electron density above the trinuclear cluster as due to peroxide anions is the one that better represents the experimental data.
The final models were inspected manually and checked with Coot and PROCHECK (52). The final refinement statistics are reported in Table S3. Figures were generated with CCP4mg (53).
Acknowledgments
We thank Prof. S. Torti (University of Connecticut) for providing us with the plasmid encoding WT HuLf and Dr. Francesco Vizza (CNR-ICCOM, Florence) for helpful discussion. We thank the Diamond Light Source for access to beamline I03 (Proposal BIOSTRUCTX_1358), Elettra (Trieste, Italy) beamline XRD1 for provision of synchrotron radiation facilities, and all the staff of the synchrotron sources for assistance in using the beamlines. We acknowledge the financial support of MIUR PRIN 2012 (Contract 2012SK7ASN) and the ESFRI Infrastructure Instruct through its Core Center, CERM, Italy. The research leading to these results has received funding from the European Community Seventh Framework Programme (FP7/2007–2013) under BioStruct-X (Grant Agreement 283570).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 5LG8 and 5LG2 for HuLf and HoLf, respectively).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1614302114/-/DCSupplemental.
References
- 1.Theil EC, Behera RK, Tosha T. Ferritins for chemistry and for life. Coord Chem Rev. 2013;257(2):579–586. doi: 10.1016/j.ccr.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theil EC. Ferritin protein nanocages use ion channels, catalytic sites, and nucleation channels to manage iron/oxygen chemistry. Curr Opin Chem Biol. 2011;15(2):304–311. doi: 10.1016/j.cbpa.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arosio P, Ingrassia R, Cavadini P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta. 2009;1790(7):589–599. doi: 10.1016/j.bbagen.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Pozzi C, et al. Time-lapse anomalous X-ray diffraction shows how Fe(2+) substrate ions move through ferritin protein nanocages to oxidoreductase sites. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 4):941–953. doi: 10.1107/S1399004715002333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X, Theil EC. Ferritins: Dynamic management of biological iron and oxygen chemistry. Acc Chem Res. 2005;38(3):167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 6.Pozzi C, et al. Iron binding to human heavy-chain ferritin. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 9):1909–1920. doi: 10.1107/S1399004715013073. [DOI] [PubMed] [Google Scholar]
- 7.Andrews SC, et al. Structure, function, and evolution of ferritins. J Inorg Biochem. 1992;47(3-4):161–174. doi: 10.1016/0162-0134(92)84062-r. [DOI] [PubMed] [Google Scholar]
- 8.Granier T, et al. Evidence of new cadmium binding sites in recombinant horse L-chain ferritin by anomalous Fourier difference map calculation. Proteins. 1998;31(4):477–485. [PubMed] [Google Scholar]
- 9.Wade VJ, et al. Influence of site-directed modifications on the formation of iron cores in ferritin. J Mol Biol. 1991;221(4):1443–1452. doi: 10.1016/0022-2836(91)90944-2. [DOI] [PubMed] [Google Scholar]
- 10.Luscieti S, et al. Mutant ferritin L-chains that cause neurodegeneration act in a dominant-negative manner to reduce ferritin iron incorporation. J Biol Chem. 2010;285(16):11948–11957. doi: 10.1074/jbc.M109.096404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, et al. Structure of human ferritin L chain. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 7):800–806. doi: 10.1107/S0907444906018294. [DOI] [PubMed] [Google Scholar]
- 12.de Val N, Declercq JP, Lim CK, Crichton RR. Structural analysis of haemin demetallation by L-chain apoferritins. J Inorg Biochem. 2012;112:77–84. doi: 10.1016/j.jinorgbio.2012.02.031. [DOI] [PubMed] [Google Scholar]
- 13.Granier T, Gallois B, Dautant A, Langlois d’Estaintot B, Précigoux G. Comparison of the structures of the cubic and tetragonal forms of horse-spleen apoferritin. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 5):580–587. doi: 10.1107/S0907444997003314. [DOI] [PubMed] [Google Scholar]
- 14.Granier T, et al. Structural description of the active sites of mouse L-chain ferritin at 1.2 A resolution. J Biol Inorg Chem. 2003;8(1-2):105–111. doi: 10.1007/s00775-002-0389-4. [DOI] [PubMed] [Google Scholar]
- 15.Shweky I, et al. A hexairon(III) complex with three nonplanar η2-μ4-peroxo ligands bridging two basic iron acetate units. J Am Chem Soc. 1997;119(5):1037–1042. [Google Scholar]
- 16.Hempstead PD, et al. Comparison of the three-dimensional structures of recombinant human H and horse L ferritins at high resolution. J Mol Biol. 1997;268(2):424–448. doi: 10.1006/jmbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 17.Bernacchioni C, Ghini V, Theil EC, Turano P. Modulating the permeability of the ferritin channels. RSC Advances. 2016;6:21219–21227. [Google Scholar]
- 18.Bernacchioni C, et al. Loop electrostatics modulates the intersubunit interactions in ferritin. ACS Chem Biol. 2014;9(11):2517–2525. doi: 10.1021/cb500431r. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki M, et al. Preparation and catalytic reaction of Au/Pd bimetallic nanoparticles in apo-ferritin. Chem Commun (Camb) 2009;32(32):4871–4873. doi: 10.1039/b908742g. [DOI] [PubMed] [Google Scholar]
- 20.Nakajima H, et al. Construction of an enterobactin analogue with symmetrically arranged monomer subunits of ferritin. Chem Commun (Camb) 2015;51(93):16609–16612. doi: 10.1039/c5cc06904a. [DOI] [PubMed] [Google Scholar]
- 21.Haldar S, Bevers LE, Tosha T, Theil EC. Moving Iron through ferritin protein nanocages depends on residues throughout each four α-helix bundle subunit. J Biol Chem. 2011;286(29):25620–25627. doi: 10.1074/jbc.M110.205278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behera RK, Theil EC. Moving Fe2+ from ferritin ion channels to catalytic OH centers depends on conserved protein cage carboxylates. Proc Natl Acad Sci USA. 2014;111(22):7925–7930. doi: 10.1073/pnas.1318417111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Högbom M, Nordlund P. A protein carboxylate coordinated oxo-centered tri-nuclear iron complex with possible implications for ferritin mineralization. FEBS Lett. 2004;567(2-3):179–182. doi: 10.1016/j.febslet.2004.04.068. [DOI] [PubMed] [Google Scholar]
- 24.Bou-Abdallah F, Biasiotto G, Arosio P, Chasteen ND. The putative “nucleation site” in human H-chain ferritin is not required for mineralization of the iron core. Biochemistry. 2004;43(14):4332–4337. doi: 10.1021/bi0498813. [DOI] [PubMed] [Google Scholar]
- 25.Santambrogio P, Levi S, Cozzi A, Corsi B, Arosio P. Evidence that the specificity of iron incorporation into homopolymers of human ferritin L- and H-chains is conferred by the nucleation and ferroxidase centres. Biochem J. 1996;314(Pt 1):139–144. doi: 10.1042/bj3140139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bou-Abdallah F, et al. μ-1,2-Peroxobridged di-iron(III) dimer formation in human H-chain ferritin. Biochem J. 2002;364(Pt 1):57–63. doi: 10.1042/bj3640057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauminger ER, et al. Iron (II) oxidation and early intermediates of iron-core formation in recombinant human H-chain ferritin. Biochem J. 1993;296(Pt 3):709–719. doi: 10.1042/bj2960709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira AS, et al. Rapid and parallel formation of Fe3+ multimers, including a trimer, during H-type subunit ferritin mineralization. Biochemistry. 1997;36(25):7917–7927. doi: 10.1021/bi970348f. [DOI] [PubMed] [Google Scholar]
- 29.Lalli D, Turano P. Solution and solid state NMR approaches to draw iron pathways in the ferritin nanocage. Acc Chem Res. 2013;46(11):2676–2685. doi: 10.1021/ar4000983. [DOI] [PubMed] [Google Scholar]
- 30.Turano P, Lalli D, Felli IC, Theil EC, Bertini I. NMR reveals pathway for ferric mineral precursors to the central cavity of ferritin. Proc Natl Acad Sci USA. 2010;107(2):545–550. doi: 10.1073/pnas.0908082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao G, et al. Multiple pathways for mineral core formation in mammalian apoferritin: The role of hydrogen peroxide. Biochemistry. 2003;42(10):3142–3150. doi: 10.1021/bi027357v. [DOI] [PubMed] [Google Scholar]
- 32.Bertini I, et al. Structural insights into the ferroxidase site of ferritins from higher eukaryotes. J Am Chem Soc. 2012;134(14):6169–6176. doi: 10.1021/ja210084n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rucker P, Torti FM, Torti SV. Recombinant ferritin: Modulation of subunit stoichiometry in bacterial expression systems. Protein Eng. 1997;10(8):967–973. doi: 10.1093/protein/10.8.967. [DOI] [PubMed] [Google Scholar]
- 34.Ravera E, et al. Solid-state NMR of PEGylated proteins. Angew Chem Int Ed Engl. 2016;55(7):2446–2449. doi: 10.1002/anie.201510148. [DOI] [PubMed] [Google Scholar]
- 35.Micklitz W, Bott SG, Bentsen JG, Lippard SJ. Characterization of a novel μ4-peroxide tetrairon unit of possible relevance to intermediates in metal-catalyzed oxidations of water to dioxygen. J Am Chem Soc. 1989;111(1):372–374. [Google Scholar]
- 36.Heath SL, Powell AK. The trapping of iron hydroxide units by the ligand ‘heidi’: Two new hydroxo(oxo)iron clusters containing 19 and 17 iron atoms. Angew Chem Int Ed Engl. 1992;31(2):191–193. [Google Scholar]
- 37.Taft KL, Papaefthymiou GC, Lippard SJ. A mixed-valent polyiron oxo complex that models the biomineralization of the ferritin core. Science. 1993;259(5099):1302–1305. doi: 10.1126/science.8446898. [DOI] [PubMed] [Google Scholar]
- 38.Taft KL, Papaefthymiou GC, Lippard SJ. Synthesis, structure, and electronic properties of a mixed-valent dodecairon oxo complex, a model for the biomineralization of ferritin. Inorg Chem. 1994;33(7):1510–1520. [Google Scholar]
- 39.Orpen G, et al. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J Chem Soc, Dalton Trans. 1989;12(Suppl):S1–S83. [Google Scholar]
- 40.Dillard BD, Demick JM, Adams MW, Lanzilotta WN. A cryo-crystallographic time course for peroxide reduction by rubrerythrin from Pyrococcus furiosus. J Biol Inorg Chem. 2011;16(6):949–959. doi: 10.1007/s00775-011-0795-6. [DOI] [PubMed] [Google Scholar]
- 41.Han Z, et al. Crystal structure of the peroxo-diiron(III) intermediate of deoxyhypusine hydroxylase, an oxygenase involved in hypusination. Structure. 2015;23(5):882–892. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 42.Rui Z, et al. Microbial biosynthesis of medium-chain 1-alkenes by a nonheme iron oxidase. Proc Natl Acad Sci USA. 2014;111(51):18237–18242. doi: 10.1073/pnas.1419701112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald W, et al. Ligand access to the active site in Thermus thermophilus ba(3) and bovine heart aa(3) cytochrome oxidases. Biochemistry. 2013;52(4):640–652. doi: 10.1021/bi301358a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benvenuti M, Mangani S. Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat Protoc. 2007;2(7):1633–1651. doi: 10.1038/nprot.2007.198. [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leslie AGW. The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 47.Evans PR. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 50.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 53.McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]