

Significance
We report a dynamical study of sweet receptor heterodimer, for which there are abundant data for various sweeteners. Because there is no crystal structure of a complete G protein-coupled receptor (GPCR) dimer of class C, we predicted the three-dimensional structure and carried out molecular dynamics with and without the sweetener agonists. We found that binding the sweetener induces major conformational changes during which the transmembrane domain (TMD) transforms from the TMD 56 interface to the TMD 6 interface, which is consistent with biophysical studies on activation of metabotropic glutamate receptor 2 homodimers, also class C. These results provide insights for how GPCRs are activated, which may have a major impact on understanding activation of other GPCR therapeutic targets.
Keywords: GPCR activation, class C GPCR, molecular dynamics, noncaloric sweetener
Abstract
The sweet taste in humans is mediated by the TAS1R2/TAS1R3 G protein-coupled receptor (GPCR), which belongs to the class C family that also includes the metabotropic glutamate and γ-aminobutyric acid receptors. We report here the predicted 3D structure of the full-length TAS1R2/TAS1R3 heterodimer, including the Venus Flytrap Domains (VFDs) [in the closed–open (co) active conformation], the cysteine-rich domains (CRDs), and the transmembrane domains (TMDs) at the TM56/TM56 interface. We observe that binding of agonists to VFD2 of TAS1R2 leads to major conformational changes to form a TM6/TM6 interface between TMDs of TAS1R2 and TAS1R3, which is consistent with the activation process observed biophysically on the metabotropic glutamate receptor 2 homodimer. We find that the initial effect of the agonist is to pull the bottom part of VFD3/TAS1R3 toward the bottom part of VFD2/TAS1R2 by ∼6 Å and that these changes get transmitted from VFD2 of TAS1R2 (where agonists bind) through the VFD3 and the CRD3 to the TMD3 of TAS1R3 (which couples to the G protein). These structural transformations provide a detailed atomistic mechanism for the activation process in GPCR, providing insights and structural details that can now be validated through mutation experiments.
G protein-coupled receptors (GPCRs) play an essential signaling function throughout all eukaryote systems, serving as the basis for detecting light, smell, nociceptive signaling, and taste along with dopamine, serotonin, adrenaline, etc. (1). Generally, binding of a signaling ligand to the exterior of a cell causes a G protein at the intracellular interface to dissociate, which then triggers a sequence of events that respond to the signal. There are now structures for ∼32 human GPCRs, including 4 that have been activated (2); however, a detailed understanding of the activation mechanisms of monomeric GPCRs is still lacking (3). This is most unfortunate because about half the drugs under development involve GPCRs and it is most important to know whether the drug will serve as an agonist to activate the G protein or as an antagonist or inverse agonist.
Particularly interesting here are the class C GPCRs, which in addition to a seven-helix transmembrane domain (TMD) include a large N-terminal segment consisting of a Venus Flytrap Domain (VFD) and a cysteine-rich domain (CRD). Biophysical measurements on the class C glutamate dimer receptor 2 (mGluR2) have shown that the inactive or resting state (R) dimer interface involves contacts between TMs 4 and 5 of each TMD, whereas formation of the fully active state is associated with motions in which the extracellular (EC) projections of the two TM6s move together to form a TM6–TM6 interface (4).
In addition, biophysical experiments on the class C sweet receptor, consisting of the TAS1R2/TAS1R3 heterodimer, indicate that sucrose and glucose bind to the VFD of both the TAS1R2 and TAS1R3 subunits (5, 6), whereas other sweeteners, such as aspartame and stevioside (Stev), interact only with the VFD2 (VFD of TAS1R2) subunit (7–9). Moreover, the allosteric binding site at the TMD3 (TMD of TAS1R3) interacts with modulators of the receptor function, such as lactisole (9, 10). Particularly interesting is that for most sweeteners binding to the VFD2 causes activation of the G protein at the intracellular region of the TMD3.
We report here dynamical studies on binding of agonists to the class C GPCR TAS1R2/TAS1R3 heterodimer (11), in which we observe that binding of an agonist to VFD2 leads to the motions of the EC projections of the two TM6s together, just as observed experimentally for mGluR2, and we observed that the dynamics involves a coupling through VFT3 through CRD3 to TMD3, which is consistent with experiments on the sweet receptor. This is an atomistic dynamical study connecting the binding of agonist to the EC surface with the changes in the TMD of GPCR.
Because there are no full heterodimer models available from either experiment or theory to explain how binding of sweetener to VFD2 elicits activation by the intracellular region for TMD3, we constructed a detailed atomistic structural model of the full sweet taste receptor heterodimer (a total of 1,598 residues) comprising all three domains: VFDs, CRDs, and TMDs. This involves three steps (see Materials and Methods and SI Appendix for details).
A: Prediction of the TMD Dimer for TAS1R2 and TAS1R3 and the Allosteric Modulator Binding Sites.
-
A1)
We predicted the ensemble of 25 stable structures for the TMD of all TAS1R1s, -2s, and -3s. Because there is no crystal structure for the sweet receptor, we used our GEnSeMBLE Complete Sampling Hierarchical Scoring (CoS-HS) technique (12). This has been successful at predicting the 3D structures of other GPCRs [such as chemokine CCR5 (13) and cannabinoid CB1 (14)].
-
A2)
We provided some validation of these TMD structures by predicting the binding pose of allosteric modulators to TMD2 and also TMD1.
-
A3)
We next constructed the TMD heterodimer for TM45/TM45 and TM56/TM56 interfaces based on GPCR dimers from recent crystal structures of class A mu opioid receptor [OPRM; Protein Data Bank (PDB) ID code 4DKL] (15) and β1 adrenergic receptor (β1AR) (16).
B: Prediction of the VFD Dimer for TAS1R2 and TAS1R3 and the Agonist Binding Sites.
-
B1)
We first constructed structures for the VFD/CRD based on homology to the X-ray structures for the VFD dimer of the metabotropic glutamate receptor (mGluR) 1, 3, and 5 using the closed–open (co) structure for the VFD dimer appropriate for the active (A) form (17). We then annealed this full VFD dimer in water with appropriate ions.
-
B2)
We provided some validation of these structures by predicting the binding pose of agonists (sucrose and Stev) bound to the VFD2. To examine the activation by other agonists, we matched nine with known sweeteners to the same binding site and annealed their structures.
C: Coupling VFD/CRD Dimer with TMD Dimer to Obtain Full TAS1R2 and TAS1R3 Heterodimer.
-
C1)
We positioned the VFDs/CRDs on top of the TMD heterodimer and coupled the bonds to construct the full-length heterodimer receptor. Then, we immersed the full heterodimer into a periodic POPC membrane containing a water box with appropriate ions (total of 192,000 atoms per periodic box).
-
C2)
Next we carried out 20 ns of molecular dynamics (MD) at 310 K for the 11 cases—10 agonists and the apo-protein—to examine the processes related to activation.
-
C3)
To probe the details of how information on binding of agonist to VFD2 gets transmitted to TMD3 and then activation, we carried out a series of constrained MD.
In step C2, we found that the agonist and the allosteric modulator-bound complexes lead to major conformational changes in the VFD3, CRD3, and TMD3 that evolve the TMD2/TMD3 interface from TM56/TM56 coupling of the co active (coA) form to a structure in which the upper parts of the TM6 helices form a TM6/TM6 interface. This result is consistent with biophysical measurements on the class C mGluR 2, where the TMD dimer interacting at the TM45/TM45 interface is associated with the resting or relaxed (R) state, whereas the TMD dimer interacting at the TM56/TM56 interface is associated with the A state, which upon activation by agonist leads to the TMD dimer migrating to a configuration in which the TMD interacts at the TM6/TM6 interface (4). Our MD simulations of the agonist-induced transformation of the TAS1R2/TAS1R3 sweet taste heterodimer lead to exactly the same transformations observed experimentally for the mGluR2 homodimer, representing an atomistic simulation that follows the GPCR dimer activation process. In contrast, the apo-dimer does not undergo this transformation.
Results
Step A: GEnSeMBLE Predictions of the Structures for the Seven-Helix TMD Bundles for All Three TASRs of Family 1.
-
Step A1)
SI Appendix, Fig. S1 shows the TMD regions and multiple sequence alignments with their homology templates of human mGluR1 and mGluR5 (SI Appendix, Fig. S1). The conserved residues in each TMD (×.50) in class C GPCRs match with the same position (N1.50, D2.50, R3.50 in D/ERY; W4.50, P5.50, P6.50 in FxxWxY; and P7.50 in NPxxY motif) as class A GPCRs:
1.50: L581 at TAS1R1, T580 at TAS1R2, and V583 at TAS1R3;
2.50: all L at TAS1Rs (L612 at TAS1R1, L611 at TAS1R2, and L614 at TAS1R3);
3.50: L656 at TAS1R1, I655 at TAS1R2, and I658 at TAS1R3;
4.50: S687 at TAS1R1, T686 at TAS1R2, and M688 at TAS1R3;
5.50: all Ls at TAS1Rs (L738 at TAS1R1, L738 at TAS1R2, and L740 at TAS1R3);
6.50: A775 at TAS1R1, S775 at TAS1R2, and S777 at TAS1R3; and
7.50: F804 at TAS1R1, S804 at TAS1R2, and L806 at TAS1R3.
The templates for the initial positions of the seven helices and the shapes of the helices were based on the X-ray structures of human mGluR1 (PDB ID code 4OR2) (18) and mGluR5 (PDB ID code 4OO9) (19), which are class C GPCRs with sequence identities of 17–25% to our two targets. As discussed in SI Appendix, we selected the most stable packing for the TMD of TAS1R1, TAS1R2, and TAS1R3, denoted as TMD1, TMD2, and TMD3.
We found a very stable salt bridge between D735 (5.47) and K689 (4.53) in the EC binding site for the allosteric agonist in TAS1R2. Other stable salt bridges in the cytoplasmic end involve (a) R651 (3.46), E758 (6.33), and K811 (7.57) among TM3, -6, and -7 in TAS1R2; (b) E751 (5.63) and K660 (3.55) between TM3–5 in TAS1R2; and (c) E657 (3.49) and R677 (4.40) between TM3 and -4 in TAS1R3.
For all TAS1Rs we find:
-
•
the conserved disulfide bridge between C3.25 in TM3 and EC2 that is observed in most class A GPCRs, and
-
•
the conserved ionic lock between R3.50 and E6.30 in the D/ERY motif of class A GPCRs is replaced with the alternative salt-bridge interaction between conserved R/K3.46 and E6.33 at the cytoplasmic end in class C GPCRs. However, for TAS1R3, the corresponding residues are Q3.46 and R6.33, leading to an H bonding, not a salt bridge.
These observations show that our structures are consistent with previously known signatures of the broader GPCR superfamily as well as the class C subfamily.
-
Step A2)
As the first validation of the predicted structures for the seven-helix TMD (SI Appendix, Fig. S2), we used DarwinDock (13) to predict the binding site for the allosteric ligands to each TAS1R TMD, with the results presented in SI Appendix, Table S10. See SI Appendix for details. Here, S819 [1-((1H-pyrrol-2-yl)methyl)-3-(4-isopropoxyphenyl)thiourea] is a sweet compound that interacts with TMD2 (20), and Lactisole is a competitive inhibitor of the sweet taste receptor that binds to TMD3 (TMD of TAS1R3) (9, 21).
-
Step A3)
To generate a starting structure for the TMD heterodimer between TAS1R2 and TAS1R3, we used structural insights about GPCR dimerization from recent crystal structures of class A OPRM (PDB ID code 4DKL) (15), kappa opioid receptor (OPRK) (22), and β1AR (16). These crystal structures consist of a parallel, symmetric dimer with symmetric dimeric interfaces (see more details in SI Appendix).
We conclude that only the TM45/TM45 or TM56/TM56 models are realistic. This is supported by the observation that TM5 and TM6 are involved in the ligand binding pocket, which might facilitate coupling of the TM5 and TM6 in one monomer through the TM56/TM56 contacts to the other monomer to affect ligand binding or signaling properties.
Step B: Predicted Structure for the VFD Dimer of TAS1R2/ TAS1R3.
-
Step B1)
X-ray structures of the VFD without the TMD have been obtained for the mGluR class C GPCRs in several conformations with and without bound agonists and antagonists (23). These results have been interpreted in terms of two configurations for each VFD: the o state and c state.
For the VFD heterodimer, the “oo” state of the VFD dimer is considered to be inactive or resting (ooR), and the “co” and “cc” states are considered to be active (coA or ccA). We used the mGluR X-ray data to generate models of the oo and co conformations for the VFD dimer of TAS1R2 and TAS1R3 heterodimer.
Using the rat mGluR1 coA structure (PDB ID code 1ewk) (17), we generated a homology model of the co56 VFD heterodimer of TAS1R2/1R3 heterodimer (SI Appendix, Fig. S3, Center). We consider this to provide a realistic dimer packing for the co VFD.
-
Step B2)
We first used DarwinDock (13) to predict the binding sites for sucrose, Stev, and MogV sweeteners binding to VFD2 (SI Appendix, Fig. S4) and equilibrated these structure with MD in explicit solvent. As described in SI Appendix, the results for sucrose and Stev are in agreement with available mutation data.
We matched in eight additional stevia or nonstevia glycoside agonists (Table 1 and SI Appendix, Table S13) into the predicted binding site at VFD2 and annealed them through 10 quench anneal cycles.
Table 1.
Characteristics of activation
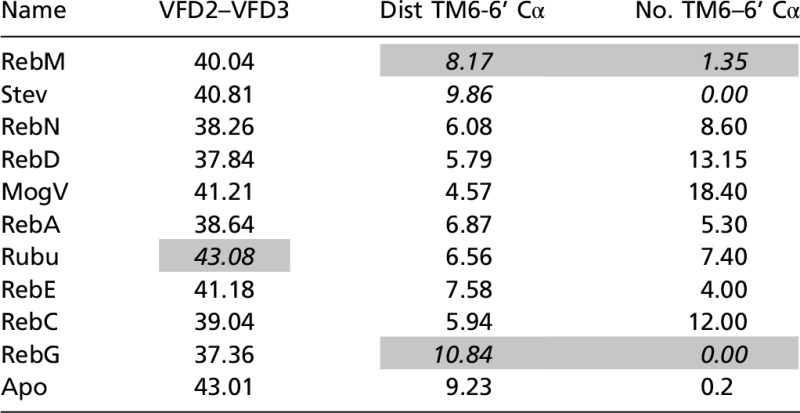 |
VFD2–VFD3 is the distance between the center of the lower VFD2 and the center of VFD3. Dist TM6-6’ is the shortest Cα distance between TM6 of TMD2 and TM6’ of TMD3. No. TM6–6 Cα is the number of Cα partners of TM6–TM6 with distances shorter than 9 Å. These quantities were averaged over the period from 8 to 10 ns MD. The cases we consider outliers in our interpretation of the binding mechanism are shaded. The outliers are displayed in italics. Mog, Mogroside; Reb, Rebaudioside; Rubu, Rubusoside; Stev, Stevioside.
We considered four Stev analogs for which experimental data (24) are available:
-
•
most sweet: Stev and Rebaudioside A (RebA);
-
•
intermediate sweetness: Steviolbioside (Stbs) and Rubusoside (Rubu); and
-
•
least sweet: 2H-Stev.
We matched each of these with the Stev binding poses at each time step and calculated the binding energy (SI Appendix, Table S12). For all six cases, the binding energy leads to similar correlations that distinguish two categories: more sweet versus less sweet (2H-Stev). The pharmacophore analysis shows that the snapshot at 8.7 ns provides the best correlation with the currently known experimental SAR data. Thus, we selected the model at 8.7 ns for further MD study, including the full heterodimer, to extract an understanding of the activation mechanism.
Based on our predicted best binding site for Stev, we also predicted the binding site of the Mogroside V (MogV) nonstevia glycoside that has an additional sugar linkage (1→6) at the R2 side of the VFD of the TAS1R2/1R3 heterodimers. MogV is found in certain plants, such as the fruit of the gourd vine luo han guo (Siraitia grosvenorii). Mogrosides are used in various natural sweetener products such as Norbu sweetener and have been investigated as possible anticancer agents (25).
We found a common binding site between MogV and Stev. The sugar of R1 (R-COO) in Stev roughly overlaps the sugar of R1’ (R-O) in MogV, and the sugar of R2 (R-O) in Stev and R2’ (R-OH) in MogV overlapped each other, as shown in Fig. 1 and SI Appendix, Fig. S4.
Fig. 1.

Predicted best binding modes for Stev and MogV bound to the VFD (VFD2) of the human sweet taste receptor (TAS1R2/1R3). The predicted pharmacophore is at the bottom. The unified cavity binding components are in SI Appendix, Table S11.
The hydroxyl groups of MogV make strong HBs with adjacent hydrophilic residues. The hydrophilic residues are D142, D218, and E302 at theR1/R’ side and K65, D278, D307, and R383 at the R2/ R2’ side. The participation of these polar residues agrees with experiments finding that D142A and E302A abolished the activities of Stev (26). Moreover, Ala mutations of residues S40, K65, Y103, D142, D278, E302, P277, and R383 all diminished the response to Stev, which is consistent with our predicted binding site (26). MogV has an extra hydrophilic interaction at the OH group with N44.
Step C: The Full-Length TAS1R2/TAS1R3 Heterodimer Sweet Taste Receptor and the Atomistic Mechanism of Activation.
-
Step C1)
We combined the equilibrated VFD dimer in the co configuration including bound agonist with the TMD dimer in the 5/6 or A configuration including bound S819 allosteric agonist at TMD2 to obtain a model for the full TAS1R2/1R3 heterodimer complex bound with or without agonists at the co VFD of TAS1R2 and with TAS1R2.
-
Step C2)
We inserted the predicted complexes from step C1 into a periodically infinite lipid POPC bilayer, solvated with 40,030 water molecules, and carried out 20 ns of MD at physiological salt concentration at 310 K (a total of 198,000 atoms) using the NAMD 2.9 (NAnoscale Molecular Dynamics) program (27). The MD results for the MogV–TAS1R2/1R3 heterodimer complex (Fig. 2) show that the binding site of MogV remains stable through 20 ns with some minor changes in HBs at the binding site of all flexible hydroxyl groups of the glucose ring. Thus, R383 and K65 of the upper lobe and D278 and D307 of the lower lobe serve as the electrostatic pincer residues to bind the agonist. These results are consistent with the loss in activity for D278K/K65D double mutations of the sweet taste receptors (26). Thus, the electrostatic pincer residues of the VFD dimer include R383 and K65 of the upper lobe and D278 and D307 of the lower lobe.
Fig. 2.

Side views of the 3D structure of the (A) apo- and (B) MogV-bound TAS1R2 (red)/1R3 (blue) heterodimer. The MogV agonist is shown in VFD2 as a yellow space-filling model, whereas the S819 agonist modulator is the yellow structure at the EC part of TMD2. SI Appendix, Fig. S2 shows a more detailed binding site for the S819 allosteric agonist. The yellow arrows between VFD2 and VFD3 show the separation (Å) between the geometric center of lower VFD2 and lower VFD3 (VFD2–VFD3 in Table 1), whereas yellow arrows between TMD2 and TMD3 show the distance (Å) between the closest Cα of TM6/TMD2 with a Cα of TM6’/TMD3 (Dist TM6-6' Cα in Table 1). These numbers are in SI Appendix, Table S13 for all 11 cases.
We found stable inter- or intra-salt bridges in the TAS1R2/1R3 heterodimer complex (SI Appendix, Fig. S9). In the co upper VFD, we observed a stable salt bridge between E145 in TAS1R2 and K155 in TAS1R3, plus a slightly stable interaction between R176 in TAS1R2 and E148 in TAS1R3 that exhibits in most MD. In the VFD of TAS1R2, we find intradomain salt bridges among K65, D278, and D307 and between D307 and R383 that stabilize the c conformation between the upper and lower ligand binding domain of VFD2. In the CRD, we found stable inter-salt bridges between D519 in TAS1R2 and R530 in TAS1R3 and between E751 in TAS1R2 and R762 in TAS1R3. In the TMD, our MD simulations identified important salt bridges between E751 (5.63) in TAS1R2 (D5.63 in TAS1R1) and R762 (6.35) in TAS1R3 that stabilize the TM5/6 interface of heterodimer complex bound with strong sweeteners but not with weak sweeteners such as RebC, RebG, or apo.
Our MD simulations with the co56 heterodimer model find that agonists bound to VFD2 lead to major conformational changes in which the TMD interaction across the heterodimer changes gradually from having a TM56/TM56 interface at the beginning to having close interactions between TM6/TM6 as shown in Fig. 3 and SI Appendix, Fig. S10. These MD results suggest that the combination of agonist and allosteric agonist facilitates the conformational rearrangement in which the TM56/TM56 co interface evolves to move the TM6 helices to much shorter distances, especially between the upper parts of the two TM6/TM6 helices. We also observed that for the TAS1R2/1R3 heterodimer with agonist and allosteric agonist, the salt bridges across the respective VFDs and TMDs are stabilized compared with MD of the apo heterodimer. We find that the TMD of TAS1R3 is highly flexible because it lacks the ionic constraints of the TAS1R2 TMD, which has many interhelical salt bridges involving TM3, -6, and -7; TM5 and -6; and TM4 and -5 as shown in SI Appendix, Fig. S9.
Fig. 3.

The conformational rearrangement at the TMD interface of heterodimers of sweet taste receptor TAS1R2 (red)/TAS1R3 (blue) from the start (Left) to the end (Right) after 20 ns of MD. Viewed from the EC view (Top) and the cytoplasmic view (Bottom). The original TM56/TM56 interface changes by moving the upper TM6 helices closer, leading to a TM6/TM6 interface mainly at the EC side. This motion is observed experimentally for the mGlu2 homodimer. The EC views for all 11 cases are shown in SI Appendix, Fig. S15.
For class C mGluR2, biophysical measurements established that the dimer interface involving TM45/TM45 is associated with the inactive or resting state (R), whereas formation of the TM6/TM6 interface is associated with the fully A state (4). This suggests an intermediate state of the receptor that may exhibit a TM56/TM56 interface. Because this interface has been observed in crystal structures of other GPCRs, we used them to build this interface to facilitate activation. The MD with the inactive oo45 heterodimer showed a very stable structure (SI Appendix, Fig. S11). The conformational rearrangement we observed in the co56 model MD, in which the TM56/TM56 TMD interface transforms to the TM6/TM6 interface, is consistent with biophysical studies on the mGluR2 homodimer (a class C GPCR), which find these changes to be associated with activation (4).
Our MD simulations show major conformational changes in the lower part of VFD2 and VFD3 (Table 1 and SI Appendix, Table S13). Here we define a geometric center for each part (SI Appendix, Fig. S12) and measure the distance across the VFD2 and VFD3. We find that 9 out of 10 agonists lead quickly to coupling of the bottom parts of VFD dimer to stabilize at 37–41 Å compared with 43 Å for the apo protein. The exception is Rubu. These distances can be compared with the crystal structures lacking the TMDs (64.7 Å for ooR, 68.1 Å for ccR, 41.3 Å for ooA, and 38.7 Å for coA), where this distance in the R (resting) structure is 25 Å larger than for the A structure (SI Appendix, Fig. S13).
We also observed that 8 out of 10 agonists show rapid rearrangement of the TMD interface from TM56/TM56 to TM6/TM6.
Our MD simulations also show major conformational changes at TM6 in TMD3 that bring it closer to the upper parts of the TM6 helix from TMD2 (SI Appendix, Fig. S14). At this point in the MD, the conformational changes in TM6 involve only the upper part, because there is a strong inter-salt bridge constraint between E751 (5.63) in TAS1R2 and R762 (6.35) in TAS1R3 that stabilizes the bottom part of the TM56/TM56 interface.
P6.53 is conserved in mGluRs and in TAS1R3 but not in TAS1R1 and TAS1R2, where residue 6.53 is Thr. Thus, the high flexibility of upper TM6 of TAS1R3 is likely due to both the weak HB network in TMD3 and the presence of Pro in the upper TM6 of TMD3.
The conformational rearrangement of TM56/TM56 to TM6/TM6 observed in the MD is consistent with the experiments involving cysteine cross-linking at the mGluR2 TMD dimer interface. Disulfide cross-linking was observed only when one amino acid in TM4 (L698C), TM5 (Y734C), or TM6 (V782C) was substituted for cysteine (4). The corresponding amino acids at 6.57 are A782 in TAS1R2 and N784 in TAS1R3. Our MD leads to an intermolecular distance of 10.5 Å between the two residues at 6.57, which we consider sufficient to allow a disulfide bridge to form.
Summarizing, the changes in the agonist-bound TAS1R2/TAS1R3 heterodimer observed in the 10 ns of MD are mostly (8 out of 10 cases) consistent with the changes associated with activation in the mGluR class C dimers.
Structural and biophysical studies of mGluR class C GPCRs reported that a major rearrangement in the TMD dimer interface from TM45/TM45 to TM6/TM6 (time constant, ∼35 ms) is essential for receptor activation by agonists, revealing a key step in mGluR activation. This change is followed by activation of a single TMD within the dimer, in which the bottom part of TM3 and TM6 of TAS1R3 separate so that there is room for G protein to insert into the membrane (time constant, ∼50 ms).
Our simulations do not include the G protein, so we cannot yet comment whether TMD3–6 separation will happen in our model.
-
Step C3)
To learn how information is transmitted from agonist binding at VFD2 to activation of the G protein at TMD3, we carried out MD simulations in which particular subdomains were fixed in the MD. We carried out 10 ns MD at 310K for the MogV-bound heterodimer (our most active agonist) with one of the domains (CRD2, TMD2, VFD3, CRD3, or TMD3) fixed. We consider the measures of activation to be (a) the shortest Cα distance between the TM6 helices of TMD2 and TMD3 and (b) total number Cα between the two TM6s within 9 Å. As shown in Fig. 4, we observed that:
•fixing VFD3 or CRD3 completely blocked activation,
•fixing CRD2 had almost has no effect, and
•fixing TMD2 or TMD3 has intermediate effects on activation, which is consistent with the higher flexibility and the major change of TAS1R3 TMD at the heterodimer interface upon activation.
Fig. 4.

Trajectory analysis of the shortest Cα distance (Å) between TM6 of TAS1R2 and TAS1R3 TMD after 10 ns of MD. We see that the fix CRD2 case is the same as no fix, indicating no effect on activation, whereas the fix-VFD3 and fix-CRD3 cases show no activation (similar to apo protein) The fix-TMD2 and fix-TMD3 cases are intermediate. The total number of Cα between the two TM6s within 9 Å is shown in SI Appendix, Fig. S16.
Based on these constrained MD results, we hypothesize that the agonist signal is transferred from VFD2 through VFD3 to CRD3 and finally to TMD3 to initiate activation. Thus, we conclude that agonist-induced coupling of the lower VFD is required for activation, followed by communication of this signal through the VFD3 and then CRD3 to impact the top part of TMD3, particularly TM6.
Our results are consistent with mutational studies of human/mouse chimeras of TAS1R3 paired with human TAS1R2, which found that CRD3 plays a critical role in sensory mechanism of the sweet receptor. Thus, the A537P mutation of TAS1R3 stopped all responses to all sweeteners, indicating that the CRD3 must couple ligand binding in VFD2 to receptor output in the TMD3 by modulating the strength of coupling (3).
Discussion
Our predictions for the heterodimer of the TAS1R2 and TAS1R3 led to several observations:
-
i)
The binding sites of sucrose and Stev have strong HBs to nearby hydrophilic residues D142 and E302, which is consistent with mutation studies. We find much stronger binding for Stev than for sucrose, perhaps explaining why Stev is 210–300 times sweeter than sucrose (24).
-
ii)
The allosteric ligands bind to the TMD of TAS1Rs (S807 at TAS1R1, S819 at TAS1R2, lactisole at TAS1R3), with the interactions remaining stable during the MD.
-
iii)
Nine out of 10 agonists lead quickly to coupling the bottom parts of VFD dimer to distances of 37–41 Å (the exception is Rubu), whereas for apo protein these bottom parts stay 43 Å apart. This has been observed experimentally for model mGluR VFD dimers not containing the TMD. It is possible that this process converts the inactive form R to the active form A.
-
iv)
Starting with the TM5/6 interface for the TAS1R2/TAS1R3 heterodimer, we find (for 8 out of 10 cases) major conformational changes in which the TM5/6 interface between the TMD dimers evolves into closer interactions between the upper parts of the TM6 helices (the two exceptions are RebG and Stev). This result agrees well with the biophysical measurements in the class C mGluR2 where the TM4/5 interface is known to be associated with the inactive state whereas the TM6 interface is associated with the fully A state.
-
v)
We find that fixing the atoms of either VFD3 or CRD3 prevents this activation, whereas fixing CRD2 has no effect. This indicates that the movement of the bottom half of the VFD causes changes in the CRD3 that communicates through CRD3 with the top part of TMD3 to induce the initial steps in activation.
Because these structures do not include the G protein that must be coupled at the intracellular interface to the EC motions of the TM6 helices, we cannot yet discuss how these motions affect the G protein activation. We hope to include such couplings in future studies. To enable others to investigate such interactions, we include 3D coordinates of the state we consider to be activated.
Our predicted 3D structures for the TAS1R2/TAS1R3 sweet taste receptor exhibit binding and activation profiles consistent with biophysical experiments on other class C mGluR2 dimers. After acceptance of our manuscript for publication, a paper appeared (28) that developed a homology model of the sweet taste receptor based on structures of other class C GPCRs. They compared the key residues in ligand binding and activation with ∼600 single-point site-directed mutations. These mutations are also in good agreement with our structure.
Most promising is that the changes known experimentally to be associated with activation are captured in our short MD studies. This is the first time that a class C GPCR dimer activation has been modeled with MD. In addition, these studies capture the structural changes related to the agonist binding at the EC VFD2 of the TAS1R2 subunit and how those changes propagate allosterically to the TMD3 of TAS1R3 subunit, which putatively couples to the G protein. We expect that these studies will be useful to gain additional understanding of the activation mechanism in both the TAS1R family and other class C GPCR dimers, including the mGluR and γ-aminobutyric acid (GABA) receptors. These structures and models provide the basis for experimental studies aimed at probing the important molecular interactions controlling activation.
Materials and Methods
We used our GEnSeMBLE CoS-HS technique (12) to generate the TMD of all TAS1Rs.
We used DarwinDock (13) to predict the binding site for the allosteric ligands to each TAS1R TMD as well as the sweeteners to each TAS1R2 VFD.
We used the NAMD 2.9 (NAnoscale Molecular Dynamics) program (27) to rub MD for the TAS1R2/1R3 heterodimer complex bound with various sweeteners.
See SI Appendix for method details.
Supplementary Material
Acknowledgments
Funding for this project was provided by a grant from Cargill Global Food Research to Caltech.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1700001114/-/DCSupplemental.
References
- 1.Julius D, Nathans J. Signaling by sensory receptors. Cold Spring Harb Perspect Biol. 2012;4(1):a005991. doi: 10.1101/cshperspect.a005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costanzi S. 2015 GPCR Structures Solved Through X-Ray Crystallography. Available at www.costanziresearch.com/p/table.html.
- 3.Cvicek V, Goddard WA, 3rd, Abrol R. Structure-based sequence alignment of the transmembrane domains of all human GPCRs: Phylogenetic, structural and functional implications. PLOS Comput Biol. 2016;12(3):e1004805. doi: 10.1371/journal.pcbi.1004805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xue L, et al. Major ligand-induced rearrangement of the heptahelical domain interface in a GPCR dimer. Nat Chem Biol. 2015;11(2):134–140. doi: 10.1038/nchembio.1711. [DOI] [PubMed] [Google Scholar]
- 5.Maîtrepierre E, Sigoillot M, Le Pessot L, Briand L. Recombinant expression, in vitro refolding, and biophysical characterization of the N-terminal domain of T1R3 taste receptor. Protein Expr Purif. 2012;83(1):75–83. doi: 10.1016/j.pep.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Nie Y, Vigues S, Hobbs JR, Conn GL, Munger SD. Distinct contributions of T1R2 and T1R3 taste receptor subunits to the detection of sweet stimuli. Curr Biol. 2005;15(21):1948–1952. doi: 10.1016/j.cub.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 7.Liu B, et al. Molecular mechanism of species-dependent sweet taste toward artificial sweeteners. J Neurosci. 2011;31(30):11070–11076. doi: 10.1523/JNEUROSCI.0791-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masuda K, et al. Characterization of the modes of binding between human sweet taste receptor and low-molecular-weight sweet compounds. PLoS One. 2012;7(4):e35380. doi: 10.1371/journal.pone.0035380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, et al. Different functional roles of T1R subunits in the heteromeric taste receptors. Proc Natl Acad Sci USA. 2004;101(39):14258–14263. doi: 10.1073/pnas.0404384101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winnig M, Bufe B, Meyerhof W. Valine 738 and lysine 735 in the fifth transmembrane domain of rTas1r3 mediate insensitivity towards lactisole of the rat sweet taste receptor. BMC Neurosci. 2005;6:22. doi: 10.1186/1471-2202-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohta M, et al. Characterization of novel steviol glycosides from leaves of stevia rebaudiana morita. J Appl Glycosci. 2010;57:199–209. [Google Scholar]
- 12.Abrol R, Bray JK, Goddard WA., 3rd Bihelix: Towards de novo structure prediction of an ensemble of G-protein coupled receptor conformations. Proteins. 2012;80(2):505–518. doi: 10.1002/prot.23216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abrol R, et al. Ligand- and mutation-induced conformational selection in the CCR5 chemokine G protein-coupled receptor. Proc Natl Acad Sci USA. 2014;111(36):13040–13045. doi: 10.1073/pnas.1413216111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott CE, Abrol R, Ahn KH, Kendall DA, Goddard WA., 3rd Molecular basis for dramatic changes in cannabinoid CB1 G protein-coupled receptor activation upon single and double point mutations. Protein Sci. 2013;22(1):101–113. doi: 10.1002/pro.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manglik A, et al. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485(7398):321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Chen S, Zhang JJ, Huang XY. Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat Struct Mol Biol. 2013;20(4):419–425. doi: 10.1038/nsmb.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunishima N, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407(6807):971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 18.Wu H, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344(6179):58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doré AS, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511(7511):557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F, et al. Molecular mechanism for the umami taste synergism. Proc Natl Acad Sci USA. 2008;105(52):20930–20934. doi: 10.1073/pnas.0810174106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang P, et al. Lactisole interacts with the transmembrane domains of human T1R3 to inhibit sweet taste. J Biol Chem. 2005;280(15):15238–15246. doi: 10.1074/jbc.M414287200. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature. 2012;485(7398):327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doumazane E, et al. Illuminating the activation mechanisms and allosteric properties of metabotropic glutamate receptors. Proc Natl Acad Sci USA. 2013;110(15):E1416–E1425. doi: 10.1073/pnas.1215615110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellfritsch C, Brockhoff A, Stähler F, Meyerhof W, Hofmann T. Human psychometric and taste receptor responses to steviol glycosides. J Agric Food Chem. 2012;60(27):6782–6793. doi: 10.1021/jf301297n. [DOI] [PubMed] [Google Scholar]
- 25.Itkin M, et al. The biosynthetic pathway of the nonsugar, high-intensity sweetener mogroside V from Siraitia grosvenorii. Proc Natl Acad Sci USA. 2016;113(47):E7619–E7628. doi: 10.1073/pnas.1604828113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang F, et al. Molecular mechanism of the sweet taste enhancers. Proc Natl Acad Sci USA. 2010;107(10):4752–4757. doi: 10.1073/pnas.0911660107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chéron J-B, Golebiowski J, Antonczak S, Fiorucci S. The anatomy of mammalian sweet taste receptors. Proteins. 2017;85(2):332–341. doi: 10.1002/prot.25228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.