

Significance
The remarkable power of enzymes as catalysts is derived from the precise spatial positioning of amino acids as a result of a polypeptide folding into its native, active fold. Protein engineers have a wide arsenal of tools available to alter the properties of enzymes but, until recently, have been limited to replacement of amino acids with one of the other naturally occurring proteogenic amino acids. Here we describe a protein engineering approach to introduce a noncanonical amino acid that results in altered substrate specificity of an aldolase to produce a novel activity that cannot be achieved by simple substitution with any of the canonical amino acids.
Keywords: protein engineering, aldolases, chemical modification
Abstract
Natural enzymes are constructed from the 20 proteogenic amino acids, which may then require posttranslational modification or the recruitment of coenzymes or metal ions to achieve catalytic function. Here, we demonstrate that expansion of the alphabet of amino acids can also enable the properties of enzymes to be extended. A chemical mutagenesis strategy allowed a wide range of noncanonical amino acids to be systematically incorporated throughout an active site to alter enzymic substrate specificity. Specifically, 13 different noncanonical side chains were incorporated at 12 different positions within the active site of N-acetylneuraminic acid lyase (NAL), and the resulting chemically modified enzymes were screened for activity with a range of aldehyde substrates. A modified enzyme containing a 2,3-dihydroxypropyl cysteine at position 190 was identified that had significantly increased activity for the aldol reaction of erythrose with pyruvate compared with the wild-type enzyme. Kinetic investigation of a saturation library of the canonical amino acids at the same position showed that this increased activity was not achievable with any of the 20 proteogenic amino acids. Structural and modeling studies revealed that the unique shape and functionality of the noncanonical side chain enabled the active site to be remodeled to enable more efficient stabilization of the transition state of the reaction. The ability to exploit an expanded amino acid alphabet can thus heighten the ambitions of protein engineers wishing to develop enzymes with new catalytic properties.
Enzymes are phenomenally powerful catalysts that increase reaction rates by up to 1018-fold (1, 2), and a new era of enzyme applications has been opened by the advancement of protein engineering and directed evolution to provide new, or improved, enzymes for industrial biocatalysis. Enzymes are attractive catalysts because they are highly selective, carrying out regio-, chemo-, and stereoselective reactions that are challenging for conventional chemistry. Moreover, enzymes are efficient catalysts, function under mild conditions with relatively nontoxic reagents, and enable the production of relatively pure products, minimizing waste generation. In recent years, there has been much success in engineering enzymes for desired reactions (3–5) using methods such as rational protein engineering (6–8), directed evolution (9–11), and, most recently, computational enzyme design (12–15).
Enzymes found in Nature achieve catalysis using active sites generally composed of only 20 canonical amino acids, which are encoded at the genetic level, plus the rarer selenocysteine and 1-pyrrolysine. However, many enzymes also rely on one or more of 27 small organic cofactor molecules and/or 13 metal ions for their function (16). In addition, in some cases, Nature has exploited noncanonical amino acids (Ncas) in catalysis to extend its catalytic repertoire: for example, the quinones TPQ, LTQ, TTQ, and CTQ, respectively, in amine oxidase, lysyl oxidase, methylamine dehydrogenase, and quinohemoprotein amine dehydrogenase (17); a pyruvoyl group in some histidine, arginine, aspartate, and S-adenosylmethionine decarboxylases (18); formyl glycine residues in type I sulfatases (19); and 4-methylideneimidazole-5-one (MIO) in ammonia lyases and 2,3-aminomutases (20). These Ncas are vital for catalysis and arise through posttranslational modifications of the polypeptide chain (21, 22), allowing access to chemistries not otherwise provided by the 20 proteogenic amino acids.
Technologies for the protein engineer to incorporate Ncas into proteins at specifically chosen sites, either by genetic means (23–25) or by chemical modification (26, 27), have recently been developed. These approaches are powerful because, unlike traditional protein engineering with the 20 canonical amino acids, protein engineering with Ncas has almost unlimited novel side-chain structures and chemistries from which to choose. This ability has been exploited to increase the stability of leucine-zipper peptides by replacing leucines with trifluoroleucine (Tfl) while maintaining the DNA-binding functions (28). Moreover, by screening random libraries with a sense codon global substitution method to replace Leu with Tfl, a mutant Tfl-containing enzyme with enhanced thermal stability (29) and a mutant Tfl-containing GFP with increased fluorescence intensity (30) were obtained. Incorporation of Ncas has also been used to generate a novel metalloenzyme by the in vivo incorporation of metal-binding Ncas into an active site (31). Furthermore, the efficiency of phosphotriesterase with the substrate paraoxon was improved by replacement of Tyr309 with the Nca l-(7-hydroxycoumarin-4-yl) ethylglycine (32), generating a 10-fold improvement of an already highly efficient enzyme.
A particular challenge is the engineering of new enzymes for carbon−carbon bond formation, for example, aldolases (33), because these can increase molecular complexity and control the formation of new stereocenters. We have taken the opportunity to incorporate Ncas into an aldolase to search for new activities that are dependent on the distinctive properties of the introduced Nca. The genetic incorporation of Ncas encodes the Nca as an amber STOP codon, which is read by an orthogonal tRNA/amino acyl tRNA synthetase (tRNAS) pair to introduce directly the Nca in vivo (25). However, this method becomes extremely laborious if the effect of many different Ncas is to be investigated, because new orthogonal tRNA/tRNAS pairs must be evolved for each Nca. We have instead used chemical mutagenesis to incorporate a diverse range of Ncas throughout the active site of an aldolase, before screening these variants for activity with new substrates. We demonstrate that using this approach, we can alter the substrate specificity of the enzyme and achieve levels of activity toward a new substrate that are otherwise unattainable by mutagenesis to any of the canonical amino acids.
Results
Production of Modified Enzymes.
N-acetylneuraminic acid lyase (NAL) catalyses the aldol reaction of N-acetyl-d-mannosamine and pyruvate to form N-acetylneuraminic acid (Fig. 1A). The substrate specificity and stereochemistry of this enzyme is highly malleable through protein engineering and directed evolution (34, 35), and we have shown that an enzyme bearing the Nca γ-thialysine (2-aminoethyl cysteine) in place of the catalytic Lys-165 retains activity (36). Here we have explored the effect of introducing Ncas throughout the active site on the reaction catalyzed.
Fig. 1.

Substrates and products of wild-type and modified NAL. (A) The wild-type enzyme catalyzes the aldol reaction of ManNAc with pyruvate to form Neu5Ac. (B) The aldol reaction of erythrose and pyruvate to form (4R, -5S, -6R) or (4S, -5S, -6R) DHA catalyzed by the modified S. aureus NAL bearing the noncanonical amino acid dihydroxypropyl cysteine at position 190.
Twelve residues in the active site of Staphylococcus aureus NAL were selected as positions for Nca incorporation (Fig. 2). The chemical modification procedure to introduce the Nca relies on the conversion of a cysteine into dehydroalanine by a bis-alkylation/elimination sequence (27, 36), followed by Michael addition of a thiol to introduce the new Nca side chain. The S. aureus NAL naturally contains no cysteines, making it an ideal target protein for chemical introduction of Ncas (36). The residues chosen for modification were picked based on their proximity to the aldehyde substrate binding pocket in the previously solved structure of an Escherichia coli NAL variant in complex with 4-epi N-acetylneuraminic acid (PDB ID code 4BWL) (37) (Fig. 3). The equivalent positions in the S. aureus NAL based on structural alignment were then mutated into cysteine residues by site-directed mutagenesis, and expressed and purified as previously described (36). The Ncas were then incorporated by first converting the cysteine to dehydroalanine (Dha) using 2,5-dibromohexan-1,6-diamide and then Michael addition with 13 different thiols resulting in installation of 13 different Ncas at each of the 12 positions (27) (Fig. 2). The thiols chosen encompass a wide variety of noncanonical side chains, some mimicking elongated versions of canonical amino acids and some incorporating functionalities that are either absent or uncommon in the canonical amino acids. Incorporation was carried out on a small (2.5 mg) scale under denaturing conditions to ensure the cysteine residue was accessible for modification before the protein was refolded. This procedure was previously shown to generate active, modified, refolded enzyme (36). The conversions of Cys→Dha and Dha→Nca were both monitored by ESI mass spectrometry (Figs. S1 and S2).
Fig. 2.

Amino acid positions modified, modifying thiols, and screening substrates. Cysteine residues were introduced individually into S. aureus NAL by site-directed mutagenesis at each position shown. After conversion into dehydroalanine, each position was modified separately with the 13 thiols shown. The resulting enzymes were screened using the thiobarbituric acid assay for activity in condensing pyruvate with the aldehydes shown.
Fig. 3.

Identification of positions for chemical modification. The crystal structure of the Y137A mutant of E. coli NAL in complex with Neu5Ac (PDB ID code 4BWL) (35) was used to select sites at which to incorporate Ncas. Residues were selected based on proximity (<8 Å) to the position of the Neu5Ac substrate (dark purple) and were separately mutated to cysteine residues. Residues at these positions are conserved between the E. coli and S. aureus enzymes, except for the following conservative changes (E. coli residue first): D170N, Y172F, Y190F, I247L, V251I, and F252Y.
Fig. S1.

ESI mass spectrum of the modified F190Dpc protein. A total of 10 mg of freeze-dried F190C NAL was dissolved in 5 mL of prewarmed (37 °C) 50-mM sodium phosphate buffer (pH 8.0) containing 6 M urea. A total of 200 µL of 2,5-dibromo-1,6-hexadiamide (0.13 mg/µL) was added and the reaction incubated for 1 h 45 min at 37 °C with shaking at 200 rpm. To this reaction 160 µL (0.1 mg/µL in 2 M Tris⋅HCl, pH 8.8) of thioglycerol was added and incubated for 2 h at 37 °C at 200 rpm. The protein was then dialyzed into 50 mM sodium phosphate buffer (pH 8.0) containing 6 M urea to remove any excess modification reagents and then into 50 mM sodium phosphate buffer (pH 7.4) to refold the protein. ESI-MS was used to check for complete conversion into the Nca-containing protein. Protein desalting, and mass analysis was performed by LC-MS using an M-class ACQUITY UPLC (Waters) interfaced to a Xevo QToF G2-XS mass spectrometer (Waters). A total of 1 µL of 5-µM sample was loaded onto a MassPREP protein desalting column (Waters) washed with 10% (vol/vol) solvent B in A for 5 min at 25 µL/min. The bound protein was eluted by a gradient of 2–40% (vol/vol) solvent B in A over 1 min at 25 µL/min. The column was subsequently washed with 95% solvent B in A for 6 min before reequilibration at 5% (vol/vol) solvent B in A ready for the next injection. Solvent A was 0.1% formic acid in water; solvent B was 0.1% formic acid in acetonitrile. The column eluent was directed into the mass spectrometer via a Z-spray electrospray source. The MS was operated in positive TOF mode using a capillary voltage of 3.0 kV, sample cone of 20 V, and source offset of 80 V. The source temperature was 100 °C and desolvation was 250 °C. Mass calibration was performed by a separate injection of [Glu]-fibrinopeptide b at a concentration of 250 fmol/µL. Data processing was performed using the MassLynx v4.1 suite of software supplied with the mass spectrometer. Small-scale (2.5 mg) modifications were used for the screening process. For detailed characterization, large-scale modifications (up to 50 mg) were performed. Modified enzymes were refolded by first dialyzing into sodium phosphate buffer (50 mM, pH 7.4) containing urea (6 M) to remove excess modification reagents, followed by dialysis into buffer without urea to refold the enzyme (36). Large-scale protein modifications were purified using size-exclusion chromatography performed using an ÄKTA Prime purification system (GE Healthcare Life Sciences) with a Superdex S200 column. Protein (8 mg/mL, 5 mL) was injected onto the column, which was run in Tris⋅HCl buffer (50 mM, pH 7.4) at 2 mL/min. (Upper) Raw (m/z) MS data. (Lower) Deconvoluted mass spectrum.
Fig. S2.


ESI-MS analysis of the modifications with various noncanonical amino acids at position 190 of S. aureus NAL. The various side-chain modifications are shown alongside the MS data (A–M). Samples were prepared for ESI-MS and analyzed as detailed in Fig. S1. The raw (m/z) MS data are shown alongside deconvoluted mass spectra (Insets).
Screening and Activity.
NAL is highly specific for its ketone donor (pyruvate) while accepting a range of aldehyde acceptors (38, 39). However, the wild-type enzyme has a strong preference for longer aldehydes (38–40): C4 aldehydes are generally poorly accepted (38) and C3 aldehydes are not substrates (39). The differently modified enzymes at each of the positions in the polypeptide chain were screened in 96-well plates for altered activity and specificity in condensing pyruvate with a range of aldehyde substrates with different lengths (C4 to C6), stereochemistry, and functional groups (Fig. 2). Enzyme activity was assessed using a variation of the established thiobarbituric acid (TBA) assay (41). Aldol reaction of pyruvate with the aldehydes in Table 1 generates 2-oxo-4,5-dihydroxy (or 2-oxo-4-acetamido-5-hydroxy) carboxylic acids. Subsequent cleavage of these products with periodate generates 1,3-dicarbonyls that react with TBA to generate an intense pink chromophore that can be quantified at 550 nm. Absorbance at 550 nm for each well was measured after 16 h condensation. The results were analyzed by comparing activity of the wild-type (unmodified) NAL with that of the Nca-containing enzyme with the same aldehyde substrate. An example analysis (Fig. 4) shows the result of screening NAL enzymes bearing Ncas at position 190 against a range of aldehydes. Whereas most modifications either generated enzymes with lower activity than the wild-type (Fig. 4, blue) or with the same level of activity (white), three modified enzymes had activities for the condensation of pyruvate with erythrose considerably greater than that of wild-type enzyme (purple). The improved enzymes had 2-hydroxypropyl cysteine (Hpc), 4-hydroxybutyl cysteine (Hbc), or 2,3-dihydroxypropyl cysteine (Dpc) at residue 190, with the Phe190Dpc enzyme having the greatest activity. Interestingly, all of the active enzymes have hydroxylated Nca side chains.
Table 1.
Steady-state kinetic parameters for the aldol condensation of erythrose and pyruvate and the aldol condensation of ManNAc and pyruvate for both wild-type and F190Dpc NAL
| Enzyme | Substrate | kcatapp, min−1 | Kmapp, mM | kcatapp/Kmapp, min−1⋅mM−1 |
| F190Dpc | Erythrose | 7.6 ± 0.56 | 4.4 ± 0.81 | 1.7 |
| F190Dpc | ManNAc | 0.22 ± 0.01 | 2.5 ± 0.43 | 0.09 |
| Wild type | Erythrose | 0.5 ± 0.05 | 3.0 ± 0.9 | 0.17 |
| Wild type | ManNAc | 0.8 ± 0.05 | 3.1 ± 0.7 | 0.26 |
Initial rates of enzyme reactions were measured in duplicate using the thiobarbituric acid assay at a fixed concentration of pyruvate of 80 mM while varying the concentration of aldehyde (0.8–15 mM). Data were fitted to the Michaelis–Menten equation to estimate the apparent kinetic parameters. Parameter values ± SE of the fit are shown.
Fig. 4.

Screening data for enzymes modified at position 190. Overnight reactions of enzyme (0.2 mg; 5.8 nmol) with aldehyde (8 mM) and pyruvate (80 mM) were assayed using the TBA assay and the absorbance read at 550 nm. The difference in A550 between the reaction with the modified enzyme and the wild-type enzyme were calculated. Values below zero (modified enzyme is less active than wild type) are blue with the intensity proportional to the activity difference. Values greater than zero (modified enzyme is more active than wild type) are similarly colored in pink. Modified enzymes are ranked by order of activity with pyruvate and erythrose.
To confirm that Phe190Dpc NAL catalyses the aldol reaction of pyruvate with erythrose to generate 3-deoxy-2-heptulosonic acid (DHA; Fig. 1B), a large-scale reaction was incubated for 48 h with Phe190Dpc NAL, and the resulting diastereomeric mixture of product (DHA) was purified by anion exchange chromatography. The 500 MHz 1H, COSY, and TOCSY NMR spectra (Fig. S3) were in agreement with reported data (42) for the expected product (DHA) and showed that a ∼75:25 mixture of C4-configured products had been formed (Table S1).
Fig. S3.

Region 1.75–2.65 ppm of the proton NMR spectrum of DHA. A sample of DHA was purified by anion exchange. Fractions containing DHA were pooled and freeze-dried. DHA was dissolved in D2O for analysis by 500 MHz 1H NMR spectroscopy. Two diastereomeric products were observed in the NMR spectrum: (4R, -5S, -6R) and (4S, -5S, -6R) DHA. The peaks in the region 1.75–2.65 ppm were used to assign these forms because these peaks correspond to the diastereotopic pair of protons formed during the aldol condensation at C3–C4. The (4R) diastereomer was seen to adopt one pyranose ring form (peaks corresponding to this form are highlighted in pink), and the (4S) diastereomer was seen to adopt three different ring forms: one pyranose (highlighted in yellow) and two distinct furanose (highlighted in gray and blue) forms.
Table S1.
NMR spectroscopic data for the forms of the two diastereomeric products
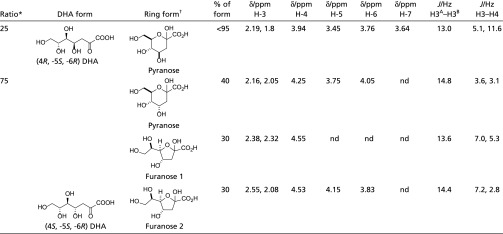 |
nd, not determined.
Ratio of the two diastereomeric products seen in the NMR spectrum, this was calculated from the integrals of the peaks in the region 1.75–2.6 ppm (shown in Fig. S3).
Ring systems observed for the forms of the two diastereomeric products, which were determined from the coupling constants between the protons on C3 and C4 J/Hz H3-H.
Characterization of Phe190Dpc NAL.
To evaluate fully the switch in substrate specificity caused by the introduction of the Dpc side chain at position 190, full kinetic characterization of the reactions involving both N-acetyl-d-mannosamine (ManNAc) and erythrose was carried out. A large quantity (∼50 mg) of Phe190Dpc NAL was produced and subjected to size-exclusion chromatography to remove any incorrectly folded protein produced during the refolding stage of the modification process. Mass spectrometry (Fig. S1) showed that the modified enzyme had the expected molecular mass for Phe190Dpc NAL. Steady-state kinetic parameters [kcat(app) and Km(app) for aldehyde] were determined for wild-type and Phe190Dpc NAL using the TBA assay (41) (Table 1 and Fig. S4).
Fig. S4.

Wild-type and Phe190Dpc NAL kinetics with erythrose and pyruvate. Kinetic parameters were determined using the TBA assay. (A) Time course of the reaction. A 600-µL reaction containing 8.3 mM erythrose, 83 mM pyruvate, and 0.07 mg (2.1 nmol) enzyme was incubated for various times at room temperature. The 50-µL samples were taken from each reaction and stopped by the addition of 10 µL trichloroacetic acid (12% wt/vol). Precipitated proteins were removed by centrifugation and samples were analyzed by the TBA assay for the wild-type enzyme (•–•) or Phe190Dpc enzyme (○–○). The time course was shown to be linear over >90 min. (B) Wild-type and (C) Phe190Dpc: 100-µL reactions were set up with a range of erythrose concentrations (0.8–15 mM) and pyruvate (80 mM). Reactions were initiated by the addition of 25 µL of enzyme (0.7 mg/mL; 0.51 nmol). Samples were incubated at room temperature for 1.5 h. Activity was determined from the absorbance at 550 nm against a standard curve of N-acetylneuraminic acid. Samples were run in duplicate, and the average value was used to estimate kinetic parameters by fitting to the Michaelis–Menten equation using nonlinear regression analysis. Parameter values ± SE of the fit are reported.
The kinetic parameters of the wild-type enzyme with ManNAc as acceptor aldehyde were in agreement with published values (43). Substitution of Phe-190 with Dpc results in an approximately threefold decrease in the specificity constant kcat/Km(app) for ManNAc as substrate compared with the wild-type enzyme, and this is almost entirely due to a decrease in kcat(app). However, when erythrose acts as the aldehyde substrate, the replacement of Phe-190 with Dpc results in a ∼10-fold increase in the kcat/Km(app) for erythrose, including a 15-fold increase in kcat(app). Taken together, these results demonstrate a switch in substrate specificity from ManNAc to erythrose of ∼30-fold brought about by the incorporation of an Nca at position 190. Interestingly the Nca-containing enzyme is ∼10× more active [kcat(app)] with the new substrate (erythrose) than the wild-type enzyme with its natural substrate (ManNAc). These results demonstrate that by replacing a hydrophobic phenylalanine with a polar Nca, 2,3-dihydroxypropyl cysteine, a new enzyme capable of the condensation of short (C4) aldehydes with high levels of catalytic activity can be generated.
Saturation Mutagenesis of Residue 190.
To compare the activity achieved by insertion of Dpc at residue 190 of NAL with that achievable by any of the 20 canonical amino acids, a saturation library at position 190 was produced. All 19 variants were produced by site-directed mutagenesis, expressed, and purified (36). Kinetic parameters were then determined for the saturation library in the same way as for the wild-type and Phe190Dpc enzymes. Fig. 5 shows that varying the canonical amino acid at position 190 of the S. aureus NAL resulted in small increases in the activity of the enzyme toward the reaction of pyruvate with erythrose. In contrast, insertion of Dpc at position 190 produced an enzyme that was ∼5× more active with erythrose [in terms of both kcat(app) and kcat/Km(app)] than the best canonical amino acid (Glu) (Fig. 5B). These data clearly highlight the benefits of exploring a widened chemical space with regards to broadening enzyme substrate specificity.
Fig. 5.

Comparison of activities for the saturation library at position 190 against F190Dpc variant. Activity of NAL variants bearing the 20 canonical amino acids at position 190 was measured using the TBA assay and compared with NAL bearing the Nca Dpc at position 190. Initial reaction rates were determined using the TBA assay under conditions where the time course of product formation is linear (Fig. S4). Steady-state kinetic parameters were determined at a fixed pyruvate concentration of 80 mM and erythrose concentrations between 0.8 and 15 mM. Each reaction contained 18 µg (0.52 nmol) of enzyme and were carried out in duplicate and data were fitted to the Michaelis–Menten equation. The fitted value ± SE of the fit is shown. (A) Comparison of kcat values; (B) Steady-state parameters.
Structural Characterization.
It was interesting that the slight changes in enzyme specificity brought about by site-directed mutagenesis to introduce other canonical amino acids at position 190 in S. aureus NAL were mainly derived from changes in the apparent Km for the aldehyde substrate (Fig. 5B). In contrast, the specificity change brought about by the introduction of the noncanonical Dpc side chain was largely generated by changes in kcat(app) rather than Km(app) (Table 1). To investigate further the specificity change, we determined the structure of the Phe190Dpc NAL in complex with pyruvate (PDB ID code 5LKY). The enzyme structure was highly similar (RMSDCα-all-chains 0.49 Å and RMSDCα-monomer 0.17–0.20 Å) to the wild-type pyruvate complex (PDB ID code 4AH7) (36) except at the modified position and around residue 167. Identical interactions between the enzyme and the pyruvate donor indicate that its binding is unaffected by the insertion of the Dpc side chain. Residue 190 is positioned toward the aldehyde-binding end of the active site, and the electron density was modeled as a (2R)-2,3-dihydroxypropyl cysteine. It is notable that the Nca has a single (L) stereochemical configuration. It is highly unlikely that the Michael addition of racemic 2,3-dihydroxy propanethiol to the unfolded protein was stereocontrolled; rather, we hypothesize, as previously (36), that only one stereoisomer of the modified protein can refold correctly and is recovered after gel filtration.
The Dpc190 side chain is positioned between Glu192 and Asp141 and points into the active site where it extends further (average 0.8 ± 0.1 Å from all four subunits) into the active site than the corresponding phenylalanine in the wild-type structure. The noncanonical side chain exhibits a high degree of flexibility in the active site, adopting different conformations in all four subunits (Fig. S5). In subunit A, the Dpc side chain was modeled in two different conformations that resulted from a rotation around the Cα–Cβ bond. In both subunits A and B, the hydroxyl groups of the Dpc side chain make hydrogen bond interactions to the side chains of Glu192 and Asp141 (Fig. 6B). Another notable difference between wild-type and Phe190Dpc NAL was at the region 167–169. In the Phe190Dpc structure, the removal of the bulky hydrophobic phenylalanine allows the backbone chain containing residues Thr167, Ala168, and Pro169 to move closer to the catalytic Lys165 in the active site (Fig. S6). This results in the Cα of Thr167 in the Phe190Dpc structure being displaced on average 1.0 ± 0.1 Å and adopting a different rotamer compared with the wild-type structure (Fig. 6 A and B and Fig. S6). Previous QMMM modeling of the reaction mechanism of the E. coli NAL (37) has suggested that Thr167 binds to the aldehyde oxygen atom and helps to stabilize the transition state of the reaction. Unfortunately, we were unable to determine the crystal structure of the modified Phe190Dpc NAL with the reaction product DHA bound, and so we turned to molecular modeling to try to elucidate the structural basis for the change of specificity.
Fig. S5.

2Fo–Fc electron density maps for the Dpc side chain found at position 190 in the Phe190Dpc crystal structure in complex with pyruvate. The 2Fo–Fc electron density for the Dpc residue at position 190 in the modified S. aureus NAL is shown contoured at 1.0 rmsd. (A–D) Density for the residue in subunits A–D, respectively, of the four subunits of the enzyme tetramer found in the asymmetric unit. The Dpc side chain adopts two conformations in chain A and single, but distinct, conformations in the other three subunits.
Fig. 6.

Structures and models of wild-type and modified NALs. Structures of (A) wild-type (PDB ID code 4AH7) (34) and (B) Phe190Dpc (PDB ID code 5LKY) NAL enzymes in complex with pyruvate were structurally aligned. B shows the network of interactions that the Nca side chain Dpc forms with Asp141 and Glu192. Energy-minimized models of product Schiff base structures [illustrated with 4(S)-DHA] were generated using Maestro and are displayed from the same view. (C) Wild-type NAL. (D) Phe190Dpc enzyme complex.
Fig. S6.

Comparison of Pro169-Ala168-Thr167 chain in the wild-type (cyan) NAL and Phe190Dpc (magenta) NAL crystal structures. The replacement of Phe190 by Dpc results in a movement of this part of the protein backbone brings Thr167 1.0 ± 0.1 Å deeper into the active site where it adopts a different rotamer compared with the wild-type structure.
Computational energy minimization experiments were carried out on the Schiff bases formed between the product DHA and both the wild-type and Phe190Dpc NALs. The models revealed that, as expected, the DHA, being shorter than the natural substrate N-acetylneuraminic acid (Neu5Ac), does not make the same interactions with Glu-192 of the enzyme (37) (Figs. S7 and S8). Because of our finding that both 4R and 4S DHA were formed by Phe190Dpc NAL, both stereoisomers were modeled into the active sites. The minimizations revealed that C1–C3 of either C4 diastereoisomer of DHA were bound in the same way in both the wild-type and Phe190Dpc models. However, there were significantly different interactions at the other end (C4–C7) of the product. Tyr252 lies slightly further from the product in both 4(R) and 4(S) DHA-Phe190Dpc NAL models, meaning that it can no longer hydrogen bond with the product C5 hydroxyl (∼4.7 Å compared with 3.1 Å in the wild-type model; Fig. 6 C and D). In addition, Thr167 lies ∼1.6 Å closer to the C4 hydroxyl of the product in the DHA-Phe190Dpc model than in the wild-type model: here, the difference in the preferred rotamer of Thr167 may enable better stabilization of the enzyme transition state. Dpc does not interact directly with DHA in the Phe190Dpc model; it forms an entirely different network of interactions between the carboxylate side chains of Asp141 and Glu192 in the wild-type model, which brings the Dpc 3.0 Å closer to the product than the phenylalanine in the wild-type model (Fig. S7). Thus, molecular modeling suggests that the newly introduced Dpc side chain at residue 190 alters the substrate specificity not by direct interaction with DHA but rather by altering the conformation and interaction network in the active site, resulting in better transition state stabilization. This observation is entirely consistent with the kinetic data (Table 1) where the switch in substrate specificity measured by kcat/Km(app) is almost entirely due to alterations in kcat(app) rather than in Km(app).
Fig. S7.

Comparison of the position of residue 190 in the Phe190Dpc and wild-type energy-minimized models. Computational energy minimization experiments were carried out using the Schrodinger Small Molecule Discovery Suite. The graphical user interface, Maestro (Schrödinger Release 2015-1: Maestro; Schrödinger, LLC), was used to model the Schiff bases formed between the product DHA and both the catalytic lysine of subunit B of the wild-type NAL and the catalytic lysine of subunit B of Phe190Dpc NAL. Energy minimization of the products formed was carried out using Macromodel with the OPLS_2005 force field (52) using water as the solvent. All side chains of amino acids within 6 Å of residues around the product were allowed to minimize, as well as the product itself.
Fig. S8.

Structures and models of substrate binding in NAL. (A) Structure of the E.coli Y137A mutant with Neu5Ac bound (PDB ID code 4BWL) (37). Neu5Ac is covalently bound as a Schiff base to Lys-165, and Glu-192 interacts with the O8 and O9 hydroxy groups of Neu5Ac. Thr-167 acts to stabilize the transition state of the reaction. (B) Structure of the pyruvate complex of the wild-type S. aureus NAL (PDB ID code 4AH7) (36). Pyruvate is bound as the Schiff base to Lys-165. Tyr-190 in the E. coli enzyme is replaced by Phe-190 in the S. aureus enzyme. In this enzyme, Phr-167 is found in a different rotamer. (C) Structure of the pyruvate complex of the F190Dpc variant of S. aureus NAL (PDB ID code 5LKY). Pyruvate is bound as the Schiff base to Lys-165. The side chain of Dpc190 is found in a number of conformations that allow Thr-167 to reorient to a similar rotamer as found in the E. coli NAL. (D) Energy-minimized model of DHA bound as the Schiff base to Lys-165. DHA is too far from Glu-192 to make any interactions. Thr-167 can act to stabilize the transition state of the reaction.
Discussion
Aldolases are an important class of biocatalysts that are finding increasing uses in the synthesis of complex compounds (33); they are particularly useful in that they can create two stereochemical centers during their reaction (34, 44–46) and the reactions can be carried out in aqueous solvent without the use of protecting groups. However, the substrate specificity of natural aldolases often has to be engineered to produce enzymes with the required substrate specificity (33, 47). Rational protein engineering and directed evolution have been successful in addressing this problem, but the protein engineer is nonetheless fundamentally limited to alterations of amino acids to any of the other 19 canonical amino acids. The recent invention of methods to allow the incorporation of noncanonical amino acids into proteins, either by genetic incorporation or by chemical modification, has opened the way to explore wider areas of structure−activity space. Chemical modification methods have particular value because many different Ncas may be incorporated easily, enabling the function of the resulting variants to be explored. Here, we have shown that by using a noncanonical amino acid, it is possible not only to alter the aldehyde acceptor of the natural enzyme, but also to increase the activity with the altered substrate to rates that are unattainable with only the 20 proteogenic amino acids.
By modifying position 190 of the S. aureus NAL to a 2,3-dihydroxypropyl cysteine, the enzyme activity for the reaction of erythrose and pyruvate to form DHA (Fig. 1) has been significantly increased. A phosphorylated form of DHA (7-phospho DHA) is an important intermediate in the shikimate pathway for the biosynthesis of aromatic amino acids (48), and there has been much interest in the preparation of DHA, and other 3-deoxy-2-ulosonic acids, due to their potential as starter units for complex oligosaccharide syntheses (42, 48–50). Our structural studies showed that the noncanonical amino acid is perfectly shaped and functionalized to interpose between Asp141 and Glu192, reducing the active site volume and causing a remodeling of the active site to stabilize better the transition state of the DHA-producing reaction. In stark contrast, none of the canonical amino acids possess the required molecular characteristics in a single residue, demonstrating that Ncas can extend the ambitions of the protein engineer. Based on the data presented here, we envisage that advances in computational (12, 13) and (semi)rational methods (6) for enzyme redesign, coupled with the development of recent powerful chemical mutagenesis methods (51) and the enormous variety of Ncas side chains and chemistries, will open the way to engineer enzymes with catalytic functions that are not found in Nature.
Materials and Methods
Further details may be found in SI Materials and Methods.
Chemical Modification Using Various Thiols.
The conversion of the cysteine residues (introduced by site-directed mutagenesis) to dehydroalanine was carried out as previously described (36) using 2,5-dibromohexan-1,6-diamide synthesized (27). ESI-MS was used to check for complete conversion into the Nca-containing protein. Small-scale (2.5 mg) modifications were used for the screening process. For detailed characterization, large-scale modifications (up to 50 mg) were performed. Modified enzymes were refolded by first dialyzing into sodium phosphate buffer (50 mM, pH 7.4) containing urea (6 M) to remove excess modification reagents, followed by dialysis into buffer without urea to refold the enzyme (36). Large-scale protein modifications were purified using size-exclusion chromatography performed using an ÄKTA Prime purification system (GE Healthcare Life Sciences) with a Superdex S200 column. Protein (8 mg/mL, 5 mL) was injected onto the column, which was run in Tris⋅HCl buffer (50 mM, pH 7.4) at 2 mL/min.
Screening Using the TBA Assay.
Modified enzymes were screened for aldol reaction activity with pyruvate and a variety of aldehydes. Reactions in 96 deep-well plates contained pyruvate (100 µL, final concentration 80 mM), aldehyde (100 µL, final concentration 8 mM), and modified enzyme [50 µL of 1.0 mg/mL (1.5 nmol)]. Reactions were incubated for 16 h at room temperature, and activity was assessed by TBA assay. Each reaction was oxidized by the addition of 11 µL of sodium periodate (0.2 M in 9 M H3PO4) and incubated for 20 min at room temperature. Oxidation was terminated by the addition of 45 µL of sodium arsenite [10% (wt/vol) in 0.5 M Na2SO4 and 0.05 M H2SO4], and reactions were agitated until all brown discoloration had dissipated. A total of 135 µL of TBA (0.6% in 0.5 M NaSO4) was added to each reaction and heated to 70 °C for 30 min. The 100-µL samples of each reaction were transferred to a flat-bottomed 96-well plate, and the absorbance was read at 550 nm.
Synthesis of DHA.
Erythrose (500 mg, 4.2 mmol) and sodium pyruvate (2.29 g, 21 mmol) were dissolved in sodium phosphate buffer (50 mM, pH 7.4, 10 mL), and Phe190Dpc (0.8 mg) was added. The reaction was incubated at room temperature for a minimum of 48 h before purification by anion exchange chromatography on AG1 × 8 resin (HCO3−, 100–200 mesh). Product was eluted using a 0–0.4 M ammonium bicarbonate linear gradient (52). Fractions containing product were identified using the TBA assay on a 20-µL sample and were pooled, freeze-dried, and redissolved in D2O before analysis by 500 MHz 1H, COSY, and TOCSY NMR spectroscopy.
Kinetic Assays.
Kinetic parameters were determined using the TBA assay. The 100-µL reactions contained erythrose (0.8–15 mM) and pyruvate (80 mM). Reactions were initiated by the addition of 25 µL of enzyme (0.7 mg/mL, 0.5 nmol) and then incubated at room temperature for 1.5 h. A 100-µL sample was taken from each reaction and stopped by the addition of 10 µL trichloroacetic acid (12% wt/vol). Precipitated proteins were removed by centrifugation and samples were analyzed. Under these conditions the rate of formation of product was linear over 90 min (Fig. S4) and the initial rate of the reaction was determined from the A550 nm against a standard curve of N-acetylneuraminic acid. Reactions were analyzed in duplicate and kinetic parameters were estimated by fitting to the Michaelis–Menten equation using nonlinear regression analysis. Parameter values ± SE of the fit are reported throughout.
Protein Crystallization, Data Collection, and Refinement.
Crystallization conditions were as previously described (36, 53). Diffraction data for the Phe190Dpc structure were collected from a single crystal at the Diamond Light Source macromolecular crystallography beam line I04-1 flash-cooled to 100 K. Data processing and refinement were carried out as previously described (36). Coordinates and restraint library files for the 2,3-dihydroxypropyl cysteine side chain [heteroatom (HET) code P9S] were generated using the PRODRG (54) server and manually edited. The models were validated using the PDB validation server. Atomic coordinates and structure factors have been deposited into the Protein Data Bank (PDB ID code 5LKY; Table S2).
Table S2.
Structural statistics for the F190Dpc X-ray crystal structure in complex with pyruvate
| Data | F190Dpc saNAL in complex with pyruvate |
| Diamond beamline | I04-1 |
| Wavelength, Å | 0.97 |
| Resolution range, Å | 75.53–1.70 (1.79–1.70) |
| Space group | P21 |
| a, Å | 55.5 |
| b, Å | 134.3 |
| c, Å | 79.5 |
| Rmerge (%)*,† | 7.1 (42.0) |
| Rpim (%)†,‡ | 3.5 (24.0) |
| Observed reflections | 565, 256 |
| Unique reflections | 120, 355 |
| Completeness (%)† | 99.4 (95.7) |
| Multiplicity† | 4.7 (4.0) |
| <I/σ(I)>† | 11.2 (3.0) |
| Refinement | |
| Rfactor (%) | 17.4 (26.1) |
| Rfree (%)§ | 20.3 (27.6) |
| No. of protein atoms | 9,298 |
| No. of solvent molecules | 278 |
| No of ligand atoms | 20 |
| Average overall B-factor, Å2 | 23 |
| Average ligand B-factor, Å2 | 22 |
| Average solvent B-factor, Å2 | 27 |
| Rms bond lengths, Ŷ | 0.019 |
| Rms bond angles, Ŷ | 2.04 |
| Ramachandran analysis, the percentage of residues in the regions of plot, %# | |
| Most favored | 98.0 |
| No. of outliers | 4|| |
Rmerge = ΣhklΣi|Ii(hkl) − <I(hkl)>|/ΣhklΣi|Ii(hkl).
Values given in parentheses correspond to those in the outermost shell of the resolution range.
Rpim, precision-indicating (multiplicity weighted); Rmerge, relative to all I+ or I−.
Rfree was calculated with 5% of the reflections set aside randomly.
Based on the ideal geometry values of Engh and Huber (56).
Ramachandran analysis using the program MolProbity (57).
The side chain of Tyr111, in all subunits, is in close proximity to Leu142, Thr143, and Phe110 from an adjacent chain causing the phi and psi angles of Tyr111 to lie in an unfavored region of the Ramachandran plot.
Computational Energy Minimizations.
Computational energy minimizations were carried out using the Schrodinger Small Molecule Discovery Suite (Schrödinger release 2015-1: Maestro; Schrödinger, LLC). Maestro was used to model the Schiff bases between the product DHA and the catalytic lysine of subunit B of the wild-type or Phe190Dpc NAL. Energy minimization of the products formed was carried out using Macromodel with the OPLS_2005 force field (55) with water as solvent. All amino acid side chains within 6 Å of the product were allowed to minimize, as well as the product itself.
SI Materials and Methods
Mutagenesis and Expression.
All mutagenesis was carried out using QuikChange Lightning kits (Stratagene) following the manufacturer’s guidelines. Expression of wild-type and variant S. aureus NAL was carried out as previously described (36).
Chemical Modification.
The conversion of the cysteine residues (introduced by site-directed mutagenesis) to dehydroalanine was carried out as previously described (36) using 2,5-dibromohexan-1,6-diamide synthesized as described (27). Modifications were carried out in sodium phosphate buffer (50 mM, pH 8.0) containing urea (6 M) at 37 °C. Successful conversion into dehydroalanine-containing protein was confirmed using electrospray ionization (ESI)-MS. The 1.25-mL aliquots of dehydroalanine-containing protein (2.0 mg/mL; 70 nmol) were combined with 40 µL of thiol (0.1 mg/µL; 250–500 nmol depending on the thiol), in either 2 M Tris⋅HCl, pH 8.8, or in DMF with the pH adjusted to 8.8, and the solution incubated for 2 h at 37 °C, with shaking at 200 rpm.
Protein Crystallization, Data Collection, and Refinement.
S. aureus NAL crystallization conditions were as previously described (36). Diffraction data for the Phe190Dpc structure were collected from a single crystal at the Diamond Light Source macromolecular crystallography beam line I04-1 flash-cooled to 100 K. Data processing and refinement were carried out as previously described (36). Coordinates and restraint library files for the lysine residue covalently bound to a pyruvoyl moiety (HET code KPI) were as previously described (53); those for the 2, 3-dihydroxypropyl cysteine side chain (HET code P9S) were generated using the PRODRG (54) server and manually edited. The models were validated using the PDB validation server. The atomic coordinates and structure factors have been deposited into the Protein Data Bank (www.wwpdb.org) with the PDB ID code 5LKY.
Acknowledgments
The authors thank Diamond Light Source for beam time (proposal mx10305), and the staff of beamline I04-1 for assistance with data collection. Mass spectrometry was performed on a Xevo G2-XS and ACQUITY UPLC M-Class funded by the Biotechnology and Biological Sciences Research Council (BBSRC) Grant BB/M012573/1, and the nanoACQUITY HPLC was kindly donated by Waters. This work was supported by BBSRC Doctoral Training Grant (DTG) PhD Studentship BB/F01614X/1 (to C.L.W.) and BBSRC Grant BB/N002091/1. Protein crystallization and X-ray facilities were funded by BBSRC Grant BB/L015056/1 and Wellcome Trust Grant WT094232MA, respectively.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.wwpdb.org (PDB ID code 5LKY).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1616816114/-/DCSupplemental.
References
- 1.Lad C, Williams NH, Wolfenden R. The rate of hydrolysis of phosphomonoester dianions and the exceptional catalytic proficiencies of protein and inositol phosphatases. Proc Natl Acad Sci USA. 2003;100(10):5607–5610. doi: 10.1073/pnas.0631607100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stockbridge RB, Lewis CA, Jr, Yuan Y, Wolfenden R. Impact of temperature on the time required for the establishment of primordial biochemistry, and for the evolution of enzymes. Proc Natl Acad Sci USA. 2010;107(51):22102–22105. doi: 10.1073/pnas.1013647107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bornscheuer UT, et al. Engineering the third wave of biocatalysis. Nature. 2012;485(7397):185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 4.Jäckel C, Hilvert D. Biocatalysts by evolution. Curr Opin Biotechnol. 2010;21(6):753–759. doi: 10.1016/j.copbio.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Davids T, Schmidt M, Böttcher D, Bornscheuer UT. Strategies for the discovery and engineering of enzymes for biocatalysis. Curr Opin Chem Biol. 2013;17(2):215–220. doi: 10.1016/j.cbpa.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 6.Lutz S. Beyond directed evolution--semi-rational protein engineering and design. Curr Opin Biotechnol. 2010;21(6):734–743. doi: 10.1016/j.copbio.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker P, Seah SYK. Rational approaches for engineering novel functionalities in carbon–carbon bond forming enzymes. Comput Struct Biotechnol J. 2012;2(3):e201209003. doi: 10.5936/csbj.201209003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chica RA, Doucet N, Pelletier JN. Semi-rational approaches to engineering enzyme activity: Combining the benefits of directed evolution and rational design. Curr Opin Biotechnol. 2005;16(4):378–384. doi: 10.1016/j.copbio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat Rev Genet. 2015;16(7):379–394. doi: 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- 10.Brustad EM, Arnold FH. Optimizing non-natural protein function with directed evolution. Curr Opin Chem Biol. 2011;15(2):201–210. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane MD, Seelig B. Advances in the directed evolution of proteins. Curr Opin Chem Biol. 2014;22:129–136. doi: 10.1016/j.cbpa.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wijma HJ, Janssen DB. Computational design gains momentum in enzyme catalysis engineering. FEBS J. 2013;280(13):2948–2960. doi: 10.1111/febs.12324. [DOI] [PubMed] [Google Scholar]
- 13.Kries H, Blomberg R, Hilvert D. De novo enzymes by computational design. Curr Opin Chem Biol. 2013;17(2):221–228. doi: 10.1016/j.cbpa.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 14.Huang PS, Boyken SE, Baker D. The coming of age of de novo protein design. Nature. 2016;537(7620):320–327. doi: 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- 15.Wijma HJ, et al. Enantioselective enzymes by computational design and in silico screening. Angew Chem Int Ed Engl. 2015;54(12):3726–3730. doi: 10.1002/anie.201411415. [DOI] [PubMed] [Google Scholar]
- 16.Holliday GL, Fischer JD, Mitchell JB, Thornton JM. Characterizing the complexity of enzymes on the basis of their mechanisms and structures with a bio-computational analysis. FEBS J. 2011;278(20):3835–3845. doi: 10.1111/j.1742-4658.2011.08190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson VL. Generation of protein-derived redox cofactors by posttranslational modification. Mol Biosyst. 2011;7(1):29–37. doi: 10.1039/c005311b. [DOI] [PubMed] [Google Scholar]
- 18.Bale S, Ealick SE. Structural biology of S-adenosylmethionine decarboxylase. Amino Acids. 2010;38(2):451–460. doi: 10.1007/s00726-009-0404-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bojarová P, Williams SJ. Sulfotransferases, sulfatases and formylglycine-generating enzymes: A sulfation fascination. Curr Opin Chem Biol. 2008;12(5):573–581. doi: 10.1016/j.cbpa.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Turner NJ. Ammonia lyases and aminomutases as biocatalysts for the synthesis of α-amino and β-amino acids. Curr Opin Chem Biol. 2011;15(2):234–240. doi: 10.1016/j.cbpa.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Yukl ET, Wilmot CM. Cofactor biosynthesis through protein post-translational modification. Curr Opin Chem Biol. 2012;16(1-2):54–59. doi: 10.1016/j.cbpa.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klinman JP, Bonnot F. Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem Rev. 2014;114(8):4343–4365. doi: 10.1021/cr400475g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang WH, Otting G, Jackson CJ. Protein engineering with unnatural amino acids. Curr Opin Struct Biol. 2013;23(4):581–587. doi: 10.1016/j.sbi.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Dumas A, Lercher L, Spicer CD, Davis BG. Designing logical codon reassignment - Expanding the chemistry in biology. Chem Sci (Camb) 2015;6(1):50–69. doi: 10.1039/c4sc01534g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young TS, Schultz PG. Beyond the canonical 20 amino acids: Expanding the genetic lexicon. J Biol Chem. 2010;285(15):11039–11044. doi: 10.1074/jbc.R109.091306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Díaz-Rodríguez A, Davis BG. Chemical modification in the creation of novel biocatalysts. Curr Opin Chem Biol. 2011;15(2):211–219. doi: 10.1016/j.cbpa.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Chalker JM, et al. Methods for converting cysteine to dehydroalanine on peptides and proteins. Chem Sci (Camb) 2011;2(9):1666–1676. [Google Scholar]
- 28.Tang Y, et al. Stabilization of coiled-coil peptide domains by introduction of trifluoroleucine. Biochemistry. 2001;40(9):2790–2796. doi: 10.1021/bi0022588. [DOI] [PubMed] [Google Scholar]
- 29.Montclare JK, Tirrell DA. Evolving proteins of novel composition. Angew Chem Int Ed Engl. 2006;45(27):4518–4521. doi: 10.1002/anie.200600088. [DOI] [PubMed] [Google Scholar]
- 30.Yoo TH, Link AJ, Tirrell DA. Evolution of a fluorinated green fluorescent protein. Proc Natl Acad Sci USA. 2007;104(35):13887–13890. doi: 10.1073/pnas.0701904104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drienovska I, Rioz-Martinez A, Draksharapu A, Roelfes G. Novel artificial metalloenzymes by in vivo incorporation of metal-binding unnatural amino acids. Chem Sci (Camb) 2015;6(1):770–776. doi: 10.1039/c4sc01525h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ugwumba IN, et al. Improving a natural enzyme activity through incorporation of unnatural amino acids. J Am Chem Soc. 2011;133(2):326–333. doi: 10.1021/ja106416g. [DOI] [PubMed] [Google Scholar]
- 33.Windle CL, Müller M, Nelson A, Berry A. Engineering aldolases as biocatalysts. Curr Opin Chem Biol. 2014;19:25–33. doi: 10.1016/j.cbpa.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams GJ, Woodhall T, Farnsworth LM, Nelson A, Berry A. Creation of a pair of stereochemically complementary biocatalysts. J Am Chem Soc. 2006;128(50):16238–16247. doi: 10.1021/ja065233q. [DOI] [PubMed] [Google Scholar]
- 35.Williams GJ, Woodhall T, Nelson A, Berry A. Structure-guided saturation mutagenesis of N-acetylneuraminic acid lyase for the synthesis of sialic acid mimetics. Protein Eng Des Sel. 2005;18(5):239–246. doi: 10.1093/protein/gzi027. [DOI] [PubMed] [Google Scholar]
- 36.Timms N, et al. Structural insights into the recovery of aldolase activity in N-acetylneuraminic acid lyase by replacement of the catalytically active lysine with γ-thialysine by using a chemical mutagenesis strategy. ChemBioChem. 2013;14(4):474–481. doi: 10.1002/cbic.201200714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daniels AD, et al. Reaction mechanism of N-acetylneuraminic acid lyase revealed by a combination of crystallography, QM/MM simulation, and mutagenesis. ACS Chem Biol. 2014;9(4):1025–1032. doi: 10.1021/cb500067z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fitz W, Schwark JR, Wong CH. Aldotetroses and C(3)-modified aldohexoses as substrates for N-acetylneuraminic acid aldolase: A model for the explanation of the normal and the inversed stereoselectivity. J Org Chem. 1995;60(12):3663–3670. [Google Scholar]
- 39.Kim MJ, Hennen WJ, Sweers HM, Wong CH. Enzymes in carbohydrate synthesis: N-acetylneuraminic acid aldolase catalyzed reactions and preparation of N-acetyl-2-deoxy-D-neuraminic acid derivatives. J Am Chem Soc. 1988;110(19):6481–6486. [Google Scholar]
- 40.Machajewski TD, Wong CH. The catalytic asymmetric aldol reaction. Angew Chem Int Ed Engl. 2000;39(8):1352–1375. doi: 10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 41.Warren L. The thiobarbituric acid assay of sialic acids. J Biol Chem. 1959;234(8):1971–1975. [PubMed] [Google Scholar]
- 42.Hekking KFW, Moelands MAH, van Delft FL, Rutjes FPJT. An in-depth study on ring-closing metathesis of carbohydrate-derived alpha-alkoxyacrylates: Efficient syntheses of DAH, KDO, and 2-deoxy-beta-KDO. J Org Chem. 2006;71(17):6444–6450. doi: 10.1021/jo060913x. [DOI] [PubMed] [Google Scholar]
- 43.Ferrero MA, Reglero A, Fernandez-Lopez M, Ordas R, Rodriguez-Aparicio LB. N-acetyl-D-neuraminic acid lyase generates the sialic acid for colominic acid biosynthesis in Escherichia coli K1. Biochem J. 1996;317(Pt 1):157–165. doi: 10.1042/bj3170157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howard JK, Müller M, Berry A, Nelson A. An enantio- and diastereoselective chemoenzymatic synthesis of α-fluoro β-hydroxy carboxylic esters. Angew Chem Int Ed Engl. 2016;55(23):6767–6770. doi: 10.1002/anie.201602852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams GJ, Domann S, Nelson A, Berry A. Modifying the stereochemistry of an enzyme-catalyzed reaction by directed evolution. Proc Natl Acad Sci USA. 2003;100(6):3143–3148. doi: 10.1073/pnas.0635924100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Royer SF, et al. Structurally informed site-directed mutagenesis of a stereochemically promiscuous aldolase to afford stereochemically complementary biocatalysts. J Am Chem Soc. 2010;132(33):11753–11758. doi: 10.1021/ja104412a. [DOI] [PubMed] [Google Scholar]
- 47.DeSantis G, et al. Structure-based mutagenesis approaches toward expanding the substrate specificity of D-2-deoxyribose-5-phosphate aldolase. Bioorg Med Chem. 2003;11(1):43–52. doi: 10.1016/s0968-0896(02)00429-7. [DOI] [PubMed] [Google Scholar]
- 48.Dondoni A, Marra A, Merino P. Installation of the pyruvate unit in glycidic aldehydes via a Wittig olefination Michael addition sequence utilizing a thiazole-armed carbonyl ylid: A new stereoselective route to 3-deoxy-2-ulosonic acids and the total synthesis of DAH, KDN and 4-epi-KDN. J Am Chem Soc. 1994;116(8):3324–3336. [Google Scholar]
- 49.Pradhan TK, Lin CC, Mong KKT. Formal synthesis of 3-deoxy-D-manno-octulosonic acid (KDO) and 3-deoxy-D-arabino-2-heptulosonic acid (DAH) Synlett. 2013;24(2):219–222. [Google Scholar]
- 50.Enders D, Gasperi T. Proline organocatalysis as a new tool for the asymmetric synthesis of ulosonic acid precursors. Chem Commun (Camb) 2007;(1):88–90. doi: 10.1039/b611265j. [DOI] [PubMed] [Google Scholar]
- 51.Wright TH, et al. Posttranslational mutagenesis: A chemical strategy for exploring protein side-chain diversity. Science. 2016 doi: 10.1126/science.aag1465. [DOI] [PubMed] [Google Scholar]
- 52.Auge C, Gautheron-Le Narvor C. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Dekker; New York: 1997. pp. 469–484. [Google Scholar]
- 53.Campeotto I, et al. Structural insights into substrate specificity in variants of N-acetylneuraminic acid lyase produced by directed evolution. J Mol Biol. 2010;404(1):56–69. doi: 10.1016/j.jmb.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schüttelkopf AW, van Aalten DMF. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 55.Banks JL, et al. Integrated modeling program, applied chemical theory (IMPACT) J Comput Chem. 2005;26(16):1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engh RA, Huber R. Accurate bond and angle parameters for X-ray protein-structure refinement. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 57.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]