

Abstract
Glycosaminoglycan (GAG) sequences that selectively target heparin cofactor II (HCII), a key serpin present in human plasma, remain unknown. Using a computational strategy on a library of 46 656 heparan sulfate hexasaccharides we identified a rare sequence consisting of consecutive glucuronic acid 2‐O‐sulfate residues as selectively targeting HCII. This and four other unique hexasaccharides were chemically synthesized. The designed sequence was found to activate HCII ca. 250‐fold, while leaving aside antithrombin, a closely related serpin, essentially unactivated. This group of rare designed hexasaccharides will help understand HCII function. More importantly, our results show for the first time that rigorous use of computational techniques can lead to discovery of unique GAG sequences that can selectively target GAG‐binding protein(s), which may lead to chemical biology or drug discovery tools.
Keywords: carbohydrates, chemical biology, glycosaminoglycans, in silico screening, serpins
Heparin cofactor II (HCII) is a serine protease inhibitor (serpin) that circulates in human plasma at high levels. Although it has been known to selectively inhibit thrombin for several decades,1, 2, 3 its true physiologic function remains to be understood.4 HCII is known to bind to glycosaminoglycans (GAGs) such as dermatan sulfate (DS) and heparan sulfate (HS), which help mediate its inhibition of thrombin. One of the key reasons for the inability to identify HCII's biologic role is the lack of knowledge on the specificity of HCII‐GAG interaction.
HCII is an interesting serpin. It bears considerable similarity to antithrombin (AT), another plasma serpin that mediates the anticoagulant action of heparin and fondaparinux,5, 6 two clinically used drugs. AT and HCII are homologous in primary, secondary and tertiary structure (Figure S1 in the Supporting Information). Yet, whereas AT binds specifically to fondaparinux sequence in heparin,7 HCII is considered to bind non‐specifically to heparin and HS. Further, although both serpins display a two‐step, induced‐fit, allosteric activation mechanism in inhibiting their target enzymes,8, 9, 10 no HS oligosaccharide has been discovered to induce robust activation of HCII.
Identifying GAG sequences that selectively target proteins is extremely challenging. A key reason for this is their structural complexity. HS, a highly anionic polymer containing variably sulfated, acetylated and epimerized residues, presents enormous structural diversity that makes comprehensive analysis of all possible sequences difficult. Thus, identifying key “needles” in this haystack, especially with synthesis11, 12 or isolation of oligosaccharides from nature nearly impossible.13
We had earlier developed a dual‐filter computational algorithm, called combinatorial virtual library screening (CVLS) strategy, to rapidly sort HS sequences into “specific” and “non‐specific” bins.14, 15 We wondered whether this tool could pinpoint HS sequences that preferentially activate HCII for inhibition of thrombin. Further, we posited that such a sequence, if any, would be different from the pentasaccharide sequence (i.e., fondaparinux) that selectively activates AT.8
Herein, we show that application of the CVLS strategy on a library of all‐possible natural HS hexasaccharides led to the identification of five distinct sequences that were predicted to differentially target the two homologous serpins. We developed a novel multi‐step synthesis of these five HS sequences and studied each for activation of the two serpins. The results led to the discovery that a unique hexasaccharide that contains two rare 2‐O‐sulfate glucuronic acid (GlcA2S) residues in tandem selectively activates HCII nearly 250‐fold, which is equivalent to AT activation induced by fondaparinux.8
To identify HCII‐specific sequences, we compared the overall tertiary structures of HCII and AT.7, 9 The HS binding site in AT is formed by helices A (hA) and D (hD), which contains R46, R47, K114, K125, R129, R132 and K133 that define the specificity of HS recognition. The corresponding region in HCII contains R103, K173, K185, R189, R192, and R193 and could be defined as the putative HS‐binding domain. Superposition of Cα atoms of these residues in the native forms of the two serpins gives a RMSD of 1.5 Å (not shown). Likewise, the structure of HCII in complex with S195A‐thrombin displays extensive similarities with that of the heparin pentasaccharide‐activated AT suggesting high degree of similarity between the activated forms of the two serpins (Cα RMSD=1.5 Å; see Figure S1). Yet, the homologous binding sites exhibit subtle differences. Whereas hA of the two serpins superpose nearly completely (except for the N‐terminus), a significant ≈26° difference is found for hD between the two serpins (Figure S1). Also, hD of HCII contains an additional electropositive residue, R184, which has no counterpart in AT. We posited that these differences may form the basis for distinct HS sequences targeting the two serpins.
We first constructed a comprehensive library of HS hexasaccharide sequences built from all possible monosaccharides reported to date in nature. These residues lead to a library of disaccharide building blocks containing glucosamines (GlcN) with substitutions at 2‐, 3‐, and 6‐positions and uronic acids (UA) with substitutions at 2‐position. Further, our library explicitly studied the iduronic acid (IdoA) residue in either 2SO or 1C4 form. This gave a library of 36 disaccharide building blocks. Combinatorial conjugation of these 36 building blocks resulted in a library of 46 656 (=36×36×36; see Figure S2) unique hexasaccharide sequences.
This library was then docked onto the homologous electropositive sites of the two serpins by implementing our CVLS algorithm (Figure 1 a). The algorithm relies on GOLDscore, a measure of in silico “affinity” (the first filter) and consistency of binding, a measure of in silico “specificity” (the second filter), as described earlier.14, 15 The algorithm rapidly identifies key sequences, i.e., “specific” sequences, from the majority that recognize the binding site in a “non‐specific” manner.
Figure 1.
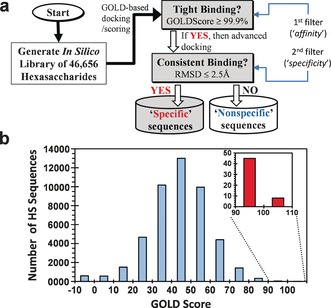
a) The dual‐filter CVLS algorithm used the library of 46 656 HS hexasaccharides. GOLD score was the first filter, while consistency of binding was the second filter. b) Results from the first filter displaying the histogram of number of HS hexasaccharides for every 10 unit change in GOLD score for HCII recognition.
Application of the first “affinity” filter showed that the majority of HS sequences (≈83.5 %) bind HCII with poor to moderate GOLDscores (−10–80 units, Figure 1 b). Only 0.8 % sequences recognize the HCII with high GOLDscores between 80–106 units (see the Supporting Information for details on methodology). In comparison, the HS‐AT system14, 16 displayed 20–30 units higher GOLDscores suggesting that the HS–HCII interaction is intrinsically likely to exhibit lower affinity than HS–AT.
We chose the top 0.1 %, i.e., 47 sequences, as candidates for the convergence (“specificity”) test. Interestingly, the ratio of IdoA‐ and GlcA‐containing disaccharides in the parent 46 656‐member library was 2.6:1, while it was 1.2:1 in the 47 hits implying a significant enrichment of GlcA‐residues with regard to HCII recognition. Further, none of the 47 sequences correspond to the AT‐binding high‐affinity pentasaccharide sequence, which is consistent with the literature.17
Application of the consistency of binding filter to the 47 sequences resulted in 3 sequences that recognized HCII with high in silico “specificity” (Figures 2 and S3–S5). These sequences, HX1, HX2 and HX3 (Figure 2 b), are structurally unique and likely to be infrequently found in natural HS. None contain GlcN2Ac, the residue most dominant in HS. Likewise, none of these sequences are heparin‐like because their IdoA composition (≈10 %) is much lower than that present in heparin (>80 %). Most interestingly, all three sequences contain one or more GlcA2S and GlcNS3S residues, which are rare in natural HS, but known to target AT in a selective manner.5, 6, 18 Our CVLS algorithm predicted HX1, HX2 and HX3 to bind to activated HCII in an essentially identical orientation with an RMSD of only 1.6 Å (Figures 2 a and S3). Yet, this orientation was dramatically different from that of heparin pentasaccharide onto AT. HX1, HX2 and HX3 orient at a ≈60° angle relative to the hD axis, whereas heparin aligns with hD axis in AT7 (Figure S6).
Figure 2.
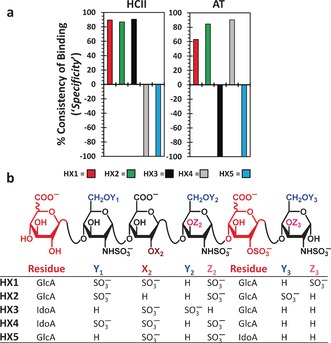
Predicted in silico “specificity” of recognition of HCII and AT by HX1–HX5 (a) and structures of five designed hexasaccharides (b). See the Supporting Information for description of calculation of in silico specificity.
To derive sequences that would serve as appropriate controls for the test of selectivity, we randomly selected 50 sequences from the library of 46 656 and subjected them to the consistency of binding test. This led to the identification of HX4 and HX5 (Figure 2 b), which displayed a poor consistency of binding and a predicted orientation some 15–20° away from that of HX1‐HX3 (Figures S3–S5).
To further assess the selectivity of recognition, we studied the interaction of the three designed sequences (HX1‐HX3) with related serpins (alpha‐1‐antitrypsin, plasminogen activator inhibitor‐1, protein C inhibitor and protease nexin‐1) as well as with other possible coagulation factor targets (thrombin, factor IXa, factor Xa and factor XIa). The consistency of binding analysis indicated that the three key sequences (HX1, HX2 and HX3) failed to recognize any other serpin or coagulation factor with selectivity higher than 80 % (Figures S7 and S8). Thus, the computational predictions support the idea that the de novo sequences present an excellent opportunity to address the challenge of differentiating two closely related HS‐binding proteins.
The synthesis of hexasaccharides containing such rare GlcA2S and GlcNS3S residues has not been reported in the literature.
Our synthetic strategy was based on 11 disaccharide building blocks (D1–D11, Figure 3) and involved two glycosylation and multiple protection/deprotection and sulfation steps. We utilized the established protecting group strategy in which acyl groups were used as temporary protection for O‐sulfation, benzyl groups as permanent protecting groups, and azides as non‐participating groups/precursors for N‐sulfation.19, 20 All the GlcA‐containing disaccharide blocks were efficiently obtained from cellobiose via a common crystalline intermediate, ethyl 3,6,2′,3′‐tetra‐O‐acetyl‐2‐azido‐4′,6′‐O‐benzylidene‐2‐deoxy‐1‐thio‐α‐d‐cellobiose.21 After deacetylation, various regioselective benzylation methods were employed to facilitate the right O‐sulfation pattern in the targets followed by acylation and hydrolysis (or reductive opening) of the benzylidene acetal. TEMPO‐oxidation and methyl ester formation then afforded GlcA disaccharide structures that could directly be utilized as terminal non‐reducing residues (D3 and D11), or after 4′‐O‐chloroacetylation as middle disaccharide units (D2, D4 and D6), or after methyl glycoside formation as reducing end moieties (D1, D5, and D7) (see the Supporting Information for description of these syntheses).
Figure 3.
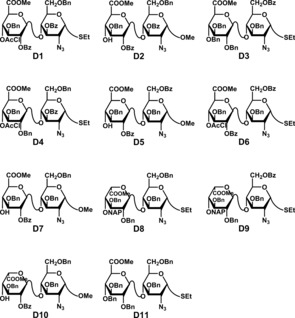
Structures of disaccharide building blocks D1–D11. Of these, D1, D3, D4, D6, D8, D9 and D11 are donor units, while acceptor disaccharides include D2, D5, D7 and D10.
To synthesize higher oligosaccharides, the key idea involved glycosylation of methyl glycoside disaccharide acceptors (D2, D5, D7 and D10) with appropriate thioglycoside disaccharide donors (D1, D4 and D6) to give tetrasaccharides (see Figure 4 for an example). These were dechloroacetylated first and then glycosylated (with D3, D8, D9 and D11) to yield target protected hexasaccharides H1–H5 (Figure 4 and S10–S14).
Figure 4.
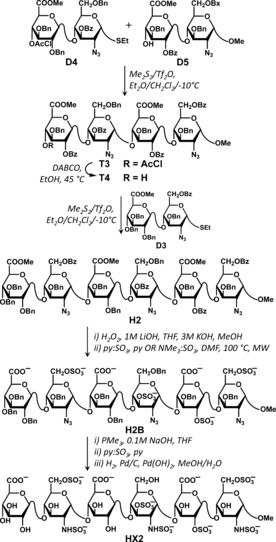
Synthesis of HX2 as a prototypical example of the thioglycoside donor methodology used in the synthesis of designed HS hexasaccharides. See Figures S10–S14 for detailed synthesis of HX1 through HX5 and associated procedures and characterization.
Although each glycosylation in the synthesis of designed hexasaccharides HX1 to HX5 was different, we reasoned that the similarities in structures of all the acceptor and donor disaccharides might make it possible to find general conditions that work well for all targeted molecules. Hence we studied HX2 synthesis in more detail and attempted optimization of glysosylation step. Of the multiple promoters studied for the coupling of acceptor D5 and donor D4 (Figure 4), Me2S/Tf2O22 gave the best result. Using a CH2Cl2/Et2O solvent system at −40 °C, a 70 % yield of tetrasaccharide T3 was obtained as a 3:1 α/β‐mixture. To improve the α‐selectivity higher temperatures were investigated, as suggested in the literature.23, 24 A temperature of −10 °C afforded T3 in similar yield (66 %) but with complete α‐stereoselectivity. These conditions were then utilized in glycosylation reactions to get intermediate, appropriately protected tetra‐ (T1, T5, and T7) and hexasaccharides (H1–H5) with good to excellent results (55–98 % yields, 3:1 to >15:1 α‐selectivity, see Table S1). Only in one glycosylation, i.e., between donor D1 and acceptor D10 to prepare tetrasaccharide T9, was further optimization required. We suspect that this is probably because D10 was the only IdoA acceptor studied. This problem was resolved by converting thioglycoside to trichloroacetimidate, i.e., donor D1 to D1TCA (see Figure S14), which afforded T9 in an 85 % yield (α/β 7:1).
The intermediate hexasaccharides H1–H5 were then sought to be transformed into the N,O‐sulfated de‐protected target structures HX1–HX5. We attempted to establish generalized conditions for these reactions by studying conversion of H2 into HX2 (Figure 4). Since the non‐reducing residue is a 4‐O‐benzyl‐protected uronic acid, care has to be exercised to avoid elimination in saponification reaction. As described earlier,25 a one pot procedure of H2O2 with LiOH efficiently reduces elimination side product and gave the deacetylated and demethylated product (H2A) in high yield (93 %). The freed hydroxyls were then sulfated using pyridine or triethylamine‐sulfur trioxide complex under microwave conditions to give H2B in 72 % yield. Microwave sulfation is known to be higher yielding than normal heating.26, 27
Finally, to remove the benzyl protecting groups, we first resorted to catalytic hydrogenolysis, which would concomitantly reduce the 2‐azides to 2‐amino groups. However, the reaction was found to be very sluggish and low yielding. By contrast, initial Staudinger reduction of the 2‐azides using trimethylphosphine followed by N‐sulfation and catalytic hydrogenolysis gave HX2 in high yield (77 % over three steps).28, 29
The conditions optimized in the synthesis of HX2 were found to work well for the deprotection/sulfation of all the other four target structures HX1, HX3–HX5 (saponification 75–99 %, O‐sulfation 82–92 %, N3‐reduction 82–98 %, N‐sulfation 72‐quant., and hydrogenolysis 81–89 % yields; see Figure S10–S14). The structural identity of each target hexasaccharide (HX1–HX5) was ascertained through combination of 1D and 2D NMR spectroscopy and ESI‐MS spectrometry. This was also greatly aided by our use of the classical, orthogonal protecting group strategy, which has been shown to work very reliably through years of heparin synthesis. Overall, we developed a general thioglycoside‐based synthetic pathway for unique hexasaccharides in eight steps from starting disaccharide building blocks with an overall yield of 10–33 % (Table S1).
To put the computational predictions to test, we measured the affinity of the designed hexasaccharides with HCII and AT utilizing intrinsic spectrofluorimetry, as reported in literature.30 All five hexasaccharides demonstrated a distinct and saturable change in fluorescence emission at pH 7.4 with both serpins from which the K Ds were calculated (Figure S15). For HCII, the K Ds were found to be in the range of 14 to 46 μm (Table S2), which are in the range of affinities reported for the polymeric entities (20–40 μm).10, 31 In comparison, the hexasaccharides bound AT with much more varied affinities (1 to 75 μm, Table S2). These affinities correlate well with the computationally predicted order of interactions. HX1, HX2 and HX3 bound HCII better than HX4 and HX5, as predicted. Likewise, HX4 displayed much better affinity for AT than HX1 and HX3, which in turn were better than HX5, as predicted. Overall, the affinities of HX1‐HX5 were not as high as that of heparin for AT. This was also predicted by our CVLS protocol, as described above (Figure 1 b).
To assess the influence of the designed HX sequences on the ability of the two serpins to inhibit their natural target proteases, we studied the kinetics of inhibition of thrombin (TH) by HCII and factor Xa (fXa) by AT in the presence of HX1–HX5, as reported earlier.8 The exponential decrease in residual protease activity as a function of time was used to derive the observed pseudo‐first order rate constant (k OBS) (Figure S16). The k OBS of inhibition was found to increase linearly with the concentration of HX–serpin complex (Figure S17), which was used to derive the second‐order rate constant for the uncatalyzed inhibition (k UNCAT) from the intercept and HX‐catalyzed inhibition from the slope (k HX) (Table S2). The k UNCAT for HCII–TH system and AT–fXa system was found to be 1.2–1.6×103 m −1 s−1 and 2.2–2.4×103 m −1 s−1, respectively, which compare favorably with the corresponding basal rates reported in the literature.8, 10 The k HX for HCII–TH reaction in the presence of HX1, HX3 and HX4 was measured to be (2.2–3.9)×105 m −1 s−1. This implies that HX1, HX3 and HX4 induce an activation of 180±70‐, 246±16‐ and 138±16‐fold, respectively (Figure 5 and Table S2), in HCII. In contrast, HX2 and HX5 induced an activation of 35±6‐ and 68±3‐fold, respectively, in HCII. We also measured fondaparinux (FPX) induced activation of HCII inhibition of thrombin and found it to be ≈57‐fold. For AT inhibition of fXa, HX1, HX2 and HX4 revealed accelerations of 380‐, 310‐ and 245‐fold, respectively, while HX3 and HX5 displayed a weaker acceleration of 5‐ and 56‐fold (Table S2). These results on activation of the two serpins correlate well with the in silico predictions. HX1 and HX3 activated HCII better than HX4 and HX5, as predicted (see Figure 1 b). The only anomaly was HX2, which displayed a weaker activation of HCII. For the AT system, HX1, HX2 and HX4 activated the serpin much better than HX3 and HX5, as predicted.
Figure 5.
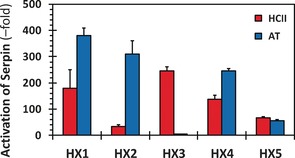
Comparison of the conformational activation of serpins HCII and AT induced by the CVLS‐designed HS hexasaccharides. Conformational activation was studied through increase in rate constant of inhibition of either thrombin or factor Xa, the two primary protease targets of HCII and AT, respectively. Activation of serpin (y‐axis) refers to the ratio of second order rate constant of inhibition in the presence of HX (k HX) to that in its absence (k UNCAT).
The seminal statement of this work is the identity of the GAG sequence that selectively targets HCII. HX3 induces HCII activation nearly 250‐fold, which is almost equal to that AT activation induced by fondaparinux, a clinically used anticoagulant.5, 8 However, HX3 is extremely poor in activating AT (only 5‐fold). The structure of HX3 includes consecutive GlcA2S residues, which are rarely found in HS. These residues are also present in HX1 and HX4, and both display high HCII activation. Yet, both HX1 and HX4 activate AT too because of the presence of 3‐O‐sulfated GlcN residue. This implies that for selective HCII activation (i.e., no AT activation), a HS sequence should contain two consecutive GlcA2S residues and be devoid of GlcN3S. This unique sequence requirement is also supported by HX2, which has only one GlcA2S residue and therefore dysfunctional in HCII activation.
A second major statement of our work is the possibility of utilizing appropriate computational tools in designing GAG sequences, especially of heparin/heparan sulfate type. To the best of our knowledge, this is the first report on the success of computationally designing de novo sequences that interact with high level of specificity. Our in silico interaction analyses presents the atomistic basis for the origin of specificity. CVLS models show that differences in activation levels probably arise from differential involvement of residues thought to be involved in binding, that is, R103, R184, K185, R189, R464, R192, and R193 (see Figures S4 and S5). In fact, R184, which occurs in HCII but not in AT, appears to be a key contributor to the recognition characteristics for HCII. A future goal of our work is to elucidate the co‐crystal structure of HCII with one or more of the designed hexasaccharides (HX1–HX5) to further advance the concept of selective HCII recognition.
Finally, we expect the HX3 sequence structure to help design advanced sequences that bind HCII with higher affinity and thereby serve as a promising leads. A simple strategy would be to add an appropriate disaccharide on either side of the HX3 sequence. Although a comprehensive library of octasaccharides would be very difficult to computational screen (1 679 616 unique sequences), we have devised an algorithm to design octasaccharide sequences based on hits identified by screening hexasaccharides.16 This dramatically enhances our CVLS technology, which in principle should help design longer oligosaccharides. We believe that this work lays a firm groundwork for discovering/designing unique GAG sequences that can selectively target GAG‐binding protein(s) with an eventual goal of deriving clinically viable candidate agents.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by grant HL107152 from the National Institutes of Health to U.R.D. and grants 08/RFP/CHE1211, 08/IN.1/B2067 and 13/IA/1959 from Science Foundation Ireland and an ERA‐Chemistry Open Initiative 2009 award to S.O. We also thank the Mizutani Foundation for Glycoscience and the computational resource provided by National Center for Research Resources (grant S10 RR027411).
N. V. Sankarayanarayanan, T. R. Strebel, R. S. Boothello, K. Sheerin, A. Raghuraman, F. Sallas, P. D. Mosier, N. D. Watermeyer, S. Oscarson, U. R. Desai, Angew. Chem. Int. Ed. 2017, 56, 2312.
Contributor Information
Prof. Dr. Stefan Oscarson, Email: stefan.oscarson@ucd.ie
Prof. Dr. Umesh R. Desai, Email: urdesai@vcu.edu.
References
- 1. Parker K. A., Tollefsen D. M., J. Biol. Chem. 1985, 260, 3501. [PubMed] [Google Scholar]
- 2. He L., Vicente C. P., Westrick R. J., Eitzman D. T., Tollefsen D. M., J. Clin. Invest. 2002, 109, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He L., Giri T. K., Vicente C. P., Tollefsen D. M., Blood 2008, 111, 4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rau J. C., Mitchell J. W., Fortenberry Y. M., Church F. C., Semin. Thromb. Hemostasis 2011, 37, 339. [DOI] [PubMed] [Google Scholar]
- 5. Desai U. R., Med. Res. Rev. 2004, 24, 151. [DOI] [PubMed] [Google Scholar]
- 6. Gettins P. G., Chem. Rev. 2002, 102, 4751. [DOI] [PubMed] [Google Scholar]
- 7. Jin L., Abrahams J. P., Skinner R., Petitou M., Pike R. N., Carrell R. N., Proc. Natl. Acad. Sci. USA 1997, 94, 14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desai U. R., Petitou M., Björk I., Olson S. T., J. Biol. Chem. 1998, 273, 7478. [DOI] [PubMed] [Google Scholar]
- 9. Baglin T. P., Carrell R. W., Church F. C., Esmon C. T., Huntington J. A., Proc. Natl. Acad. Sci. USA 2002, 99, 11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Keeffe D., Olson S. T., Gasiunas N., Gallagher J., Baglin T. P., Huntington J. A., J. Biol. Chem. 2004, 279, 50267. [DOI] [PubMed] [Google Scholar]
- 11. Zulueta M. M. L., Lin S.-Y., Hu Y.-P., Hung S.-C., Curr. Opin. Chem. Biol. 2013, 17, 1023. [DOI] [PubMed] [Google Scholar]
- 12. Liu J., Linhardt R. J., Nat. Prod. Rep. 2014, 31, 1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keiser N., Venkataraman G., Shriver Z., Sasisekharan R., Nat. Med. 2001, 7, 123. [DOI] [PubMed] [Google Scholar]
- 14. Raghuraman A., Mosier P. D., Desai U. R., J. Med. Chem. 2006, 49, 3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sankaranarayanan N. V., Sarkar A., Desai U. R., Mosier P. D., Methods Mol. Biol. 2015, 1229, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sankaranarayanan N. V., Desai U. R., Glycobiology 2014, 24, 1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maimone M. M., Tollefsen D. M., Biochem. Biophys. Res. Commun. 1988, 152, 1056. [DOI] [PubMed] [Google Scholar]
- 18. Boothello R. S., Sarkar A., Tran V. M., Nguyen T. K., Sankaranarayanan N. V., Mehta A. Y., Alabbas A., Brown S., Rossi A., Joice A. C., Mencio C. P., Quintero M. V., Kuberan B., Desai U. R., ACS Chem. Biol. 2015, 10, 1485. [DOI] [PubMed] [Google Scholar]
- 19. van Boeckel C. A. A., Petitou M., Angew. Chem. Int. Ed. Engl. 1993, 32, 1671; [Google Scholar]; Angew. Chem. 1993, 105, 1741. [Google Scholar]
- 20.K. Sheerin, The synthesis of structures related to heparin. A disaccharide approach starting from D-cellobiose, PhD thesis, University College Dublin, 2013.
- 21. Petitou M., Duchaussoy P., Lederman I., Choay J., Sinay P., Jacquinet J. C., Torri G., Carbohydr. Res. 1986, 147, 221. [DOI] [PubMed] [Google Scholar]
- 22. Tatai J., Fügedi P., Org. Lett. 2007, 9, 4647. [DOI] [PubMed] [Google Scholar]
- 23. Chao C.-S., Li C.-W., Chen M.-C., Chang S.-S., Mong K.-K. T., Chem. Eur. J. 2009, 15, 10972. [DOI] [PubMed] [Google Scholar]
- 24. Mong K.-K. T., Yen Y.-F., Hung W.-C., Lai Y.-H., Chen J.-H., Eur. J. Org. Chem. 2012, 3009. [Google Scholar]
- 25. Vogel C., Boye H., Kristen H., J. Prakt. Chem. 1990, 332, 28. [Google Scholar]
- 26. Raghuraman A., Riaz M., Hindle M., Desai U. R., Tetrahedron Lett. 2007, 48, 6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Al-Horani R. A., Desai U. R., Tetrahedron 2010, 66, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maza S., Macchione G., Ojeda R., Lopez-Prados J., Angulo J., de Paz J. L., Nieto P. M., Org. Biomol. Chem. 2012, 10, 2146. [DOI] [PubMed] [Google Scholar]
- 29. Noti C., de Paz J. L., Polito L., Seeberger P. H., Chem. Eur. J. 2006, 12, 8664. [DOI] [PubMed] [Google Scholar]
- 30. Boothello R. S., Al-Horani R. A., Desai U. R., Methods Mol. Biol. 2015, 1229, 335. [DOI] [PubMed] [Google Scholar]
- 31. Maimone M. M., Tollefsen D. M., J. Biol. Chem. 1990, 265, 18263. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary