

Abstract
Lignin structural studies play an essential role both in understanding the development of plant cell walls and for valorizing lignocellulosics as renewable biomaterials. Dimeric products released by selectively cleaving β–aryl ether linkages between lignin units reflect the distribution of recalcitrant lignin units, but have been neither absolutely defined nor quantitatively determined. Here, 12 guaiacyl‐type thioacidolysis dimers were identified and quantified using newly synthesized standards. One product previously attributed to deriving from β–1‐coupled units was established as resulting from β–5 units, correcting an analytical quandary. Another longstanding dilemma, that no β–β dimers were recognized in thioacidolysis products from gymnosperms, was resolved with the discovery of two such authenticated compounds. Individual GC response factors for each standard compound allowed rigorous quantification of dimeric products released from softwood lignins, affording insight into the various interunit‐linkage distributions in lignins and thereby guiding the valorization of lignocellulosics.
Keywords: analytical methods, GC-FID, lignin dimers, response factor, synthesis design
Lignin, a major component of plant cell‐wall (CW) complexes, plays an essential role in their development and strongly affects the utilization of sustainable lignocellulosic biomass.1 Lignin valorization is becoming essential for the overall economics of a lignocellulosic biorefinery.2 However, lignins have complex and heterogeneous structures that are neither absolutely definable nor determinable. The analysis of degradation products released by lignin depolymerization, especially dimeric and larger fragments, is valuable for characterizing lignins but is currently imperfect owing to the lack of authentic compounds and the resulting uncertainty regarding their structures.2b, 3 Such a status, to some extent, hinders the utilization of lignocellulosic biomass, especially lignins.
In softwood CWs, lignin consists primarily of guaiacyl (G) units, derived from coniferyl alcohol, along with a small proportion of p‐hydroxyphenyl (H) units, derived from p‐coumaryl alcohol.4 It has been well documented that the majority of interunit linkages in softwood lignins are, in order of decreasing abundance: β–O–4′ (β‐aryl ether, henceforth just termed β‐ether), β–5′ (phenylcoumaran), β–β′ (pinoresinol), and 5–5′ (biaryl, usually present in lignins as dibenzodioxocin units).5 Also present at relatively minor levels are β–1′ (spirodienone), and 4–O–5′ (diaryl ether) units. Degradative methods, including acidolysis, thioacetolysis, hydrogenolysis, and the widely used thioacidolysis and derivatization followed by reductive cleavage (DFRC) methods, have contributed extensively to our current knowledge of lignin structure.6 All of these methods rely primarily on β‐ether cleavage, such that any units or substructures linked by β‐ethers (at either their 4–O‐ or β‐positions) are released as low‐molecular‐weight compounds suitable for separation by gas or liquid chromatography (GC or LC), and analysis by mass (MS) or nuclear magnetic resonance (NMR) spectroscopy.
As an analytical method for the characterization of lignins, thioacidolysis has been successfully applied to a wide array of lignocellulosic materials and isolated lignins in various studies related to the biosynthesis of lignins, biological processing of plant biomass, and the pulping industry, as reviewed.7 By capitalizing on its selective cleavage of β‐ether units, diagnostic monomers and—after Raney‐Ni desulfurization to reduce the number of products and improve the volatility—dimeric fragments can be analyzed by GC or GC–MS. However, although this approach has provided insight into lignin structure, particularly with regard to carbon–carbon and diaryl ether linkages between units,8 it has also produced some dilemmas such as the large proportion of dimers that are β–1 (although only 1–2 % of lignin units are typically so‐linked) and the total absence, seemingly, of β–β‐units that are known to be quite prominent in softwood G‐lignins. Structural authentication, along with accurate quantification of lignin‐derived thioacidolysis products, normally accomplished using flame‐ionization detection (GC–FID) or GC–MS, would provide improved structural information. Historically, the most common thioacidolysis‐released dimers recovered after Raney‐Ni desulfurization were quantified based on the single GC response factor (RF) for the 5–5 (biphenyl) dimer XII (Figure 1), using docosane, tetracosane, or octadecane as the internal standard.9
Figure 1.
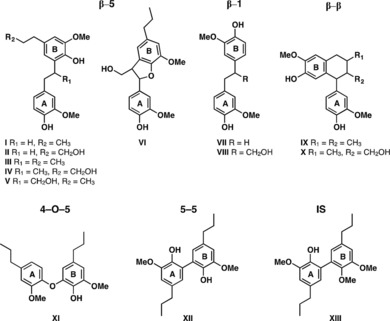
Structures of synthetic G‐type standard authentic compounds and the internal standard (IS) used in this study.
Among the many releasable thioacidolysis dimers, only the 5–5 (biphenyl) dimer has been authenticated by independent synthesis; the remaining dimers have been assigned based solely on analysis of their mass spectra. Accurate quantification of degradation fragments by GC strictly requires individual RFs between each analyte and the internal standard itself, but the authentic compounds required to obtain response factors have not been available. Thus, of the many laboratories using GC or GC–MS for analysis of thioacidolysis dimers, all have reported quantitative results based on the assumption that all dimers share the same GC–FID or GC–MS RF, which seems unlikely in light of their considerable structural diversity. Clearly, although lignin analysis by thioacidolysis has been improved over several decades by various researchers and, primarily, by the originator of the method, the complexity of lignins and the lack of authentic compounds still limits its efficacy to some degree.9a, 10 In particular, it has been difficult to quantify dimeric and oligomeric products owing to this lack of reference standards. Models for the original structures in lignin can be difficult to synthesize, as are the direct thioacidolysis products, the thioethyl ethers, owing to the abundance of regio‐ and stereoisomers that thioacidolysis releases. Unexpected side reactions also complicate the results. In some cases, as we show here, the synthesis of the products of thioacidolysis followed by Raney‐Ni desulfurization can be somewhat easier.
Recognizing a need for identification and quantification of dimeric degradation products, here we have synthesized a variety of G‐type dimeric thioacidolysis products (the same dimers can come from, for example, hydrogenolysis) and then used these G‐type lignin‐derived dimers I–XII (Figure 1) as standard authentic reference compounds to verify and quantify lignin dimers released by thioacidolysis. We have also synthesized a mono‐methylated 5–5‐linked dimer (XIII) to be used as an internal standard that, in comparison with the standards used previously, will have an equilibrium partitioning coefficient in organic and aqueous phases closer to those of the analytes and a more similar chemical response.
This newly synthesized library of compounds I–XII (but not yet compound VI, see below) contained the majority of lignin‐derived dimeric products known, or expected (in the case of the previously unreported β–β‐dimers IX and X), to be released from thioacidolysis of G‐type lignin. To confirm the presence of these products in thioacidolysis‐processed biomass, representative softwood (white spruce and loblolly pine) samples were subjected to the standard thioacidolysis method, followed by Raney‐Ni desulfurization. After silylating the reaction mixture using N,O‐bis(trimethylsilyl)trifluoroacetamide (BSTFA), the recovered degradation products underwent GC–FID and GC–MS analysis. Model compounds I–XII were also subjected to GC–MS and GC–FID analysis under the same conditions, and the resulting mass spectral data (Table S1 and Figure S5 in the Supporting Information) and GC retention times match those obtained from the white spruce sample (Figure 2), thus confirming these twelve synthesized compounds as authentic thioacidolysis products. All analyses were conducted in at least triplicate, with standard deviations all below 5 %.
Figure 2.
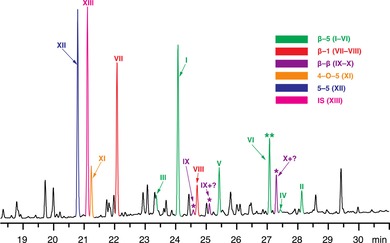
Partial (dimer region) GC–FID chromatogram of thioacidolysis products (after Raney‐Ni desulfurization) released from white spruce, showing determined peak identities. * β–β‐linked dimers are expected but have escaped detection among the degradation products from acidolysis and thioacidolysis in the past. ** VI is the newly identified β–5‐linked dimer that corrects an analytical quandary.
The quantification of individual “condensed” thioacidolysis‐released products, with their characteristic interunit linkages, is used to detail differences among various units in lignins. In the past, however, the required reference compounds for such a quantification were not available. Consequently, individual GC–FID RFs of synthesized dimers I–XII versus compound XIII were measured. Compared to the long‐chain hydrocarbons for which RFs have been reported,9 the synthesized mono‐methylated 5–5‐linked dimer XIII is structurally closer to the desulfurized lignin‐derived thioacidolysis dimers (I–XII) so that they have similar equilibrium partitioning concentrations between organic and water phases, thus providing more accurate results. The RFs determined for dimers I–XII varied from 0.87 to 1.17 (Table 1), rather than being invariant as previously assumed.9a These data also suggest that compound XIII is an appropriate internal standard for thioacidolysis dimers, as most of the RF values were close to 1. In other words, all of the dimers had appropriate RFs relative to the new internal standard by GC–FID, but displayed significant differences that, when taken into account, allow for more accurate quantification.
Table 1.
GC‐FID RFs of thioacidolysis/Raney‐Ni dimers (I–XII) and their yields (μmol g−1 KL[a] or CEL lignin) from softwood samples.
| Dimers | RFs | Loblolly pine CW | White spruce CW | Loblolly pine CEL | White spruce CEL | Spruce MWL[c] | |
|---|---|---|---|---|---|---|---|
| β–5 | I | 0.92 | 55.7 | 49.8 | 40.0 | 45.0 | |
| II | 0.92 | 5.4 | 6.8 | 3.6 | 5.4 | ||
| III | 0.87 | 7.8 | 7.1 | 3.2 | 4.9 | ||
| IV | 1.07 | 2.0 | 1.6 | 1.1 | 1.1 | ||
| V | 0.95 | 36.8 | 13.3 | 15.9 | 23.8 | ||
| VI | 1.05 | 1.1 | 25.1 | 15.0 | 2.1 | ||
| total | I–VI | 108.8 | 103.7 | 78.6 | 82.3 | 65 | |
| RR [%] | 44.5 | 45.2 | 44.7 | 46.5 | 31 | ||
| β–1 | VII | 0.92 | 51.2 | 48.3 | 35.0 | 32.5 | |
| VIII | 0.94 | 8.8 | 10.3 | 11.1 | 7.4 | ||
| total | VII–VIII | 60.0 | 58.6 | 46.1 | 39.9 | 66 | |
| RR [%] | 24.5 | 25.5 | 26.2 | 22.5 | 31 | ||
| β–β | IX | 1.15 (1.26)[b] | 0.9 | 1.2 | 0.9 | 1.1 | – |
| X | 1.17 (1.02)[b] | 0.3 | 0.4 | 0.1 | 0.2 | – | |
| total | IX–X | 1.2 | 1.6 | 1.0 | 1.3 | – | |
| RR [%] | 0.5 | 0.7 | 0.6 | 0.7 | – | ||
| 4–O–5 | XI | 1.05 | 19.4 | 15.6 | 15.2 | 16.1 | 16 |
| RR [%] | 8.0 | 6.8 | 8.6 | 9.1 | 8 | ||
| 5–5 | XII | 0.87 | 55.1 | 49.9 | 35.1 | 37.5 | 62 |
| RR [%] | 22.5 | 21.8 | 19.9 | 21.2 | 30 | ||
| total | I–XII | 244.5 | 229.4 | 176.0 | 177.1 | 209 | |
[a] KL=Klason (acid‐insoluble) Lignin; KL contents for loblolly pine CW, white spruce CW, loblolly pine CEL, and white spruce CEL are 27.5, 28.6, 90, and 88 %, respectively. [b] RFs of IX and X were determined from their selected‐ion chromatograms; m/z 385, 439, and 386 were chosen for IX, X, and XIII, respectively. [c] Yields are from reported data.7b Relative ratios (RR) for each dimer are their percentages relative to the total identified dimers in this study. In addition, the GC–FID RF of XIII versus docosane is 1.12.
With these RFs in hand, four softwood samples, including two extract‐free whole‐CW samples and two cellulolytic enzyme lignins (CELs) from loblolly pine and white spruce, were analyzed by thioacidolysis (followed by Raney‐Ni desulfurization), again using XIII as the internal standard. The yields of dimers I–XII (μmol g−1 Klason lignin or μmol g−1 CEL lignin) obtained from each sample were adjusted based on each dimer's experimentally determined RF (Table 1).
The β–5, β–1, and 5–5 dimers (in order of decreasing abundance) are the three main dimeric products released by thioacidolysis from softwood CWs or CELs (Table 1). Although 2D HSQC NMR spectroscopy (Figure 3) showed that the contents of β–5 units B in both white spruce and loblolly pine lignins are close to each other, the total yield of β–5 dimers (I–V) obtained by thioacidolysis of the white spruce sample was much lower than that obtained from the loblolly pine sample (78.6 and 107.7 μmol g−1 Klason lignin, respectively), suggesting a possible issue with the current thioacidolysis product assignments. The total yield of β–5 dimers appeared to vary based on the yields of V, which was authenticated by our model compound, and compound VI (m/z=488) (Figure 2), which was at that time reported to be a β–6 dimer (but see below).9a Indeed, the data suggested an inverse relationship between the yields of V and VI, with a similar combined yield for the two in loblolly pine and white spruce (37.9 and 38.4 μmol g−1 Klason lignin, respectively). In other words, if peak VI was included as a β–5 product (consistent with our NMR analysis), the total yield of β–5 products obtained from the white spruce sample was close to that of loblolly pine. As shown in Table 1, similar results could be observed from the thioacidolysis of CELs. Based on these results and the mass spectral characteristics of VI, this compound appeared to have originated from β–5 structures (somehow related to V) during thioacidolysis or desulfurization. The peak for this compound VI (Figure 2) was originally assigned to be a β–6 dimer but, because such coupling is chemically impossible, it was later suggested to be consistent with a β–1 isochroman structure.8b, 9a, 11 From MS analysis and the herein established derivation of compound VI from a β–5 dimeric model compound, it is now clear that the structure shown in Figure 1 and Figure S1 is simply derived from an unopened phenylcoumaran, as will be described more fully elsewhere. Its more logical assignment as the β–5 dimer VI shown in Figure 1 not only partially helps to reduce the level of compounds purporting to be β–1‐derived, but also solves the apparent discrepancy in yields for β–5 thioacidolysis products between softwood samples. Accordingly, we included the yield of compound VI in the total yield of β–5 dimers for all samples.
Figure 3.

Partial short‐range 1H–13C (HSQC) NMR spectra (oxygenated aliphatic regions) of CELs isolated from loblolly pine and white spruce (acetylated, [D6]acetone); percentages are from volume integrals of α‐proton/carbon correlations, A+B+C+D+E=100 %.
The β–5 products released from loblolly pine CW, white spruce CW, loblolly pine CEL, and white spruce CEL accounted for approximately 44.5, 45.2, 44.7, and 46.5 % of the total identified dimeric products, respectively, although the yield of individual β–5 products varied between samples, especially for V and VI (Table 1). The concordance in relative abundances is remarkable, but the diversity of products released indicates that thioacidolysis of β–5 structures involves many complicated and competing pathways that might be difficult to exactly replicate between sample runs.9a The only measurement of real value is the total amount of product derived from a given structure in lignin, and the sum of the β–5 units now has a rather consistent value across these four samples, providing confidence in both the assignment and in the quantification of total β–5 dimers released.
Similar results were observed for the β–1 products VII and VIII. β–1 dimers, the second most abundant of the thioacidolysis dimeric products, arise from the breakdown of the native lignin spirodienones during the reaction. They accounted for 24.5, 25.5, 26.2, and 22.5 % of the total identified dimers in loblolly pine CW, white spruce CW, loblolly pine CEL, and white spruce CEL, respectively. Explaining the prevalence of β–1 products in the dimer fraction from various acidolytic degradative methods (acidolysis, thioacidolysis, and DFRC) has always been somewhat difficult especially because it was shown that traditional β–1 dimeric units cannot be found in the lignin polymer,12 and because the levels of the more recently discovered spirodienones that explain how the products of such β–1‐coupling arise in the polymer are typically <2 % of the units in softwoods.13 Without knowing the relative propensities for the various coupling modes of the various phenolic end‐units in lignin, a partial explanation is that β–1 units in spirodienones can only be etherified at one (latent) phenolic end during lignification,14 so the chance of that being a 4–O–β‐ether is high, and release of such β–1 units might be expected to be higher than from the other condensed units in which, typically, linkages to two other lignin units are present, thus requiring that they both must be β‐ethers to release the dimer.
It is well documented that β–β (resinol) structures are present in lignins and can be readily observed by NMR spectroscopy (Figure 3). It has been estimated that pinoresinol structures comprise at least 2 % of softwood lignins, based on the total C9 units.5a, 15 In the past, failure to detect the expected degradation products by acidolysis and thioacidolysis (e.g., dimer IX from thioacidolysis) led to the suggestion that pinoresinol structures were connected to the rest of the lignin chain by at least one 5‐linkage, which can be explained to some extent,16 but could mean that they formed in a manner different from the traditional theory of lignification.15b, 17 The non‐observation of (any) β–β dimers from acidolysis or thioacidolysis became one of the cornerstones for the championed dismissal of the current combinatorial coupling theory of lignification and was used in strong support of a notion of absolute structural control over lignin assembly by dirigent proteins (or dirigent sites on a putative protein),18 an over‐zealous hypothesis that has now lost a basis for its support.16 The dilemma from the apparent non‐release of β–β dimers by ether‐cleaving analytical procedures had already been refuted by studies of DFRC products,5a but it was never clear why DFRC would release such dimers whereas thioacidolysis would not. In this study we detected the expected β–β degradation product IX, further supporting the idea that either both phenolic ends on pinoresinol units can be involved in 4–O–β‐ethers, or that one can while the other phenol remains free, according to the traditional free‐radical theory of lignification in softwoods.5c Furthermore, dimer X, another pinoresinol degradation product, is identified here for the first time. In total, then, β–β dimers (IX and X) accounted for below 1 % (0.5, 0.7, 0.6, and 0.7 % of the dimers released from loblolly pine CW, white spruce CW, loblolly pine CEL, and white spruce CEL, respectively), but can no longer be claimed to be absent from the array of thioacidolysis dimers. As shown in Table 1, the total‐ion chromatogram and its selected‐ion spectrum was used for a better calculation of β–β dimers (IX and X), owing to the possible peak overlap with other products in the GC chromatogram.
The 4–O–5 dimer XI accounted for 8.0, 6.8, 8.6, and 9.1 of the total identified dimers in loblolly pine CW, white spruce CW, loblolly pine CEL, and white spruce CEL, respectively. Until recently, it was not clear why such products were detected at such moderate levels from the various degradative methods although 4–O–5‐linked structures had never been detected in NMR spectra of softwood lignins. Two studies have now reported their detection and authentication at low levels, estimated to be no more than approximately 1 % of lignin units.19 These studies also now partially explain why they are prevalent in the thioacidolysis dimers beyond their actual proportion in the lignin; both research groups reporting on the findings could find no evidence that 4–O–5 dimeric units in lignin were further etherified, which also implies that they do not form the branching structure typically envisioned.19
The 5–5‐linked dimer XII comprised 22.5, 21.8, 19.9, and 21.2 %, of the total identified dimers in loblolly pine CW, white spruce CW, loblolly pine CEL, and white spruce CEL. Although other biphenyl structures have been reported in lignin,5a, 11b, 20 dibenzodioxocins D (Figure 3) are evidenced to be the main biphenyl structures present,5a, 21 so the 5–5 dimers can be considered mostly as dibenzodioxocin degradation products. No estimate of the conversion yield from the eight‐membered ring dibenzodioxocins is yet available, and this might not even be accessible from model compounds given that attempted acidolytic cleavage of model dibenzodioxocins can yield stable seven‐membered‐ring oxepine products.21a, 22
CELs from white spruce and loblolly pine were isolated and used for this study. We chose this isolation method instead of using milled wood lignin (MWL), which has commonly been used in previous studies, because CEL preparations have higher yields (ca. 55 %),19a, 23 and should therefore be more representative. In contrast, MWLs obtained from softwoods are typically obtained in approximately 15 % yield based on Klason Lignin.23b Interestingly, the total relative yields of 5–5 and β–1 products released from MWL (as measured in previous studies) were much higher (31 and 30 %, respectively) than those from the CELs (19.9–22.5 and 22.5–26.2 %, respectively) we studied in this work (Table 1).7b With regard to 5–5 dimers, this discrepancy could be explained by the fact that we have included only dimer XII as a 5–5 product, whereas previous studies have included an additional (although unidentified/unauthenticated) 5–5 thioacidolysis dimer.7b, 10a As for the β–1 products, the difference in yields could be attributed to the fact that β–1 structures can be selectively enriched in MWLs, as previously reported.10a Nevertheless, the relatively lower abundance of β–1 products detected in CELs may suggest that the CELs used in this study are more representative than the MWLs used in previous studies, as hypothesized above.9a, 10a Finally, the total relative yield of β–5 products released from MWL was much lower (31 %) than the level we found from the CELs (44.5–46.5 %, Table 1); a discrepancy that likely arose either from the omission of dimers III and V in the calculation of β–5 products or the lack of compound‐specific RFs in previous studies. It should also be noted that the results obtained for the CELs were calculated based on the CEL weight, rather than on the actual lignin content of the isolate, which may have led to an underestimation of the yields reported in Table 1. The similar relative ratios of dimers recovered after thioacidolysis between the CEL and whole cell wall samples suggests that CEL is indeed representative,23b and that either material can be used in future studies.
In summary, a total of twelve guaiacyl‐type dimeric compounds, including six β–5, two β–1, two β–β, one 4–O–5, and one 5–5, released by thioacidolysis from softwood lignins have been verified and quantified with synthesized standard compounds. With the use of these authenticated compounds, β–β products from pinoresinol structures in softwood lignins were verified and quantified for the first time, supporting the traditional theory of lignification that β–β (resinol) structures of softwood lignins are formed through the usual combinatorial β–β‐coupling of coniferyl alcohol. Additionally, we have assigned VI as a β–5 product, in contrast to previous studies that assumed it to be a β–6 dimer or a β–1 isochroman. Moreover, the quantification of various thioacidolysis‐released dimeric products by using individual response factors (RFs) obtained in this work provides the most accurate information about carbon–carbon and diaryl ether units in various lignins that can be released by β‐ether cleaving reactions, and is also beneficial to our efforts toward lignocellulosics valorization.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank the China Scholarship Council, State Education Department for supporting F.Y. as a visiting student in the Department of Biochemistry and the Great Lakes Bioenergy Research Center (GLBRC) at the University of Wisconsin. This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science, DE‐FC02‐07ER64494).
F. Yue, F. Lu, M. Regner, R. Sun, J. Ralph, ChemSusChem 2017, 10, 830.
Contributor Information
Prof. Fachuang Lu, Email: fachuanglu@wisc.edu.
Prof. John Ralph, Email: jralph@wisc.edu.
References
- 1.
- 1a. Adler E., Wood Sci. Technol. 1977, 11, 169–218; [Google Scholar]
- 1b. Whetten R., Sederoff R., Plant Cell 1995, 7, 1001–1013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Sette M., Wechselberger R., Crestini C., Chem. Eur. J. 2011, 17, 9529–9535. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Shuai L., Amiri M. T., Questell-Santiago Y. M., Héroguel F., Li Y., Kim H., Meilan R., Chapple C., Ralph J., Luterbacher J. S., Science 2016, 354, 329–333; [DOI] [PubMed] [Google Scholar]
- 2b. Van den Bosch S., Schutyser W., Vanholme R., Driessen T., Koelewijn S.-F., Renders T., De Meester B., Huijgen W. J. J., Dehaen W., Courtin C. M., Lagrain B., Boerjan W., Sels B. F., Energy Environ. Sci. 2015, 8, 1748–1763; [Google Scholar]
- 2c. Lahive C. W., Deuss P. J., Lancefield C. S., Sun Z. H., Cordes D. B., Young C. M., Tran F., Slawin A. M. Z., de Vries J. G., Kamer P. C. J., Westwood N. J., Barta K., J. Am. Chem. Soc. 2016, 138, 8900–8911. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Li C. Z., Zhao X. C., Wang A. Q., Huber G. W., Zhang T., Chem. Rev. 2015, 115, 11559–11624; [DOI] [PubMed] [Google Scholar]
- 3b. Upton B. M., Kasko A. M., Chem. Rev. 2016, 116, 2275–2306. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Whetten R. W., MacKay J. J., Sederoff R. R., Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 585–609; [DOI] [PubMed] [Google Scholar]
- 4b. Ralph J., Phytochem. Rev. 2010, 9, 65–83; [Google Scholar]
- 4c. Ralph J., Bunzel M., Marita J. M., Hatfield R. D., Lu F., Kim H., Schatz P. F., Grabber J. H., Steinhart H., Phytochem. Rev. 2004, 3, 79–96. [Google Scholar]
- 5.
- 5a. Ralph J., Lundquist K., Brunow G., Lu F., Kim H., Schatz P. F., Marita J. M., Hatfield R. D., Ralph S. A., Christensen J. H., Boerjan W., Phytochem. Rev. 2004, 3, 29–60; [Google Scholar]
- 5b. Rinaldi R., Jastrzebshi R., Clough M. T., Ralph J., Kennema M., Bruijnincx P. C. A., Weckhuysen B. M., Angew. Chem. 2016, 55, 8164–8215; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8296–8354; [Google Scholar]
- 5c. Lignins: Occurrence, Formation, Structure and Reactions (Eds.: K. V. Sarkanen, C. H. Ludwig), John Wiley & Sons, New York, 1971. [Google Scholar]
- 6.
- 6a. Brunow G. in Lignin, Humic Substances and Coal, Vol. 1 (Eds.: M. Hofrichter, A. Steinbüchel), Wiley-VCH, Weinheim, 2001, pp. 89–116; [Google Scholar]
- 6b. Lapierre C. in Forage Cell Wall Structure and Digestibility (Eds.: H. G. Jung, D. R. Buxton, R. D. Hatfield, J. Ralph), American Society of Agronomy, Crop Science Society of America, Soil Science Society of America, Madison, 1993, pp. 133–166; [Google Scholar]
- 6c. Lu F. C., Ralph J., J. Agric. Food Chem. 1997, 45, 2590–2592. [Google Scholar]
- 7.
- 7a. Lapierre C., Pollet B., Petit-Conil M., Toval G., Romero J., Pilate G., Leple J. C., Boerjan W., Ferret V., De Nadai V., Jouanin L., Plant Physiol. 1999, 119, 153–163; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Lignin and Lignans: Advances in Chemistry (Eds.: C. Heitner, D. R. Dimmel, J. A. Schmidt), CRC Press, Boca Raton, 2010; [Google Scholar]
- 7c. Rolando C., Monties B., Lapierre C. in Methods in Lignin Chemistry (Eds.: S. Y. Lin, C. W. Dence), Springer, Berlin Heidelberg, 1992, pp. 334–349; [Google Scholar]
- 7d. Kim H., Ralph J., Lu F., Pilate G., Leplé J. C., Pollet B., Lapierre C., J. Biol. Chem. 2002, 277, 47412–47419; [DOI] [PubMed] [Google Scholar]
- 7e. Yue F., Lu F., Sun R.-C., Ralph J., J. Agric. Food Chem. 2012, 60, 922–928. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Rencoret J., Marques G., Gutiérrez A., Ibarra D., Li J., Gellerstedt G., Ignacio Santos J., Jiménez-Barbero J., Martínez A. T., del Río J. C., Holzforschung 2008, 62, 514–526; [Google Scholar]
- 8b. Rencoret J., Gutiérrez A., Nieto L., Jiménez-Barbero J., Faulds C. B., Kim H., Ralph J., Martínez Á. T., del Río J. C., Plant Physiol. 2011, 155, 667–682; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Choi J. W., Faix O., J. Wood Sci. 2010, 56, 242–249. [Google Scholar]
- 9.
- 9a. Lapierre C., Pollet B., Monties B., Rolando C., Holzforschung 1991, 45, 61–68; [Google Scholar]
- 9b. Lapierre C., Monties B., Rolando C., Holzforschung 1986, 40, 113–118; [Google Scholar]
- 9c. Lapierre C., Monties B., Rolando C., Holzforschung 1986, 40, 47–50. [Google Scholar]
- 10.
- 10a. Lapierre C., Pollet B., Monties B., Phytochemistry 1991, 30, 659–662; [Google Scholar]
- 10b. Lapierre C., Pollet B., MacKay J. J., Sederoff R. R., J. Agric. Food Chem. 2000, 48, 2326–2331. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. del Río J. C., Rencoret J., Marques G., Li J., Gellerstedt G., Jiménez-Barbero J., Martínez A. T., Gutiérrez A., J. Agric. Food Chem. 2009, 57, 10271–10281; [DOI] [PubMed] [Google Scholar]
- 11b. del Río J. C., Rencoret J., Gutiérrez A., Nieto L., Jiménez-Barbero J., Martínez A. T., J. Agric. Food Chem. 2011, 59, 11088–11099. [DOI] [PubMed] [Google Scholar]
- 12. Ede R. M., Ralph J., Torr K. M., Dawson B. S. W., Holzforschung 1996, 50, 161–164. [Google Scholar]
- 13.
- 13a. Zhang L., Gellerstedt G., Ralph J., Lu F., J. Wood Chem. Technol. 2006, 26, 65–79; [Google Scholar]
- 13b. Zhang L., Gellerstedt G., J. Chem. Soc. Chem. Commun. 2001, 2744–2745. [Google Scholar]
- 14. Gellerstedt G., Zhang L., Nord. Pulp Pap. Res. J. 1991, 6, 136–139. [Google Scholar]
- 15.
- 15a. Capanema E. A., Balakshin M. Y., Kadla J. F., J. Agric. Food Chem. 2004, 52, 1850–1860; [DOI] [PubMed] [Google Scholar]
- 15b. Yue F., Lu F., Sun R., Ralph J., Chem. Eur. J. 2012, 18, 16402–16410. [DOI] [PubMed] [Google Scholar]
- 16. Ralph J., Brunow G., Harris P. J., Dixon R. A., Schatz P. F., Boerjan W. in Recent Advances in Polyphenol Research, Vol. 1 (Eds.: F. Daayf, V. Lattanzio), Wiley-Blackwell, Oxford, 2008, pp. 36–66. [Google Scholar]
- 17. Zhang L. M., Henriksson G., Gellerstedt G., Org. Biomol. Chem. 2003, 1, 3621–3624. [DOI] [PubMed] [Google Scholar]
- 18. Davin L. B., Lewis N. G., Curr. Opin. Biotechnol. 2005, 16, 407–415. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Yue F., Lu F., Ralph S., Ralph J., Biomacromolecules 2016, 17, 1909–1920; [DOI] [PubMed] [Google Scholar]
- 19b. Li Y., Akiyama T., Yokoyama T., Matsumoto Y., Biomacromolecules 2016, 17, 1921–1929. [DOI] [PubMed] [Google Scholar]
- 20. Karhunen P., Rummakko P., Sipilä J., Brunow G., Kilpeläinen I., Tetrahedron Lett. 1995, 36, 169–170. [Google Scholar]
- 21.
- 21a. Argyropoulos D. S., Jurasek L., Kristofova L., Xia Z. C., Sun Y. J., Palus E., J. Agric. Food Chem. 2002, 50, 658–666; [DOI] [PubMed] [Google Scholar]
- 21b. Kukkola E. M., Koutaniemi S., Pollanen E., Gustafsson M., Karhunen P., Lundell T. K., Saranpaa P., Kilpelainen I., Teeri T. H., Fagerstedt K. V., Planta 2004, 218, 497–500; [DOI] [PubMed] [Google Scholar]
- 21c. Ralph J., Brunow G., Boerjan W. in Encyclopedia of Life Sciences (Eds.: F. Rose, K. Osborne), John Wiley & Sons, Chichester, 2007. [Google Scholar]
- 22. Karhunen P., Mikkola J., Parjunen A., Brunow G., Nord. Pulp Pap. Res. J. 1999, 14, 123–128. [Google Scholar]
- 23.
- 23a. Pew J. C., Tappi 1957, 40, 553–558; [Google Scholar]
- 23b. Capanema E., Balakshin M., Katahira R., Chang H. M., Jameel H., J. Wood Chem. Technol. 2014, 35, 17–26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary