

Abstract
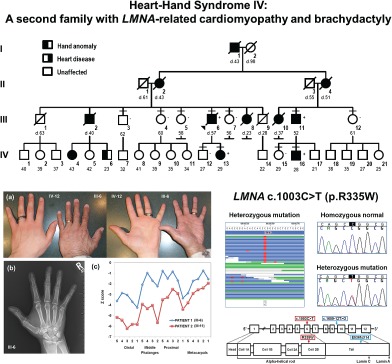
To the Editor:
Heart‐hand syndromes (HHS) include the most common type – HHS I: Holt–Oram syndrome (congenital heart disease and radial ray anomalies) and several rare types – HHS II: Tabatznik syndrome (arrhythmias and brachytelephalangy), HHS III: Spanish type (arrhythmia and brachydactyly type C), and a potential HHS IV: Slovenian type [arrhythmia, dilated cardiomyopathy (DCM), and brachydactyly] 1, 2. TBX5 for HHS I: Holt–Oram syndrome and LMNA for HHS‐Slovenian type (due to a cryptic splice‐site mutation) are the only two genes found to contribute to HHS 1, 2, 3.
We report a second HHS IV family due to a previously reported LMNA mutation (p.Arg335Trp). All procedures were approved by the UCI institutional review board. Written informed consent was obtained for all participants. The proband, who recently died at age 57, had DCM and ‘small hands.’ His family had German and Irish ancestry and multiple individuals born with ‘small hands’ and later developed heart disease. Eleven individuals were classified as affected or unaffected for heart disease 4 and hand anomalies (Fig. S1 and Table S1, Supporting Information). Characterization of the hands and feet for the proband (III‐6) and his affected first‐cousin (III‐11) revealed small hands with short digital rays (Fig. 1a). Hand radiographs showed shortened digital rays with no carpal anomalies (Fig. 1b), and metacarpophalangeal pattern (MCPP) profile analysis 5 revealed a consistent wavy pattern with brachydactyly of all tubular bones with marked shortening of the first and third distal phalanges, second middle phalanx and first proximal phalanx (Fig. 1c). For both patients, the feet were normal in appearance with normal radiographs (not shown). In addition, there were no signs of myopathy or lipodystrophy.
Figure 1.
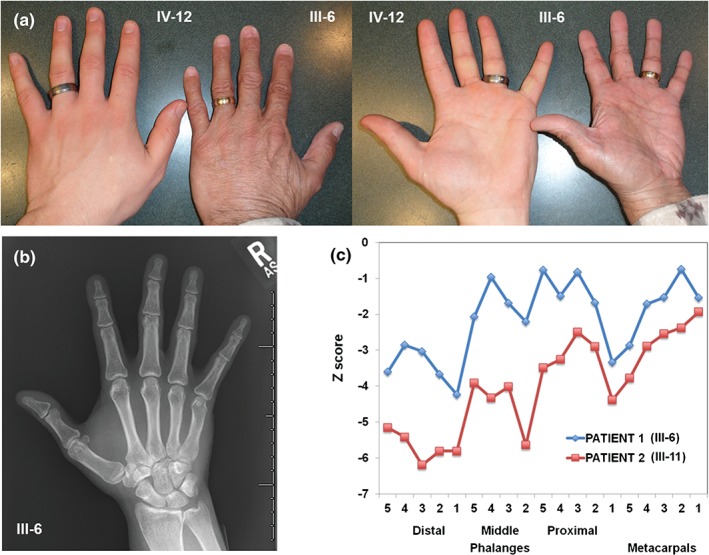
Characterization of hands. (a) Photographs showing the ‘small hand’ of the proband, Patient 1 (III‐6) compared to hand of his unaffected son (IV‐12). (b) Radiograph of the right hand for Patient 1 demonstrating short rays with no carpal anomalies. (c) Metacarpophalangeal pattern profiles of the right hands for Patient 1 and Patient 2 (III‐11) demonstrating a consistent wavy pattern with brachydactyly of all tubular bones and more pronounced shortening of the first and third distal phalanges, second middle phalanx and first proximal phalanx.
To identify the causative variant(s) for the cardiac and/or limb phenotypes, exome sequencing for the proband was conducted 4. The results included 14 gigabases of data from 164 million reads, 91% of mapped reads to hg19, and 134X average read coverage of each exon. To identify high‐priority candidate variants from 10,395 variants predicted to impact the protein, we focused on variants located in candidate genes associated with cardiomyopathy, arrhythmia, ion channels, or limb anomalies (Table S2). Ten candidates were identified (Table S3) and validated by Sanger sequencing. Family studies to determine co‐segregation identified LMNA c.1003C>T, p.Arg335Trp [RefSeq NM_170707.3], a heterozygous C>T missense variant in exon 6 as the primary mutation for both the cardiac and limb phenotypes (Figs S2 and S3).
With similar cardiac and skeletal features as the HHS‐Slovenian family 2, we provide further evidence for a fourth type of HHS due to a LMNA mutation. In both families, the cardiac features are typical for LMNA‐related cardiac disease 2, and the skeletal features evaluated by MCPP profile analysis displayed a similar wavy pattern with brachydactyly affecting all the tubular bones of the hands with more severely affected first and third distal phalanges and first proximal phalanx 2. Differences between the families were noted. Patients in the Slovenian family presented in the fourth to fifth decades with cardiac disease 2 while patients in our family often presented younger: daughter (IV‐13) with syncope at age 12, nephew (IV‐16) with cardiogenic shock at age 27, and sister with sudden death at age 23. Differences in skeletal features included severe foot involvement as short or absent phalanges, short metatarsal bones, and IV–V syndactyly in the Slovenian family 2 while our family showed no significant foot abnormalities.
Patients with LMNA c.1003C>T (p.Arg335Trp) and cardiac disease have been published; however, limb anomalies were not reported (ClinVar: Variation ID:36473 and Leiden Open Variation Database DB‐ID LMNA_00127). This includes a heterozygous patient with atrial fibrillation, hypertriglyceridemia, muscle weakness, and acro‐osteolysis. LMNA‐associated acro‐osteoylsis has been seen in autosomal recessive mandibuloacral dysplasia (OMIM 248370). The more severe involvement of the distal phalanges seen in both families and acro‐osteolysis seen in other LMNA patients suggests an increased susceptibility of the distal limb elements to disturbances in LMNA dosage.
The reasons for the clinical variability are not known. In the Slovenian family, a heterozygous cryptic splice‐site mutation LMNA c.1609‐12T>G was found; expression studies showed abnormal mRNA splicing and a truncated Lamin A/C protein with 14 new amino acids at the C‐terminus (p.E536fsX14) consistent with dominant negative or gain of function mechanism for disease 3. For the LMNA p.Arg335Trp missense mutation, a different molecular mechanism is possible. We also consider brachydactyly may be overlooked, and highlights the importance of a clinical genetics evaluation for these families. Further studies are needed to elucidate the mechanisms that influence the complex genotype–phenotype relationships in laminopathies and to determine the role of the LMNA to both cardiac function and limb development.
Supporting information
Table S1. Clinical features Table S2. Candidate genes Table S3. Candidate DNA variants Fig. S1. Pedigree. Squares denote males and circles females with current age below. Short horizontal lines mark consented individuals. Diagonal lines indicate deceased individuals with age of death (d). The proband is designated with an arrow. Left half‐filled and right half‐filled symbols indicate hand anomaly (small hands/brachydactyly) and cardiac disease (cardiomyopathy/arrhythmia), respectively. The presence (+) or absence (−) of LMNA c.1003C>T (p. Arg335Trp) is provided. Fig. S2. Family studies. Co‐segregation studies for 10 candidates (first column) were done by Sanger sequencing for eleven family members (top row). Results depicted as the presence (+) or absence (−) of the DNA variant. LMNA c.1003C>T (p. Arg335Trp) (highlighted) was present in all four affected individuals and absent in all seven unaffected family members. Fig. S3. LMNA c.1003C>T (p.Arg335Trp). Left panel shows the exome sequencing reads of the proband represented as horizontal lines. Variant nucleotides are shown in red. Right top panel shows the sequencing chromatograms: normal allele for unaffected individuals and heterozygous mutation for patients. Right bottom panel depicts the location of the mutation and the HHS‐Slovenian type mutation in the mRNA and Lamin A/C protein.
Acknowledgements
We are grateful to the study family. This work was supported by the U.S. Department of Health and Human Services: NIH NHLBI grant 1R01HL129008 (M. V. Z.).
References
- 1. McDermott DA, Fong JC, Basson CT. Holt‐Oram syndrome In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. University of Washington, Seattle, WA, 1993. [PubMed] [Google Scholar]
- 2. Sinkovec M, Petrovic D, Volk M, Peterlin B. Familial progressive sinoatrial and atrioventricular conduction disease of adult onset with sudden death, dilated cardiomyopathy, and brachydactyly. A new type of heart‐hand syndrome? Clin Genet 2005: 68: 155–160. [DOI] [PubMed] [Google Scholar]
- 3. Renou L, Stora S, Yaou RB et al. Heart‐hand syndrome of Slovenian type: a new kind of laminopathy. J Med Genet 2008: 45: 666–671. [DOI] [PubMed] [Google Scholar]
- 4. Zaragoza MV, Fung L, Jensen E et al. Exome sequencing identifies a novel LMNA splice‐site mutation and multigenic heterozygosity of potential modifiers in a family with sick sinus syndrome, dilated cardiomyopathy, and sudden cardiac death. PLoS One 2016: 11: e0155421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garn SM, Hertzog KP, Poznanski AK, Nagy JM. Metacarpophalangeal length in the evaluation of skeletal malformation. Radiology 1972: 105: 375–381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical features Table S2. Candidate genes Table S3. Candidate DNA variants Fig. S1. Pedigree. Squares denote males and circles females with current age below. Short horizontal lines mark consented individuals. Diagonal lines indicate deceased individuals with age of death (d). The proband is designated with an arrow. Left half‐filled and right half‐filled symbols indicate hand anomaly (small hands/brachydactyly) and cardiac disease (cardiomyopathy/arrhythmia), respectively. The presence (+) or absence (−) of LMNA c.1003C>T (p. Arg335Trp) is provided. Fig. S2. Family studies. Co‐segregation studies for 10 candidates (first column) were done by Sanger sequencing for eleven family members (top row). Results depicted as the presence (+) or absence (−) of the DNA variant. LMNA c.1003C>T (p. Arg335Trp) (highlighted) was present in all four affected individuals and absent in all seven unaffected family members. Fig. S3. LMNA c.1003C>T (p.Arg335Trp). Left panel shows the exome sequencing reads of the proband represented as horizontal lines. Variant nucleotides are shown in red. Right top panel shows the sequencing chromatograms: normal allele for unaffected individuals and heterozygous mutation for patients. Right bottom panel depicts the location of the mutation and the HHS‐Slovenian type mutation in the mRNA and Lamin A/C protein.