

Introduction
It was initially envisaged that upon completion of the Human Genome Project, our understanding of human disease would lead to the development of abundant new targeted therapies with which to modify disease progression or prevent disease onset. Furthermore, advances in next‐generation sequencing (NGS) have allowed for large‐scale DNA and RNA transcriptome analysis of multiple tissue and cell types encompassing a myriad of disease states, leading to an increased understanding of the composition and regulation of the human genome. However, as a result of such studies, it has become increasingly apparent that many chronic diseases are likely to be the result of the interaction between intrinsic genes and the external environment, which can result in modification of the DNA sequence and have an impact on gene expression.
This understanding has led to the emergence of the research field known as epigenetics, which includes the analysis of DNA modifications (including methylation and histone acetylation) as well as the transcription of noncoding RNAs (ncRNAs). Indeed, the protein‐coding elements of the human genome are now known to be restricted to only 2% of the total genetic material present. Initially, the remaining DNA was thought to be mostly “junk,” but much of this so‐called “genomic dark matter” is now known to transcribe multiple families of ncRNAs 1, many of which have been shown to modulate gene expression (Figure 1).
Figure 1.
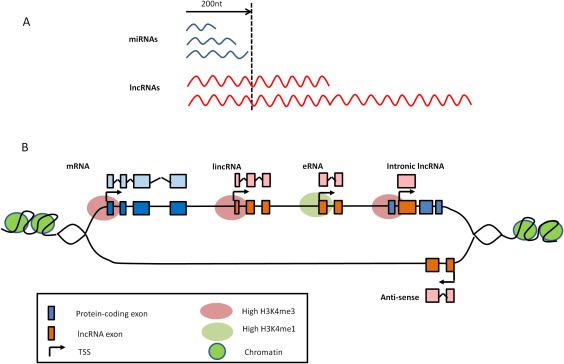
Classification of noncoding RNAs (ncRNAs) and long noncoding RNA (lncRNA) subtypes. A, LncRNAs are classified as being >200 nucleotides (nt) in length, in contrast to microRNAs (miRNAs), which are <200 nucleotides long. B, LncRNAs are further classified by their position relative to protein‐coding genes. Intronic lncRNAs are transcribed from intronic transcription start sites (TSS) and can be in either sense or antisense orientation. They do not intersect any exons. In contrast, antisense lncRNAs are transcribed across protein‐coding exons on the opposite strand. Long intergenic noncoding RNAs (lincRNAs) are transcribed from TSS between protein‐coding genes. Pseudogenes can be classified as a type of lincRNA and originate when a gene loses its coding ability (e.g., through a mutation). Enhancer RNAs (eRNAs) can also be considered a type of lincRNA but are transcribed from enhancer regions (identified by the presence of high H3K4me1 histone marks). In contrast, other lncRNAs are transcribed from regions associated with the presence of high H3K4me3 histone marks, similar to protein‐coding genes.
One such family of ncRNAs is the microRNA (miRNA) family 2, which binds to and prevents the translation of target messenger RNAs (mRNAs) (Figure 2A). Importantly, there is now overwhelming evidence that miRNAs are regulators of the inflammatory response 3, and several have been associated with multiple inflammatory diseases including osteoarthritis (OA) 3 and rheumatoid arthritis (RA) 4.
Figure 2.
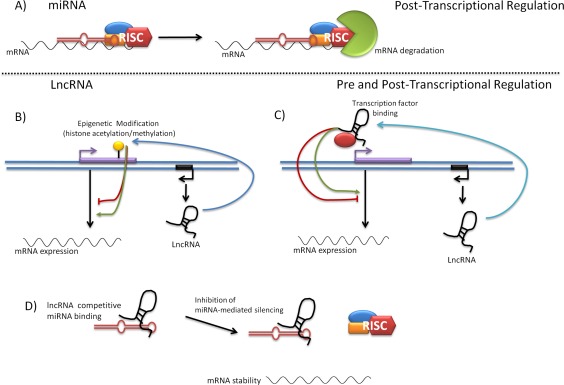
Functional diversity attributed to long noncoding RNAs (lncRNAs). A, Noncoding microRNAs (miRNAs) are involved in posttranscriptional regulation of gene expression. MiRNAs bind to the 3′‐untranslated region of target mRNAs in the cytoplasm. Assembly of an RNA‐induced silencing complex (RISC), composed of Ago2, Dicer, and R2D2, leads to mRNA degradation. Conversely, lncRNAs can modulate gene expression through both pretranscriptional and posttranscriptional regulation mechanisms. B, LncRNAs can act as scaffolds for the assembly of complexes that epigenetically modify the DNA (e.g., histone acetylation and methylation), leading to modulation of transcription. C, LncRNAs can bind to transcription factors and mediate assemblage of transcription initiation complexes, leading to modulation of transcription. D, LncRNAs can bind to miRNAs, acting as sponges to block their activity or affecting miRNA stability.
However, recent NGS studies have now identified a new family of ncRNAs known as long noncoding RNAs (lncRNAs) 5, which are defined as transcripts >200 nucleotides in length (Figure 1A). More than 14,000 lncRNAs have thus far been identified in humans 6, and they are currently subclassified, based on their position relative to protein‐coding genes (Figure 1B), as antisense lncRNAs, pseudogene lncRNAs, enhancer RNAs, intronic lncRNAs, and long intergenic noncoding RNAs (lincRNAs). These multiple variants may in part explain the functional diversity that has thus far been assigned to lncRNAs 7, including inhibiting transcriptional machinery, functioning as miRNA sponges, affecting miRNA stability, and epigenetically modifying DNA (Figures 2B–D). Many of these functions have been attributed to the theory that lncRNAs form flexible scaffolds that can bind proteins and RNAs.
Notably, increasing evidence suggests that lncRNAs are important regulators of pathologic and physiologic processes, and evidence is now emerging to suggest that lncRNAs are central regulators of the inflammatory response. Therefore, we examined evidence for the role of lncRNAs as regulators of inflammatory joint disease, by highlighting studies that have demonstrated differential expression of lncRNAs in the inflamed joint tissue of patients with RA and those with OA, as well as studies in which lncRNAs have been implicated as regulators of known inflammatory pathways relevant to joint pathology (Tables 1 and 2). LncRNAs are poorly conserved across species, which has been a limitation of studies performed to determine their function. This poor conservation places great importance on the requirement to validate the function of lncRNAs in disease‐relevant human cells and tissues 8. For this reason, our review focuses on lncRNA data generated from analysis of human cells and tissues.
Table 1.
Involvement of lncRNA in inflammatory pathwaysa
| Pathway, lncRNA | Tissue/cell | Chromosome locus | Function | Author, year (ref.) |
|---|---|---|---|---|
| NF‐κB signaling | ||||
| lincRNA‐p21 | T cells | 6p21.2 | Inhibition of NF‐κB following methotrexate treatment | Spurlock et al, 2014 18 |
| NKILA | Epithelial cells | 20q13.31 | Inhibition of IκB phosphorylation | Liu et al, 2015 20 |
| Lethe | Fibroblasts | NR | Inhibition of RelA DNA binding | Rapicavoli et al, 2013 19 |
| p38 MAPK | ||||
| MALAT1 | Endothelial cells | 11q13.1 | Reduced p38 phosphorylation and inaction of p38 MAPK signaling |
Liu et al, 2014 32
Puthanveetil et al, 2015 33 |
| Arachidonic acid | ||||
| PACER | Epithelial cells | 1q31.1 | Inhibition of COX‐2 | Krawczyk et al, 2014 25 |
| Toll‐like receptor | ||||
| THRIL | Macrophages | 12q24.31 | Complex formation with hnRNPL to induce TNF expression | Li et al, 2014 34 |
| IL‐1β–RBT46 | Monocytes | 2q14 | Inhibition of IL‐1β and IL‐8 secretion | Ilott et al, 2014 37 |
| LincRNA–COX‐2 | BMDCs | 1q31.1 | Up‐regulation of IL‐6 and down‐regulation of CCL5 | Carpenter et al, 2013 35 |
| NEAT1 | HeLa | 11q13.1 | Activated by p38 leading to expression of IL‐8 | Imamura et al, 2014 36 |
LncRNA = long‐noncoding RNA; lincRNA‐p21 = long intergenic noncoding RNA p21; NKILA = NF‐κB–interacting lncRNA; NR = not reported; MALAT1 = metastasis‐associated lung adenocarcinoma transcript 1; PACER = p50‐associated cyclooxygenase 2 (COX‐2) extragenic lncRNA; THRIL = tumor necrosis factor (TNF) and heterogeneous nuclear RNP L (hnRNPL)–related immunoregulatory lincRNA; IL‐1β–RBT46 = interleukin‐1β regions of bidirectional transcription 46; BMDCs = bone marrow–derived dendritic cells; NEAT1 = nuclear‐enriched abundant transcript 1.
Table 2.
Evidence for the differential expression of lncRNAs in osteoarthritis and rheumatoid arthritis joint tissuea
| Disease, lncRNA | Tissue/cell | Chromosome locus | Disease expression | Function | Author, year (ref.) |
|---|---|---|---|---|---|
| Osteoarthritis | |||||
| Hivep2‐AS (CILinc01) | Chondrocytes | 6q23‐q24 | Up‐regulated | Suppresses IL‐6, IL‐8, TNF, G‐CSF, MIP‐1β | Pearson et al, 2016 26 |
| IL‐7AS (CILinc02) | Chondrocytes | 8q12‐q13 | Up‐regulated | Suppresses IL‐6 | Pearson et al, 2016 26 |
| H19 | Chondrocytes | 11p15.5 | Up‐regulated | Anabolic functions | Steck et al, 2012 44 |
| LncRNA‐CIR | Chondrocytes | 6q22.32 | Up‐regulated | Promotes expression of MMP‐13 and ADAMTS‐5 while inhibiting collagen and aggrecan production | Liu et al, 2014 51 |
| MEG3 | Cartilage | 14q32.2 | Down‐regulated | Inversely correlated with VEGF | Su et al, 2015 49 |
| HOTAIR | Cartilage | 12q13.13 | Down‐regulated | Unknown | Xing et al, 2014 41 |
| GAS‐5 | Cartilage | 1q25.1 | Down‐regulated | Increases expression of MMPs and ADAMTS‐4; induces apoptosis | Song et al, 2014 48, Xing et al, 2014 41 |
| LncRNA uc.343 | Cartilage | NR | Up‐regulated | Regulation of HOXC8 | Fu et al 2015 (50) |
| Rheumatoid arthritis | |||||
| H19 | Synovium | 11p15.5 | Up‐regulated | Increases expression of NF‐κB, IL‐6, TNF | Stuhlmüller et al, 2003 43 |
| HOTAIR | Synovial fibroblasts, osteoclasts | 12q13.13 | Down‐regulated | Increases MMP‐2 and MMP‐13 expression | Song et al, 2015 42 |
| HOTAIR | Bone mononuclear cells | 12q13.13 | Up‐regulated | Migration and activation of macrophages | Song et al, 2015 42 |
| LincRNA‐p21 | T cells | 6p21.2 | Down‐regulated (up‐regulated following methotrexate treatment) | Inhibition of NF‐κB following methotrexate treatment | Spurlock et al, 2014 18 |
LncRNA = long noncoding RNA; Hivep2‐AS = human immunodeficiency virus type I enhancer binding protein 2AS; CILinc01 = chondrocyte inflammation–associated lincRNA 01; IL‐6 = interleukin‐6; TNF = tumor necrosis factor; G‐CSF = granulocyte colony‐stimulating factor; MIP‐1β = macrophage inflammatory protein 1β; lncRNA‐CIR = cartilage injury–related lncRNA; MMP‐13 = matrix metalloproteinase 13; MEG3 = maternally expressed gene 3; VEGF = vascular endothelial growth factor; HOTAIR = homeobox antisense intergenic RNA; GAS‐5 = grow arrest–specific 5; NR = not reported; HOXC8 = homeobox C8.
LncRNA regulation of inflammatory pathways
Inflammation is a central hallmark of RA joint disease, and evidence for inflammatory pathway dysregulation and cell dysfunction has been observed in almost all innate and adaptive immune cells. In addition, despite OA historically being seen as a “wear and tear” disease of cartilage, there is now substantial evidence that inflammation is also a key contributor to OA joint pathology 9. OA synovial inflammation (synovitis) has been shown histologically as well as by ultrasonography and magnetic resonance imaging 10, 11, and there is evidence for increased cellular infiltration of activated B cells and T lymphocytes and elevated levels of proinflammatory cytokines in synovial fluid 12. Furthermore, ex vivo stimulation of cartilage tissue with proinflammatory cytokines mimics several pathologic features of the OA joint 13, and inhibitors of inflammatory pathways have modified OA disease in preclinical models 14.
As described below, we examined current evidence for the role of lncRNAs in regulating canonical inflammatory pathways that are relevant to inflammatory joint pathology. Of the major signaling pathways that are known to govern joint inflammation, 4 pathways were highlighted as having lncRNAs associated with their regulation, namely NF‐κB signaling, the p38 MAPK pathway, the arachidonic acid pathway, and Toll‐like receptor (TLR) signaling.
NF‐κB pathway
NF‐κB is considered to be a master regulator of inflammation, and its activity has been shown to regulate the production of several proinflammatory cytokines implicated in OA and RA joint pathology, including interleukin‐1β (IL‐1β), IL‐6, IL‐17, and tumor necrosis factor (TNF) 12, 15. Therefore, inhibition of NF‐κB signaling has long been considered to be an attractive pathway for developing an effective therapy for chronic inflammatory disorders 15. However, therapeutic targeting of NF‐κB presents a challenge, because NF‐κB may also have an antiinflammatory role and be important for the resolution of inflammation 16. Therefore, it is important to identify new approaches to appropriately target the proinflammatory activity of NF‐κB within the joint 17, so that systemic effects are avoided and patients are not immunocompromised.
Thus far, several lncRNAs have been identified and implicated in targeting different components of the NF‐κB signaling pathway (Figure 3A). Of most immediate interest with regard to inflammatory joint disease is lincRNA‐p21, whose ability to inhibit NF‐κB signaling by sequestering RelA (the p65 subunit of NF‐κB) in T cells has been demonstrated following methotrexate treatment in patients with RA. The expression of LincRNA‐p21 was shown to be up‐regulated by methotrexate via a DNA‐dependent protein kinase catalytic subunit–dependent mechanism 18.
Figure 3.
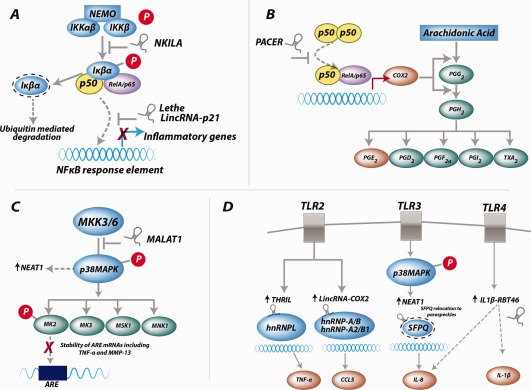
Functional long noncoding RNAs (lncRNAs) in inflammatory pathways. A, Canonical NF‐κB signaling. The lncRNA NKILA (NF‐κB–interacting lncRNA) blocks the Iκβα phosphorylation site, preventing its activation. The lncRNAs Lethe and long intergenic noncoding RNA p21 (lincRNA‐p21) both bind to RelA and prevent NF‐κB binding to NF‐κB response elements. B, Arachidonic acid pathway. The lncRNA p50‐associated cyclooxygenase 2 (COX‐2)–extragenic RNA (PACER) binds to and removes the repressive action of the p50 homodimer at the COX‐2 promoter, leading to activation of COX‐2 gene expression. C, P38 MAPK signaling. The lncRNA MALAT1 (metastasis‐associated lung adenocarcinoma transcript 1) inhibits p38 phosphorylation. Adenine‐rich elements (AREs) in mRNA transcripts are stabilized via downstream MAPK‐activated protein kinase 2 (MK‐2) activation. D, Toll‐like receptor (TLR) signaling. The lncRNAs THRIL (tumor necrosis factor [TNF] and heterogeneous nuclear RNP L [hnRNPL]–related immunoregulatory lincRNA) and lincRNA–COX‐2 form complexes with hnRNPLs, which then bind to and activate the promoters of proinflammatory cytokines TNF and CCL5, respectively. The lncRNA NEAT1 (nuclear‐enriched abundant transcript 1) binds to SFPQ (splicing factor proline/glutamine‐rich) and relocates it to paraspeckle bodies, relieving its repression of the interleukin‐8 (IL‐8) promoter. The lncRNA IL‐1β–RBT46 (IL‐1β regions of bidirectional transcription 46) is induced by TLR‐4 signaling and promotes induction of IL‐8 and IL‐1β via an as‐yet unknown mechanism. NEMO = NF‐κB essential modulator; PGG2 = prostaglandin G2; TXA2 = thromboxane A2; MMP‐13 = matrix metalloproteinase 13.
Another lncRNA that has been implicated as regulating NF‐κB activity is the pseudogene Lethe, which has been shown to be expressed in fibroblasts. Functional studies have demonstrated that Lethe interacts with and blocks the DNA binding of RelA (p65). It is believed that Lethe acts as part of a negative feedback loop that acts to regulate the inflammatory response, whereby following NF‐κB activation by proinflammatory cytokines, up‐regulation of Lethe inhibits NF‐κB signaling. Of interest, the tissue expression of Lethe has been shown to be down‐regulated in aged mice 19. If the same is true in human aging, the reduction in the expression of this potential brake on NF‐κB activity could contribute to age‐associated inflammatory joint disorders such as RA and OA. Finally, studies conducted in human inflammation‐stimulated breast epithelial cells have shown that the lncRNA NKILA (NF‐κB–interacting lncRNA) binds to and blocks phosphorylation sites on IκB, thereby inhibiting IKK‐induced IκB phosphorylation and NF‐κB activation 20. Thus far, there are no studies that demonstrated the functional role of lincRNA‐p21, Lethe, or NKILA in RA or OA joint tissues or cells.
Arachidonic acid pathway
Arachidonic acid metabolites (prostaglandins and leukotrienes) are known mediators of the inflammatory response 21, and several have been shown to be differentially expressed in the joints of patients with either OA or RA 22. Therefore, targeted inhibition of the arachidonic acid pathway has long been considered to be an important area of research for developing a therapeutic agent that combats joint inflammation and pain. Indeed, inhibition of the prostaglandin synthase (cyclooxygenase [COX]) enzymes is the basis for the mode of action of nonsteroidal antiinflammatory drugs (NSAIDs) such as ibuprofen 23 and selective PTGS‐2 (prostaglandin‐endoperoxide synthase 2)/COX‐2 inhibitors such as valdecoxib 24, which have shown efficacy in reducing joint inflammation in several clinical trials.
Of importance, therefore, was the recent finding that the expression of PTGS‐2 was positively regulated by the lncRNA p50‐associated COX‐2–extragenic RNA (PACER) 25 (Figure 3B). PACER was shown to bind to and remove the repressive action of the p50 homodimer (of NF‐κB) at the COX‐2 promoter, leading to activation of COX‐2 gene expression. Furthermore, we recently observed that the expression of PACER was associated with the IL‐1β–mediated inflammatory response in primary human OA chondrocytes and was differentially expressed in human OA cartilage compared with non‐OA cartilage 26.
Unfortunately, long‐term administration of nonselective NSAIDs is associated with gastrointestinal toxicity 27, while selective COX‐2 inhibitors increase the risk of cardiovascular adverse events 28, attributed to a resulting imbalance between thromboxane T and prostacyclin levels. Therefore, identifying new regulators of prostaglandin production such as PACER is important, because it may lead to new targeted approaches that are capable of selectively inhibiting the production of inflammation‐associated arachidonic acid metabolites without any resulting toxicity.
MAPK signaling
Several studies have provided evidence that the p38 MAPK pathway is central to mediating both the production of proinflammatory cytokines and their signal transduction in the joint 13. Furthermore, p38 small molecule inhibitors have shown efficacy in both reducing cartilage degeneration in OA preclinical models 29, 30 and reducing inflammatory pain in RA models 14, 31, although clinical trials of p38 inhibitors in patients with RA have thus far failed to meet clinical end points.
With regard to lncRNA regulation of the p38 pathway, recent studies suggest the involvement of MALAT1 (metastasis‐associated lung adenocarcinoma transcript 1) 32 (Figure 3C). MALAT1 is one of the most abundantly expressed lncRNAs, with high levels of conservation across multiple tissue types. It has previously been well‐characterized in cancer, in which it is a transcription regulator implicated in cell cycle control and metastasis. However, Liu and colleagues have now highlighted MALAT1 cross‐talk with p38 MAPK as being pathogenic in diabetes mellitus. MALAT1 is believed to activate p38 MAPK, because small interfering RNA (siRNA)–mediated knockdown of MALAT1 reduced p38 phosphorylation 32. Furthermore, MALAT1 expression has been shown to be implicated in inflammation, again in diabetes mellitus under hyperglycemic conditions; in this setting, it was shown to contribute to increased IL‐6 expression and reactive oxygen species generation 33.
TLR signaling
Innate immune cells such as macrophages and neutrophils are present in the inflamed joints of patients with RA and those with OA, and it is known that TLR signaling plays a key role in the recruitment and activation of these cells. Therefore, understanding the regulation of cytokine and chemokine signaling by these cells will provide greater clues regarding the onset and maintenance of chronic inflammation within the joint.
THRIL (TNF and heterogeneous nuclear RNP L [hnRNPL]–related immunoregulatory lincRNA) was recently identified in macrophages as an lncRNA that could form a complex with hnRNPL within the nucleoplasm (Figure 3D). HnRNPs are known to be important for the processing, function, and stabilization of mRNAs. Critically, it was demonstrated that the THRIL–hnRNPL complex binds to the promotor of TNF and induces its expression following TLR‐2 activation 34.
Carpenter and colleagues 35 identified an lncRNA (lincRNA–COX‐2) that is located in close proximity to the PTGS‐2 gene but, unlike the aforementioned PACER, this lncRNA had no functional interaction with PTGS‐2. Instead, lncRNA–COX‐2 was shown to be up‐regulated following activation of the TLR‐2 pathway (Figure 3D). Furthermore, following knockdown of lincRNA–COX2 and under the challenge of a synthetic TLR‐2 antagonist, expression of IL‐6 became undetectable, while CCL5 expression increased. Similarly to THRIL, lincRNA–COX‐2 forms complexes with hnRNPs, specifically hnRNP A/B and hnRNP A2/B1, and the formation of such complexes has been shown to modulate target gene expression.
Activation of TLR‐3 has been shown to induce an lncRNA referred to as NEAT1 (nuclear‐enriched abundant transcript 1). NEAT1 was shown to bind to a repressor of IL‐8 transcription known as SFPQ (splicing factor proline/glutamine‐rich) and relocate it to paraspeckle bodies (Figure 3D), resulting in IL‐8 transcriptional activation 36. Ilott and colleagues 37 demonstrated that TLR‐4 activation in monocytes up‐regulated expression of an lncRNA known as IL‐1β–RBT46 (IL‐1β regions of bidirectional transcription 46). IL‐1β–RBT46 shares a promoter with the IL‐1β–coding gene, and following pathway activation, expression of both the lincRNA and the IL‐1β gene is induced (Figure 3D). Locked nucleic acid (LNA)–mediated knockdown of IL‐1β–RBT46 led to a decrease in IL‐1β mRNA expression, suggesting that the lincRNA promotes expression of the gene. A further consequence of IL‐1β–RBT46 knockdown was a reduction in the expression of IL‐8, a well‐defined neutrophil chemoattractant.
The lncRNAs described above represent a pool of lncRNAs that have not yet been associated with inflammatory joint disease. However, given their prevalence in the regulation of central pathways of inflammation, studies determining their expression in joint tissue, and their relationship to OA and/or RA joint pathology would appear to be important.
The significance of genetic variations in the sequences of lncRNAs is currently not well understood and to date has been little studied. There is evidence that large alterations such as chromosomal rearrangements can affect lncRNA expression 38. However, as illustrated by the functional studies highlighted in this review, lncRNA function may be determined largely by its secondary structure, through binding to proteins and RNA. Therefore, smaller mutations and genetic variations such as single‐nucleotide polymorphisms (SNPs) may have little effect on lncRNA function. It is conceivable that SNPs could alter lncRNA function by altering the lncRNA secondary structure or causing alternative splicing of the lncRNA. Indeed, there is evidence that SNPs in the chromosomal region that harbor the lncRNA ANRIL are associated with susceptibility to cardiovascular disorders 39. However, it is not currently known whether these SNPs have an impact on the functional role of the ANRIL lncRNA or simply affect its expression. Predominantly, therefore, studies have thus far linked lncRNA to disease by expression analysis.
LncRNA expression in inflammatory OA and RA joint tissue
Transcriptional analyses by both NGS and microarray have identified several lncRNAs that are differentially expressed in inflamed joint tissue. Thus far, studies using OA patient samples have focused on cartilage tissue, while in RA disease other perijoint tissues such as synovium and subchondral bone have been interrogated.
One lncRNA that has been shown to be differentially expressed in both OA and RA joint tissue is HOTAIR (homeobox antisense intergenic RNA). Located on chromosome 12, HOTAIR was the first lncRNA to be characterized and has been shown to interact with polycomb repressive complex 2, which regulates the chromatin state, and the histone demethylase, lysine‐specific demethylase 1. As such, it regulates epigenetic changes within its target transcripts 40.
In OA cartilage, compared with non‐OA cartilage, HOTAIR was shown to be up‐regulated 41, although as yet its function in cartilage tissue or in isolated chondrocytes has not been reported. Song et al 42 have also identified differential expression of HOTAIR in RA perijoint tissues, namely synovial fibroblasts, osteoclasts, and bone mononuclear cells. Of note, Song et al found that the function of HOTAIR differed depending on the RA cell studied. For example, in RA synovial fibroblasts and osteoclasts, HOTAIR expression was lower than in controls, and its overexpression decreased the expression of matrix metalloproteinase 2 (MMP‐2) and MMP‐13. In contrast, HOTAIR was up‐regulated in RA bone mononuclear cells, where it functioned to facilitate macrophage activation and migration toward the joint.
Another lncRNA for which differential expression has been reported in inflamed joint tissue is H19. H19 lncRNA has been demonstrated to be highly expressed in RA synovial tissue 43 and in OA cartilage 44. Previous studies have demonstrated that H19 is a developmental reservoir of the 2 miRNA‐675 (miR‐675) family members, miR‐675‐p3 and miR‐675‐p5, which have been shown to regulate type II collagen expression and mediate cellular development in multiple tissues 45, 46, 47. Of interest, numerous studies have demonstrated that H19 controls the expression of several genes that are part of the imprinted gene network, a series of parentally inherited genes the expression of which is epigenetically determined within the sperm or oocyte. Such “imprinting disorders” have been linked to a number of human pathologies. Currently, inflammatory disease is not known to be affected by imprinting disorders, but given the inheritability of RA and other inflammatory diseases, studies that investigate the role of H19 in determining epigenetic susceptibility to disease could be of interest.
As previously mentioned, lncRNA analysis in OA has focused primarily on cartilage tissue, and the lncRNA GAS‐5 (growth arrest–specific 5) 48 and maternally expressed gene 3 (49) have been reported to be down‐regulated in OA‐diseased cartilage. Fu and colleagues 50 performed microarray analysis and identified 4,714 lncRNAs that were differentially expressed in OA cartilage compared with non‐OA cartilage. Although in the current study we did not perform any modulation of lncRNA expression to determine lncRNA function, expression correlation patterns were used to predict candidate lncRNA‐regulated genes. This analysis highlighted lncRNA uc.343, which was up‐regulated in OA cartilage, as regulating in cis the expression of homeobox C8 (50), and that many of the differentially expressed lncRNAs worked in concert with the transcription factor Sp1 to modulate expression of trans target genes.
In a separate study, microarray analysis was performed to identify a lncRNA with a specific role in the degradation of the cartilage matrix, named cartilage injury–related lncRNA (lncRNA‐CIR). LncRNA‐CIR was shown to be up‐regulated in OA cartilage compared with non‐OA cartilage, and siRNA‐mediated knockdown of its expression resulted in increased expression of collagen and aggrecan accompanied by reduced expression of MMP‐13 and ADAMTS‐5 (51).
Furthermore, we recently identified lncRNAs associated with the IL‐1β–mediated inflammatory response in primary human OA chondrocytes that were also differentially expressed in diseased OA cartilage. Of particular note were human immunodeficiency virus type I enhancer binding protein 2S (Hive2pAS) and IL‐7AS, 2 intergenic lncRNAs proximal to the Hivep2 and IL‐7 protein‐coding genes, respectively 26. Both Hive2pAS and IL‐7 AS were observed to be up‐regulated in both knee OA and hip OA cartilage compared with non‐OA control cartilage 26. Furthermore, locked nucleic acid (LNA)–mediated knockdown of Hive2pAS and IL‐7AS gene expression in human chondrocytes resulted in an increase in the production of proinflammatory cytokines, including IL‐6 (26), suggesting that these lncRNAs may function to control aberrant joint inflammation.
Conclusions
LncRNA research in inflammatory joint disease is very much a nascent field. However, several lncRNAs have now been identified as being either differentially expressed in diseased joint tissue or as candidate central regulators of inflammatory pathways relevant to joint pathology. Therefore, future studies to determine the functional role and mode of action of these disease‐associated lncRNAs will be insightful, as will joint tissue expression profiling of lncRNAs for which functional roles within key inflammatory pathways have been determined. To this end, it will be important to conduct further transcriptomic analysis of joint tissues (including subchondral bone, cartilage, and synovium) in which diseased joint tissues are compared with age‐matched noninflammatory control joint tissue and to conduct functional mode‐of‐action studies in disease‐relevant cells.
It should be noted that, currently, expression analysis of lncRNAs in the joint tissue of patients with other inflammatory joint diseases including gout, psoriatic arthritis, and juvenile idiopathic arthritis has not yet been reported. Clearly, such studies could also prove to be informative in building a broader pool of inflammatory joint disease–associated lncRNAs. Indeed, because inflammatory joint diseases share several common pathways, such studies are likely to identify lncRNAs that are dysregulated across multiple inflammatory joint disorders. Such findings will add to the recent paradigm shift in our understanding of the dysregulation in chronic disease, whereby the disease is no longer classified solely by its gross pathology but by the involvement of a particular dysregulated pathway.
It is currently too early to determine whether disease‐modifying therapeutic agents could be developed that are designed to directly target and modulate lncRNA function. Recent progress has been achieved using siRNA‐mediated RNA interference–based therapeutics, whereby targeted knockdown of TNF 52 or NF‐ĸB 53 has modified disease progression in animal models of RA and OA. Therefore, it is conceivable that lncRNAs could provide a new class of targets for RNA interference–based therapeutics. Regardless, ultimately determining the mode of action of lncRNAs that regulate inflammatory pathways and determining their relationship to joint pathology will provide a better understanding of how inflammation is epigenetically regulated within the joint. This improved understanding will likely be important in aiding the identification of new targets for therapeutic intervention as well as in identifying at‐risk patient populations.
AUTHOR CONTRIBUTIONS
Drs. Pearson and Jones were involved in drafting the article or revising it critically for important intellectual content, and both authors approved the final version to be published.
REFERENCES
- 1.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Okada Y, Muramatsu T, Suita N, Kanai M, Kawakami E, Iotchkova V, et al. Significant impact of miRNA‐target gene networks on genetics of human complex traits. Sci Rep 2016;6:22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones SW, Watkins G, Le Good N, Roberts S, Murphy CL, Brockbank SM, et al. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF‐α and MMP13. Osteoarthritis Cartilage 2009;17:464–72. [DOI] [PubMed] [Google Scholar]
- 4. Smigielska‐Czepiel K, van den Berg A, Jellema P, van der Lei RJ, Bijzet J, Kluiver J, et al. Comprehensive analysis of miRNA expression in T‐cell subsets of rheumatoid arthritis patients reveals defined signatures of naive and memory Tregs. Genes Immun 2014;15:115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin‐modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 2009;106:11667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 2012;22:1775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell 2011;43:904–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 2015;47:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tonge DP, Pearson MJ, Jones SW. The hallmarks of osteoarthritis and the potential to develop personalised disease‐modifying pharmacological therapeutics. Osteoarthritis Cartilage 2014;22:609–21. [DOI] [PubMed] [Google Scholar]
- 10. Oehler S, Neureiter D, Meyer‐Scholten C, Aigner T. Subtyping of osteoarthritic synoviopathy. Clin Exp Rheumatol 2002;20:633–40. [PubMed] [Google Scholar]
- 11. Rhodes LA, Conaghan PG, Radjenovic A, Grainger AJ, Emery P, McGonagle D. Further evidence that a cartilage‐pannus junction synovitis predilection is not a specific feature of rheumatoid arthritis. Ann Rheum Dis 2005;64:1347–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sohn DH, Sokolove J, Sharpe O, Erhart JC, Chandra PE, Lahey LJ, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll‐like receptor 4. Arthritis Res Ther 2012;14:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones SW, Brockbank SM, Clements KM, Le Good N, Campbell D, Read SJ, et al. Mitogen‐activated protein kinase‐activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology. Osteoarthritis Cartilage 2009;17:124–31. [DOI] [PubMed] [Google Scholar]
- 14. Badger AM, Griswold DE, Kapadia R, Blake S, Swift BA, Hoffman SJ, et al. Disease‐modifying activity of SB 242235, a selective inhibitor of p38 mitogen–activated protein kinase, in rat adjuvant‐induced arthritis. Arthritis Rheum 2000;43:175–83. [DOI] [PubMed] [Google Scholar]
- 15. Roman‐Blas JA, Jimenez SA. NF‐κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage 2006;14:839–48. [DOI] [PubMed] [Google Scholar]
- 16. Lawrence T, Fong C. The resolution of inflammation: anti‐inflammatory roles for NF‐κB. Int J Biochem Cell Biol 2010;42:519–23. [DOI] [PubMed] [Google Scholar]
- 17. Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF‐κB signaling: multiple angles to target OA. Curr Drug Targets 2010;11:599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spurlock CF 3rd, Tossberg JT, Matlock BK, Olsen NJ, Aune TM. Methotrexate inhibits NF‐κB activity via long intergenic (noncoding) RNA‐p21 induction. Arthritis Rheumatol 2014;66:2947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti‐inflammatory therapeutics. Elife 2013;2:e00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X, et al. A cytoplasmic NF‐κB interacting long noncoding RNA blocks IκB phosphorylation and suppresses breast cancer metastasis. Cancer Cell 2015;27:370–81. [DOI] [PubMed] [Google Scholar]
- 21. Samuelsson B. Arachidonic acid metabolism: role in inflammation. Z Rheumatol 1991;50 Suppl 1:3–6. [PubMed] [Google Scholar]
- 22. Kojima F, Kapoor M, Kawai S, Crofford LJ. New insights into eicosanoid biosynthetic pathways: implications for arthritis. Expert Rev Clin Immunol 2006;2:277–91. [DOI] [PubMed] [Google Scholar]
- 23. Bradley JD, Brandt KD, Katz BP, Kalasinski LA, Ryan SI. Comparison of an antiinflammatory dose of ibuprofen, an analgesic dose of ibuprofen, and acetaminophen in the treatment of patients with osteoarthritis of the knee. N Engl J Med 1991;325:87–91. [DOI] [PubMed] [Google Scholar]
- 24. Kivitz A, Eisen G, Zhao WW, Bevirt T, Recker DP. Randomized placebo‐controlled trial comparing efficacy and safety of valdecoxib with naproxen in patients with osteoarthritis. J Fam Pract 2002;51:530–7. [PubMed] [Google Scholar]
- 25. Krawczyk M, Emerson BM. p50‐associated COX‐2 extragenic RNA (PACER) activates COX‐2 gene expression by occluding repressive NF‐κB complexes. Elife 2014;3:e01776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pearson MJ, Philp AM, Heward JA, Roux BT, Walsh DA, Davis ET, et al. Long intergenic noncoding RNAs mediate the human chondrocyte inflammatory response and are differentially expressed in osteoarthritis cartilage. Arthritis Rheumatol 2016;68:845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med 1999;340:1888–99. [DOI] [PubMed] [Google Scholar]
- 28. Catella‐Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, et al. Effects of specific inhibition of cyclooxygenase‐2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther 1999;289:735–41. [PubMed] [Google Scholar]
- 29. Brown KK, Heitmeyer SA, Hookfin EB, Hsieh L, Buchalova M, Taiwo YO, et al. P38 MAP kinase inhibitors as potential therapeutics for the treatment of joint degeneration and pain associated with osteoarthritis. J Inflamm (Lond) 2008;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pelletier JP, Fernandes JC, Brunet J, Moldovan F, Schrier D, Flory C, et al. In vivo selective inhibition of mitogen‐activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum 2003;48:1582–93. [DOI] [PubMed] [Google Scholar]
- 31. Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther 1996;279:1453–61. [PubMed] [Google Scholar]
- 32. Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li YJ, et al. Pathogenic role of lncRNA‐MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis 2014;5:e1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non‐coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med 2015;19:1418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Z, Chao TC, Chang KY, Lin N, Patil VS, Shimizu C, et al. The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc Natl Acad Sci U S A 2014;111:1002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science 2013;341:789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, et al. Long noncoding RNA NEAT1‐dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell 2014;53:393–406. [DOI] [PubMed] [Google Scholar]
- 37. IIott NE, Heward JA, Roux B, Tsitsiou E, Fenwick PS, Lenzi L, et al. Long non‐coding RNAs and enhancer RNAs regulate the lipopolysaccharide‐induced inflammatory response in human monocytes. Nat Commun 2014;5:3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maass PG, Rump A, Schulz H, Stricker S, Schulze L, Platzer K, et al. A misplaced lncRNA causes brachydactyly in humans. J Clin Invest 2012;122:3990–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pasmant E, Sabbagh A, Vidaud M, Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J 2011;25:444–8. [DOI] [PubMed] [Google Scholar]
- 40. Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non‐coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010;464:1071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xing D, Liang JQ, Li Y, Lu J, Jia HB, Xu LY, et al. Identification of long noncoding RNA associated with osteoarthritis in humans. Orthop Surg 2014;6:288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song J, Kim D, Han J, Kim Y, Lee M, Jin EJ. PBMC and exosome‐derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin Exp Med 2015;15:121–6. [DOI] [PubMed] [Google Scholar]
- 43. Stuhlmüller B, Kunisch E, Franz J, Martinez‐Gamboa L, Hernandez MM, Pruss A, et al. Detection of oncofetal h19 RNA in rheumatoid arthritis synovial tissue. Am J Pathol 2003;163:901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steck E, Boeuf S, Gabler J, Werth N, Schnatzer P, Diederichs S, et al. Regulation of H19 and its encoded microRNA‐675 in osteoarthritis and under anabolic and catabolic in vitro conditions. J Mol Med 2012;90:1185–95. [DOI] [PubMed] [Google Scholar]
- 45. Ariel I, de Groot N, Hochberg A. Imprinted H19 gene expression in embryogenesis and human cancer: the oncofetal connection. Am J Med Genet 2000;91:46–50. [DOI] [PubMed] [Google Scholar]
- 46. Berteaux N, Lottin S, Monte D, Pinte S, Quatannens B, Coll J, et al. H19 mRNA‐like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J Biol Chem 2005;280:29625–36. [DOI] [PubMed] [Google Scholar]
- 47. Dey BK, Pfeifer K, Dutta A. The H19 long noncoding RNA gives rise to microRNAs miR‐675‐3p and miR‐675‐5p to promote skeletal muscle differentiation and regeneration. Genes Dev 2014;28:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Song J, Ahn C, Chun CH, Jin EJ. A long non‐coding RNA, GAS5, plays a critical role in the regulation of miR‐21 during osteoarthritis. J Orthop Res 2014;32:1628–35. [DOI] [PubMed] [Google Scholar]
- 49. Su W, Xie W, Shang Q, Su B. The long noncoding RNA MEG3 is downregulated and inversely associated with VEGF levels in osteoarthritis. Biomed Res Int 2015;2015:356893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fu M, Huang G, Zhang Z, Liu J, Zhang Z, Huang Z, et al. Expression profile of long noncoding RNAs in cartilage from knee osteoarthritis patients. Osteoarthritis Cartilage 2015;23:423–32. [DOI] [PubMed] [Google Scholar]
- 51. Liu Q, Zhang X, Dai L, Hu X, Zhu J, Li L, et al. Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis Rheumatol 2014;66:969–78. [DOI] [PubMed] [Google Scholar]
- 52. Komano Y, Yagi N, Onoue I, Kaneko K, Miyasaka N, Nanki T. Arthritic joint‐targeting small interfering RNA‐encapsulated liposome: implication for treatment strategy for rheumatoid arthritis. J Pharmacol Exp Ther 2012;340:109–13. [DOI] [PubMed] [Google Scholar]
- 53. Chen LX, Lin L, Wang HJ, Wei XL, Fu X, Zhang JY, et al. Suppression of early experimental osteoarthritis by in vivo delivery of the adenoviral vector‐mediated NF‐κBp65‐specific siRNA. Osteoarthritis Cartilage 2008;16:174–84. [DOI] [PubMed] [Google Scholar]