

Summary
This work investigates the extent of translational regulation during seed germination.
The polysome occupancy of each gene is determined by genome‐wide profiling of total mRNA and polysome‐associated mRNA. This reveals extensive translational regulation during Arabidopsis thaliana seed germination.
The polysome occupancy of thousands of individual mRNAs changes to a large extent during the germination process. Intriguingly, these changes are restricted to two temporal phases (shifts) during germination, seed hydration and germination. Sequence features, such as upstream open reading frame number, transcript length, mRNA stability, secondary structures, and the presence and location of specific motifs correlated with this translational regulation. These features differed significantly between the two shifts, indicating that independent mechanisms regulate translation during seed germination.
This study reveals substantial translational dynamics during seed germination and identifies development‐dependent sequence features and cis elements that correlate with the translation control, uncovering a novel and important layer of gene regulation during seed germination.
Keywords: Arabidopsis, germination, imbibition, polysomal profiling, ribosome, RNA structure, seedling establishment, translatomics
Introduction
Seed germination represents the start of a new plant life cycle. In seed plants, it involves the switch from a quiescent (dry seed) state to a metabolically active embryo which breaks through the encapsulating structures (endosperm and testa) to establish a young seedling. These early stages are critical for plant establishment and crop production. Arabidopsis seed germination is characterized by two visible events. First, the testa (seed coat) ruptures exposing the underlying endosperm layer, and second the endosperm ruptures which occurs when the root tip protrudes through the endosperm thereby completing germination sensu stricto.
In Arabidopsis, seed germination is triphasic starting with fast water uptake (imbibition, phase I). Genes encoding ribosomal proteins (r‐proteins) are not transcribed at this developmental stage (Jimenez‐Lopez et al., 2011). The first phase ends with a plateau phase (phase II) featuring the activation of a series of metabolic processes facilitating energy production and reserve mobilization. During this process, ribosomal protein gene expression and ribosomal activity increase dramatically, facilitating the de novo synthesis of proteins important for seed germination (Fu et al., 2005; Dekkers et al., 2013; Galland et al., 2014). The last phase (III) is characterized by testa and endosperm rupture (germination) followed by radicle extension associated with post‐germination events controlling seed to seedling transition. In the end, germinated seeds are equipped with the machineries and substrates necessary for autotrophic growth (Bewley, 1997). Intensive studies have elucidated the molecular changes during early seed imbibition and seed to seedling transition, including transcriptome (Yu et al., 2014), proteome (Gallardo et al., 2001) and metabolome (Fait et al., 2006) analyses. However, the steady state mRNA pool may not reflect the protein output due to lack of the linearity between transcription and translation (Gibon et al., 2006; Baerenfaller et al., 2008; Fernie & Stitt, 2012). Polysomal profiling using sucrose gradient‐based fractionation allows the separation of mRNAs based on their association with polysomes and thus the identification of mRNAs actively involved in translation. With high‐throughput mRNA profiling techniques such as microarray analysis and RNA sequencing, thousands of translated mRNAs can be quantified (Mustroph et al., 2009b; Layat et al., 2014; Lin et al., 2014; Vragovic et al., 2015). Generating datasets of both the total mRNA and the polysomal bound mRNA allows calculation of the ratio between the abundance of an individual mRNA in the total mRNA fraction and the abundance in the polysomal mRNA fraction. Changes in this ratio between time points or between different treatments indicate changes in the polysome occupancy, showing that a certain mRNA is under translational regulation. Polysome occupancy is partly synonymous with the term polysome loading and is used as a proxy of translational efficiency in the literature. This system has been successfully applied to investigate translational control in, for example, Saccharomyces cerevisiae (Arava et al., 2003; Halbeisen & Gerber, 2009; Ingolia et al., 2009), Aspergillus fumigatus (Krishnan et al., 2014), a mammalian cell line (de Klerk et al., 2015) and Arabidopsis thaliana (Jiao & Meyerowitz, 2010; Liu et al., 2012, 2013; Juntawong et al., 2014; Basbouss‐Serhal et al., 2015).
To investigate the degree and dynamics of translational regulation as well as to identify gene sets under translational regulation during germination, total mRNA and polysomal mRNA changes were investigated using microarray analysis of five consecutive stages during Arabidopsis seed germination. Thousands of individual mRNAs whose polysome occupancy was affected were identified. Intriguingly, changes in polysome occupancy were not uniformly present throughout the germination process but were restricted to two temporal phases, one encompassing seed hydration and one seed germination. Using bioinformatic analysis, we were able to correlate the translational regulation to mRNA structure and the presence of sequence motifs present in these mRNAs. Thus, next to the strong transcriptional regulation observed previously during Arabidopsis seed germination, this study identified large sets of genes that are regulated on the translational level, revealing an additional layer of gene expression regulation and its dynamics during germination.
Materials and Methods
Plant material and growth conditions
Seeds of the Arabidopsis thaliana (L.) Heynh accession Columbia‐0 (Col‐0) were used for all assays described (NASC N60000). The timing of testa and endosperm rupture and seedling greening of fully after‐ripened seeds was determined as described previously (Joosen et al., 2010). In brief, two layers of blue blotter paper (Anchor Paper Co., St Paul, MN, USA) were equilibrated with 48 ml of demineralized water in plastic trays (15 × 21 cm). For germination assays, c. 50–150 seeds were spread on wetted papers in the germination trays using a mask to ensure accurate spacing. Germination trays were stacked and wrapped in a closed transparent plastic bag to ensure equal illumination from the sides to each plate. The experiment was carried out in a 22°C incubator under continuous light (143 μmol m−2 s−1). Germination parameters were manually counted.
For ribosome analyses, dry seeds were imbibed as mentioned earlier using three independent biological replicates. Seeds and seedlings were harvested at each physiological state during the seed to seedling transition, frozen in liquid nitrogen followed by freeze‐drying. The dry material was stored at −80°C until further analysed.
Isolation polysomal mRNA
Polysomal RNA was isolated according to Subramanian (1978) and Mustroph et al. (2009a) with some modification. In detail, 400 mg (DW) of freeze‐dried tissue was extracted with 8 ml of polysome extraction buffer (PEB: 400 mM Tris pH 9.0, 0.25 M sucrose, 200 mM KCl, 35 mM MgCl2, 5 mM EGTA, 1 mM phenylmethane sulfonyl fluoride, 5 mM dithiothreitol (DTT), 50 μg ml−1 cycloheximide, 50 μg ml−1 chloramphenicol). The extracts were loaded on top of a sucrose cushion (1.75 M sucrose in PEB) and centrifuged (18 h, 90 000 g) using a Beckman Ti70 rotor for 18 h at 4°C (Beckman Coulter, Brea, CA, USA). The resulting pellet was resuspended in wash buffer (200 mM Tris pH 9.0, 200 mM KCl, 0.025 M EGTA, 35 mM MgCl2, 5 mM DTT, 50 μg ml−1 cycloheximide, 50 μg ml−1 chloramphenicol). Optical density at 260 nm (OD260) was measured for the samples which were loaded on a 20–60% linear sucrose gradient, and centrifuged at 190 000 g for 1.5 h at 4°C using a Beckman SW55 rotor (Beckman Coulter) either according to equal DW or equal OD260 values. After ultracentrifugation, the gradients were fractionated into 20 fractions using a Teledyne Isco Density Gradient Fractionation System (Teledyne Isco, Lincoln, NE, USA) with online spectrophotometric detection (at 254 nm). The fractions corresponding to polysomes (mRNAs with two or more ribosomes) were pooled for further analysis. This was performed using at least three independent biological repeats.
The ribosome abundance is reflected by the total area under the curve and was calculated after subtracting the baseline obtained by measuring a blank gradient and normalizing to total area under the curve to account for possible uneven amount of plant material in the samples. Polysomal loading of samples was calculated by comparing the area corresponding to two or more ribosomes (polysomes) after background subtraction and normalization with the total area under the curve.
Isolation and analysis of RNA species
The relative ratio of ribosomal types was calculated by determining the relative amounts of the small subunits of cytosolic, plastidic and mitochondrial rRNAs by quantitative real‐time PCR (qRT‐PCR) normalized to the geometric mean of the spike‐in standards assuming no presence of naked rRNA species (Piques et al., 2009). Aliquots of 300 μl total extract and 800 μl pooled polysome fraction were spiked with a mix of the four eukaryotic poly(A) RNAs including lys, phe, thr and dap (Affymetrix, Santa Clara, CA, USA; Ambion, P/N900433), with relative final concentrations of 1 : 100 000, 1 : 50 000, 1 : 25 000 and 1 : 6667, respectively, and purified with TriPure Isolation Reagent (Roche, Basel, Switzerland), followed by clean‐up using RNeasy Mini spin columns (Qiagen, Hilden, Germany) and dissolved in RNase‐free H2O. cDNA was synthesized using the iScript™ cDNA synthesis kit (Bio‐Rad, Hercules, CA, USA) according to the manufacturer's protocol. After DNase1 treatment (Thermo Scientific, Waltham, MA, USA), qRT‐PCR was performed using Power SYBR Green (Applied Biosystems, Waltham MA, USA) in a 5 μl reaction using the standard program of the ViiA™ 7 instrument (Applied Biosystems). Data were analysed using ViiA™ 7 Software v.1.1 (Applied BioSystems). Primer amplification efficiency was calculated using linregPCR (Ruijter et al., 2009). The quantification of the poly(A) RNA control spikes were used to normalize qRT‐PCR data. All primers used are provided in the Supporting Information, Table S1(a).
Data analysis
Affymetrix Arabidopsis Gene 1.1 ST Arrays (Affymetrix) were hybridized using the GeneChip® 3′ IVT Express kit (cat. no. 901229; Affymetrix, Santa Clara,CA, USA) according to instructions from the manufacturer. Hybridization data were analysed and gene‐specific signal intensities were computed using the R statistical programming environment (http://www.R-project.com), the BioConductor package affy (Gautier et al., 2004) and the Brainarray cdf file v.17.1.0 (http://brainarray.mbni.med.umich.edu/). DNA microarray data are available in the Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE65780. The limma and affy packages were used for RMA normalization (Irizarry et al., 2003). Probe set intensity signals that never exceeded the noise threshold (log2Exprs ≤ 4 in all samples) were removed. A linear model and empirical Bayes methods were applied for assessing differential expression (Smyth, 2004) and genes are only considered as differentially expressed when log2 fold change (FC) > 1 and P < 0.05 adjusted by the false discovery rate (FDR) according to the method of Benjamini & Hochberg (1995). Correlation between RMA normalized biological replicates averaged 0.98 (Pearson's correlation) and ranged between 0.96 and 0.99 (Fig. S1f). Relative RNA levels were validated with qRT‐PCR experiments with four spikes as internal standard for the normalization (Vandesompele et al., 2002). DNA sequences and efficiencies of primer pairs used for qRT‐PCR experiments and comparison of relative mRNA levels determined in GeneChip and qRT‐PCR experiments are given in Table S1(a). Principal component analysis (PCA) was performed using TM4 (Saeed et al., 2003). Polysome occupancy for each mRNA was calculated by comparison of normalized levels from the polysomal and total RNA. The same criterion for a significant change was used for polysome occupancy as for changes in total and polysomal levels.
Analysis of identified mRNA sequences
gene trail (http://genetrail.bioinf.uni-sb.de/) and revigo (http://revigo.irb.hr/) were used for over‐representation analysis using default parameters to characterize the dominant transcriptional and translational processes related to seed germination. Geneset enrichment analysis was performed via agriGO (http://bioinfo.cau.edu.cn/agriGO/; TAIR10) using a Fisher test followed by the Hochberg method at a significance level of 0.05. The generated GO lists related to biological processes with a P‐value for FDR were put into Revigo with a dispensability cut‐off of 0.05 to remove GO term redundancy.
Sequence feature analysis
Genes with significantly increased and decreased polysome occupancy at each translational shift were compared with the microarray background that represents total gene sets on the microarray for several sequence features using custom scripts. The distributions of sequence lengths and GC content were evaluated separately for coding DNA sequence (CDS), 5′ untranslated region (5′UTR), 3′UTR and full transcript. CDSs were also analysed for GC levels in third positions of codons (GC3), after removing sequences missing the start codon and/or containing premature stop codons; CDSs shorter than 100 codons were further removed for the codon bias analysis, measured using the effective number of codons (N c) index (Sun et al., 2013). The same analyses were performed separately for the CDSs of protein‐coding genes having both or no annotated UTR (UTRs called present when having length > 1 nucleotide). Given the nonnormality of the distributions of values, a Wilcoxon signed‐rank test was adopted for all statistical comparisons (median as the test statistic).
RNA structural analysis
Experimentally determined structure scores per nucleotide, as provided by Li et al. (2012), were used to calculate average structure scores of the genes with significantly increased and decreased ribosomal association at each developmental switch. Relative scaling was achieved by averaging the structure scores per region (5′UTR, CDS and 3′UTR) in 100 bins. Standard errors were determined and Student's t‐tests were performed using the python scipy module (http://www.scipy.org/).
Motif analysis
DNA motif analyses were performed using the MEME suite (Bailey et al., 2009), for full transcript, 5′UTR, CDS and 3′UTR sequences, extracted from the TAIR10 database (http://www.arabidopsis.org/). The minimum and maximum motif width was set to 6 and 10, respectively. If a gene had multiple isoforms, only the TAIR10 representative splice form was used. Background dinucleotide frequencies were provided separately for each sequence region of the transcript. To test the specificity of the resulting motifs, FIMO (Bailey et al., 2009) was used to scan all genes represented on the microarray for motif hits in the corresponding sequence type. Motifs with a P‐value ≤ 0.001 were considered significant hits. Obtained motif counts were used to compute the enrichment P‐value for the gene lists vs the background by means of a one‐tailed Fisher's exact test, performed with a custom script and the R software package (http://www.r-project.org/). For each motif, the positions on the transcripts, as provided by the FIMO output, were used to calculate the relative number of motifs per (relative) position along the mRNA. Relative scaling was performed in a similar fashion as for the structure scores.
Results
Translational activation precedes ribosome biogenesis during Arabidopsis seed germination
Monitoring the seed to seedling transition of fully after‐ripened Arabidopsis seeds was performed by scoring the percentage of testa rupture (TR), radicle protrusion (RP) and seedling greening (SG) over time. TR started c. 26 h after the start of imbibition (HAI). RP was first observed 35 HAI, and at 48 HAI 80% of the seeds showed radicle protrusion. By 72 HAI, 80% of the seedlings had reached the SG stage and at 82 HAI all the seedlings had turned green (Fig. 1a). The time points (0, 6, 26, 48 and 72 HAI) that mark different physiological stages (dry seeds, early imbibition, and initiation of TR, 80% RP and 80% SG, respectively) were selected for polysome profiling based on both equal DW (Fig. 1b) and equal RNA level in the samples (Fig. 1c). Ribosome profiles changed dramatically during the seed to seedling transition. In dry seeds, ribosomes were mainly present in the monosome form (Fig. 1b,c). Following imbibition ribosome profiles changed. This was first visible by an increase in the polysome peak from dry to 6 HAI (Fig. 1b) concurrent with a decrease of the monosome peak, followed by an increased total area that represents the increase in ribosome abundance (Fig. 1b–d). These newly synthesized ribosomes mostly represent organellar ribosomes, especially plastid ribosomes after 48 HAI, as shown by the relative quantity of ribosomal RNA specific to each organelle (Fig. 1e).
Figure 1.
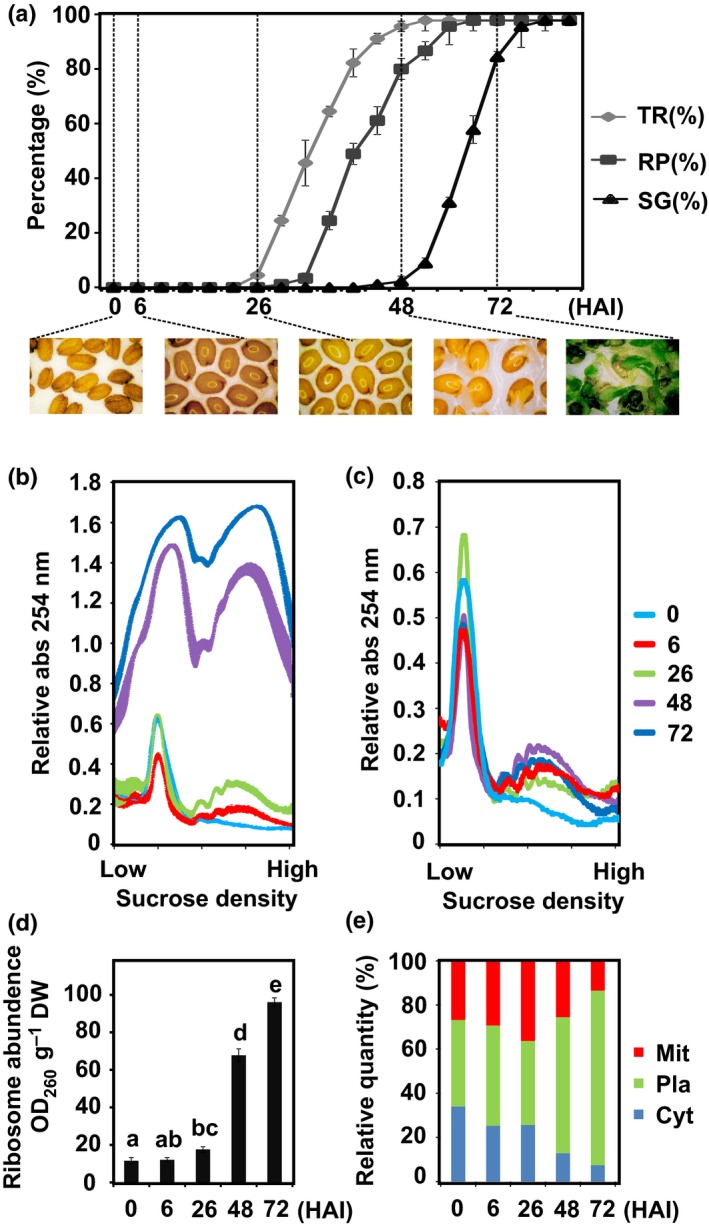
Polysome profiling during seed germination. (a) Frequency of testa rupture (TR), radicle protrusion (RP) and seedling greening (SG) during the Arabidopsis Col‐0 seed to seedling transition. Time points during seed germination include dry seeds, 6, 26 (TR initiation), 48 (80% RP) and 72 h after the start of imbibition (HAI) (80% GS). Data are presented as mean ± SD of three independent replicates. (b) Absorbance profiles of sucrose density gradient fractionated ribosomes for the five time points during the seed to seedling transition. Data are presented as mean ± SD of three independent replicates. SD is indicated by the width of the line. Gradient loading according to equal dry weight. (c) Representative absorbance profiles of sucrose density gradient fractionated ribosomes for the five time points during the seed to seedling transition; gradient loading according to identical RNA loading. (d) Ribosome abundance for the five time points during the seed to seedling transition. Data are presented as mean ± SD of three independent replicates and the letters above each bar indicate the significance (P < 0.05). (e) Relative abundance of different ribosomes (Cyt, cytosolic; Pla, plastidic; Mit, mitochondrial).
Transcriptional changes are reflected in polysomal mRNA levels during seed germination
Total mRNA (T) and polysomal mRNA (P) expression levels were analysed to investigate the translational dynamics during the seed to seedling transition using the high‐throughput Gene ST1.1 Array. No obvious RNA degradation was observed in any of the samples and the sample preparation was robust based on the same signal distribution after normalization and the high similarity between sample replicates (r > 0.96 and r = 0.98 on average, Fig. S1). The 19 781 genes with intensities that passed the noise filter (log2Exprs > 4) based on the intensity distribution (Fig. S1a,b) in all three independent biological replicate samples for at least one time point during seed germination were subjected to further analysis (Table S1b). The change in mRNA abundance between each stage was determined by comparing RNA levels to the preceding stage (Fig. S2a) as well as to the dry state (Fig. S2b) during seed germination. mRNA levels changed similarly in the total RNA and polysomal RNA (Figs S2, S3). Similar GO functions were over‐represented for both RNA preparations (Fig. S4). The changes at the transcriptional level are in agreement (≥ 50% overlap, FC = 2, FDR = 0.05) with recent data describing transcriptional changes during the seed to seedling transition (Silva et al., 2016; Table S1c). The upregulated genes were over‐representing processes involved in protein localization, oxygen and reactive oxygen species metabolic process, ribosome biogenesis, translation, stress responses, cell wall organization, photosynthesis, and lipid transport and localization (Fig. S4b). By contrast, chitin response, abscisic acid response, defence, secondary metabolism, seed development, RNA processing and ribosome biogenesis (both cytosolic and organellar) were over‐represented in the downregulated gene set (Fig. S4c). The genes encoding ribosomal protein genes were strikingly differentially expressed during the seed to seedling transition (Fig. S5). Generally, a strong correlation between transcription and translation across seed germination was observed (Figs 2, S3).
Figure 2.
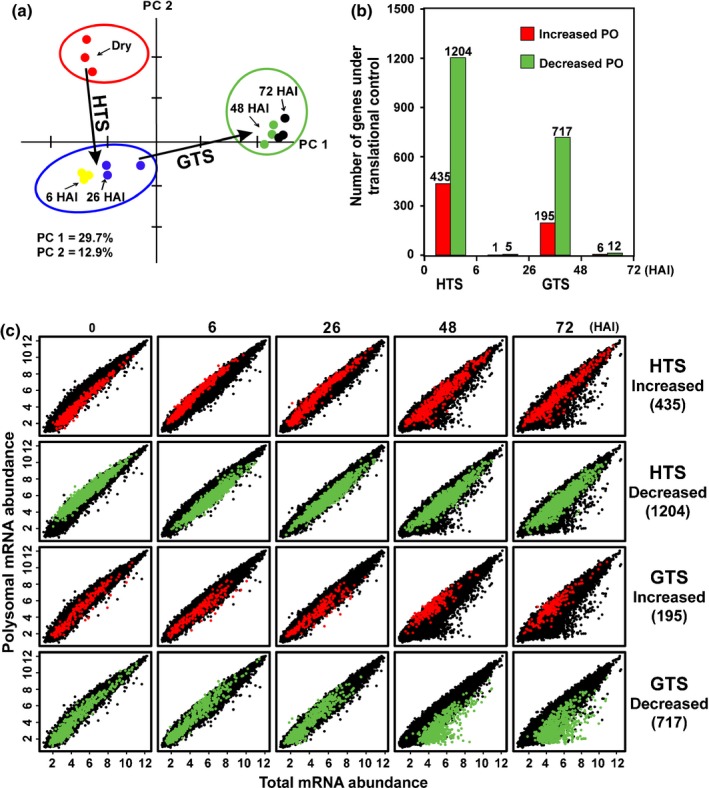
Arabidopsis seed germination is characterized by two translational shifts. (a) Principal component (PC) analysis of polysome occupancy (PO) changes (polysomal mRNA levels/total mRNA levels) during seed germination. The first two components (PC1 and PC2) explain 30 and 13% of the total variation, respectively. (b) The number of mRNAs with changed PO at the two translational shifts, at 6 h after the start of imbibition (HAI) the hydration translational shift (HTS) and 48 HAI, the germination translational shift (GTS). (c) Dynamics of genes underlying the two translational shifts. The levels of total (x‐axis) and polysomal (y‐axis) mRNAs following seed imbibition (dry seeds, 6, 26, 48 and 72 HAI) are plotted (black). Genes identified with increased (red dots) or decreased (green dots) polysome occupancy are indicated for both the HTS (upper panels) and the GTS (lower panels).
Polysomal profiling reveals two phases of translational control
To identify genes that are under translational control, we assessed the polysome occupancy of each mRNA species, which is defined as the ratio between the mRNA in the polysome pool and the total mRNA (Bailey‐Serres, 1999; Branco‐Price et al., 2005, 2008; Gamm et al., 2014). By comparing the polysome occupancy between each stage and the preceding time point, we identified two temporal phases with extensive changes in translational control: between dry seeds and 6 HAI seeds and between 26 and 48 HAI seeds (Fig. 2). Changes exceeding twofold and associated with a corrected P‐value of 0.05 were considered significant. We refer to these phases as the hydration and the germination translational shifts (HTS and GTS). In total 1204 genes were downregulated in the HTS (HTS down) and 435 genes were upregulated (HTS up). For the GTS the numbers were 717 (GTS down) and 195 (GTS up). Minor significant translational changes were identified between 6 and 26 HAI and between 48 and 72 HAI (Fig. 2b). The genes identified in two translational shifts during seed germination were largely non‐redundant (Fig. S6a). There was a large overlap with genes subjected to translational control under hypoxia stress (Fig. S6b,c,h), but no significant overlap with genes under translational regulation in the sucrose feeding and starvation experiments or seed dormancy (Fig. S6e–g).
Dynamics of genes under translational control
To visualize the transcriptional and translational dynamics of genes under translational control, the four different gene sets were highlighted in correlation plots of the different time points either by transcript abundance or by fold change (Figs 2c, S3). This showed that the translationally upregulated genes in HTS were relatively lowly expressed and similarly weakly associated to the polysomes in dry seeds. At 6 HAI these genes were associated with polysomes at higher levels than expected based on their expression, followed by similar levels in both pools during the later imbibition phases. The opposite pattern was shown for the downregulated genes. These genes were generally highly associated with the polysome in dry seeds, and decreased in polysome association at 6 HAI. The translationally upregulated genes in the GTS up gene set were specifically highly associated with the polysomes at 48 HAI and this continues in the later time point, while the downregulated genes were represented by mRNAs associated with polysomes at levels corresponding to total mRNA levels at early time points but specifically less associated with polysomes at the two later time points. In principle, changes in polysomal occupancy can be caused both by changes in the transcript level and by changed association of the mRNA to ribosomes (or combinations thereof). By comparing these effects separately, different patterns emerge. The group of genes downregulated in the HTS seem primarily affected negatively on the polysomal mRNA level, while most of the genes downregulated in GTS down are characterized by a dramatic upregulation on the total mRNA level (Fig. 3). The different patterns between the downregulated genes in the two shifts indicate different regulatory mechanisms at different stages of seed germination.
Figure 3.
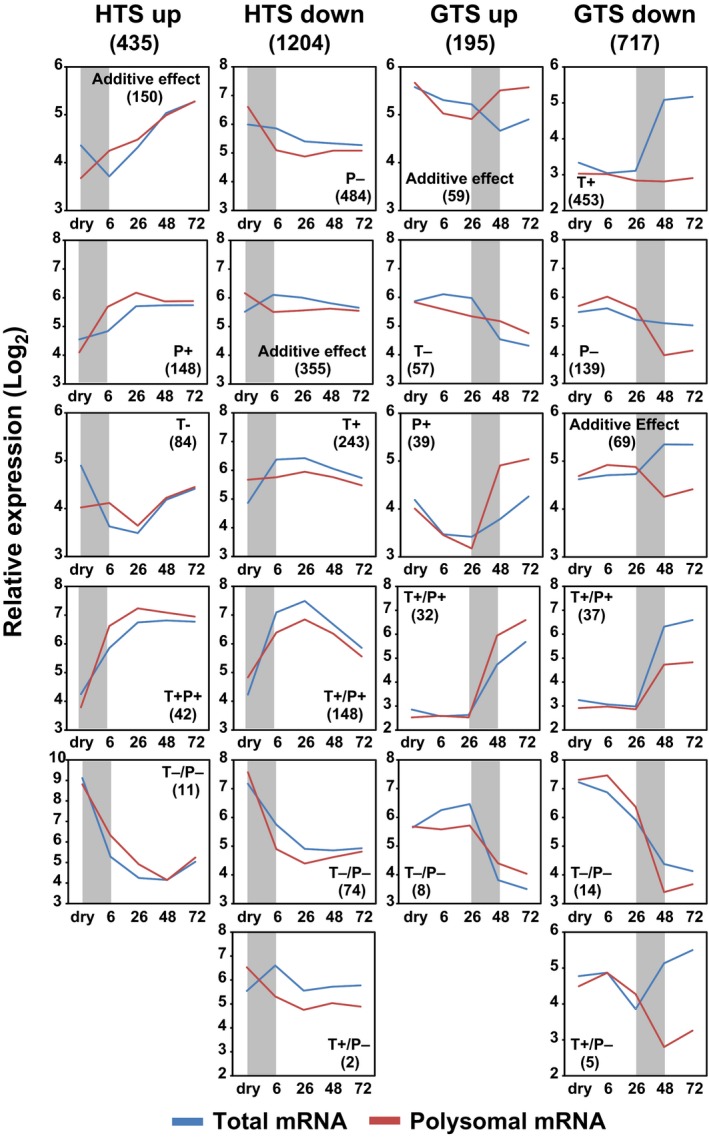
Total RNA and polysomal RNA profile of translational regulated genes during the seed to seedling transition in Arabidopsis. Translational regulated genes are divided into subgroups based on their total RNA and polysomal RNA changes. In total, 22 subgroups under nine categories are identified with their corresponding expression profiles following seed germination and genes numbers under each subgroup in parentheses. T+ represents that the transcription is increased while the translation is unchanged under a specific shift. P+ indicates translation is increased while the total mRNA abundance is unchanged. T+/P+ indicates both transcription and translation are enhanced and an additive effect means that the change of gene expression in either transcription or translation is not significant while the additive effect of the two contributes to the significant change of polysome occupancy. The opposite effect is represented by T−, P− and T−/P− in the corresponding subgroups. HTS, hydration translational shift; GTS, germination translational shift; HAI, hours after the start of imbibition.
Examples of genes upregulated in the HTS are the wounding responsive gene (AT4G10270, log2 FC = 2.6, FDR = 0.05), HSP‐20 LIKE chaperon super family protein gene (AT4G21870) and glycine rich protein gene (AT3G29075, log2 FC = 2.3, FDR = 0.05; Table S1d). The first of these was also reported as highly translationally repressed in response to hypoxia (Branco‐Price et al., 2005). By contrast, a dramatic translational reduction was observed for a pyruvate kinase family protein gene (AT3G04050, log2 FC = −2.1, FDR = 0.05) while no significant change at total mRNA level was detected. This gene product is important for the conversion of photosynthates into oil in the developing seeds (Andre et al., 2007), which is an essential developmental programme during seed maturation. At the GTS, translation of 39 and 139 transcripts specifically increased or decreased independent of transcription (Fig. 3; Table S1d). Representatives of these genes include CYCLIN‐DEPENDENT KINASE B2 (AT1G20930, log2 FC = 2.4, FDR = 0.05), RmlC‐like cupins superfamily protein gene (AT2G18540, log2 FC = −2.9, FDR = 0.05) and WUSHEL RELATED HOMEOBOX2 (AT5G59340, log2 FC = −2.4, FDR = 0.05). Furthermore, genes related to seed development such as embryo development genes, seed storage protein genes, late embryogenesis abundant genes, stress response genes such as heat shock protein genes, hormone related abscisic acid (ABA) and auxin response genes, metabolic genes related to lipid and sucrose metabolism, cell wall related genes, chloroplast related genes and ribosomal protein genes were identified as dominant gene groups that were under intensive translational control (Table S1e,f).
Transcript features correlate with translational regulation
To investigate whether transcript features correlate with translational regulation, we determined transcript length and GC content of the translationally regulated mRNAs. It is established that short transcripts and transcripts with low GC content are in general more efficiently translated than long transcripts (Qu et al., 2011; Valleriani et al., 2011; Liu et al., 2012). For the HTS we found significantly longer genes in the downregulated set compared with the upregulated and background gene sets (all genes present on the array). However, the GTS showed an opposite pattern (Figs 4a, S7; Table S1g). This indicates that translation at the shifts is regulated by distinct mechanisms. Significantly higher GC contents were identified in the 5′UTR and 3′UTR of the HTS and in the CDS of the GTS downregulated genes, which correlates with suppression of translation (as determined by changed polysome occupancy). Due to redundancy in the genetic code, most amino acids are encoded by several synonymous codons, although it is thought that some codons are translated more efficiently than others. This codon bias is calculated based on the effective number of codons (N c), the number of codons used in a gene, ranging from 20 (extreme bias) to 61 (all codons used; Hershberg & Petrov, 2008, 2009). Codon degeneracy is nearly completely related to the third‐base position (Crick, 1966) and related to the GC content at this position. Interestingly, polysome occupancy changes during seed hydration correlate negatively with N c and positively with GC3 (guanine and cytosine content in the third codon position; synonymous sites) (Figs 4, S7). Highly translated transcripts often use the optimal codons, as observed in several species (Ikemura, 1985; Bulmer, 1987; Akashi, 1994; Duret, 2000; Drummond & Wilke, 2008; Shabalina et al., 2013).
Figure 4.
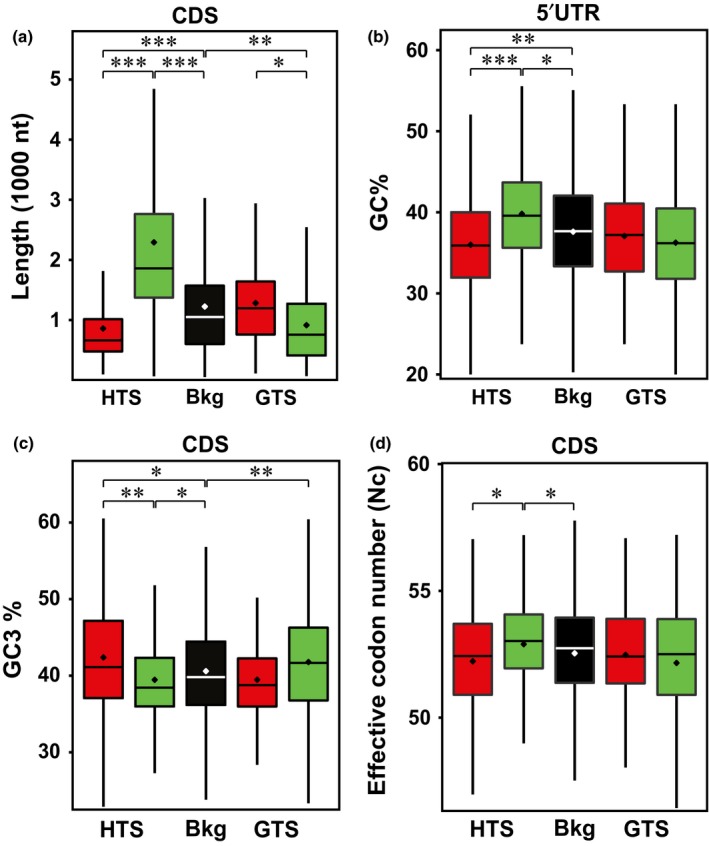
mRNAs of the two shifts during the seed to seedling transition in Arabidopsis are characterized by distinct sequence features. (a) Length of coding DNA sequence (CDS), (b) GC content of 5′UTR, (c) GC3 content and (d) effective codon number (N c) of the CDS are shown. Colour scheme: black, microarray background (Bkg); red or green, translationally up‐ or downregulated genes at the hydration translational shift (HTS) and germination translational shift (GTS), respectively. Error bars represent SE (*, P < E‐10; **, P < E‐20; ***, P < E‐50). P‐values are calculated by a Wilcoxon nonparametric test.
Upstream open reading frames (uORFs) are commonly involved in regulating translation. We tested whether genes with uORFs are over‐represented in the translationally regulated gene list during seed germination by a χ2‐test, using the published 2020 uORF containing genes identified in the Arabidopsis genome (Juntawong et al., 2014). At the HTS, 15 out of 435 genes were identified to harbour uORFs in the translationally upregulated genes, significantly lower than the uORF occurrence in the genome (expected = 31.32, P < 0.005); by contrast, significantly more genes (110 out of 1204) with uORFs (expected = 75.60, P < 0.005) were present in the downregulated genes. For the GTS there was no enrichment for uORFs in the translationally upregulated genes (20/195) while a significantly low occurrence was detected for the downregulated genes (23/717) (expected = 51.32, P < 0.005).
We mapped the genes shown to be translationally regulated to the dataset of mRNA decay profiles and associated sequence features of mRNAs of cultured cells (Narsai et al., 2007). The genes translationally regulated in the HTS differed significantly in transcript stability. Translationally downregulated genes of the shift were significantly less stable than expected by random and the upregulated genes were more stable as judged from the mRNA stability measurements in Arabidopsis cell culture. There were significantly high numbers of introns especially in the CDS and 3′UTR, while other mRNA characteristics of the genes were not different from expected values (Table S1h).
The role of the mRNA's secondary structure on translational control was tested by mapping the translationally controlled genes to the experimentally derived structural score defined by Li et al. (2012). This score is an indicator of transcript complexity, in which high structure scores are equivalent to more double‐stranded (ds) than single‐stranded (ss) RNA at a certain position in a transcript and vice versa for low structure scores. Average structure scores were plotted over the 5′UTR, CDS and 3′UTR for both translational shifts (Fig. 5a,b) and compared with the average score of all genes present on the array (background). In general, mRNAs in vitro have been shown to have a steep decrease in structure at the start and stop codon, which is a conserved structural feature for eukaryotes facilitating accessibility of the ribosome for translational initiation (Kozak, 2005; Kertesz et al., 2010; Li et al., 2012). At the HTS, upregulated mRNAs are less structured in the 5′UTR and CDS than those downregulated, which suggests that mRNAs with lower structure scores are translationally favoured over more structured transcripts at specific stages. The GTS downregulated mRNAs have an overall higher structure score in the CDS, and the opposite trend was found in the 3′UTR. The high structure of the CDS may attenuate the progression of the ribosomes and thereby inhibit translation of these mRNAs at later stages.
Figure 5.
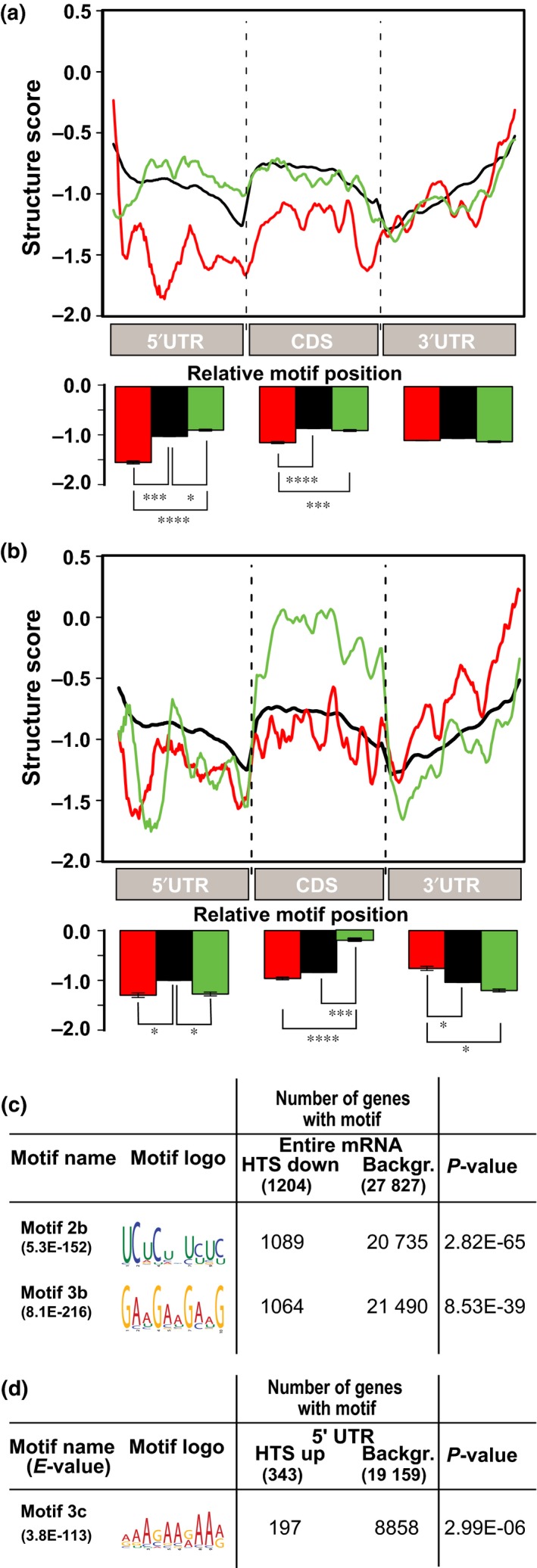
Secondary structures and motif features correlate with polysome association. The average structure score is plotted over the 5′UTR, coding DNA sequence (CDS) and 3′UTR of the (a) hydration translational shift (HTS) and (b) germination translational shift (GTS) affected transcripts. Background: all transcripts on the array (black line), translational up (red line) or translational down (green line). P‐values are calculated by a Student's test according to Li et al. (2012). The bar plots represent the mean structure scores per transcript region (5′UTR, CDS and 3′UTR, respectively). Error bars represent SE (*, P < E‐10; ***, P < E‐50; ****, P < E‐100 ). Significantly enriched motifs are detected across (c) the whole transcript length (cDNA) and (d) 5′UTR. P‐values are calculated by a Fisher's t‐test. Note: several sequence motifs overlapping with motif 2b were identified. Only the most significant is depicted.
Motif analysis on four regions (5′UTR, CDS, 3′UTR and the whole transcript) for both shifts revealed three significantly (P < E‐5) enriched motifs (Figs 5, S8), all for the HTS. One was present in the 5′UTR of the HTS up transcripts, and two in the entire transcript RNA sequence of the HTS down set.
Motif 3c was significantly enriched in the 5′UTR of translationally enhanced transcripts in the HTS. This adenosine‐enriched tract is mainly localized in the 50 nucleotide region upstream of the start codon which could potentially bind to translation initiation factors and enhance translation initiation (Fig. S8) (Xia et al., 2011).
Discussion
Translation is essential for seed germination and apparently mRNAs stored in the dry seed are translated during germination (Rajjou et al., 2004; Narsai et al., 2011; Galland et al., 2014; Layat et al., 2014). However, an accurate temporal evaluation of the extent of translational regulation during seed germination is missing. Here we present a time course study on the translational dynamics, which reveals that translational activation precedes ribosome biogenesis during the seed to seedling transition. Polysome profiling identified thousands of genes differentially transcribed and translated with temporal resolution. Thus, polysome profiling efficiently helps in identifying genes regulated during seed germination. Our data demonstrated a large overlap of differentially regulated genes on both the transcriptional and the translational level, a natural consequence of mRNA‐dependent translation (Figs S2a, S3). However, by analysing polysome occupancy, defined as the ratio of polysome‐associated and total mRNA levels, across the course of germination we found two phases where the polysome occupancy of thousands of mRNAs changes. This extensive translational regulation during seed germination exceeds what has been shown for translational regulation in other studies both in the numbers of affected genes and in the extent of the effect (Nicolai et al., 2006; Gamm et al., 2014; Lin et al., 2014). These two major shifts are here referred to as the HTS and GTS (Fig. 6). The shifts occur in temporal correlation with key stages of the seed to seedling transition and might refer to physiological control points (Fig. 6), and distinct genes are affected in the two shifts (Fig. S5a). Thus, translational control during seed germination is stage specific and possibly represents different development‐dependent regulatory mechanisms as revealed by the different secondary structures and motifs identified in the two shifts. (Fig. S6b–f; Nicolai et al., 2006; Branco‐Price et al., 2008; Gamm et al., 2014; Juntawong et al., 2014; Lin et al., 2014; Sorenson & Bailey‐Serres, 2014). Overlap is the largest with translationally regulated genes under hypoxia stress (c. 25% of the total identified genes under translational control during seed germination). This may be related to low oxygen responses during the seed to seedling transition.
Figure 6.
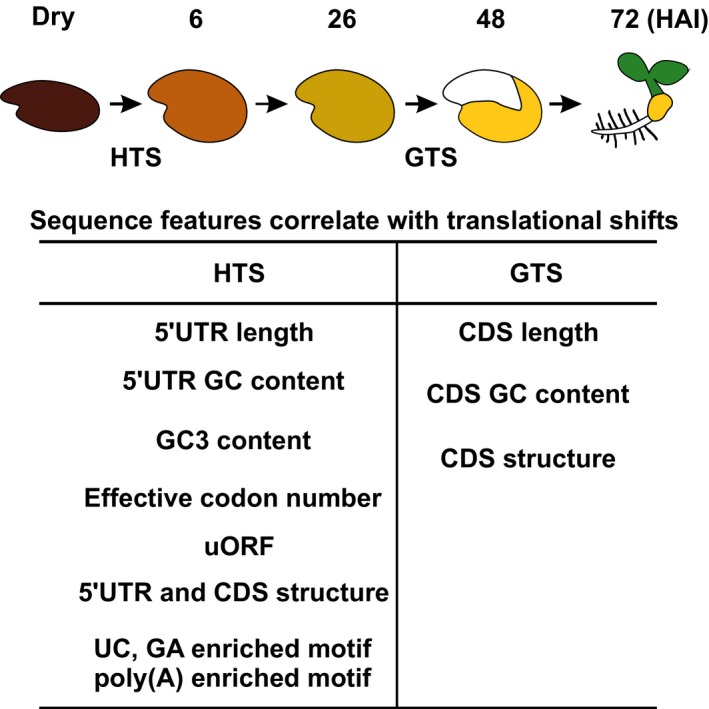
Summary of the sequence features in mRNAs identified as translationally regulated during germination. The upper panel shows a schematic presentation of the transition from a dry seed to a seedling at 72 h after the start of imbibition (HAI). The two phases where large sets of mRNAs are under translational control, the hydration translational shift (HTS) and germination translational shift (GTS), are indicated. The identified features that correlate with these translational shifts are listed. CDS, coding DNA sequence; uORF, upstream open reading frame; UC, pyrimidine; GA, purine.
We have identified sequence features that correlate with translational regulation (Fig. 4). Different features correlate with different translational shifts. For the HTS we found that a reduced number of uORFs, transcript length, GC content and secondary structure correlate with an upregulation of translation. The translationally regulated genes differ in secondary structure, as judged by comparison to the in vitro structure of the mRNAs (Fig. 4a,b). The different level of secondary structure of the translationally regulated mRNAs indicates the possibility that structural features are important for this regulation. Since the translationally regulated transcripts identified differ between the two shifts, there should be additional factors that affect the translational regulation. One can envisage a model in which sensitivity to structure at different stages of the seed to seedling transition is mediated by differential activity of RNA helicases, which are dedicated to unpacking the annealed nucleic acid strands such as secondary structures of the RNA complex. Whether helicases play a role in translational regulation of the two shifts remains to be investigated. Other factors that might affect translation are RNA binding proteins specifically interacting with the identified elements in translationally regulated mRNAs. The Arabidopsis genome encodes hundreds of RNA binding proteins and for one class, PUF (proteins that are characterized by the presence of a conserved Pumilio homology domain) proteins, a role in germination has recently been proposed (Xiang et al., 2014). The pyrimidine (UC) and purine (GA) enriched motifs identified among the transcripts in the HTS may represent binding sites for polypyrimidine tract‐binding protein, PTB (Singh et al., 1995; Perez et al., 1997; Oberstrass et al., 2005). Motif 3b GAAGAAGAAG is similar to the target sequence (GAAGAAGAAGCUC) of SERINE/ARGININE‐RICH PROTEIN SPLICING FACTOR 40, which acts as an exon enhancer mediated by a complex of nuclear proteins (Yeakley et al., 1996). The Arabidopsis SR paralogue SERINE/ARGININE‐RICH SC35‐LIKE SPLICING FACTOR 33 has been identified and plays a role in regulating alternative splicing (Thomas et al., 2012). Although not investigated here, splicing might play a role in the HTS as introns are specifically more frequent in HTS mRNAs (Table S1h). Interestingly, the ribosomal protein genes are differentially expressed during the seed to seedling transition (Fig. S5). This may be affecting the composition of the translating ribosomes and thereby the selection of translated mRNAs.
Overall, our data reveal a model of changing translational regulation during seed germination and seedling establishment (Fig. 6). The extensive translational regulation during germination and the changes therein are unlikely to be regulated by a single mechanism. The diversity of sequence features identified favours a multifactorial model. Further research will focus on how these identified features are recognized and thus mediate the translation control. The Arabidopsis genome harbours hundreds of mRNA binding proteins, of which a large majority have no assigned function. The regulators of translation during seed germination are likely to be found in this group of interesting proteins.
Author contributions
B.B., J.H. and L.B. designed the experiments. B.B. performed the ribosome fractionation and conducted the gene expression microarray experiments. B.B., L.B., J.H., A.P., M.G., S.v.d.H. and B.S. directed the design of analysis approaches. B.B., A.P. and S.v.d.H. conducted the analyses. B.B., L.B. and J.H. drafted the manuscript. All authors participated in revising and editing the manuscript.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Gene 1.1 ST GeneChip quality assessment and reproducibility.
Fig. S2 Coordinated transcriptional and translational expression changes during the seed to seedling transition.
Fig. S3 Arabidopsis seed to seedling transition is characterized by two translational shifts.
Fig. S4 Temporal functional changes of total RNA and polysomal RNA during the seed to seedling transition using over‐representation analysis.
Fig. S5 Ribosomal protein genes are differentially expressed during the seed to seedling transition.
Fig. S6 Dataset comparison of the translational shift genes during the seed to seedling transition.
Fig. S7 Comparison of sequence features between genes regulated at the hydration and germination translational shift and the background.
Fig. S8 Spatial distribution of enriched motifs in genes translationally regulated during the Arabidopsis seed to seedling transition.
Table S1a qRT‐PCR data for gene expression analysis and primers used
Table S1b Total gene set with fold change (FC), adjusted P‐value, for total RNA, polysomal RNA and polysomal occupancy changes across seed germination
Table S1c Comparison of the transcriptional difference in the current data set and dataset from Silva et al. (2016)
Table S1d Translational regulated genes during seed germination
Table S1e Dominant gene groups among the translational regulated shift genes
Table S1f Gene set enrichment analysis for the translational shift genes
Table S1g Sequence feature analysis for the translational shifts during seed germination
Table S1h Literature derived sequence feature analysis for the translational shifts during seed germination
Table S1i Ribosomal protein gene transcriptional profile during seed germination
Acknowledgements
This work was supported by the Netherlands Organization for Scientific Research (project number 821.02.010). Financial support came from the Marie Curie ITN project MERIT, Grant Agreement number 264474 (A.P.), and Bio4Energy, a Strategic Research Environment appointed by the Swedish government. We thank Service XS, Leiden, the Netherlands, for performing the microarray hybridizations. We also thank Dr Bas Dekkers for proofreading and suggested changes to the manuscript.
Contributor Information
Leónie Bentsink, Email: leonie.bentsink@wur.nl.
Johannes Hanson, Email: johannes.hanson@umu.se.
References
- Akashi H. 1994. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics 136: 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre C, Froehlich JE, Moll MR, Benning C. 2007. A heteromeric plastidic pyruvate kinase complex involved in seed oil biosynthesis in Arabidopsis. The Plant Cell 19: 2006–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. 2003. Genome‐wide analysis of mRNA translation profiles in Saccharomyces cerevisiae . Proceedings of the National Academy of Sciences, USA 100: 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller K, Grossmann J, Grobei MA, Hull R, Hirsch‐Hoffmann M, Yalovsky S, Zimmermann P, Grossniklaus U, Gruissem W, Baginsky S. 2008. Genome‐scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320: 938–941. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research 37: W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey‐Serres J. 1999. Selective translation of cytoplasmic mRNAs in plants. Trends in Plant Science 4: 142–148. [DOI] [PubMed] [Google Scholar]
- Basbouss‐Serhal I, Soubigou‐Taconnat L, Bailly C, Leymarie J. 2015. Germination potential of dormant and nondormant Arabidopsis seeds is driven by distinct recruitment of messenger RNAs to polysomes. Plant Physiology 168: 1049–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate – a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 57: 289–300. [Google Scholar]
- Bewley JD. 1997. Seed germination and dormancy. The Plant Cell 9: 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco‐Price C, Kaiser KA, Jang CJ, Larive CK, Bailey‐Serres J. 2008. Selective mRNA translation coordinates energetic and metabolic adjustments to cellular oxygen deprivation and reoxygenation in Arabidopsis thaliana . Plant Journal 56: 743–755. [DOI] [PubMed] [Google Scholar]
- Branco‐Price C, Kawaguchi R, Ferreira RB, Bailey‐Serres J. 2005. Genome‐wide analysis of transcript abundance and translation in Arabidopsis seedlings subjected to oxygen deprivation. Annals of Botany 96: 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M. 1987. Coevolution of codon usage and transfer RNA abundance. Nature 325: 728–730. [DOI] [PubMed] [Google Scholar]
- Crick FHC. 1966. Codon‐anticodon pairing: the wobble hypothesis. Journal of Molecular Biology 19: 548–555. [DOI] [PubMed] [Google Scholar]
- Dekkers BJ, Pearce S, van Bolderen‐Veldkamp RP, Marshall A, Widera P, Gilbert J, Drost HG, Bassel GW, Muller K, King JR et al 2013. Transcriptional dynamics of two seed compartments with opposing roles in Arabidopsis seed germination. Plant Physiology 163: 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation‐induced protein misfolding as a dominant constraint on coding‐sequence evolution. Cell 134: 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L. 2000. tRNA gene number and codon usage in the C. elegans genome are co‐adapted for optimal translation of highly expressed genes. Trends in Genetics 16: 287–289. [DOI] [PubMed] [Google Scholar]
- Fait A, Angelovici R, Less H, Ohad I, Urbanczyk‐Wochniak E, Fernie AR, Galili G. 2006. Arabidopsis seed development and germination is associated with temporally distinct metabolic switches. Plant Physiology 142: 839–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernie AR, Stitt M. 2012. On the discordance of metabolomics with proteomics and transcriptomics: coping with increasing complexity in logic, chemistry, and network interactions scientific correspondence. Plant Physiology 158: 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Wang BC, Jin X, Li HB, Han P, Wei KH, Zhang XM, Zhu YX. 2005. Proteomic analysis and extensive protein identification from dry, germinating Arabidopsis seeds and young seedlings. Journal of Biochemistry and Molecular Biology 38: 650–660. [DOI] [PubMed] [Google Scholar]
- Galland M, Huguet R, Arc E, Cueff G, Job D, Rajjou L. 2014. Dynamic proteomics emphasizes the importance of selective mRNA translation and protein turnover during Arabidopsis seed germination. Molecular and Cellular Proteomics 13: 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo K, Job C, Groot SP, Puype M, Demol H, Vandekerckhove J, Job D. 2001. Proteomic analysis of Arabidopsis seed germination and priming. Plant Physiology 126: 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamm M, Peviani A, Honsel A, Snel B, Smeekens S, Hanson J. 2014. Increased sucrose levels mediate selective mRNA translation in Arabidopsis. BMC Plant Biology 14: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. 2004. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315. [DOI] [PubMed] [Google Scholar]
- Gibon Y, Usadel B, Blaesing OE, Kamlage B, Hoehne M, Trethewey R, Stitt M. 2006. Integration of metabolite with transcript and enzyme activity profiling during diurnal cycles in Arabidopsis rosettes. Genome Biology 7: R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbeisen RE, Gerber AP. 2009. Stress‐dependent coordination of transcriptome and translatome in yeast. PLoS Biology 7: e1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. 2008. Selection on codon bias. Annual Review of Genetics 42: 287–299. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. 2009. General rules for optimal codon choice. PLoS Genetics 5: e1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Molecular Biology and Evolution 2: 13–34. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Meyerowitz EM. 2010. Cell‐type specific analysis of translating RNAs in developing flowers reveals new levels of control. Molecular Systems Biology 6: 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez‐Lopez S, Mancera‐Martinez E, Donayre‐Torres A, Rangel C, Uribe L, March S, Jimenez‐Sanchez G, Sanchez de Jimenez E. 2011. Expression profile of maize (Zea mays L.) embryonic axes during germination: translational regulation of ribosomal protein mRNAs. Plant & Cell Physiology 52: 1719–1733. [DOI] [PubMed] [Google Scholar]
- Joosen RV, Kodde J, Willems LA, Ligterink W, van der Plas LH, Hilhorst HW. 2010. GERMINATOR: a software package for high‐throughput scoring and curve fitting of Arabidopsis seed germination. Plant Journal 62: 148–159. [DOI] [PubMed] [Google Scholar]
- Juntawong P, Girke T, Bazin J, Bailey‐Serres J. 2014. Translational dynamics revealed by genome‐wide profiling of ribosome footprints in Arabidopsis. Proceedings of the National Academy of Sciences, USA 111: E203–E212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. 2010. Genome‐wide measurement of RNA secondary structure in yeast. Nature 467: 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Klerk E, Fokkema IF, Thiadens KA, Goeman JJ, Palmblad M, den Dunnen JT, von Lindern M, t Hoen PA. 2015. Assessing the translational landscape of myogenic differentiation by ribosome profiling. Nucleic Acids Research 43: 4408–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. 2005. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361: 13–37. [DOI] [PubMed] [Google Scholar]
- Krishnan K, Ren Z, Losada L, Nierman WC, Lu LJ, Askew DS. 2014. Polysome profiling reveals broad translatome remodeling during endoplasmic reticulum (ER) stress in the pathogenic fungus Aspergillus fumigatus . BMC Genomics 15: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layat E, Leymarie J, El‐Maarouf‐Bouteau H, Caius J, Langlade N, Bailly C. 2014. Translatome profiling in dormant and nondormant sunflower (Helianthus annuus) seeds highlights post‐transcriptional regulation of germination. New Phytologist 204: 864–872. [DOI] [PubMed] [Google Scholar]
- Li F, Zheng Q, Vandivier LE, Willmann MR, Chen Y, Gregory BD. 2012. Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. The Plant Cell 24: 4346–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Chen PW, Chuang MH, Juntawong P, Bailey‐Serres J, Jauh GY. 2014. Profiling of translatomes of in vivo‐grown pollen tubes reveals genes with roles in micropylar guidance during pollination in Arabidopsis. The Plant Cell 26: 602–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MJ, Wu SH, Chen HM. 2012. Widespread translational control contributes to the regulation of Arabidopsis photomorphogenesis. Molecular Systems Biology 8: 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MJ, Wu SH, Wu JF, Lin WD, Wu YC, Tsai TY, Tsai HL, Wu SH. 2013. Translational landscape of photomorphogenic Arabidopsis. The Plant Cell 25: 3699–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustroph A, Juntawong P, Bailey‐Serres J. 2009a. Isolation of plant polysomal mRNA by differential centrifugation and ribosome immunopurification methods. Methods in Molecular Biology 553: 109–126. [DOI] [PubMed] [Google Scholar]
- Mustroph A, Zanetti ME, Jang CJ, Holtan HE, Repetti PP, Galbraith DW, Girke T, Bailey‐Serres J. 2009b. Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proceedings of the National Academy of Sciences, USA 106: 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsai R, Howell KA, Millar AH, O'Toole N, Small I, Whelan J. 2007. Genome‐wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana . The Plant Cell 19: 3418–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsai R, Law SR, Carrie C, Xu L, Whelan J. 2011. In‐depth temporal transcriptome profiling reveals a crucial developmental switch with roles for RNA processing and organelle metabolism that are essential for germination in Arabidopsis. Plant Physiology 157: 1342–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai M, Roncato MA, Canoy AS, Rouquie D, Sarda X, Freyssinet G, Robaglia C. 2006. Large‐scale analysis of mRNA translation states during sucrose starvation in Arabidopsis cells identifies cell proliferation and chromatin structure as targets of translational control. Plant Physiology 141: 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir‐Ahmady B, Pitsch S, Black DL et al 2005. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309: 2054–2057. [DOI] [PubMed] [Google Scholar]
- Perez I, Lin CH, McAfee JG, Patton JG. 1997. Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo . RNA 3: 764–778. [PMC free article] [PubMed] [Google Scholar]
- Piques M, Schulze WX, Hohne M, Usadel B, Gibon Y, Rohwer J, Stitt M. 2009. Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Molecular Systems Biology 5: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu XH, Wen JD, Lancaster L, Noller HF, Bustamante C, Tinoco I. 2011. The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature 475: 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajjou L, Gallardo K, Debeaujon I, Vandekerckhove J, Job C, Job D. 2004. The effect of alpha‐amanitin on the Arabidopsis seed proteome highlights the distinct roles of stored and neosynthesized mRNAs during germination. Plant Physiology 134: 1598–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Research 37: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M et al 2003. TM4: a free, open‐source system for microarray data management and analysis. BioTechniques 34: 374–378. [DOI] [PubMed] [Google Scholar]
- Shabalina SA, Spiridonov NA, Kashina A. 2013. Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Research 41: 2073–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AT, Ribone PA, Chan RL, Ligterink W, Hilhorst HW. 2016. A predictive co‐expression network identifies novel genes controlling the seed‐to‐seedling phase transition in Arabidopsis thaliana . Plant Physiology 170: 2218–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Valcarcel J, Green MR. 1995. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract‐binding proteins. Science 268: 1173–1176. [DOI] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology 3: article3. [DOI] [PubMed] [Google Scholar]
- Sorenson R, Bailey‐Serres J. 2014. Selective mRNA sequestration by OLIGOURIDYLATE‐BINDING PROTEIN 1 contributes to translational control during hypoxia in Arabidopsis. Proceedings of the National Academy of Sciences, USA 111: 2373–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian V. 1978. Polyribosome formation during early germination of bean embryonic axes. Indian Journal of Biochemistry & Biophysics 15: 235–237. [PubMed] [Google Scholar]
- Sun X, Yang Q, Xia X. 2013. An improved implementation of effective number of codons (nc). Molecular Biology and Evolution 30: 191–196. [DOI] [PubMed] [Google Scholar]
- Thomas J, Palusa SG, Prasad KV, Ali GS, Surabhi GK, Ben‐Hur A, Abdel‐Ghany SE, Reddy AS. 2012. Identification of an intronic splicing regulatory element involved in auto‐regulation of alternative splicing of SCL33 pre‐mRNA. Plant Journal 72: 935–946. [DOI] [PubMed] [Google Scholar]
- Valleriani A, Zhang G, Nagar A, Ignatova Z, Lipowsky R. 2011. Length‐dependent translation of messenger RNA by ribosomes. Physical Review E: Statistical, Nonlinear, and Soft Matter Physics 83: 042903. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biology 3: 0034.1–0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vragovic K, Sela A, Friedlander‐Shani L, Fridman Y, Hacham Y, Holland N, Bartom E, Mockler TC, Savaldi‐Goldstein S. 2015. Translatome analyses capture of opposing tissue‐specific brassinosteroid signals orchestrating root meristem differentiation. Proceedings of the National Academy of Sciences, USA 112: 923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, MacKay V, Yao X, Wu J, Miura F, Ito T, Morris DR. 2011. Translation initiation: a regulatory role for poly(A) tracts in front of the AUG codon in Saccharomyces cerevisiae . Genetics 189: 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Nakabayashi K, Ding J, He F, Bentsink L, Soppe WJ. 2014. REDUCED DORMANCY5 encodes a protein phosphatase 2C that is required for seed dormancy in Arabidopsis. The Plant Cell 26: 4362–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeakley JM, Morfin JP, Rosenfeld MG, Fu XD. 1996. A complex of nuclear proteins mediates SR protein binding to a purine‐rich splicing enhancer. Proceedings of the National Academy of Sciences, USA 93: 7582–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Guo G, Lv D, Hu Y, Li J, Li X, Yan Y. 2014. Transcriptome analysis during seed germination of elite Chinese bread wheat cultivar Jimai 20. BMC Plant Biology 14: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Gene 1.1 ST GeneChip quality assessment and reproducibility.
Fig. S2 Coordinated transcriptional and translational expression changes during the seed to seedling transition.
Fig. S3 Arabidopsis seed to seedling transition is characterized by two translational shifts.
Fig. S4 Temporal functional changes of total RNA and polysomal RNA during the seed to seedling transition using over‐representation analysis.
Fig. S5 Ribosomal protein genes are differentially expressed during the seed to seedling transition.
Fig. S6 Dataset comparison of the translational shift genes during the seed to seedling transition.
Fig. S7 Comparison of sequence features between genes regulated at the hydration and germination translational shift and the background.
Fig. S8 Spatial distribution of enriched motifs in genes translationally regulated during the Arabidopsis seed to seedling transition.
Table S1a qRT‐PCR data for gene expression analysis and primers used
Table S1b Total gene set with fold change (FC), adjusted P‐value, for total RNA, polysomal RNA and polysomal occupancy changes across seed germination
Table S1c Comparison of the transcriptional difference in the current data set and dataset from Silva et al. (2016)
Table S1d Translational regulated genes during seed germination
Table S1e Dominant gene groups among the translational regulated shift genes
Table S1f Gene set enrichment analysis for the translational shift genes
Table S1g Sequence feature analysis for the translational shifts during seed germination
Table S1h Literature derived sequence feature analysis for the translational shifts during seed germination
Table S1i Ribosomal protein gene transcriptional profile during seed germination