

Abstract
Ibrutinib (Imbruvica™) is an irreversible, potent inhibitor of Bruton's tyrosine kinase (BTK). Over the last few years, ibrutinib has developed from a promising drug candidate to being approved by FDA for the treatment of three B cell malignancies, a truly remarkable feat. Few, if any medicines are monospecific and ibrutinib is no exception; already during ibrutinib's initial characterization, it was found that it could bind also to other kinases. In this review, we discuss the implications of such interactions, which go beyond the selective effect on BTK in B cell malignancies. In certain cases, the outcome of ibrutinib treatment likely results from the combined inhibition of BTK and other kinases, causing additive or synergistic, effects. Conversely, there are also examples when the clinical outcome seems unrelated to inhibition of BTK. Thus, more specifically, adverse effects such as enhanced bleeding or arrhythmias could potentially be explained by different interactions. We also predict that during long‐term treatment bone homoeostasis might be affected due to the inhibition of osteoclasts. Moreover, the binding of ibrutinib to molecular targets other than BTK or effects on cells other than B cell‐derived malignancies could be beneficial and result in new indications for clinical applications.
Introduction
Ibrutinib is a potent, orally available Bruton's tyrosine kinase (BTK)1 inhibitor belonging to a class of therapeutics termed ‘targeted covalent drugs’ and demonstrates promising preclinical and clinical activity in several B cell malignancies 1, 2. BTK is a non‐receptor tyrosine kinase that belongs to the TEC family kinases (TFK), which is the second largest family of non‐receptor kinases in humans 3. BTK is known to be an essential component of B cell receptor (BCR) signalling and is involved in B cell differentiation, proliferation and survival 4, 5. Many studies have reported an essential role of BCR signalling in the pathogenesis of several B cell malignancies 6. As a key molecule for this pathway, BTK became an important target for the treatment of B cell‐derived tumours.
Ibrutinib inactivates BTK by binding covalently to Cysteine 481 in the ATP‐binding site in an irreversible manner 7. This active site occupancy inhibits the subsequent phosphorylation of BTK, phospholipase Cγ2 (PLCγ2), AKT and ERK and abolishes the BCR signalling downstream of BTK both in vitro and in vivo 8. Furthermore, inhibition of BTK impairs proliferation, survival and induces apoptosis of malignant B cells 9.
Thus, many preclinical and clinical studies have shown a profound antitumour activity of ibrutinib in different B cell malignancies, including chronic lymphocytic leukaemia (CLL), mantle cell lymphoma (MCL), multiple myeloma, diffuse large B cell lymphoma (DLBCL) and Waldenstrӧm's macroglobulinemia (WM) 10, 11, 12.
Ibrutinib (Imbruvica™; Pharmacyclics, Inc., Sunnyvale, CA, USA) received approval from the U.S. Pharmacyclics, Inc. 999 East Arques Avenue Sunnyvale, California 94085 Food and Drug Administration (FDA) for MCL in November 2013, for CLL in July 2014 and for WM in January 2015 1, 12, 13. The use of the drug has also been approved by the European Medicines Agency (EMA) for the treatment of CLL and MCL. Ongoing studies are also evaluating ibrutinib as a therapy for other B cell malignancies as well as inflammatory and autoimmune diseases, as the results in animal models provided strong evidence of its effectiveness 14, 15.
Kinase selectivity is an important issue for small molecule inhibitors affecting their efficacy and safety. Ibrutinib exhibits remarkable selectivity for BTK. However, nine other kinases have a corresponding cysteine residue in the ATP‐binding site. These include four TFK members (ITK, TEC, BMX and RLK/TXK), three EGFR family kinases (EGFR, ErbB2/HER2 and ErbB4/HER4) and two other kinases, BLK and JAK3 2 (Fig. 1). Ibrutinib shows different affinity for these kinases (Table 1). Even among TFKs, it shows differential binding. The half maximal inhibitory concentration (IC50) of ibrutinib for inhibition of BTK enzymatic activity is 0.5 nm, while the corresponding value for the weakest binder TEC, is 78 nm. It has also been shown by in vitro assays that ibrutinib binds to and inhibits other proteins that lack the cysteine residue. The binding of the drug to these kinases is reversible. However, it is of importance that the affinity of binding for the proteins such as BRK, CSK, FRG and HCK is in the low nanomolar range, that is not very different from that of BTK 15. Another molecule with high binding affinity (IC50 0.5 nm) and ibrutinib binding site is BLK (Table 1). Interestingly, this protein was recently reported to be involved in regulation of pro‐inflammatory cytokine production and its reduced activity could contribute to the development and pathogenesis of autoimmune disorders 16.
Figure 1.
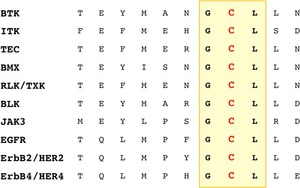
Alignment of kinases having a cysteine residue in the ATP‐binding site corresponding to cysteine 481 in Bruton's tyrosine kinase.
Table 1.
The expression and biological functions of proteins with ibrutinib binding sites
| Proteins | Expression | Biological function | Binding of ibrutinib IC50 (nm) 15 |
|---|---|---|---|
| TEC family kinases | |||
| BTK | B cells, platelets, erythrocytes, macrophages, neutrophils, mast cells, dendritic cells, NK cells 27, 28 | TEC family kinases, non‐receptor tyrosine kinases playing an important role in signalling pathways in hematopoietic cells 29 | 0.5 |
| TEC | B cells, T cells, platelets, erythrocytes, macrophages, neutrophils, mast cells, liver, heart 27, 29, 30 | 78 | |
| ITK | T cells, NK cells, mast cells 27 | 10.7 | |
| BMX | Macrophages, neutrophils, endothelial cells, arterial endothelium 27, 31 | 0.8 | |
| RLK/TXK | T cells, NK cells, mast cells 27, 29, 32 | Not reported | |
| Other kinases | |||
| BLK | B cells, thymocytes, plasmacytoid dendritic cells 16, 33 | Non‐receptor tyrosine kinase of the SRC‐family kinases, involved in B‐lymphocyte development, differentiation and signalling 16 | 0.5 |
| EGFR | Epithelial cells 34 | Epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases involved in cell growth, proliferation and differentiation 34 | 5.6 |
| ErbB2/HER2 | 9.4 | ||
| ErbB4/HER4 | Not reported | ||
| JAK3 | B cells, T cells, NK cells, myeloid cells, vascular smooth muscle cells, endothelium 35 | Janus family of kinases, involved in cytokine receptor‐mediated intracellular signal transduction, cell proliferation and differentiation 35 | 16.1 |
Ibrutinib is generally well tolerated, and long‐term therapy with ibrutinib is associated with modest toxicity 17. The majority of the adverse events are grade 1 or 2 in severity and typically resolving without additional therapy 18. The most common adverse events are diarrhoea, fatigue, bleeding and infections 17, 19. Some of these events are off‐target‐related adverse effects. For instance, recent studies on human platelets suggested that the increased risk of bleeding associated with ibrutinib treatment was due to platelet dysfunction caused by inhibition of both BTK and TEC 20.
The off‐target effects of ibrutinib on cells inside as well as outside the haematopoietic system could be useful in treatment of other diseases. In HER2‐positive breast cancer, the aberrant expression of HER2 is associated with poor prognosis of the disease. A recent study showed that ibrutinib is an effective inhibitor of HER2 and that treatment with ibrutinib completely abrogated the phosphorylation of HER2 and potently inhibited the cell viability of HER2‐positive breast cancer cells 21. Ibrutinib treatment also leads to dose‐dependent inhibition of EGFR phosphorylation and it is speculated that it could be a candidate drug for the treatment of EGFR‐mutant, non‐small cell lung cancer 22. Other studies show that ibrutinib is an effective mast cell inhibitor and treatment of insulinoma‐bearing mice with this drug blocks mast cell degranulation and triggers collapse of tumour vasculature, tumour hypoxia and regression 23. In a preclinical model of pancreatic ductal adenocarcinoma as well as in patient‐derived xenograft‐models, ibrutinib showed mast cell dependent, antifibrotic activity and improved survival 24. Based on this finding, it was also suggested that the use of ibrutinib could potentially be extended to the treatment of other fibrotic diseases or chronic pancreatitis. Furthermore, other studies have shown that ibrutinib can inhibit inflammasome activation by BTK and TEC and attenuate inflammatory responses. Inhibition of TEC in a murine model of fungal infection protected the treated mice from fungal sepsis suggesting the drug could be used to combat invasive microbial infections 25. Targeting BTK could also have a therapeutic potential in ischaemic stroke, as ibrutinib suppresses the inflammasome in the ischaemic brain 26.
TEC family kinases
TFKs are expressed preferentially in different lineages of haematopoietic cells but also in other organs, such as liver (TEC) or in endothelial cells (BMX), as previously reviewed 29 and as summarized in Table 1. Loss‐of‐function mutations of the family members BTK and ITK, as well as elevated expression of TXK and ITK are associated with human disease. All members of TFKs have been knocked out in mice and their phenotypes have been studied, as summarized in Table 2. There, we focus on B and T cell compartments and compare KO mouse phenotypes with human diseases related to the dysfunction of corresponding genes.
Table 2.
TEC family kinases: the human diseases and the phenotype of KO mice
| Proteins | Human disease | Knock‐out mouse phenotype (B and T lymphocyte compartments) |
|---|---|---|
| BTK |
X‐linked agammaglobulinemia: Lack of mature B cells and gammaglobulin production (<1% CD19+ cells in peripheral blood) 36, 37 |
X‐linked immunodeficient (XID) and BTK KO mouse: milder B cell deficiency than in XLA patients with 50% of the peripheral B cells remaining, loss of B1 B cells in the peritoneal cavity, immature phenotype of peripheral B cells, low levels of secretory IgM and IgG3, absent responses to T cell independent type II antigens 39, 40 |
| TEC | Not reported | No overt B cell phenotype 42 |
| Tec KO mouse: enhanced generation of CD44highCD62L− Th17 subset of CD4+ T cells 43 | ||
| ITK | ITK mutation: EBV‐associated lymphoproliferative disorder 44, 45 | ITK KO mouse: increased percentage of memory phenotype CD44highCD62L−CD4+ T cells and CD8+ T cells of almost exclusively CD44highCD62L+ innate‐like phenotype 46, 58, defective Th2 and Th17 differentiation and function, increase in regulatory T cell (Treg) numbers and function 50, 51, defective NKT 59 |
| ITK increased expression: patients with atopic dermatitis 49 | ||
| BMX | Not reported | No overt phenotype 53 |
| RLK/TXK | TXK increased expression: patients with Behcet's disease 56 | No overt phenotype, marginal T cell defect 60 |
Thus, mutations in the BTK gene in humans cause a severe B cell deficiency X‐linked, agammaglobulinemia (XLA) 36, 37. In affected individuals, a defect in B cell maturation and function leads to the profound reduction of serum immunoglobulins and increased susceptibility to infections 38. A less severe, B cell disorder manifested at later stages of B cell development is observed in mice carrying BTK mutations 39, 40. It seems that in mice TEC partially compensates for the loss of BTK, while this does not occur in humans, suggesting that the corresponding signal transduction pathways are not identical in these species (Table 3). Of interest is also the fact that BTK seems to be required for NK cell activation and cytokine production 28, 41. Surprisingly, patients treated with ibrutinib do not develop an XLA‐like disease and the immunoglobulin (Ig) blood levels in patients receiving this drug were not reduced even after 12 cycles (35‐day cycle) of ibrutinib, suggesting that drug activity can be limited to immature B cells 18 or that antibody levels are maintained due to the fact that Ig producing plasma cells are not BTK dependent. However, in the mouse models for rheumatoid arthritis and systemic lupus erythematosus, the production of autoantibodies was reduced after ibrutinib treatment 14, 15. This suggests that ibrutinib would influence antibody production towards immunogens acquired during the treatment.
Table 3.
The severity of the phenotype in mice defective for multiple TFKs
| Cell type | More severe double KO phenotype when compared to single KO mice |
|---|---|
| B lymphocytes | BTK/TEC 42 |
| T lymphocytes | ITK/TXK 59, 61 |
| Platelets | BTK/TEC 65 |
| Osteoclasts | BTK/TEC 64 |
| Macrophages | BTK/TEC 62, 63 |
| Mast cells | BTK/TEC, BTK/ITK 66, 67 |
| Dendritic cells | Not reported |
| Erythroblasts/Erythrocytes | Not reported |
| Neutrophils | Not reported |
TEC does not appear to be crucial for B cell maturation, as TEC KO mice do not have any altered phenotype in the B cell compartment 42. However, other studies show that TEC deficiency leads to the enhanced generation of Th17 effector/memory subsets in vivo. Moreover, CD44highCD62L−CD4+ T cells are slightly increased in TEC KO mice, and they produce more IL‐17 upon activation when compared to WT mice 43. ITK mutations in humans have been reported in several cases of a fatal Epstein–Barr virus (EBV)‐associated lymphoproliferative disorder 44, 45. ITK KO mice show altered T cell development and mature T cell effector function, affecting in particular conventional T cells, thus leading to increased numbers of CD4+ T cells with a CD44highCD62L− memory phenotype and CD8+ T cells, with a CD44highCD62L+ innate‐like phenotype, that upon stimulation rapidly secrete high levels of effector cytokines such as IFN‐γ 46. The Th2 deficiency related to the lack of the ITK function, has made this kinase an interesting therapeutic target in asthma as ITK KO mice are resistant to ovalbumin‐induced asthma 47, 48. Furthermore, studies in 59 patients with atopic dermatitis, which is a Th2 mediated disease, showed highly elevated levels of ITK in peripheral blood T lymphocytes 49. It was also shown that ITK deletion affects the function of Th17 cells and that there is an increase in regulatory T cell (Treg) numbers and function in the absence of this kinase. This makes ITK a promising drug target for autoimmune disorders 50, 51. It was also found that in human NK cells ITK plays a role in regulating cell‐mediated cytotoxicity 52.
Human disease related to mutations in the BMX gene has not been reported as yet and the BMX KO mouse does not show an overt phenotype. Nonetheless, studies in the BMX KO mice suggest that this tyrosine kinase is involved in ischaemia‐induced arteriogenesis and angiogenesis 53. In addition to the role of BMX in the ischaemic response, BMX KO mice show reduced cardiac hypertrophy in a model of transverse aortic constriction, suggesting that BMX is required for the morphological response to pressure overload in the heart 54. Furthermore, it was shown that BMX participates in the wound healing process as observed in BMX transgenic mice 55.
Mutations affecting TXK have not been reported in humans as yet. TXK is preferentially expressed in Th1 cells. Its elevated expression was observed in patients with Behcet's disease, an inflammatory disorder associated with Th1 cytokine production and inflammation 56, 57, 58.
Many cell types express more than one TFK (Table 1) and loss‐of‐function mutants show additive functions. Mice deficient for various members of TEC family kinases give an important insight into the function of these proteins in different cell types and reveal biological processes for which these molecules are essential. It has been demonstrated for several of the tyrosine kinases that combined deletions (double KO mice) result in more severe phenotypes when compared to individual gene disruptions (Table 3). This can be exemplified by concomitant absence of BTK and TEC in B cells 42, ITK and TXK in both T cells 61 and NKT cells 59.
The dependence on more than a single TEC family kinase was also reported for BTK and TEC in macrophages 62, 63, osteoclasts 64 and platelets 65 (Table 3). Interestingly, compensatory pathways were described for BTK and TEC as well as for BTK and ITK in mast cells 66, 67, which will be described in more detail in the next sections of this review.
Even if multiple mutations affecting different TFK genes are unlikely to occur in the same individual, there is a risk that drugs binding to several target proteins and blocking their function could mediate such an outcome in treated patients. Here, studies in animals simultaneously defective in multiple TFKs would be of great value as demonstrated for SRC‐family protein kinases. There, only the BLK, FYN and LYN triple‐deficient mice have shown the profound impairment of the B cell development 68.
Thus, blocking the function of other/multiple TFKs would lead to ibrutinib off‐BTK target effects, as discussed below. Importantly, such effects could have either positive treatment outcomes, for example irreversible binding to ITK in T cells has been suggested as a treatment strategy for T cell malignancies 69 or negative, for example ibrutinib binding to both BTK and TEC in platelets could potentially lead to impaired coagulation and bleedings as observed in a subgroup of ibrutinib‐treated patients 20 (Table 3).
Consequences of ibrutinib treatment not related to B cells
Ibrutinib affects ITK in T cells and NK cells
ITK is highly expressed in T cells. Upon T cell receptor (TCR) ligation ITK and RLK/TXK activate a signal cascade ultimately leading to cellular activation, proliferation and cytokine secretion 58. In CD8+ and Th1 CD4+ T cells, ITK plays a non‐essential supportive role to RLK/TXK. However, ITK retains a singular dominant role for the function of Th2 CD4+ T cells 50, 57, which do not express RLK/TXK 70 and for this reason, loss of ITK function impairs Th2 immunity.
Despite the fact that ITK shares significant sequence and functional homology with BTK, the possibility that it could be targeted by ibrutinib had been initially neglected due to a number of in vitro observations 15. Nevertheless, other in vitro observations pointed out that ibrutinib could affect T cell functions, for example production of certain cytokines 71.
In 2013, it was finally confirmed 69 that ibrutinib is capable to irreversibly bind to endogenous ITK in a T cell leukaemia cell line and in PBMC samples obtained from ibrutinib‐treated CLL patients. In the latter, the percentage of ITK bound to ibrutinib after 8 days of the treatment was found to be 40–80%, which was in line with in vitro data.
Furthermore, it was shown that ibrutinib could inhibit TCR‐induced ITK signalling and activation in both primary CD4+ and in Jurkat T cells as well as in CD4+ T cells collected from CLL patients receiving ibrutinib. Such inhibition neither affected TCR‐mediated cell proliferation nor T cell subsets (naïve, central, effector, and terminal memory). However, ibrutinib treatment affected T helper cell polarization as seen in the decrease of serum Th2‐type cytokines IL‐10, IL‐4 and IL‐13 as well as increase of IFN‐γ levels in CLL patients after 28 days of treatment. Accordingly, ibrutinib favoured Th1 polarization of human CD4+ cells in vitro and induced Th1/Th2 skewing of Ig subclasses in treated mice 69. It was concluded that ibrutinib selectively inhibits Th2 cells, because RLK/TXK is not expressed in these cells and cannot compensate for the ITK inhibited by ibrutinib 70. Nevertheless, the fact that ibrutinib seems to not bind to RLK/TXK is somewhat unexpected, as both kinases have ibrutinib binding sites. There is a possibility that the structure of RLK/TXK makes this protein less accessible for the drug. Still, this would need to be further investigated.
It is well known that one of the immune escape mechanisms exploited by tumours and by some pathogens is the subversion of adaptive immunity by promoting a Th2‐dominant helper T cell response favouring antibody production. This, in turn, has a detrimental effect on the ability of the immune system to trigger robust Th1 and CD8+ responses inducing direct effector cell cytotoxicity, thereby suppressing antitumour immune responses 72, 73.
The ability of ibrutinib to target ITK and therefore to modulate T cell responses suggested the possibility to combine this drug with other immune modulating therapies such as checkpoint inhibitors, that is agents interfering with either the CD80/CTLA‐4 or the PD‐1/PD‐L1 axis. Recently, the combination of ibrutinib with anti‐PD‐L1 antibody was tested in a mouse model of lymphoma that is intrinsically insensitive to ibrutinib but highly expresses PD‐L1 74. Treatment of animals with established tumours by the anti‐PD‐L1 antibody alone resulted in delayed tumour growth and slightly prolonged overall survival, but was not curative. In contrast, the combined treatment with anti‐PD‐L1 and ibrutinib resulted in the cure of approximately 50% of the mice. Tumour‐specific IFN‐γ producing T cells were found in the mice treated with the combination of ibrutinib and anti‐PD‐L1 antibody, but not with either agent alone. A similar effect was observed in models of solid tumours lacking BTK and expressing PD‐L1 at very low levels, such as 4T1, a breast cancer model and CT26, a colon cancer model. The authors of the study therefore conclude that ibrutinib is able to potentiate the antitumour effect induced by anti‐PD‐L1 therapy and that this effect is mediated by inhibition of ITK, which provides the rationale for the use of ibrutinib in combination with T cell therapies 74.
Still, the effects of ibrutinib could also be detrimental to antitumour immune responses depending on, for example. NK cell functions. In a preclinical study, an antagonistic effect was observed when ibrutinib was combined with rituximab, an anti‐CD20 antibody widely used in lymphoma treatment 75. Rituximab has various mechanisms of action, for instance antibody‐dependent cell‐mediated cytotoxicity (ADCC). In activated human NK cells, ITK positively regulates Fc receptor (FcR)‐initiated cytotoxicity 52, suggesting that treatment with ibrutinib could impair NK cell‐mediated ADCC. Indeed, the presence of the drug in co‐culture of NK cells with rituximab‐coated lymphoma cells impaired NK cell cytokine secretion in a dose‐dependent manner as well as FcR‐stimulated NK cell degranulation and inhibited NK cell‐mediated cytotoxicity 75. Nevertheless, the direct cytotoxic effect of BTK inhibition on lymphoma cells in vitro outweighed the inhibition of NK cell cytotoxicity, making this finding less of a concern for antilymphoma therapy.
Ibrutinib inhibits BTK and possibly TEC in platelets
Bleeding is one of the common side effects of treatment with ibrutinib. A study including 111 patients with mantle cell lymphoma reported that 17% of the treated patients had contusions and 4.5% had more severe bleedings 76 and a recent 3‐year follow‐up of two other clinical studies reported that 61% of CLL and SLL patients treated with ibrutinib experienced bleeding events 19. The majority of these events were not severe, with contusion and petechiae being the most commonly reported terms. The subcutaneous bleedings observed in ibrutinib‐treated patients are usually not associated with thrombocytopenia 17 indicating a defect in platelet function.
Ibrutinib has been shown to affect platelet function in vitro. Platelets from treated patients display an impaired aggregation response to collagen and reduced adhesion to von Willebrand Factor (vWF)‐coated surfaces under high shear conditions, while aggregation responses to other stimuli, such as ADP, thrombin or thromboxane A2 are unaffected 20, 77. The aggregation defect in response to collagen correlates with clinical observations as platelets from patients that experienced bleedings had a significantly lower aggregation response compared to patients without haemostatic complications 20, 77.
The two TFKs expressed by platelets, namely BTK and TEC, are both involved in collagen‐induced platelet activation 78, 79. Binding of collagen to the receptor glycoprotein VI (GPVI) leads to PI3K‐dependent phosphorylation of both molecules independently from each other 65, 79, and they in turn activate PLCγ2 65, 78. Autophosphorylation of BTK and phosphorylation of PLCγ2 in response to collagen are both reduced in ibrutinib‐treated platelets 20. The collagen response of platelets without functional BTK (i.e. from XLA patients or BTK KO mice) is reduced, but almost normal at high collagen concentrations 65, 78. It is thought that TEC compensates for BTK in these platelets, because platelets from BTK/TEC double KO mice are completely unresponsive to stimulation of the collagen receptor GPVI and show strongly reduced aggregation responses to collagen even at high concentrations 65. The contribution of TEC to collagen signalling in the presence of BTK seems to be minor, as platelets from TEC KO mice have only a slight defect in the aggregation response to collagen 65. Platelet activation by vWF, the other activation pathway that is affected by ibrutinib treatment 20, also involves BTK 80. So far, no role for TEC has been described in this process, but it does not seem to compensate for absent BTK, because no phosphorylation of PLCγ2 was found in platelets from BTK‐deficient mice after vWF stimulation, and no stable vWF‐dependent thrombus formation was observed in vivo in these mice 81. A recent report showed the importance of BTK and TEC in CLEC‐2 receptor‐mediated platelet activation 82. Activation of platelets by binding of CLEC‐2 to its ligand podoplanin on lymphatic vessel endothelial cells has been found to be a major mechanism for maintaining blood‐lymphatic separation 83. Inhibition of TFKs in murine platelets with ibrutinib abolishes aggregation responses to CLEC‐2 stimulation completely, and embryos of BTK/TEC KO mice exhibit cutaneous oedema with blood‐filled vessels with a lymphatic pattern similar to CLEC‐2‐deficient mice 82. Furthermore, engagement of the fibrinogen receptor integrin α IIb β 3 has been reported to induce BTK and TEC phosphorylation 84, 85, indicating that the kinases are not only involved in the initial activation of platelets at sites where the extracellular matrix is exposed to the blood stream, but also in later stages of thrombus formation.
These findings suggest that the platelet aggregation defects observed in ibrutinib‐treated patients are caused by the inhibition of both BTK and TEC in platelets. The impaired responses to collagen and vWF, as well as defective blood‐lymph separation can explain the spontaneous bleedings of these patients. The bleeding events are mostly mild, probably because other activation pathways are not affected by ibrutinib 20, 77, and collagen can still induce very weak responses in the absence of BTK and TEC 65. However, ibrutinib is suspected to have more severe effects in combination with other anticoagulant drugs, and for example, warfarin usage has been excluded from ibrutinib clinical trials 76.
One factor that could possibly play an additional role in the bleeding events under ibrutinib treatment is an effect of the drug on endothelial cells. Endothelial cells express the TEC family kinase BMX 31, 86. BMX has been reported to be involved in angiogenesis and wound healing 55. One could speculate that inhibition of BMX by ibrutinib might affect the integrity of the endothelium. However, mice deficient for BMX have no obvious phenotype and no disturbance of the vascular endothelium 87. The expression in mice is the strongest in big arteries, weaker in small arteries and absent in capillaries 87. The fact that most of the bleeding events in ibrutinib‐treated patients are subcutaneous, that is in places where the vasculature expresses only little or no BMX, makes it less likely that this is an effect of BMX inhibition by ibrutinib.
Potential Ibrutinib binding to BTK and TEC in osteoclasts
Bone tissue homoeostasis is regulated by bone‐forming osteoblasts and bone‐resorbing osteoclasts. The imbalance of the bone turnover causes various bone disorders 88. Thus, skewing the balance towards osteoclasts leads to the pathologic osteolysis and diseases with low bone mass, while impaired osteoclastic function causes pathologies characterized by a high bone mass, for example osteopetrosis. Both BTK and TEC are expressed in osteoclasts and were shown to play an important role in osteoclastogenesis. In these cells, they are activated by the receptor activator of nuclear factor kappa‐B ligand (RANKL). BTK and TEC kinases link the RANKL and immunoreceptor tyrosine‐based activation motif (ITAM) signals to phosphorylate PLCγ2, which leads to stimulation of calcium signalling and activation of NFATc1 (nuclear factor of activated T cells c1), which is the key transcription factor of osteoclastogenesis 89. Mice deficient in both BTK and TEC have a severe osteopetrotic phenotype characterized by increased bone volume and reduced numbers of osteoclasts in the epiphyseal region 64. BTK‐deficient mice have defective osteoclast differentiation 90 but the phenotype of BTK/TEC double KO mice is far more obvious suggesting that TEC has a compensatory function. Furthermore, it was shown that human BTK‐deficient osteoclasts have defective resorption activity in vitro but in 20 studied XLA patients abnormal bone metabolism has not been observed, indicating the existence of compensatory mechanisms in vivo 91. Thus, it could be speculated that during long‐term use of ibrutinib, binding to both BTK and TEC could have an effect on the bone homoeostasis and possibly lead to pathological conditions due to the lack of bone‐resorbing activity of osteoclasts. To the best of our knowledge, studies of bone density have not been performed as yet in ibrutinib‐treated patients, even if they could be of clinical importance. Nevertheless, ibrutinib was suggested as a treatment option for bone diseases associated with an increased activity of osteoclasts and enhanced bone resorption, as in osteoporosis and rheumatoid arthritis (RA) 92. Ibrutinib's therapeutic effect was shown in an RA mouse model, and it is attributed to the fact that ibrutinib affects both B lymphocytes and inflammatory cells, which are BTK‐expressing effector cells involved in the pathology of arthritis 14, 93.
Ibrutinib effects on macrophages, neutrophils, dendritic cells and mast cells
TFKs are prominently expressed in haematopoietic cells, where they play crucial roles in lymphocyte development and activation 29. They are also expressed in cells of the myeloid lineage, including monocytes, macrophages, neutrophils, mast cells, erythrocytes and dendritic cells 27. It is known that TFKs play important roles within these cells. Targeting these kinases with ibrutinib will therefore interfere with such functions.
Macrophages contain four TEC family members BTK, TEC, ITK and BMX 27, 94. Several studies show that BTK‐deficient macrophages have impaired phagocytic function and chemotaxis. The murine macrophages that lack BTK have reduced secretion of the proinflammatory cytokines TNF‐α and IL‐1β after stimulation 27. In monocytes and macrophages, ibrutinib treatment inhibited the BTK auto‐phosphorylation as well as phosphorylation of the BTK substrate PLCγ2 and also blocked calcium mobilization and dose dependently inhibited TNF‐α and IL‐1β production following Fc receptor stimulation 14.
Neutrophils represent the major group of phagocytes. They play a key role in innate immune responses. Human neutrophils express BMX, BTK and TEC 27, 95. These TFKs are crucial for functional activation of neutrophils. Neutrophil adhesion and migration were reduced after treatment with other BTK inhibitors. Following ibrutinib treatment, there was a nearly complete inhibition of neutrophil infiltration into the synovial joints in mice with collagen‐induced arthritis 14.
Dendritic cells (DC) are the most potent antigen‐presenting cells. They express two kinases with an ibrutinib binding site, BTK and BLK (Table 1). It was shown that BTK is essential for human DC function. Signalling through several Toll‐like receptors is impaired in DCs from XLA patients resulting in reduced production of pro‐inflammatory cytokines, and it has been suggested that this may contribute to the susceptibility to infections in these patients. Furthermore, in vitro treatment of healthy DCs with ibrutinib resulted in a similar defect 96, 97.
The SRC‐family kinase BLK is expressed in plasmacytoid DCs (pDCs), an interferon type I‐producing subset of DCs that is important for the response to viral infections. In mice with reduced BLK expression, plasma levels of IFNα were found to be lower compared to wild type animals 16. Thus, ibrutinib‐mediated activity in DCs could be of clinical relevance for side effects observed in some of the treated patients.
Mast cells play crucial roles in a variety of normal and pathophysiological processes and they are important for the initiation of allergic reactions. In mast cells, BTK, ITK, TEC and RLK/TXK are expressed 27. Among these, the roles for BTK, ITK, and TEC have been investigated. The dependence of mast cells on more than one of TFKs was reported. BTK‐deficient mast cells are defective in degranulation, histamine release and cytokine secretion following FcR stimulation 67. ITK‐deficient mice have been reported to display decreased mast cell degranulation and histamine release. In ITK/BTK double KO mice, these defects were significantly more severe 67. A role for TEC during mast cell activation has also been reported. Studies show that TEC deficient mast cells have impaired production of leukotriene C4 and IL‐4 and that they require both TEC and BTK for the efficient production of TNF‐α, IL‐13 and GM‐CSF 66. Thus, it could be assumed that binding of ibrutinib to three of the TFKs and inhibiting their function would have even more severe consequences.
In human cultured mast cells, ibrutinib potently inhibited histamine release following FcR activation. The drug also dose dependently reduced the release of the inflammatory cytokines TNF‐α and IL‐8 14, indicating that the drug interferes and impairs mast cell function. Conversely, mast cell inhibition may be clinically advantageous in certain tumour types, in which functional mast cells are required for maintenance of malignant cells 23, 24.
Ibrutinib and atrial fibrillation
Recently, atrial fibrillation was reported as an adverse effect of ibrutinib treatment, occurring in a few percentages of the patients. The effect was attributed to the binding of ibrutinib to BTK and TEC in the heart and subsequent inhibition of cardioprotective PI3K‐AKT signalling 30. The mentioning that BTK transcripts are expressed in heart tissue is difficult to reconcile as, to our knowledge, this kinase has never been convincingly detected at the protein level in this organ. However, TEC has been detected in rat neonatal cardiomyocytes 98 and in adult mouse cardiac myocytes, where it plays a role in myocardial ischaemia 99, Interestingly, it was also shown that blocking another protein containing an ibrutinib binding site ErbB2/HER2 (Table 1) results in cardiomyocyte dysfunction and reduced heart contractile efficiency. This was observed as an adverse side effect in patients with breast cancer treated by HER2 inhibitors 100. It was also shown that conditional ErbB2/HER2 mutation in ventricular cardiomyocytes leads to impaired cardiac conduction 101 and the protein is required for atrial electrical activity during development 102. Furthermore, another member of the EGFR family, ErbB4/HER4, is also expressed in the myocardiocytes. Heterodimerization of HER4 with HER2 and subsequent downstream signalling, including the PI3K‐AKT pathway, plays a crucial role in heart physiology 103. As described in the previous section, also the ibrutinib‐binding protein BMX was shown to be of importance for the cardiovascular system 53. Thus, there are several possibilities for that ibrutinib's interaction with target proteins other than BTK could cause cardiac dysfunction and atrial fibrillation, and we favour the idea of an off‐BTK target mechanism.
Conclusions
The therapeutic off‐target effects of ibrutinib studied until now in the haematopoietic system mainly relate to its activity as an ITK inhibitor, which affects helper T cells differentiation, thereby influencing the immune response against tumours and infections, but also potentially harness other immune functions. The final therapeutic outcome of ibrutinib treatment in ‘BTK‐dependent’ diseases will anyhow result from the combination of its on‐target with its off‐target effects. For instance, it may be that clinically relevant adverse manifestations in bone tissue could result from both on‐ and off‐target effects, and that such potential outcomes only appear after long‐term treatment given the rather slow turnover of the bone. Learning more about the influence of ibrutinib on different target proteins, will undoubtedly contribute to the development of strategies for adequate follow‐up of patients and efficient correction of adverse effects. Moreover, there is no doubt that further knowledge of its off‐targets effects likely will pave the way to a broader clinical application of this drug. Thus, such knowledge is useful when considering new indications, where the ‘off‐target’ activity or combination of on‐ and off‐target effects may form the basis for new treatments. To this end, the history of drug development repeatedly teaches us that, what was initially considered a single‐target compound, may later turn out to be a less restricted medicine, but also that an adverse effect may later contribute to the definition of a highly relevant drug target.
Acknowledgment
This work was supported by the Swedish Cancer Society (CAN2013/389), Swedish Medical Research Council (K2015‐68X‐11247‐21‐3) and Swedish County Council (ALF‐project 2012006).
The copyright line for this article was changed on 6 March 2017 after the original publication.
Note
For simplification we have used capital letters for all kinases throughout this review.
References
- 1. Cameron F, Sanford M. Ibrutinib: first global approval. Drugs 2014;74:263–71. [DOI] [PubMed] [Google Scholar]
- 2. Singh J, Petter RC, Kluge AF. Targeted covalent drugs of the kinase family. Curr Opin Chem Biol 2010;14:475–80. [DOI] [PubMed] [Google Scholar]
- 3. Mohamed AJ, Vargas L, Nore BF, Backesjo CM, Christensson B, Smith CI. Nucleocytoplasmic shuttling of Bruton's tyrosine kinase. J Biol Chem 2000;275:40614–9. [DOI] [PubMed] [Google Scholar]
- 4. Lindvall JM, Blomberg KE, Valiaho J et al Bruton's tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol Rev 2005;203:200–15. [DOI] [PubMed] [Google Scholar]
- 5. Berglof A, Turunen JJ, Gissberg O, Bestas B, Blomberg KE, Smith CI. Agammaglobulinemia: causative mutations and their implications for novel therapies. Exp Rev Clin Immunol 2013;9:1205–21. [DOI] [PubMed] [Google Scholar]
- 6. Seda V, Mraz M. B‐cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol 2015;94:193–205. [DOI] [PubMed] [Google Scholar]
- 7. Pan Z, Scheerens H, Li SJ et al Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem 2007;2:58–61. [DOI] [PubMed] [Google Scholar]
- 8. Aalipour A, Advani RH. Bruton's tyrosine kinase inhibitors and their clinical potential in the treatment of B‐cell malignancies: focus on ibrutinib. Ther Adv Hematol 2014;5:121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vargas L, Hamasy A, Nore BF, Smith CI. Inhibitors of BTK and ITK: state of the new drugs for cancer, autoimmunity and inflammatory diseases. Scand J Immunol 2013;78:130–9. [DOI] [PubMed] [Google Scholar]
- 10. Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton's tyrosine kinase. Future Oncol 2014;10:957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Treon SP, Tripsas CK, Meid K et al Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med 2015;372:1430–40. [DOI] [PubMed] [Google Scholar]
- 12. Kim ES, Dhillon S. Ibrutinib: a review of its use in patients with mantle cell lymphoma or chronic lymphocytic leukaemia. Drugs 2015;75:769–76. [DOI] [PubMed] [Google Scholar]
- 13. Zhang SQ, Smith SM, Zhang SY, Lynn Wang Y. Mechanisms of ibrutinib resistance in chronic lymphocytic leukaemia and non‐Hodgkin lymphoma. Br J Haematol 2015. doi: 10.1111/bjh.13427. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 14. Chang BY, Huang MM, Francesco M et al The Bruton tyrosine kinase inhibitor PCI‐32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther 2011;13:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Honigberg LA, Smith AM, Sirisawad M et al The Bruton tyrosine kinase inhibitor PCI‐32765 blocks B‐cell activation and is efficacious in models of autoimmune disease and B‐cell malignancy. Proc Natl Acad Sci U S A 2010;107:13075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Samuelson EM, Laird RM, Papillion AM, Tatum AH, Princiotta MF, Hayes SM. Reduced B lymphoid kinase (Blk) expression enhances proinflammatory cytokine production and induces nephrosis in C57BL/6‐lpr/lpr mice. PLoS One 2014;9:e92054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Byrd JC, Furman RR, Coutre SE et al Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Advani RH, Buggy JJ, Sharman JP et al Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol 2013;1 (31):88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Byrd JC, Furman RR, Coutre SE et al Three‐year follow‐up of treatment‐naive and previously treated patients with CLL and SLL receiving single‐agent ibrutinib. Blood 2015;125:2497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levade M, David E, Garcia C et al Ibrutinib treatment affects collagen and von Willebrand factor‐dependent platelet functions. Blood 2014;124:3991–5. [DOI] [PubMed] [Google Scholar]
- 21. Grabinski N, Ewald F. Ibrutinib (ImbruvicaTM) potently inhibits ErbB receptor phosphorylation and cell viability of ErbB2‐positive breast cancer cells. Invest New Drugs 2014;32:1096–104. [DOI] [PubMed] [Google Scholar]
- 22. Gao W, Wang M, Wang L et al Selective antitumor activity of ibrutinib in EGFR‐mutant non‐small cell lung cancer cells. J Natl Cancer Inst. 2014;106. pii: dju204. doi: 10.1093/jnci/dju204. Print 2014 Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soucek L, Buggy JJ, Kortlever R et al Modeling pharmacological inhibition of mast cell degranulation as a therapy for insulinoma. Neoplasia 2011;13:1093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Masso‐Valles D, Jauset T, Serrano E et al Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res 2015;75:1675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zwolanek F, Riedelberger M, Stolz V et al The non‐receptor tyrosine kinase Tec controls assembly and activity of the noncanonical caspase‐8 inflammasome. PLoS Pathog 2014;10:e1004525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ito M, Shichita T, Okada M et al Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 2015;6:7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmidt U, Boucheron N, Unger B, Ellmeier W. The role of Tec family kinases in myeloid cells. Int Arch Allergy Immunol 2004;134:65–78. [DOI] [PubMed] [Google Scholar]
- 28. Bao Y, Zheng J, Han C et al Tyrosine kinase Btk is required for NK cell activation. J Biol Chem 2012;287:23769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. BioEssays 2001;23:436–46. [DOI] [PubMed] [Google Scholar]
- 30. McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K‐Akt signaling. Blood 2014;124:3829–30. [DOI] [PubMed] [Google Scholar]
- 31. Ekman N, Lymboussaki A, Vastrik I, Sarvas K, Kaipainen A, Alitalo K. Bmx tyrosine kinase is specifically expressed in the endocardium and the endothelium of large arteries. Circulation 1997;96:1729–32. [DOI] [PubMed] [Google Scholar]
- 32. Prince AL, Yin CC, Enos ME, Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate conventional versus innate T‐cell development. Immunol Rev 2009;228:115–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Islam KB, Rabbani H, Larsson C, Sanders R, Smith CI. Molecular cloning, characterization, and chromosomal localization of a human lymphoid tyrosine kinase related to murine Blk. J Immunol 1995;154:1265–72. [PubMed] [Google Scholar]
- 34. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127–37. [DOI] [PubMed] [Google Scholar]
- 35. Leonard WJ, O'Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol 1998;16:293–322. [DOI] [PubMed] [Google Scholar]
- 36. Vetrie D, Vorechovsky I, Sideras P et al The gene involved in X‐linked agammaglobulinaemia is a member of the src family of protein‐tyrosine kinases. Nature 1993;361:226–33. [DOI] [PubMed] [Google Scholar]
- 37. Tsukada S, Saffran DC, Rawlings DJ et al Deficient expression of a B cell cytoplasmic tyrosine kinase in human X‐linked agammaglobulinemia. Cell 1993;72:279–90. [DOI] [PubMed] [Google Scholar]
- 38. Winkelstein JA, Marino MC, Lederman HM et al X‐linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine 2006;85:193–202. [DOI] [PubMed] [Google Scholar]
- 39. Rawlings DJ, Saffran DC, Tsukada S et al Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science 1993;261:358–61. [DOI] [PubMed] [Google Scholar]
- 40. Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X‐linked agammaglobulinemia and X‐linked immunodeficiency genes. Science 1993;16 (261):355–8. [DOI] [PubMed] [Google Scholar]
- 41. Ni Gabhann J, Spence S, Wynne C et al Defects in acute responses to TLR4 in Btk‐deficient mice result in impaired dendritic cell‐induced IFN‐gamma production by natural killer cells. Clin Immunol 2012;142:373–82. [DOI] [PubMed] [Google Scholar]
- 42. Ellmeier W, Jung S, Sunshine MJ et al Severe B cell deficiency in mice lacking the tec kinase family members Tec and Btk. J Exp Med 2000;192:1611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boucheron N, Sharif O, Schebesta A et al The protein tyrosine kinase Tec regulates a CD44highCD62L‐ Th17 subset. J Immunol 2010;185:5111–9. [DOI] [PubMed] [Google Scholar]
- 44. Linka RM, Risse SL, Bienemann K et al Loss‐of‐function mutations within the IL‐2 inducible kinase ITK in patients with EBV‐associated lymphoproliferative diseases. Leukemia 2012;26:963–71. [DOI] [PubMed] [Google Scholar]
- 45. Mansouri D, Mahdaviani SA, Khalilzadeh S et al IL‐2‐inducible T‐cell kinase deficiency with pulmonary manifestations due to disseminated Epstein‐Barr virus infection. Int Arch Allergy Immunol 2012;158:418–22. [DOI] [PubMed] [Google Scholar]
- 46. August A, Ragin MJ. Regulation of T‐cell responses and disease by tec kinase Itk. Int Rev Immunol 2012;31:155–65. [DOI] [PubMed] [Google Scholar]
- 47. Mueller C, August A. Attenuation of immunological symptoms of allergic asthma in mice lacking the tyrosine kinase ITK. J Immunol 2003;170:5056–63. [DOI] [PubMed] [Google Scholar]
- 48. Sahu N, August A. ITK inhibitors in inflammation and immune‐mediated disorders. Curr Top Med Chem 2009;9:690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matsumoto Y, Oshida T, Obayashi I et al Identification of highly expressed genes in peripheral blood T cells from patients with atopic dermatitis. Int Arch Allergy Immunol 2002;129:327–40. [DOI] [PubMed] [Google Scholar]
- 50. Fowell DJ, Shinkai K, Liao XC et al Impaired NFATc translocation and failure of Th2 development in Itk‐deficient CD4+ T cells. Immunity 1999;11:399–409. [DOI] [PubMed] [Google Scholar]
- 51. Gomez‐Rodriguez J, Wohlfert EA, Handon R et al Itk‐mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med 2014;211:529–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khurana D, Arneson LN, Schoon RA, Dick CJ, Leibson PJ. Differential regulation of human NK cell‐mediated cytotoxicity by the tyrosine kinase Itk. J Immunol 2007;178:3575–82. [DOI] [PubMed] [Google Scholar]
- 53. Holopainen T, Lopez‐Alpuche V, Zheng W et al Deletion of the endothelial Bmx tyrosine kinase decreases tumor angiogenesis and growth. Cancer Res 2012;72:3512–21. [DOI] [PubMed] [Google Scholar]
- 54. Cenni B, Gutmann S, Gottar‐Guillier M. BMX and its role in inflammation, cardiovascular disease, and cancer. Int Rev Immunol 2012;31:166–73. [DOI] [PubMed] [Google Scholar]
- 55. Paavonen K, Ekman N, Wirzenius M, Rajantie I, Poutanen M, Alitalo K. Bmx tyrosine kinase transgene induces skin hyperplasia, inflammatory angiogenesis, and accelerated wound healing. Mol Biol Cell 2004;15:4226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Suzuki N, Nara K, Suzuki T. Skewed Th1 responses caused by excessive expression of Txk, a member of the Tec family of tyrosine kinases, in patients with Behcet's disease. Clin Med Res 2006;4:147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sahu N, Venegas AM, Jankovic D et al Selective expression rather than specific function of Txk and Itk regulate Th1 and Th2 responses. J Immunol 2008;181:6125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gomez‐Rodriguez J, Kraus ZJ, Schwartzberg PL. Tec family kinases Itk and Rlk/Txk in T lymphocytes: cross‐regulation of cytokine production and T‐cell fates. FEBS J 2011;278:1980–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate NKT cell maturation, cytokine production, and survival. J Immunol 2008;180:3007–18. [DOI] [PubMed] [Google Scholar]
- 60. Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol 2005;23:549–600. [DOI] [PubMed] [Google Scholar]
- 61. Schaeffer EM, Debnath J, Yap G et al Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science 1999;284:638–41. [DOI] [PubMed] [Google Scholar]
- 62. Melcher M, Unger B, Schmidt U, Rajantie IA, Alitalo K, Ellmeier W. Essential roles for the Tec family kinases Tec and Btk in M‐CSF receptor signaling pathways that regulate macrophage survival. J Immunol 2008;180:8048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jongstra‐Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton's tyrosine kinase and Tec kinase during Fcgamma receptor‐induced signaling and phagocytosis. J Immunol 2008;181:288–98. [DOI] [PubMed] [Google Scholar]
- 64. Shinohara M, Koga T, Okamoto K et al Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008;132:794–806. [DOI] [PubMed] [Google Scholar]
- 65. Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood 2003;102:3592–9. [DOI] [PubMed] [Google Scholar]
- 66. Schmidt U, Abramova A, Boucheron N et al The protein tyrosine kinase Tec regulates mast cell function. Eur J Immunol 2009;39:3228–38. [DOI] [PubMed] [Google Scholar]
- 67. Iyer AS, Morales JL, Huang W et al Absence of Tec family kinases interleukin‐2 inducible T cell kinase (Itk) and Bruton's tyrosine kinase (Btk) severely impairs Fc epsilonRI‐dependent mast cell responses. J Biol Chem 2011;286:9503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saijo K, Schmedt C, Su IH et al Essential role of Src‐family protein tyrosine kinases in NF‐kappaB activation during B cell development. Nat Immunol 2003;4:274–9. [DOI] [PubMed] [Google Scholar]
- 69. Dubovsky JA, Beckwith KA, Natarajan G et al Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1‐selective pressure in T lymphocytes. Blood 2013;122:2539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miller AT, Wilcox HM, Lai Z, Berg LJ. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T‐bet. Immunity 2004;21:67–80. [DOI] [PubMed] [Google Scholar]
- 71. Herman SE, Gordon AL, Hertlein E et al Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI‐32765. Blood 2011;117:6287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Horna P, Sotomayor EM. Cellular and molecular mechanisms of tumor‐induced T‐cell tolerance. Curr Cancer Drug Targets 2007;7:41–53. [DOI] [PubMed] [Google Scholar]
- 73. Loose D, Van de Wiele C. The immune system and cancer. Cancer biother Radiopharm 2009;24:369–76. [DOI] [PubMed] [Google Scholar]
- 74. Sagiv‐Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A 2015;112:E966–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kohrt HE, Sagiv‐Barfi I, Rafiq S et al Ibrutinib antagonizes rituximab‐dependent NK cell‐mediated cytotoxicity. Blood 2014;123:1957–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang ML, Rule S, Martin P et al Targeting BTK with ibrutinib in relapsed or refractory mantle‐cell lymphoma. N Engl J Med 2013;369:507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kamel S, Horton L, Ysebaert L et al Ibrutinib inhibits collagen‐mediated but not ADP‐mediated platelet aggregation. Leukemia 2015;29:783–7. [DOI] [PubMed] [Google Scholar]
- 78. Quek LS, Bolen J, Watson SP. A role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Current Biol 1998;8:1137–40. [DOI] [PubMed] [Google Scholar]
- 79. Oda A, Ikeda Y, Ochs HD et al Rapid tyrosine phosphorylation and activation of Bruton's tyrosine/Tec kinases in platelets induced by collagen binding or CD32 cross‐linking. Blood 2000;95:1663–70. [PubMed] [Google Scholar]
- 80. Crosby D, Poole AW. Interaction of Bruton's tyrosine kinase and protein kinase Ctheta in platelets. Cross‐talk between tyrosine and serine/threonine kinases. J Biol Chem 2002;277:9958–65. [DOI] [PubMed] [Google Scholar]
- 81. Liu J, Fitzgerald ME, Berndt MC, Jackson CW, Gartner TK. Bruton tyrosine kinase is essential for botrocetin/VWF‐induced signaling and GPIb‐dependent thrombus formation in vivo. Blood 2006;108:2596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Manne BK, Badolia R, Dangelmaier C et al Distinct pathways regulate Syk activation downstream of ITAM and hemITAM receptors in platelets. J Biol Chem. 2015;290:11557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hess PR, Rawnsley DR, Jakus Z et al Platelets mediate lymphovenous hemostasis to maintain blood‐lymphatic separation throughout life. J Clin Investig 2014;124:273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Laffargue M, Monnereau L, Tuech J et al Integrin‐dependent tyrosine phoshorylation and cytoskeletal translocation of Tec in thrombin‐activated platelets. Biochem Biophys Res Commun 1997;238:247–51. [DOI] [PubMed] [Google Scholar]
- 85. Laffargue M, Ragab‐Thomas JM, Ragab A et al Phosphoinositide 3‐kinase and integrin signalling are involved in activation of Bruton tyrosine kinase in thrombin‐stimulated platelets. FEBS Lett 1999;443:66–70. [DOI] [PubMed] [Google Scholar]
- 86. Tamagnone L, Lahtinen I, Mustonen T et al BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene 1994;9:3683–8. [PubMed] [Google Scholar]
- 87. Rajantie I, Ekman N, Iljin K et al Bmx tyrosine kinase has a redundant function downstream of angiopoietin and vascular endothelial growth factor receptors in arterial endothelium. Mol Cell Biol 2001;21:4647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2002;2:389–406. [DOI] [PubMed] [Google Scholar]
- 89. Takayanagi H, Kim S, Koga T et al Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 2002;3:889–901. [DOI] [PubMed] [Google Scholar]
- 90. Lee SH, Kim T, Jeong D, Kim N, Choi Y. The tec family tyrosine kinase Btk regulates RANKL‐induced osteoclast maturation. J Biol Chem 2008;283:11526–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Danks L, Workman S, Webster D, Horwood NJ. Elevated cytokine production restores bone resorption by human Btk‐deficient osteoclasts. J Bone Miner Res 2011;26:182–92. [DOI] [PubMed] [Google Scholar]
- 92. Shinohara M, Chang BY, Buggy JJ et al The orally available Btk inhibitor ibrutinib (PCI‐32765) protects against osteoclast‐mediated bone loss. Bone 2014;60:8–15. [DOI] [PubMed] [Google Scholar]
- 93. Burger JA, Buggy JJ. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765). Leuk Lymphoma 2013;54:2385–91. [DOI] [PubMed] [Google Scholar]
- 94. Weil D, Power MA, Smith SI, Li CL. Predominant expression of murine Bmx tyrosine kinase in the granulo‐monocytic lineage. Blood 1997;90:4332–40. [PubMed] [Google Scholar]
- 95. Lachance G, Levasseur S, Naccache PH. Chemotactic factor‐induced recruitment and activation of Tec family kinases in human neutrophils. Implication of phosphatidynositol 3‐kinases. J Biol Chem 2002;277:21537–41. [DOI] [PubMed] [Google Scholar]
- 96. Taneichi H, Kanegane H, Sira MM et al Toll‐like receptor signaling is impaired in dendritic cells from patients with X‐linked agammaglobulinemia. Clin Immunol 2008;126:148–54. [DOI] [PubMed] [Google Scholar]
- 97. Lougaris V, Baronio M, Vitali M et al Bruton tyrosine kinase mediates TLR9‐dependent human dendritic cell activation. J Allergy Clin Immunol 2014;133:1644–50.e4. [DOI] [PubMed] [Google Scholar]
- 98. Bony C, Roche S, Shuichi U et al A specific role of phosphatidylinositol 3‐kinase gamma. A regulation of autonomic Ca(2)+ oscillations in cardiac cells. J Cell Biol 2001;152:717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhang MJ, Franklin S, Li Y et al Stress signaling by Tec tyrosine kinase in the ischemic myocardium. Am J Physiol Heart Circ Physiol 2010;299:H713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Albini A, Cesana E, Donatelli F et al Cardio‐oncology in targeting the HER receptor family: the puzzle of different cardiotoxicities of HER2 inhibitors. Future Cardiol 2011;7:693–704. [DOI] [PubMed] [Google Scholar]
- 101. Ozcelik C, Erdmann B, Pilz B et al Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci U S A 2002;99:8880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tenin G, Clowes C, Wolton K et al Erbb2 is required for cardiac atrial electrical activity during development. PLoS One 2014;9:e107041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Milano GA, Serres E, Ferrero JM, Ciccolini J. Trastuzumab‐induced cardiotoxicity: is it a personalized risk? Curr Drug Targets 2014;15:1200–4. [DOI] [PubMed] [Google Scholar]