

Abstract
Solid organ transplant patients are vulnerable to suffering neurologic complications from a wide array of viral infections and can be sentinels in the population who are first to get serious complications from emerging infections like the recent waves of arboviruses, including West Nile virus, Chikungunya virus, Zika virus, and Dengue virus. The diverse and rapidly changing landscape of possible causes of viral encephalitis poses great challenges for traditional candidate‐based infectious disease diagnostics that already fail to identify a causative pathogen in approximately 50% of encephalitis cases. We present the case of a 14‐year‐old girl on immunosuppression for a renal transplant who presented with acute meningoencephalitis. Traditional diagnostics failed to identify an etiology. RNA extracted from her cerebrospinal fluid was subjected to unbiased metagenomic deep sequencing, enhanced with the use of a Cas9‐based technique for host depletion. This analysis identified West Nile virus (WNV). Convalescent serum serologies subsequently confirmed WNV seroconversion. These results support a clear clinical role for metagenomic deep sequencing in the setting of suspected viral encephalitis, especially in the context of the high‐risk transplant patient population.
Keywords: basic (laboratory) research/science; clinical research/practice; infectious disease; kidney transplantation/nephrology; genetics; diagnostic techniques and imaging; genomics; infection and infectious agents; viral: West Nile, infection and infectious agents; viral: Epstein‐Barr Virus (EBV); kidney (allograft) function/dysfunction
Short abstract
Unbiased metagenomic deep sequencing identifies West Nile virus in the cerebrospinal fluid of a child who had a renal transplant and presented with acute meningoencephalitis.
Abbreviations
- bp
base pair
- cDNA
complementary DNA
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- CSF
cerebrospinal fluid
- CT
computed tomography
- DASH
depletion of abundant species by hybridization
- ds
double stranded
- EBV
Epstein‐Barr virus
- IVIg
IV immunoglobulin
- IV
intravenous
- MDS
metagenomic deep sequencing
- MRI
magnetic resonance imaging
- NCBI
National Center for Biotechnology Information
- nt
nucleotide
- PCR
polymerase chain reaction
- rpm
reads per million
- RT‐PCR
reverse transcription polymerase chain reaction
- UCSF
University of California San Francisco
- WNV
West Nile virus
Introduction
Solid organ transplant patients are particularly susceptible to unusual infections and are more likely to suffer from neuroinvasive disease, including viral encephalitis 1, 2, 3. Candidate‐based infectious disease diagnostics already fail to identify a causative pathogen in approximately 50% of encephalitis cases 4, 5, 6. Diagnosing a neurologic infection is even more challenging in the transplant population as patients may lack the classic signs and symptoms of meningitis because of their immunosuppressed state, and they are susceptible to atypical complications of common infections like JC virus causing progressive multifocal leukoencephalopathy 3. Transplant patients are also susceptible to more unusual infections such as lymphocytic choriomeningitis virus either from environmental exposures or from their donor organ 3. In addition, the transplant population can serve as a sentinel group in whom new and emerging infections first appear. Indeed, deaths of transplant patients have been noted during recent outbreaks of West Nile virus (WNV) and Dengue virus 7, 8, 9. Comprehensive, unbiased, and “hypothesis‐free” diagnostic strategies that can simultaneously identify both common and rare pathogens are urgently needed for this population.
Metagenomic deep sequencing (MDS) has been used to investigate infections of unknown etiology in a wide variety of contexts 10, 11, 12, 13, 14, 15. We previously demonstrated the utility of MDS to detect unusual eukaryotic and bacterial organisms in the cerebrospinal fluid (CSF) of patients with idiopathic subacute and chronic meningoencephalitis 13, 14. Here, we report on the ability of MDS to identify an arboviral infection in the CSF of an adolescent girl, presenting with acute meningoencephalitis, who was immunosuppressed for a renal transplant. Conventional diagnostics had failed to detect the infectious etiology.
Case History
In the summer of 2015, a 14‐year‐old girl with a history of renal transplant managed with mycophenolic acid, tacrolimus, and prednisone presented to an emergency department with 2 days of fevers, chills, and upper back and neck pain. She denied headache, sensory changes, or motor deficits. One month prior to presentation she attended summer camp by a lake in the Angeles National Forest, California. She developed a rash on her legs that was attributed to mosquito bites and persisted at the time of presentation. Her past medical history was significant for perinatal acute tubular necrosis status‐post directed donor renal transplant at 8 years old. Her transplant was complicated by systemic infections with cytomegalovirus and BK virus as well as low‐grade clear cell carcinoma in the transplanted kidney. She underwent a partial nephrectomy with clear margins when she was 13 years old.
On initial presentation her vital signs were as follows: temperature 39.9°C, heart rate 148 beats per minute, blood pressure 126/75 mmHg, respiratory rate 22 breaths per minute, and an oxygen saturation of 100% on room air. Her mental status was normal. She had no nuchal rigidity or focal neurological deficits. There was an erythematous blanching maculopapular rash on her legs. As part of a sepsis protocol, she was given 2 L of intravenous (IV) normal saline and was started on broad‐spectrum antibiotic coverage with IV vancomycin and meropenem.
Her initial laboratory evaluation demonstrated a white blood cell (WBC) count of 9.08 × 103 cells/μL (normal range 4.16–9.95 × 103/μL), hemoglobin of 12.9 g/dL (11.6–15.2 g/dL), platelets 163 × 103 cells/μL (143–398 × 103/μL), sodium 137 mEq/L (135–146 mmol/L), blood urea nitrogen 36 mg/dL (7–22 mg/dL), creatinine 2.4 mg/dL (baseline 1.2 mg/dL) (0.6–1.3 mg/dL), and glucose 123 mg/dL (65–99 mg/dL).
After 36 h, she remained febrile (maximal temperature 40.5°C) and developed severe headaches, agitation, confusion, and language impairment (spoke in one‐to‐three‐word phrases and was unable to follow commands) as well as diarrhea and urinary retention. She was transferred to the pediatric intensive care unit and was started on increased doses of antimicrobials for empiric treatment of bacterial meningitis (cefepime and linezolid followed by ceftriaxone and IV vancomycin) and herpes simplex virus encephalitis (acyclovir). Noncontrast head computed tomography (CT) and noncontrast magnetic resonance imaging (MRI) of the thoracic and lumbar spine were unremarkable. CSF analysis demonstrated 49 red blood cells/mm3 (0–10 cells/mm3), 87 WBC/mm3 (0–5 cells/mm3) with a differential of 50% neutrophils, 49% lymphocytes, and 1% monocytes, glucose 59 mg/dL (43–74 mg/dL), and protein 54 mg/dL (15–45 mg/dL) (Table 1).
Table 1.
Clinical laboratory results
| Test | Site | Day #2 | Day #4 | Day #24 | Day #37 | Convalescenta |
|---|---|---|---|---|---|---|
| RBC (cells/mm3) | CSF | 49 | 27 | 2 | ||
| WBC (cells/mm3) | CSF | 87 | 48 | 18 | ||
| Neutrophils | CSF | 50% | – | – | ||
| Lymphocytes | CSF | 49% | 98% | 100% | ||
| Monocytes | CSF | 1% | 2% | – | ||
| Protein (mg/dL) | CSF | 54 | 89 | 82 | ||
| Glucose (mg/dL) | CSF | 59 | 64 | 67 | ||
| Oligoclonal bands | CSF | – | 11 | 13 | ||
| EBV PCR (copies of viral DNA per PCR reaction) | CSF | 9 copies (ref < 5) | 90 copies | 1.9 copies | ||
| WNV IgM (reference <0.90 Ab not detected) | CSF | <0.90 | ||||
| WNV IgG (<1.30 Ab not detected) | CSF | <1.30 | ||||
| WNV IgM (<0.90 Ab not detected, >1.10 Ab detected)) | Serum | <0.90 | 5.11 (positive) | |||
| WNV IgG (<1.30 Ab not detected, ≥1.5 Ab detected) | Serum | <1.30 | 3.41 (positive) | |||
| WNV RT‐PCR | Serum | – | ||||
| HSV‐1, 2 IgM, IgG | CSF | – | ||||
| LCMV IgM, IgG | CSF | – | ||||
| Adenovirus Ab | CSF | – | ||||
| Influenza Type A, B Ab | CSF | – | ||||
| MV IgM, IgG | CSF | – | ||||
| MuV IgM, IgG | CSF | – | ||||
| VZV Ab CF | CSF | – | ||||
| Coxsackie A Ab 2,4,7,9,10,16 | CSF | – | ||||
| Coxsackie B Ab 1–6 | CSF | – | ||||
| Echovirus 4,7,9,11,30 Ab | CSF | – | ||||
| CMV IgM, IgG | CSF | – | ||||
| CrAg | CSF | – |
Day, hospital day; RBC, red blood cell; CSF, cerebrospinal fluid; WBC, white blood cell; EBV, Epstein‐Barr virus; PCR, polymerase chain reaction; WNV, West Nile virus; IgM, immunoglobulin M; Ab, antibody; IgG, immunoglobulin G; RT‐PCR, reverse transcription polymerase chain reaction; HSV‐1, 2, herpes simplex virus type 1 and 2; LCMV, lymphocytic choriomeningitis virus; Ab, antibody; MV, measles virus; MuV, mumps virus; CF, complement fixation; VZV, varicella zoster virus; CMV, cytomegalovirus; CrAg, cryptococcal antigen.
114 days after symptom onset.
She developed progressive leukopenia (nadir WBC 1.68 × 103 cells/μL on hospital day 5), normocytic anemia (nadir 6.4 on hospital day 12), and thrombocytopenia (nadir platelets 72 × 103 cells/μL on hospital day 2). Clostridium difficile polymerase chain reaction (PCR) was positive in the stool. Empiric treatment with broad‐spectrum antimicrobials at meningeal doses was continued for 5 days without improvement. She was given metronidazole for treatment of C. difficile. Mycophenolic acid was discontinued on hospital day 3, and tacrolimus was discontinued on hospital day 12. She was continued on prednisone 10 mg daily.
MRI of the brain with and without gadolinium on hospital day 5 demonstrated symmetric T2 hyperintensities and edema in the thalami and basal ganglia bilaterally (Figure 1A), most pronounced in the dorsomedial and pulvinar nuclei as well as prominent enhancement in the thalami and in the leptomeninges along the cerebellum and brainstem. A repeat MRI of the whole spine with and without gadolinium on hospital day 37 demonstrated leptomeningeal enhancement along the brainstem and cervical spine (Figures 1B and C) as well as smooth enhancement of the nerve roots of the cauda equina.
Figure 1.
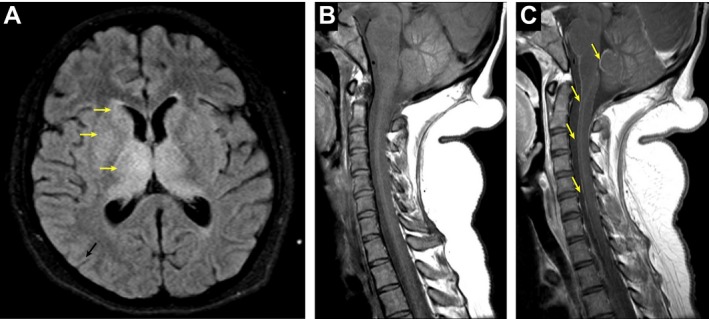
Neuroimaging. A, Axial T2‐weighted/fluid attenuated inversion recovery (FLAIR) brain MRI demonstrating symmetric hyperintensities in the basal ganglia and thalami (arrows). B and C, Pre‐ (B) and Post‐ (C) contrast T1‐weighted cervical spine MRI demonstrating leptomeningeal enhancement (arrows).
Given the enhancing lesions on MRI and the lack of response to antimicrobials, she was treated for possible acute disseminated encephalomyelitis with two doses of IV immunoglobulins (IVIg) 1 mg/kg on hospital days 5 and 6. IVIg was repeated on hospital days 38 and 40 due to a perceived clinical response. Her hospital course was complicated by clinical seizures characterized by episodes of generalized stiffness and extension of the right arm followed by shaking in the arms and legs. Electrographically these episodes correlated with a diffuse background change with high‐amplitude rhythmic slowing (1–4 Hz) in the bifrontal regions consistent with electrographic seizures. The seizures resolved after treatment with levetiracetam.
As detailed in Table 1, an extensive diagnostic work‐up for infectious causes of encephalitis was performed during her 41‐day hospitalization, including additional testing on CSF obtained from two repeat lumbar punctures. The only organism identified on the patient's CSF examination from hospital day 24 was Epstein‐Barr virus (EBV) via PCR (Table 1). As a result, the patient was treated with IV ganciclovir for 12 days and then transitioned to oral valganciclovir. Her renal function improved over the course of 2 weeks with hydration, and there was no evidence of transplant rejection. Her leukopenia and anemia also slowly resolved and were attributed to viral suppression of bone marrow.
The patient was discharged on hospital day 41 to an acute rehabilitation center. She returned home 3 weeks later. She has persistent neurologic deficits including dysarthria, horizontal nystagmus, and gait instability, but she is now able to ambulate independently. Her mental status is normal and she has returned to school where she is doing well. Since discharge, her renal function has remained stable (serum creatinine 1.2 mg/dL) on prednisone 10 mg PO daily.
Materials and Methods
Sequencing library preparation
A 500‐μL sample of CSF obtained from the study subject's initial lumbar puncture was submitted for unbiased MDS under a research protocol (#13‐12236) for the identification of potential pathogens approved by the Institutional Review Board of the University of California, San Francisco (UCSF). A common issue in metagenomic sequencing is that essentially all reagents used during the library preparation process contain some residual nucleic acid. The process of acquiring a human sample also introduces unwanted nucleic acid from skin or environmental contamination. To control for irrelevant sequences, an unrelated, uninfected human CSF sample (“control”) was prepared in parallel and sequenced on the same run. Samples were processed for MDS analysis as previously described 14. Briefly, total RNA was extracted. The RNA was reverse transcribed to single‐stranded complementary DNA (cDNA) using random hexamer primers and amplified to double‐stranded (ds) cDNA using the NuGEN Ovation v2 kit (NuGEN, San Carlos, CA) for low nucleic acid content samples. Two MDS libraries were constructed from the ds cDNA using the Nextera protocol (Illumina, San Diego, CA). Prior to the Nextera reaction, one of the libraries was subjected to an additional step to selectively deplete library amplicons whose sequences corresponded to the human mitochondrial genome. This was accomplished via depletion of abundant sequences by hybridization (DASH), a novel molecular technique using the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)–associated nuclease Cas9 in vitro 16. In addition to the 54 DASH guide RNAs targeting the 12S and 16S mitochondrial rRNA genes as previously described 16, 212 new DASH guide RNAs targeting the remaining portion of the mitochondrial chromosome were also utilized (Table S1). The pooled library was size‐selected using a Blue Pippin (Sage Science, Beverly, MA), and concentration was determined using a Kapa Universal quantitative PCR kit (Kapa Biosystems, Woburn, MA). Samples were sequenced on an Illumina HiSeq 2500 instrument using 135/135 base pair (bp) paired‐end sequencing.
Bioinformatics
Sequences were analyzed using a rapid computational pipeline developed by the DeRisi Laboratory to classify MDS reads and identify potential pathogens by comparison to the entire National Center for Biotechnology Information (NCBI) nucleotide (nt) reference database 14. First, all paired‐end reads were aligned to the human reference genome 38 (hg38) and the Pan troglodytes genome (panTro4, 2011, UCSC), using the Spliced Transcripts Alignment to a Reference aligner (v2.5.1b) 17. Unaligned (i.e. nonhuman) reads were quality filtered using PriceSeqFilter 18 with the “‐rnf 90” and “‐rqf 85 0.98” settings. Quality filtered reads were then compressed by cd‐hit‐dup (v4.6.1) if they were more than 95% identical 19. Paired‐end reads were then assessed for complexity by compression with the Lempel‐Ziv‐Welch algorithm 20. Read‐pairs with a compression score less than 0.45 were removed. Next, a second phase of human removal was conducted using the “–very‐sensitive‐local” mode of Bowtie2 (v2.2.4) with the same hg38 and PanTro4 reference as described above 21. The remaining nonhuman read pairs were processed with GSNAPL (v2015‐12‐31) 22, which was used to align the reads to the NCBI nt database (downloaded July 2015, indexed with k = 16mers), and preprocessed to remove known repetitive sequences with RepeatMasker (vOpen‐4.0) (www.repeatmasker.org). The same reads were also aligned to the NCBI nonredundant protein database (July 2015) using the Rapsearch2 algorithm 23. The resulting sequence hits identified at both the nucleotide and protein (translated) level from the control sample were subtracted from each patient sample by matching genus level taxonomic identifications. To further control for rare spurious sequence reads, a minimum read count per taxonomic category of two unique reads per million (rpm) reads mapped was further imposed.
Results
A total of 7 777 470 paired‐end reads and 12 829 879 paired‐end reads were obtained for the two libraries built from the study subject's CSF sample (with and without DASH, respectively). The third sample, an uninfected CSF control, yielded 12 750 348 paired‐end reads.
As described above, the paired‐end sequences were processed through a custom bioinformatics pipeline. The runtime for the bioinformatics pipeline described above was 10–15 min per sample on a single 24‐core server. After filtering, there were four genera remaining in our study subject's MDS dataset: Flavivirus, Tobacco mosaic virus, Microcoleus, and Cupriavidus, of which only Flavivirus was considered to be a credible pathogen. Finally, Flavivirus was the only taxonomic identification that was in common between the two library preparations (i.e. with and without DASH) from the study subject. Thus, the very simple and conservative algorithm described above resulted in a single taxonomic category in our patient's sample, corresponding to WNV, a flavivirus known to cause encephalitis. These nonhuman sequence reads corresponding to the libraries from this patient have been deposited at the NCBI Sequence Read Archive (SRA), BioProject PRJNA338853.
In the study subject's sample not subjected to DASH, 57 sequence read pairs corresponding to WNV were present, 52 of which were unique. In the sample subjected to the DASH technique, there was a 29‐fold decrease in the percentage of total reads that aligned to the human mitochondrial genome (29% vs. 1%) and a corresponding increase in the total number of read pairs aligning to WNV to 142, 101 of which were unique. This translates to a fourfold improvement in the limit of detection in the sample subjected to DASH (18.3 rpm mapping to WNV) compared to the sample prepared without DASH (4.4 rpm mapping to WNV). The combined WNV sequences from both samples covered 19% of the WNV genome, including portions of the matrix, envelope, and nonstructural proteins 1, 2A, 3, and 5. No sequences aligning to WNV were present in the 2 serum, 1 brain biopsy, and 13 other CSF samples, including the autoimmune control, sequenced on the same run. No WNV reads have ever been detected previously in this laboratory. A 354‐bp contig of the gene encoding the envelope protein had 99.4% identity to a WNV strain isolated in the 2012 WNV outbreak in Texas (GenBank accession number KJ501530.1) while there was only 87% similarity to the next most closely related flavivirus, Kunjin virus (GenBank accession number KT9348041). The patient's markedly positive convalescent WNV serologies confirmed the WNV infection (Table 1).
The patient was admitted for a total of 41 days. CSF from her initial lumbar puncture was received for MDS on hospital day 19. The sequencing library was prepared and put on an Illumina HiSeq 2500 27 days later (5 days after discharge from the hospital). Sequencing data were available 2 days later (7 days after discharge), and preliminary results were reported to the treating clinicians 9 days after discharge from the hospital (31 days after receipt of the CSF sample).
Discussion
It has long been recognized that solid organ transplant patients are particularly susceptible to new and unusual infections and can also have atypical presentations of common infections. As a result, the traditional candidate‐based diagnostic approach to infectious disease diagnosis is even more likely to fail in this vulnerable patient population 2. Thus, there is a need for a more comprehensive, unbiased approach to detect pathogens, especially in the context of acute encephalitis in which existing diagnostic tests are already imperfect.
Here we report on the ability of MDS to identify WNV in the initial CSF sample obtained from a 14‐year‐old girl immunosuppressed for a renal transplant who presented with an idiopathic acute meningoencephalitis. We also show that DASH, a novel molecular method for selectively depleting unwanted sequences from next‐generation sequencing libraries, can be utilized to enhance detection of a virus in the CSF 16. DASH was developed for this protocol because all other depletion techniques remove input RNA and require multiple nanograms of input material, whereas DASH operates on the cDNA present in the final sequencing library. Therefore, DASH is agnostic to the amount of original material or type of library preparation protocol, a key advantage given that patient CSF typically only yields RNA in femtogram to picogram quantities, in our experience.
Likely because of our subject's immunosuppressed status, her acute WNV antibody testing was falsely negative in both the CSF and blood. The WNV reverse transcription PCR (RT‐PCR) assay was not sent, given that the RT‐PCR for WNV in CSF has inferior sensitivity to serology, though the superiority of the WNV serology may have been hampered in this case due to the patient's immunosuppressed state 24. The patient's convalescent WNV serologies later confirmed the WNV infection (Table 1).
In addition to WNV being missed by traditional diagnostics, the patient also had a positive EBV CSF PCR. In the absence of an alternate diagnosis, this test result led to treatment with ganciclovir, an antiviral medication that causes significant pancytopenia, a particularly harmful side effect for an already immunosuppressed transplant patient suffering from a neuroinvasive arboviral infection. However, the patient's maculopapular rash, recent mosquito exposure, basal ganglia and thalamic involvement on MRI, as well as her early neutrophilic‐predominant CSF pleocytosis were all more consistent with WNV meningoencephalitis 24. In this case, MDS provided an alternate diagnostic possibility that fit much better with the patient's acute presentation 25, 26.
While there is no approved antiviral treatment for WNV, earlier knowledge of the infectious etiology would have had consequences for the patient. Although in this case there was a 31‐day turnaround for the MDS assay as a result of research laboratory staffing levels, we have previously shown that this assay can be performed in a 48‐h timeframe 13. During the patient's 41‐day hospitalization, she was subjected to a wide array of antimicrobial medications, many of which would have been avoided had MDS been performed as a frontline test. In addition, WNV has previously been associated with an increased risk of acute and, more controversially, chronic kidney disease 27. In a patient with a renal transplant, knowledge that interval worsening in her renal function may have been due to a viral infection and not necessarily due to rejection of the donor organ could influence if and when the treating physicians decide to enhance the degree of immunosuppression to stave off presumed rejection.
MDS is increasingly available for clinical use, and its performance characteristics are being evaluated in an ongoing prospective, multicenter study (http://www.ciapm.org/project/precision-diagnosis-acute-infectious-diseases). As MDS gains wider adoption and the performance characteristics of the test become better defined, even a negative MDS result may provide useful information in the transplant population by helping to rule out an infection in a transplant patient suffering from ill‐defined signs and symptoms that may or may not be infectious in origin.
Disclosure
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. J.L.D., L.L.Z., and M.R.W. are Co‐Investigators of the Precision Diagnosis of Acute Infectious Diseases study funded by the CA Initiative to Advance Precision Medicine mentioned in the Discussion section, and H.A.S. is the Program Manager of the study. The other authors of this manuscript have no conflicts of interest to disclose.
Supporting information
Table S1: Sequences targeted with DASH (depletion of abundant sequences by hybridization). Mitochondrial ribosomal RNA‐1 (mt‐rRNA‐1) through mt‐rRNA‐54 targets the human 12S and 16S mitochondrial rRNA genes, as described in 16; ChrM‐1 through chrM‐212 target the full human mitochondrial chromosome.
Acknowledgments
Research reported in this publication was supported by the UCSF Center for Next‐Gen Precision Medicine supported by the Sandler and William K. Bowes, Jr. Foundations (J.L.D., H.A.S, L.M.K., and M.R.W.); Howard Hughes Medical Institute (J.L.D.); and the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under Award Number KL2TR000143 (M.R.W.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. We thank Derek Bogdanoff and Eric Chow in the UCSF Center for Advanced Technology for their expertise and assistance operating the Illumina sequencer. We thank the Sandler and William K. Bowes, Jr. Foundations for their generous philanthropic support. Lastly, we thank the patient and her family for their participation in this research program.
Wilson MR, Zimmermann LL, Crawford ED, Sample HA, Soni PR, Baker AN, Khan LM & DeRisi JL. Acute West Nile Virus Meningoencephalitis Diagnosed Via Metagenomic Deep Sequencing of Cerebrospinal Fluid in a Renal Transplant Patient. Am J Transplant 2017; 17: 803–808
See also: Blumberg and Fishman, Kuppachi et al.
References
- 1. Pastula DM, Smith DE, Beckham JD, Tyler KL. Four emerging arboviral diseases in North America: Jamestown Canyon, Powassan, chikungunya, and Zika virus diseases. J Neurovirol 2016; 22: 257–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pruitt AA, Graus F, Rosenfeld MR. Neurological complications of solid organ transplantation. Neurohospitalist 2013; 3: 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wright AJ, Fishman JA. Central nervous system syndromes in solid organ transplant recipients. Clin Infect Dis 2014; 59: 1001–1011. [DOI] [PubMed] [Google Scholar]
- 4. Glaser CA, Gilliam S, Schnurr D, et al. In search of encephalitis etiologies: Diagnostic challenges in the California Encephalitis Project, 1998–2000. Clin Infect Dis 2003; 36: 731–742. [DOI] [PubMed] [Google Scholar]
- 5. Granerod J, Tam CC, Crowcroft NS, Davies NW, Borchert M, Thomas SL. Challenge of the unknown. A systematic review of acute encephalitis in non‐outbreak situations. Neurology 2010; 75: 924–932. [DOI] [PubMed] [Google Scholar]
- 6. Vora NM, Holman RC, Mehal JM, Steiner CA, Blanton J, Sejvar J. Burden of encephalitis‐associated hospitalizations in the United States, 1998‐2010. Neurology 2014; 82: 443–451. [DOI] [PubMed] [Google Scholar]
- 7. Yango AF, Fischbach BV, Levy M, et al. West Nile virus infection in kidney and pancreas transplant recipients in the Dallas‐Fort Worth Metroplex during the 2012 Texas epidemic. Transplantation 2014; 97: 953–957. [DOI] [PubMed] [Google Scholar]
- 8. Prasad N, Bhadauria D, Sharma RK, Gupta A, Kaul A, Srivastava A. Dengue virus infection in renal allograft recipients: A case series during 2010 outbreak. Transpl Infect Dis 2012; 14: 163–168. [DOI] [PubMed] [Google Scholar]
- 9. Iwamoto M, Jernigan DB, Guasch A, et al. Transmission of West Nile virus from an organ donor to four transplant recipients. N Engl J Med 2003; 348: 2196–2203. [DOI] [PubMed] [Google Scholar]
- 10. Greninger AL, Runckel C, Chiu CY, et al. The complete genome of klassevirus—A novel picornavirus in pediatric stool. Virol J 2009; 6: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kistler AL, Smith JM, Greninger AL, Derisi JL, Ganem D. Analysis of naturally occurring avian bornavirus infection and transmission during an outbreak of proventricular dilatation disease among captive psittacine birds. J Virol 2010; 84: 2176–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stenglein MD, Sanders C, Kistler AL, et al. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: Candidate etiological agents for snake inclusion body disease. MBio 2012; 3: e00180‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilson MR, Naccache SN, Samayoa E, et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med 2014; 370: 2408–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson MR, Shanbhag NM, Reid MJ, et al. Diagnosing balamuthia mandrillaris encephalitis with metagenomic deep sequencing. Ann Neurol 2015; 78: 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yozwiak NL, Skewes‐Cox P, Gordon A, et al. Human enterovirus 109: A novel interspecies recombinant enterovirus isolated from a case of acute pediatric respiratory illness in Nicaragua. J Virol 2010; 84: 9047–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gu W, Crawford ED, O'Donovan BD, et al. Depletion of abundant sequences by hybridization (DASH): Using Cas9 to remove unwanted high‐abundance species in sequencing libraries and molecular counting applications. Genome Biol 2016; 17: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dobin A, Davis CA, Schlesinger F, et al. STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruby JG, Bellare P, Derisi JL. PRICE: Software for the targeted assembly of components of (Meta) genomic sequence data. G3 (Bethesda) 2013; 3: 865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu L, Niu B, Zhu Z, Wu S, Li W. CD‐HIT: Accelerated for clustering the next‐generation sequencing data. Bioinformatics 2012; 28: 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ziv J, Lempel A. A universal algorithm for sequential data compression. IEEE Trans Inf Theory 1977; 23: 337–343. [Google Scholar]
- 21. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods 2012; 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu TD, Nacu S. Fast and SNP‐tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010; 26: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y, Tang H, Ye Y. RAPSearch2: A fast and memory‐efficient protein similarity search tool for next‐generation sequencing data. Bioinformatics 2012; 28: 125–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davis LE, DeBiasi R, Goade DE, et al. West Nile virus neuroinvasive disease. Ann Neurol 2006; 60: 286–300. [DOI] [PubMed] [Google Scholar]
- 25. Wang J, Ozzard A, Nathan M, et al. The significance of Epstein‐Barr virus detected in the cerebrospinal fluid of people with HIV infection. HIV Med 2007; 8: 306–311. [DOI] [PubMed] [Google Scholar]
- 26. Xuan L, Jiang X, Sun J, et al. Spectrum of Epstein‐Barr virus‐associated diseases in recipients of allogeneic hematopoietic stem cell transplantation. Transplantation 2013; 96: 560–566. [DOI] [PubMed] [Google Scholar]
- 27. Barzon L, Pacenti M, Palu G. West Nile virus and kidney disease. Expert Rev Anti Infect Ther 2013; 11: 479–487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Sequences targeted with DASH (depletion of abundant sequences by hybridization). Mitochondrial ribosomal RNA‐1 (mt‐rRNA‐1) through mt‐rRNA‐54 targets the human 12S and 16S mitochondrial rRNA genes, as described in 16; ChrM‐1 through chrM‐212 target the full human mitochondrial chromosome.