

Abstract
Bacterial pathogens can interfere during infection with host cell organelles, such as mitochondria, the endoplasmic reticulum‐Golgi system or nuclei. As important cellular functions are often compartmentalized in these organelles, their targeting allows pathogens to manipulate key host functions during infection. Here, we identify lysosomes as a new class of organelles targeted by the pathogenic bacterium Listeria monocytogenes. We demonstrate that extracellular Listeria, via secretion of the pore‐forming toxin listeriolysin O, alters lysosomal integrity in epithelial cells but not in macrophages. Listeriolysin O induces lysosomal membrane permeabilization and release of lysosomal content, such as cathepsins proteases, which remain transiently active in the host cytosol. We furthermore show that other bacterial pore‐forming toxins, such as perfringolysin O and pneumolysin, also induce lysosomes alteration. Together, our data unveil a novel activity of bacterial cholesterol‐dependent cytolysins.
1. INTRODUCTION
Listeria monocytogenes is a Gram‐positive pathogen responsible for human listeriosis, a leading cause of deaths due to food‐transmitted bacterial pathogens. After ingestion of contaminated food, Listeria can breach the intestinal and blood–brain barriers, leading to febrile gastroenteritis, septicemia, and meningitis. In pregnant women, Listeria can furthermore breach the placental barrier leading to abortion or neonatal infections.
At the cell level, Listeria has the ability to enter and replicate in both phagocytic and nonphagocytic cells (Cossart, 2011; Pizarro‐Cerda, Kuhbacher, & Cossart, 2012). Listeria's intracellular life cycle involves hijacking of host‐cell pathways and interference with host cell organelles (Lebreton, Stavru, & Cossart, 2015). Indeed, it has been reported that Listeria alters the dynamic of mitochondria fission/fusion events (Stavru, Bouillaud, Sartori, Ricquier, & Cossart, 2011; Stavru, Palmer, Wang, Youle, & Cossart, 2013), activates the endoplasmic reticulum (ER) stress responses (Pillich, Loose, Zimmer, & Chakraborty, 2012), and reshapes host nuclear functions by altering histone modifications and chromatin condensation (Eskandarian et al., 2013; Hamon et al., 2007; Hamon & Cossart, 2011; Lebreton et al., 2011). Among the different virulence factors of Listeria involved in these alterations of host organelle functions, the listeriolysin O (LLO) toxin plays a central role (reviewed in Hamon, Ribet, Stavru, & Cossart, 2012). This pore‐forming toxin was first reported for its role in the destabilization of the internalization vacuole and escape of bacteria in the host cell cytosol (Cossart et al., 1989; Gaillard, Berche, & Sansonetti, 1986; Kathariou, Metz, Hof, & Goebel, 1987; Portnoy, Jacks, & Hinrichs, 1988). Several studies have now established that LLO can also be secreted by extracellular Listeria and forms pores in the host plasma membranes (reviewed in Hamon et al., 2012). These pores allow potassium efflux and calcium influx, which alter the ionic balance of the host cell and trigger several signaling pathways leading to inflammasome activation and IL‐1β secretion, mitochondria fragmentation, or histone posttranslational modifications (Hamon & Cossart, 2011; Meixenberger et al., 2010; Stavru et al., 2011; Stavru et al., 2013). Pores formed at the plasma membrane can also alter other cellular processes, independently of ion fluxes, such as SUMOylation (Impens, Radoshevich, Cossart, & Ribet, 2014; Ribet et al., 2010).
The endomembrane system is a privileged target of Listeria during infection (Lebreton et al., 2015). In nonphagocytic cells, internalization of Listeria and its escape from the internalization vacuole requires tight control of the host endocytic compartments (Hamon et al., 2012; Pizarro‐Cerda et al., 2012). In professional phagocytes such as macrophages, survival of Listeria is promoted by intracellular secretion of LLO. This toxin alters phagosome integrity, delays their acidification, inhibits their fusion with lysosomes, and eventually participates in the disruption of phagosomal membranes (Henry et al., 2006; Shaughnessy, Hoppe, Christensen, & Swanson, 2006).
In contrast to the reported effect of LLO on host endosomes or phagosomes, the consequences of Listeria infection on lysosomes remain poorly characterized. Lysosomes are single membrane‐bound cytoplasmic organelles specialized in the degradation and recycling of macromolecules. These dynamic vacuoles are characterized by low pH and contain numerous hydrolases, such as cathepsins, as well as specific membrane proteins. Lysosomes are able to fuse and thus to degrade the content of a wide range of vesicles, including endocytic and phagocytic vacuoles, autophagosomes, or post‐Golgi originating vacuoles. Besides their catabolic properties, lysosomes have been shown to have broader functions in cell homeostasis and are involved in secretion, membrane repair, cell growth, or cell death (Aits & Jaattela, 2013; Andrews, Almeida, & Corrotte, 2014; Luzio, Hackmann, Dieckmann, & Griffiths, 2014; Settembre, Fraldi, Medina, & Ballabio, 2013).
Since their first description by Christian DeDuve in 1950s, lysosomes were often referred to as “suicide bags”, as lysosomal membrane damage results in leakage of lysosomal content to the cytosol, which can then trigger apoptosis, pyroptosis, or necrosis (Boya & Kroemer, 2008; Repnik, Stoka, Turk, & Turk, 2012). Indeed, various components, such as H2O2 or sphingosine, can increase lysosomal membrane permeability, leading to the neutralization of lysosomal lumen and the release of cathepsins and other hydrolases into the cytosol (Boya & Kroemer, 2008). The consequences of lysosomal membrane permeabilization (LMP) vary according to the extent of lysosomal damage and the cell type. While extensive LMP is often linked to necrosis or apoptosis, moderate LMP may trigger oxidative stress, due to the release of protons from the lysosomes into the cytosol and reduction of lysosomal catabolic capacities (Appelqvist, Waster, Kagedal, & Ollinger, 2013; Boya & Kroemer, 2008; Repnik, Hafner Cesen, & Turk, 2014).
Here, we investigated whether Listeria interferes with host lysosome functions during infection. We demonstrated that the bacterial pore‐forming toxin LLO triggers host lysosomes permeabilization and release of lysosomal proteases such as cathepsins in the cytosol. Our results identify host lysosomes as new targets of Listeria during infection.
2. RESULTS
2.1. Listeria infection neutralizes host cell acidic compartments
In order to determine whether Listeria alters host lysosome integrity during infection, we stained cells with acridine orange (AO), a lysosomotropic dye that emits red fluorescence when accumulating in acidic compartments such as lysosomes. AO‐stained HeLa cells were infected with L. monocytogenes for 1 or 5 hr, fixed and analyzed by flow cytometry. We observed that infection by L. monocytogenes induces a significant decrease in AO fluorescence intensity, both at 1 or 5 hr of infection, which indicates a neutralization of host acidic compartments (Figure 1). To decipher whether the pore‐forming toxin LLO was involved in this process, we infected HeLa cells with a Listeria mutant deleted for hly, the gene coding for LLO. Of note, in HeLa cells, LLO is not strictly required for bacterial escape from the internalization vacuole and Δhly Listeria are thus able to reach the host cytosol and replicate intracellularly in this cell type (Gründling, Gonzalez, & Higgins, 2003). Strikingly, infection of cells with a Δhly Listeria mutant does not affect AO fluorescence intensity, suggesting that LLO is the virulence factor responsible for the observed loss of acidity of host compartments (Figure 1).
Figure 1.
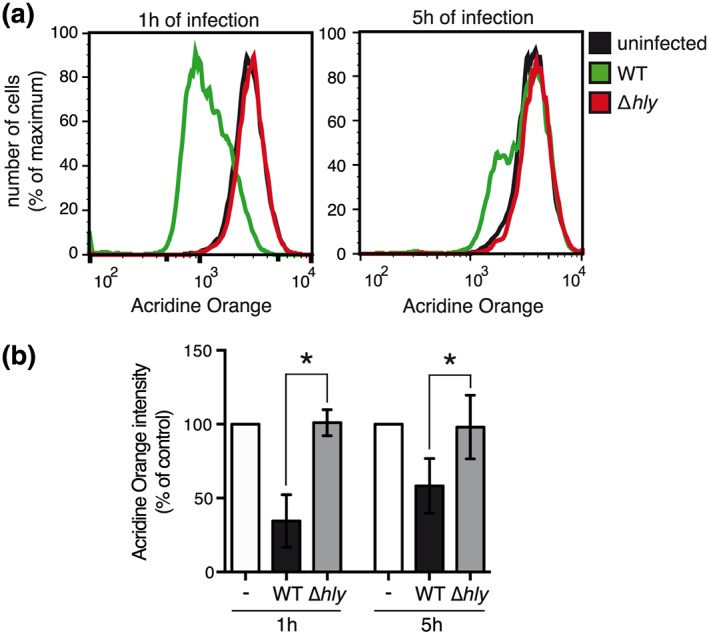
Neutralization of host acidic compartments during Listeria infection. Acridine orange‐stained HeLa cells were infected for 1 or 5 hr with wild‐type (WT) or Δhly Listeria monocytogenes . (a) Representative flow cytometry analysis of uninfected or infected acridine orange‐stained HeLa cells. (b) Data represent the geometric mean of acridine orange fluorescence in infected cells normalized to that of uninfected cells, as measured by flow cytometry analysis (mean ± SD from at least three independent experiments; *, p < 0.05; unpaired two‐tailed Student's t test)
To confirm the involvement of LLO in this process, we treated AO‐stained HeLa cells with sublytic concentrations of purified LLO (from 0.3 to 3 nM). Flow cytometry analysis showed that purified LLO can trigger a dose‐dependent and time‐dependent decrease of AO fluorescence intensity of treated cells (Figure 2a,b). Neutralization of acidic compartments in response to LLO was further confirmed by microscopic analysis of cells stained with Lysotracker, another acidophilic dye (Figure 2c).
Figure 2.
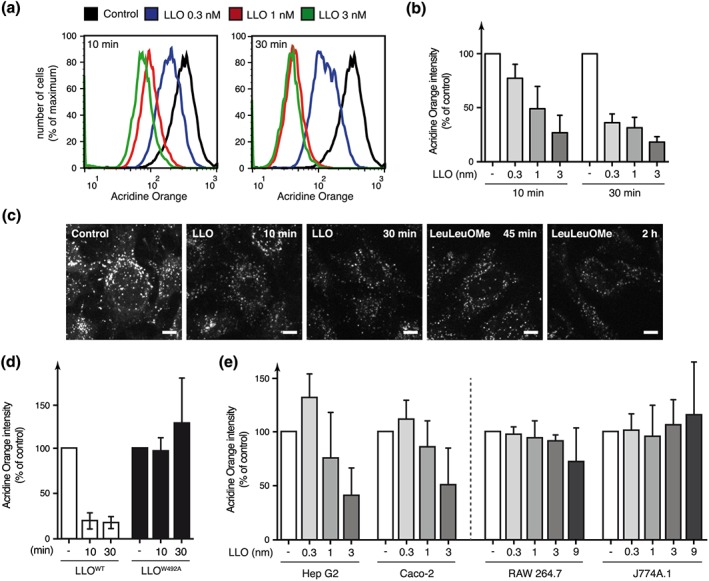
Neutralization of host acidic compartments in response to extracellular LLO. (a) Representative flow cytometry analysis of acridine orange‐stained HeLa cells treated with different concentrations of purified LLO, for 10 or 30 min. (b, d) Geometric mean of acridine orange fluorescence in HeLa cells treated with various concentrations of LLO (b) or with 3 nM wild‐type LLO or the pore‐deficient LLOW492A mutant (d), normalized to that of untreated cells, as measured by flow cytometry analysis (mean ± SD from at least three independent experiments). (c) Microscopic analysis of HeLa cells stained with Lysotracker and treated with 3 nM LLO or 2 mM of the LMP inducer l‐leucyl‐l‐leucine‐methyl ester (LeuLeuOMe) (scale bar; 2 μm). (e) Geometric mean of acridine orange fluorescence in Hep G2, Caco‐2, RAW264.7, or J774A.1 cells after 30 min of treatment with increasing concentrations of LLO, normalized to that of untreated cells (mean ± SD from at least three independent experiments)
We further assessed whether LLO‐induced loss of AO fluorescence intensity is pore‐dependent using an LLO mutant, LLOW492A, able to bind to cell membrane but unable to form pores (Ribet et al., 2010). HeLa cells treated with LLOW492A do not show a decrease in AO fluorescence intensity indicating that pore formation is critical to induce changes in host compartments acidity (Figure 2d).
We then determined whether LLO alters the acidity of intracellular compartments in other cell lines. Hep G2, a human liver epithelial cell line; Caco‐2, a human intestinal cell line; RAW 264.7 and J774A.1, two murine macrophage‐like cell lines were stained with AO and treated with various concentrations of LLO. We observed a dose‐dependent decrease in AO fluorescence intensity in Hep G2 and Caco‐2 cells lines in response to LLO, indicating that this toxin alters intracellular compartments acidity in different epithelial cell types (Figure 2e). Interestingly, no variation in AO fluorescence was observed in macrophages treated with LLO (even at a high LLO concentration of 9 nM), suggesting that alteration of acidic compartments in response to LLO is cell‐type specific and does not occur in macrophages (Figure 2e).
Previous reports established that host plasma membrane wounding by pore‐forming toxins such as LLO induces a calcium‐dependent fusion of membrane proximal lysosomes with the damaged membrane, which is critical for the resealing of wounded cells (Andrews et al., 2014; Jaiswal, Andrews, & Simon, 2002; Reddy, Caler, & Andrews, 2001; Tam et al., 2010). To confirm that our observed LLO‐dependent loss of AO fluorescence intensity is not due to lysosomal fusion with the plasma membrane, and thus to the release of lysosomal content in the extracellular milieu, we repeated our experiments in the presence of the calcium chelator EGTA to block LLO‐dependent calcium influx and lysosomes fusion to the plasma membrane. We validated that EGTA efficiently inhibits LLO‐induced lysosomes fusion with the plasma membrane by monitoring the release of lysosomal acid sphingomyelinase in the supernatant of cells treated with LLO (Figure S1). Whereas EGTA inhibited lysosomes fusion with the plasma membrane, it did not block the decrease in AO fluorescence intensity observed in HeLa cells treated with LLO (Figure 3). This result confirms that LLO triggers the neutralization of intracytoplasmic lysosomes. To definitely rule out a role for calcium in LLO‐induced neutralization of host lysosomes, we repeated our experiments in the presence of the cell permeant calcium chelator BAPTA‐AM. Pre‐incubation with BAPTA‐AM alone, or with BAPTA‐AM and EGTA, does not inhibit the decrease in AO fluorescence triggered by LLO, indicating that neither extra‐cellular calcium influx nor calcium mobilized from intracellular stores is involved in this process (Figure 3).
Figure 3.
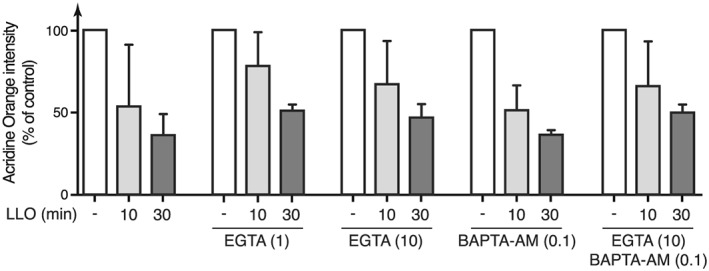
LLO‐induced neutralization of acidic compartments is calcium‐independent. Geometric mean of acridine orange fluorescence, as measured by flow cytometry analysis, in HeLa cells pre‐incubated with 1 or 10 mM EGTA, or with 0.1 mM BAPTA‐AM, and treated with 3 nM LLO (mean ± SD from at least three independent experiments)
Altogether, our results indicate that LLO, secreted by Listeria during infection, modifies the acidity of intracellular compartments such as lysosomes, which may reflect LMP (Boya et al., 2003; Erdal et al., 2005).
2.2. Listeria triggers host cell lysosomes permeabilization during infection
To assess whether host lysosomes are permeabilized in response to LLO, we monitored if intralysosomal proteins such as Cathepsin D (CtsD), a lysosomal aspartyl‐protease, can be translocated from the lysosomes to the cytosol after exposure to the toxin. HeLa cells were first treated or not with LLO, lysed mechanically, and fractionated to obtain lysosome‐depleted cytosolic fractions. The absence of lysosomal components in the cytosolic fractions was controlled by immunoblot analysis of LAMP1, a lysosomal transmembrane glycoprotein (Figure 4a). Compared to non treated cells, we observed a significant increase in the level of the mature CtsD heavy chain (27 kDa) in the cytosolic fractions of cells treated with LLO, indicating that mature CtsD has been released from lysosomes to the cytosol, and demonstrating that LLO does trigger LMP (Figure 4a). Quantification of CtsD signal showed that only a fraction of lysosomes are permeabilized in response to LLO, or that only a fraction of intralysosomal Cathepsin D is transferred to the cytosol.
Figure 4.
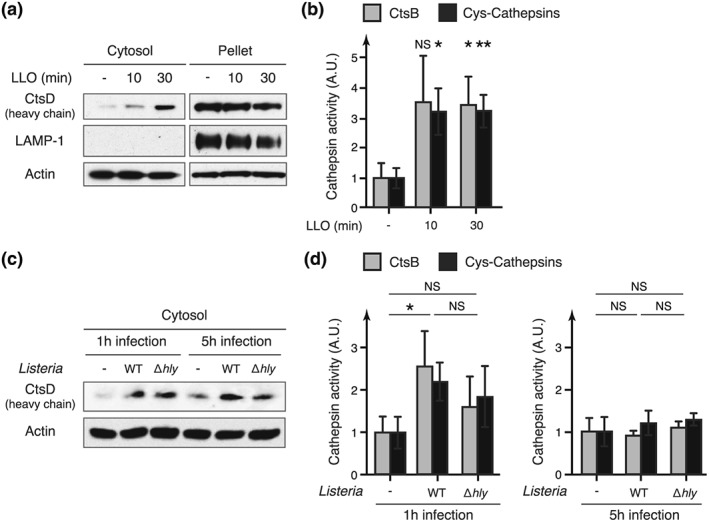
Release of active lysosomal proteases in the host cytosol in response to LLO. HeLa cells were treated with 3 nM purified LLO, or infected with wild‐type (WT) or Δhly Listeria monocytogenes for 1 or 5 hr. (a, c) Immunoblot analysis using anti‐cathepsin D (CtsD), anti‐lysosomal‐associated membrane protein‐1 (LAMP‐1) and anti‐actin antibodies of cell fractions from HeLa cells treated with LLO or infected with Listeria (“cytosol” = cytosolic fraction devoid of lysosomes; “pellet” = fraction containing lysosomes). (b, d) Quantification of cathepsin activity in cytosolic fractions from HeLa cells, treated with LLO or infected with Listeria, normalized to that of control cells (“CtsB” = substrate specific for cathepsin B; “cys‐cathepsins” = substrate specific for cathepsins belonging to cysteine proteases) (mean ± SD from three independent experiments; *, p < 0.05; **, p < 0.01; NS = not significant; unpaired two‐tailed Student's t test)
Cathepsins, once released from the lysosomes, can be inactivated either by the neutral pH of the cytosol or by intracellular inhibitors such as stefins and serpins (Lee, Gulnik, & Erickson, 1998; Repnik et al., 2012; Turk et al., 1995). Some cathepsins, however, were shown to retain their proteolytic activity at neutral pH for several hours (Kirschke, Wiederanders, Bromme, & Rinne, 1989; Pratt, Sekedat, Chiang, & Muir, 2009). Thus, cathepsins released from lysosomes may be transiently active in the cytosol. To assess if cytosolic cathepsins released in response to LLO retain some proteolytic activity, we monitored cathepsins activity in cytosolic fractions of LLO‐treated cells using cathepsins‐specific fluorogenic substrates. We observed a significant threefold increase in cathepsins activity in cytosolic fractions of LLO‐treated cells compared to control cells (Figure 4b). This confirms that cathepsins can be translocated from lysosomes to the cytosol and that these cathepsins remain, at least transiently, proteolytically active even at the neutral pH of the cytosol.
In order to show that the LMP observed in response to purified LLO is also induced during bacterial infection, we performed the same experiments on HeLa cells infected for 1 or 5 hr with Listeria. Again, we could detect an increase in cathepsin activity in the cytosolic fractions of cells infected for 1 hr with WT Listeria, which correlates with an increased level of CtsD in these fractions (Figure 4c,d). These results confirm that host cell lysosomes are permeabilized during infection. Infection with the Δhly mutant also showed a slight increase in cytosolic cathepsin levels and activity compared to uninfected cells, suggesting that LLO‐independent LMP may occur during infection in HeLa cells (Figure 4c,d). Interestingly, cytosolic cathepsins activity could not be detected after 5 hr of infection, suggesting that cathespins released to the cytosol are only transiently active and loose their activity after a few hours (Figure 4d).
As LMP may be a downstream consequence of cell death (Boya & Kroemer, 2008; Vanden Berghe et al., 2010), we finally monitored cell mortality after treatment with purified LLO or infection with Listeria. HeLa cells were treated with 3 nM LLO for 30 min or infected for 5 hr with WT and Δhly Listeria mutant. Cells were then labeled with a fluorescent dye allowing quantification of dead cells (see Experimental Procedures). Less than 10% of dead cells were observed in all tested conditions, indicating that neither LLO nor Listeria infection induces cell death in our experimental settings (Figure S2). Thus, the observed LLO‐induced LMP is not a downstream consequence of cell death.
2.3. LLO‐dependent Ubc9 degradation is independent of lysosomal permeabilization
We have previously demonstrated that plasma membrane perforation by LLO triggers the degradation of Ubc9, an essential enzyme of the host SUMOylation machinery, and a remodeling of the host cell SUMOylome (Impens et al., 2014; Ribet et al., 2010). This degradation can be partially blocked by an aspartyl‐protease inhibitor (Ribet et al., 2010). As several lysosomal proteases, such as Cathepsin D and Cathespin E, belong to the family of aspartyl‐proteases, we assessed whether lysosomal proteases released by LMP might be involved in LLO‐induced Ubc9 degradation. To do so, we inactivated lysosomal proteases by using a well‐established protocol based on horseradish peroxidase (HRP)‐mediated chemical crosslinking, which selectively targets endosomes and lysosomes (Laulagnier et al., 2011; Stoorvogel, 1998). HeLa cells were incubated with HRP at 37 °C for 15 min (pulse) to internalize HRP into endosomes by fluid‐phase endocytosis. After a chase period, allowing HRP to reach late endosome and lysosomes through the endocytic pathway, cells were incubated with H2O2 and 3,3′‐diaminobenzidine (DAB) that diffuses through cell membranes. The peroxidase activity of HRP‐containing compartments triggers polymerization of DAB monomers and selective fixation of these organelles through chemical crosslinking of the luminal and integral membrane protein to the DAB polymer.
We quantified lysosomal cathepsins activity after HRP/DAB‐mediated crosslinking of lysosomes and demonstrated that these proteases were indeed inactivated by this chemical treatment (Figure 5a). We then monitored Ubc9 degradation in response to LLO in HeLa cells pretreated or not with HRP/DAB. No significant differences in Ubc9 degradation could be observed in HeLa cells pre‐incubated with HRP/DAB compared to control cells (Figure 5b). These results suggest that Ubc9 degradation is independent of lysosomal proteases released into the cytosol after exposure to LLO.
Figure 5.
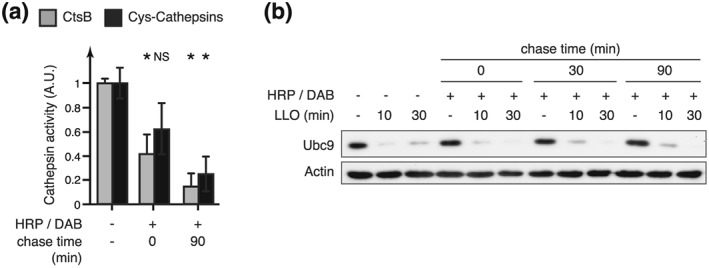
Inhibition of lysosomal proteases does not block LLO‐dependent Ubc9 degradation. HRP was endocytosed in HeLa cells for 15 min. After different chasing period (0, 30, or 90 min), cells were incubated with DAB and H2O2 to cross‐link lysosomal content. (a) Quantification of cathespin activity in HeLa whole cell extracts treated or not with HRP/DAB (“CtsB” = substrate specific for cathepsin B; “Cys‐cathepsins” = substrate specific for cathepsins belonging to cysteine‐proteases) (mean ± SD from at least three independent experiments; *, p < 0.05; NS, not significant; unpaired two‐tailed Student's t test). (b) Immunoblot analysis using anti‐Ubc9 and anti‐actin antibodies of whole cell extracts from HeLa cells pretreated with HRP/DAB and incubated with 3 nM LLO for 10 or 30 min
2.4. Host lysosomal permeabilization is triggered by other bacterial pore‐forming toxins
Listeriolysin O belongs to the family of cholesterol‐dependent cytolysins (CDCs), which includes toxins secreted by several extracellular pathogens (Hamon et al., 2012; Peraro & van der Goot, 2016). To decipher whether other bacterial pore‐forming toxins may also induce host‐cell lysosomal permeabilization, we treated AO‐stained HeLa cells with Perfringolysin O (PFO; secreted by Clostridium perfringens) or Pneumolysin (PLY; secreted by Streptococcus pneumoniae). We observed that these two other CDCs also lead to a concentration‐dependent decrease in acridine‐orange fluorescence intensity, as observed with LLO (Figure 6a). To assess whether pore‐forming activity of PFO and PLY was required to trigger LMP, as observed for LLO, we purified mutated versions of these toxins that are unable to form pores (PFOW467A and PLYW436A, designed by homology to LLOW492A. Treatment of AO‐stained HeLa cells with these nonhaemolytic toxins does not induce a decrease in fluorescence intensity, demonstrating that pores formation is strictly required for these CDCs to trigger lysosomal permeabilization in epithelial cells (Figure 6b). These results suggest that alteration of host lysosomes integrity may occur in response to infection by different pathogenic bacteria secreting pore‐forming toxins in the extracellular milieu.
Figure 6.
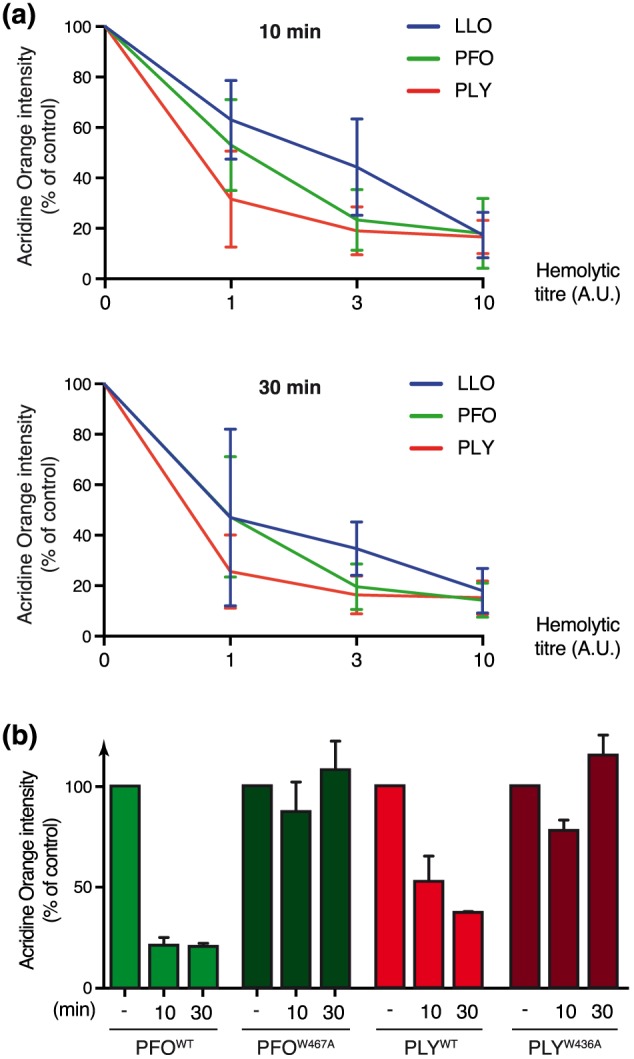
Neutralization of host acidic compartments induced by PFO and PLY toxins. Acridine orange‐stained HeLa cells were treated with wild‐type or pore‐deficient LLO, PFO, or PLY toxins for 10 or 30 min. (a) Wild‐type toxins were used at various hemolytic titres (a titre of 10 A.U. corresponds to the hemolytic activity of 3 nM LLO). (b) Wild‐type toxins were used at a hemolytic titre of 10 A.U. Mutant toxins were used at similar protein concentration than the corresponding wild‐type. Geometric means of acridine orange fluorescence, as measured by flow cytometry analysis are represented, normalized to that of untreated cells (mean ± SD from three independent experiments)
3. DISCUSSION
In this study, we identify lysosomes as a new host cell organelle targeted by L. monocytogenes during infection. We show that secretion of LLO by Listeria triggers host lysosome loss of integrity, leading to their neutralization and the translocation of active lysosomal proteases in the host cytosol.
Many bacterial pathogens have evolved the ability to interfere with host cell organelles (Escoll, Mondino, Rolando, & Buchrieser, 2016; Lebreton et al., 2015). Important cellular functions are compartmentalized in these organelles, such as DNA maintenance and gene expression in the nucleus, sorting of newly synthetized proteins and lipids in the ER and in the Golgi or bioenergetics and programmed cell death in mitochondria. Targeting of these organelles allows bacteria to manipulate key functions of the host cell in order to promote infection. Besides the numerous examples of bacteria targeting either mitochondria, the ER–Golgi system or the nucleus, only few pathogenic bacteria were previously reported to target lysosomes. Vibrio parahaemolyticus constitutes one such example, as it can translocate into host cells the cytotoxic VepA effector protein, which induces LMP through the targeting of the subunit C of lysosomal V‐ATPase (Matsuda, Okada, Kodama, Honda, & Iida, 2012). Pseudomonas aeruginosa is another example that secretes a toxic metabolite called pyocyanin, inducing LMP and apoptosis in neutrophils (Prince et al., 2008). Infection with S. pneumoniae was also shown to trigger LMP and apoptosis of host macrophages (Bewley et al., 2011). The involved mechanism is complex and partially requires the pneumolysin toxin but not its pore‐forming activity, as well as additional bacterial factors (Bewley et al., 2014). This mechanism differs from the one observed in our study in epithelial cells, which strictly requires PLY pore‐forming activity (Figure 6b). Finally, the α‐toxin from Clostridium septicum, which belongs to a different family of pore forming toxins than CDCs, was shown to trigger programmed necrosis in myoblast cells and cathepsins release from lysosomes (Kennedy, Smith, Lyras, Chakravorty, & Rodd, 2009). The mechanism involves pores‐mediated calcium influx and is associated with cell necrosis, which, again, differs from the mechanism observed here with LLO (Figure 3 and Figure S2).
Here, in the case of L. monocytogenes, we identified that LLO induces lysosomes permeabilization in epithelial cells but not in macrophages. In epithelial cells, it remains to be determined whether LLO and other CDCs directly target lysosomes and form active pores in lysosomal membranes or if pore formation at the level of the plasma membrane triggers signaling cascades leading, indirectly, to LMP. Indeed, large pores such as those formed by LLO and other CDCs are rapidly repaired, compared to smaller pores formed by other class of toxins (Gonzalez et al., 2011; Idone et al., 2008). Endocytosis of membrane‐bound toxins is one possible repair mechanism of perforated plasma membranes (Andrews et al., 2014; Corotte et al., 2013; Corotte, Fernandes, Tam, & Andrews, 2012). One could envisage that endocytosed CDCs pores traffic through the cell and fuse with lysosomal membranes leading to their permeabilization. Pores formed at the plasma membrane may also constitute an entry gate for toxin monomers to reach intracellular targets such as lysosomes. On the other hand, pores in the plasma membrane are known to trigger several signaling cascades. One of these cascades may indirectly lead to lysosome permeabilization, which can be triggered in response for example to oxidative stress or DNA damage (Boya & Kroemer, 2008).
While release of cathepsins during LMP was shown to induce apoptosis in some instances (Boya & Kroemer, 2008), we could not detect activation of caspase 3 nor induction of cell death in HeLa cells treated with LLO or infected with Listeria (Figure S2). These results are consistent with previous studies showing that exposure to LLO or infection by Listeria does not trigger classical apoptosis in HeLa cells, in contrast to infection by other bacterial pathogens such as Shigella flexneri or Salmonella Typhimurium (Stavru et al., 2011; Tattoli et al., 2008). This absence of cell death induction, despite LMP, may result from a limited amount of destabilized lysosomes, and thus a limited amount of released cathepsins, or from the only transient activity of cathepsins detected in the cytosol after infection. The rapid resealing of LLO pores in epithelial cells may also contribute to the lack of accumulation of permeabilized lysosomes over time (Andrews et al., 2014; Gonzalez et al., 2011; Idone et al., 2008; Peraro & van der Goot, 2016).
One interesting hypothesis would be that LLO‐induced LMP has different consequences depending on the cell type and may participate to cell death induction in only a specific subset of host cells. Apoptosis induced in response to Listeria infection has indeed been previously reported in vivo (Carrero & Unanue, 2012). At the liver level, hepatocytes can die from apoptosis following infection, independently of circulating immune cells, which then triggers the release of neutrophils chemoattractants (Rogers, Callery, Deck, & Unanue, 1996). Intracerebral infection leads to apoptosis of specific neurons from the hippocampal region (Schluter et al., 1998). Finally, LLO can induce apoptosis of T lymphocytes at infective loci by both caspase‐dependent and caspase‐independent pathways. One proposed mechanism is that LLO triggers the release of granzyme from lytic granules to the lymphocyte cytosol (Carrero, Vivanco‐Cid, & Unanue, 2008). As lytic granules are functionally related to lysosomal compartments, this mechanism interestingly echoes our observed LLO‐dependent release of lysosomal cathepsins in epithelial cells.
Previous studies have reported a role for LLO in the alteration of Listeria internalization vacuole integrity, following bacterial entry (Henry et al., 2006; Shaughnessy et al., 2006). These effects are mediated by LLO secreted intracellulary by engulfed bacteria and are restricted to the internalization vacuoles. In this study, we report an independent role of LLO, which, when secreted extracellularly, triggers host lysosomes permeabilization. The fact that other toxins, secreted by strictly extracellular pathogens, also induce lysosomes alterations strengthens our conclusions that LLO, as other CDCs, act in this case from the outside of host cells.
In conclusion, we have identified lysosomes as a previously overlooked target of Listeria infection. As lysosomes play a central role in the cell homeostasis, our study suggests that Listeria infection may affect important cellular pathways linked to lysosomal functions, which may be cell type‐dependent. This alteration of host lysosomal functions may either be beneficial for Listeria and thus participates to the strong attenuated virulence of the Listeria Δhly mutant, or, conversely, be sensed as a danger signal triggering host antibacterial responses. This study provides another example of the role of extracellular LLO on the modulation of host cell activities and possibly of cells that are not invaded by Listeria, in addition to the initially described role of this toxin in bacterial escape from the internalization vacuole. Our results also suggest that other pore‐forming toxin secreting pathogens may target host lysosomes and trigger host lysosomal permeabilization during infection. Finally, our study identifies LLO as an interesting tool to study mechanisms and consequences of lysosomal permeabilization in cell physiology and diseases.
4. EXPERIMENTAL PROCEDURES
4.1. Cell culture
HeLa cells (American Type Culture Collection (ATCC) CCL‐2), Hep G2 (ATCC HB‐8065), Caco‐2 (ATCC HTB‐37), RAW 264.7 (ATCC TIB‐71), and J774 A.1 (ATCC TIB‐67) were cultivated at 37 °C in a 10% CO2 atmosphere in Minimum Essential Medium (MEM) (Invitrogen). Culture media for HeLa and Hep G2 cells were supplemented with 2 mM Glutamax (Invitrogen), 10% Fetal Bovine Serum (FBS), MEM nonessential aminoacids (Invitrogen), and 1 mM sodium pyruvate. Culture medium for Caco‐2 cells was supplemented with 2 mM Glutamax, 20% FBS, MEM nonessential aminoacids, and 1 mM sodium pyruvate. Culture medium for RAW 264.7 and J774A.1 cells was supplemented with 4 mM Glutamax, 10% FBS, and 1 mM sodium pyruvate.
4.2. Bacterial strains
Listerias strains were grown in brain heart infusion broth or agar plates (BD Difco) at 37 °C. Strains used in this study were L. monocytogenes EGD (BUG 600), the corresponding isogenic deletion mutants EGD Δhly (BUG 3650; see below), and chromosomally GFP‐tagged EGD (EGD‐GFP; BUG 2539) (Balestrino et al., 2010) and EGD Δhly (EGD Δhly‐GFP; BUG 2786; see below).
4.3. Generation of Δhly mutant strains
To generate L. monocytogenes EGD Δhly strain (BUG 3650), two ~1,000 pb fragments flanking hly gene were PCR amplified from EGD chromosomal DNA. The primers used for the hly 5′ flanking fragment were 5′‐ATACAGTCGACTTATTGTCCGCT‐3′ and 5′‐TACAAACGCGTGGGTTTCACTC‐3′, and the primers used for the 3′ fragment were 5′‐AACCCACGCGTTTGTAAAAGTAA‐3′ and 5′‐TGTATAGATCTAAGCGCTTGAAA‐3′. After restriction of the amplified 5′ and 3′ fragments with SalI and MluI, and MluI and BglII, respectively, 5′ and 3′ fragments were coligated in the thermosensitive pMAD plasmid (Arnaud, Chastanet, & Debarbouille, 2004) digested by SalI and BglII, yielding the pMAD‐Δhly plasmid (BUG3621). This plasmid was electroporated into L. monocytogenes strain EGD, and gene deletion was performed as described in (Arnaud et al., 2004). Deletion of the entire hly gene was confirmed by sequencing.
Chromosomally GFP‐tagged EGD Δhly strain (BUG 2786) was obtained by electroporating pAD‐cGFP plasmid into EGD Δhly strain as described in Balestrino et al., 2010.
4.4. Bacterial infections
HeLa cells were seeded at a density of 5 × 105 cells per 960 mm2 wells the day before infection. Bacteria were cultured overnight at 37 °C, then subcultured 1:20 in brain heart infusion until exponential‐phase (OD600 nm of 1.0), and washed 4 times in PBS. HeLa cells were serum‐starved for 2 hr before infection. Bacteria were added to cells at a multiplicity of infection of 50 and centrifuged on cells for 5 min at 200×g. After 1 hr of infection, cells were washed and harvested, or incubated for four additional hours with fresh medium supplemented with 10% FBS and 50 μg/ml gentamicin (Euromedex) to kill extracellular bacteria.
4.5. Pore‐forming toxins treatment
Bacterial expression vectors for wild type of pore‐deficient toxins were generated by inserting genes coding for C‐terminally His6‐tagged LLO, PFO, and PLY toxins in pET29b plasmid (Novagen) (pET29b‐His6‐LLOWT, BUG1965 (Glomski, Gedde, Tsang, Swanson, & Portnoy, 2002); pET29b‐His6‐PFOWT, BUG3888; pET29b‐His6‐PLYWT, BUG3889). Punctual mutations were introduced in these plasmids by PCR mutagenesis to generate expression vectors for pore‐deficient toxins (pET29b‐His6‐LLOW492A, BUG2664 (Ribet et al., 2010); pET29b‐His6‐PFOW467A, BUG4081; pET29b‐His6‐PLYW436A, BUG4082). These vectors were then used to purify pore‐forming toxins from E. coli cell extracts as previously described (Glomski et al., 2002). Purified pore‐forming toxins were then added directly in culture medium of cells serum‐starved for 2 hr as indicated in the text.
4.6. Fluorescent staining of lysosomes
For LLO or LeuLeuOMe treatments, cells were incubated with either 5 mM Acridine Orange (ThermoFisher) for 15 min or 500 nM Lysotracker Red (ThermoFisher) for 30 min, then washed 3 times with MEM before treatment with 3 nM LLO or 2 mM LeuLeuOMe (H‐Leu‐Leu‐OMe·HBr; Bachem). Cells infected for 1 or 5 hr with chromosomally GFP‐tagged Listeria were stained for 15 min with Acridine Orange 2 hr before infection or 2 hr postinfection, respectively. Cells stained with Acridine Orange were harvested using a 1 mM EDTA‐1 mM EGTA‐PBS solution, pelleted, and fixed for 20 min using Cytofix/cytoperm solutions (BD Biosciences). Acridine Orange fluorescence was quantified by flow cytometry (excitation 488 nm; detection window: 665–715 nm). Forward‐ and side‐scatter light was used to identify viable cells, and at least 5,000 events were captured and analyzed using FlowJo software. Cells stained with Lysotracker were incubated in phenol red‐free DMEM and directly imaged on a Axiobserver Z1 confocal microscope (Zeiss) in prewarmed culture chamber for live‐cell imaging. Images were analyzed with the ImageJ software. All displayed images are representative fields of at least three independent experiments.
4.7. Chelation of extra‐ and intracellular calcium
HeLa cells were pre‐incubated, 20 min before LLO treatment, in MEM supplemented with 1 or 10 mM EGTA, or with 0.1 mM BAPTA‐AM (Sigma‐Aldrich). After removal of the medium, cells were incubated again in MEM supplemented with EGTA or BAPTA‐AM, and with 3 nM LLO.
4.8. Cell fractionation
Fractionation of cells was carried out by mechanical lysis and ultracentrifugation. Cells were seeded at 5 × 106 cells per 100 mm dishes the day before LLO treatment or Listeria infection. After treatment, cells were gently scrapped in 0.5% BSA‐PBS and centrifuged at 300×g for 5 min at 4 °C. Pellets were then washed with chilled homogenization buffer (8% sucrose, 3 mM imidazole, 1 mM MgCl2) and pelleted at 2000×g for 10 min at 4 °C. Cells were resuspended again in homogenization buffer supplemented with either 0.05% gelatine for cathepsins activity assays or 0.05% gelatine, 0.5 mM EGTA, and protease inhibitors (complete protease inhibitor cocktail tablets; Roche) for immunoblot analysis. Cell disruption was carried out by multiple passages through a 25G 5/8 in. needle. Cell lysates were then centrifuged at 200×g for 5 min at 4 °C. The supernatant was additionally centrifuged for 1 hr at 55,000×g at 4 °C. The final supernatants correspond to cytosolic fractions devoid of lysosomes. The corresponding pellets, containing lysosomes, were kept for further immunoblot analysis.
4.9. Immunoblot analysis
For immunoblot analysis, HeLa cells or subcellular fractions were mixed with Laemmli buffer (125 mM Tris–HCl [pH 6.8], 4% SDS, 20% glycerol, 100 mM dithiothreitol [DTT], 0.02% bromophenol blue), sonicated, boiled for 5 min, and protein content was resolved by SDS polyacrylamide gel electrophoresis. Proteins were then transferred on PVDF membranes (GE Healthcare) and detected after incubation with specific antibodies using Pierce ECL 2 Western Blotting Substrate (Fisher Scientific). The following primary antibodies were used for immunoblot analysis: mouse anti‐actin (A5441; Sigma‐Aldrich; 1:10,000 dilution), mouse anti‐LAMP1 (555798 ; BD Pharmingen; 1:500 dilution), mouse anti‐Ubc9 (610748 ; BD Transduction Lab; 1:1,000 dilution), and goat anti‐cathepsin D (sc‐6486 ; Santa‐Cruz; 1:500 dilution). Anti‐mouse and anti‐goat HRP‐conjugated antibodies were used as secondary antibodies (Abliance; 1:8,000 and 1:4,000 dilution, respectively). All displayed immunoblots are representative of at least three independent experiments.
4.10. HRP‐mediated chemical crosslinking of lysosomes
HeLa cells, seeded at 2.105 cells per 400 mm2 wells the day before treatment, were serum‐starved for 2 hr in MEM and then incubated with 5 mg/ml HRP in MEM, buffered at pH 7.5 with 12 mM Hepes, for 15 min at 37 °C. Cells were then washed 3 times with 5% BSA‐PBS, followed or not by a 30 or 90 min chase period in HRP‐free MEM without FBS at 37 °C. Cells were incubated on ice with PBS containing 100 μg/ml DAB and 0.003% H2O2 for 30 min. After three washes with PBS and one with MEM without FBS, cells were transferred again at 37 °C for 5 min and then treated with purified LLO. Cells were finally lysed either in Laemmli buffer for immunoblot analysis, or in 8.6% Sucrose; 20 mM Hepes; 10 mM KCl; 1.5 mM MgCl2; 1 mM EDTA; 8 mM DTT; 1% NP‐40 for quantification of cathepsins activity.
4.11. Cathepsins activity assay
Proteolytic activity of cathespins was determined using fluorogenic substrates specific for cysteine cathepsins (Z‐FR‐AMC) or for Cathepsin B (Z‐RR‐AMC) (Enzo Life Science). Fifty microliter of whole cell lysates or cytosolic fractions were transfered to microtitre plates and mixed with 50 μL Cathepsin Reaction Buffer (100 mM Sodium Acetate, 16 mM DTT, 16 mM EDTA, and 50 μM fluorogenic substrate; pH 7.0). Cathepsins activity was determined by measuring absorbance at 450 nm using a TriStar LB 941 system (Berthold) and by calculating the maximum rate of AMC release in each condition.
4.12. Caspase 3 activity assay
Measurements of caspase‐3 activity were performed using Colorimetric Caspase 3 Assay Kit (Sigma) according to manufacturer's instruction. After indicated treatments, HeLa cells were lysed at 4 °C and centrifuged. Supernatants were incubated with caspase 3‐specific substrate (Ac‐DEVD‐pNA substrate) overnight. P‐Nitroaniline (pNa) production was determined by measuring absorbance at 405 nm using a Tristar LB 941 system (Berthold), and pNa calibration curves were used to calculate caspase activities.
4.13. Acid sphingomyelinase activity assay
HeLa cells, seeded at 1.105 cells per 200 mm2 wells the day before treatment, were serum‐starved for 2 hr in MEM before treatment with EGTA or BAPTA‐AM. Supernatants, recovered after 30 min of 3 nM LLO treatment, were centrifuged for 5 min at 13,000×g to remove cell remnants. Quantification of acid sphingomyelinase activity in 50 μL of these supernatants was then performed using the Acid Sphingomyelinase Assay Kit (Abcam), following manufacturer's instruction and by measuring fluorescence on a Tristar LB 491 system (excitation 530 nm/emission 590 nm; Berthold).
4.14. Quantification of cell death
Cell viability was determined using LIVE/DEAD® Fixable Red Dead Cell Staining Kit (ThermoFischer). HeLa cells were seeded at 3.2 × 105 cells per 960 mm2 2 days before LLO treatment or Listeria infection. After treatment, cells were detached with 0.05% Trypsin–EDTA (Life Technologies), washed in PBS and stained for 30 min with LIVE/DEAD® fluorescent dye. Cells were then washed in PBS, fixed for 30 min in IC Fixation Buffer (eBioscience), and washed again. Quantification of dead cells was performed by flow cytometry analysis (excitation 561 nm; detection window: 590–630 nm). Forward‐ and side‐scatter light was used to exclude cellular fragments, and at least 10,000 events were captured and analyzed using FlowJo software.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTION
D. R. and P. C. conceived and designed the study. D. R., P. C., and J. M. wrote the paper. D. R. and J. M. performed and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Supporting information
Figure S1. Supporting info item
Figure S2. Supporting info item
ACKNOWLEDGEMENTS
We thank J. Gruenberg and R. Schekman for the helpful discussion, M. Hamon for the help with flow cytometry experiments, J. J. Quereda for the construction of the Listeria EGD Δhly strain and F. Stavru for the construction of the GFP‐tagged Listeria EGD Δhly strain. Work in P.C.'s laboratory received financial support from Institut Pasteur, INSERM, INRA, National Research Agency (ANR; ERANET Infect‐ERA PROANTILIS ANR‐13‐IFEC‐0004‐02), the French Government's Investissement d'Avenir program, Laboratoire d'Excellence “Integrative Biology of Emerging Infectious Diseases” (ANR‐10‐LABX‐62‐IBEID), the European Research Council (ERC) (Advanced Grant #233348 MODELIST, H2020‐ERC‐2014‐ADG 670823‐BacCellEpi), the Fondation le Roch les Mousquetaires and the Fondation Louis‐Jeantet. J. M. is supported by a fellowship from Paris Diderot University; P. C. is a Senior International Research Scholar of the Howard Hughes Medical Institute and D. R. is a Research Associate from INSERM.
Malet JK, Cossart P, Ribet D. Alteration of epithelial cell lysosomal integrity induced by bacterial cholesterol‐dependent cytolysins. Cellular Microbiology. 2017;19:e12682 https://doi.org/10.1111/cmi.12682
Contributor Information
Pascale Cossart, Email: pascale.cossart@pasteur.fr.
David Ribet, Email: david.ribet@inserm.fr.
REFERENCES
- Aits, S. , & Jaattela, M. (2013). Lysosomal cell death at a glance. Journal of Cell Science, 126, 1905–1912. [DOI] [PubMed] [Google Scholar]
- Andrews, N. W. , Almeida, P. E. , & Corrotte, M. (2014). Damage control: Cellular mechanisms of plasma membrane repair. Trends in Cell Biology, 24, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelqvist, H. , Waster, P. , Kagedal, K. , & Ollinger, K. (2013). The lysosome: From waste bag to potential therapeutic target. Journal of Molecular Cell Biology, 5, 214–226. [DOI] [PubMed] [Google Scholar]
- Arnaud, M. , Chastanet, A. , & Debarbouille, M. (2004). New vector for efficient allelic replacement in naturally nontransformable, low‐GC‐content, gram‐positive bacteria. Applied and Environmental Microbiology, 70, 6887–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino, D. , Hamon, M. A. , Dortet, L. , Nahori, M. A. , Pizarro‐Cerda, J. , Alignani, D. , … Toledo‐Arana, A. (2010). Single‐cell techniques using chromosomally tagged fluorescent bacteria to study Listeria monocytogenes infection processes. Applied and Environmental Microbiology, 76, 3625–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewley, M. A. , Marriott, H. M. , Tulone, C. , Francis, S. E. , Mitchell, T. J. , Read, R. C. , … Dockrell, D. H. (2011). A cardinal role for cathepsin d in co‐ordinating the host‐mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathogens, 7, e1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewley, M. A. , Naughton, M. , Preston, J. , Mitchell, A. , Holmes, A. , Marriott, H. M. , … Dockrell, D. H. (2014). Pneumolysin activates macrophage lysosomal membrane permeabilization and executes apoptosis by distinct mechanisms without membrane pore formation. MBio, 5, e01710–e01714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya, P. , & Kroemer, G. (2008). Lysosomal membrane permeabilization in cell death. Oncogene, 27, 6434–6451. [DOI] [PubMed] [Google Scholar]
- Boya, P. , Andreau, K. , Poncet, D. , Zamzami, N. , Perfettini, J. L. , Metivier, D. , … Kroemer, G. (2003). Lysosomal membrane permeabilization induces cell death in a mitochondrion‐dependent fashion. The Journal of Experimental Medicine, 197, 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero, J. A. , & Unanue, E. R. (2012). Mechanisms and immunological effects of apoptosis caused by Listeria monocytogenes . Advances in Immunology, 113, 157–174. [DOI] [PubMed] [Google Scholar]
- Carrero, J. A. , Vivanco‐Cid, H. , & Unanue, E. R. (2008). Granzymes drive a rapid listeriolysin O‐induced T cell apoptosis. Journal of Immunology, 181, 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corotte, M. , Almeida, P. E. , Tam, C. , Castro‐Gomes, T. , Fernandes, M. C. , Millis, B. A. , … Andresw, N. W. (2013). Caveolae internalization repairs wounded cells and muscle fibers. eLife, 2, e00926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corotte, M. , Fernandes, M. C. , Tam, C. , & Andrews, N. W. (2012). Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic, 13, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart, P. (2011). Illuminating the landscape of host‐pathogen interactions with the bacterium Listeria monocytogenes . Proceedings of the National Academy of Sciences of the United States of America, 108, 19484–19491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart, P. , Vicente, M. F. , Mengaud, J. , Baquero, F. , Perez‐Diaz, J. C. , & Berche, P. (1989). Listeriolysin O is essential for virulence of Listeria monocytogenes: Direct evidence obtained by gene complementation. Infection and Immunity, 57, 3629–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdal, H. , Berndtsson, M. , Castro, J. , Brunk, U. , Shoshan, M. C. , & Linder, S. (2005). Induction of lysosomal membrane permeabilization by compounds that activate p53‐independent apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 102, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoll, P. , Mondino, S. , Rolando, M. , & Buchrieser, C. (2016). Targeting of host organelles by pathogenic bacteria: A sophisticated subversion strategy. Nature Reviews. Microbiology, 14, 5–19. [DOI] [PubMed] [Google Scholar]
- Eskandarian, H. A. , Impens, F. , Nahori, M. A. , Soubigou, G. , Coppee, J. Y. , Cossart, P. , & Hamon, M. A. (2013). A role for SIRT2‐dependent histone H3K18 deacetylation in bacterial infection. Science, 341, 1238858. [DOI] [PubMed] [Google Scholar]
- Gaillard, J. L. , Berche, P. , & Sansonetti, P. (1986). Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes . Infection and Immunity, 52, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glomski, I. J. , Gedde, M. M. , Tsang, A. W. , Swanson, J. A. , & Portnoy, D. A. (2002). The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. The Journal of Cell Biology, 156, 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, M. R. , Bischofberger, M. , Frêche, B. , Ho, S. , Parton, R. G. , & van der Goot, F. G. (2011). Pore forming toxins induce multiple cellular responses promoting survival. Cellular Microbiology, 13, 1026–1043. [DOI] [PubMed] [Google Scholar]
- Gründling, A. , Gonzalez, M. D. , & Higgins, D. E. (2003). Requirement of the Listeria monocytogenes broad‐range phospholipase PC‐PLC during infection of human epithelial cells. Journal of Bacteriology, 185, 6295–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon, M. A. , & Cossart, P. (2011). K+ efflux is required for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin O and other pore‐forming toxins. Infection and Immunity, 79, 2839–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon, M. A. , Batsche, E. , Regnault, B. , Tham, T. N. , Seveau, S. , Muchardt, C. , & Cossart, P. (2007). Histone modifications induced by a family of bacterial toxins. Proceedings of the National Academy of Sciences of the United States of America, 104, 13467–13472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon, M. A. , Ribet, D. , Stavru, F. , & Cossart, P. (2012). Listeriolysin O: the Swiss army knife of Listeria . Trends in Microbiology, 20, 360–368. [DOI] [PubMed] [Google Scholar]
- Henry, R. , Shaughnessy, L. , Loessner, M. J. , Alberti‐Segui, C. , Higgins, D. E. , & Swanson, J. A. (2006). Cytolysin‐dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes . Cellular Microbiology, 8, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idone, V. , Tam, C. , Goss, J. W. , Toomre, D. , Pypaert, M. , & Andrews, N. W. (2008). Repair of injured plasma membrane by rapid Ca2 +‐dependent endocytosis. The Journal of Cell Biology, 180, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impens, F. , Radoshevich, L. , Cossart, P. , & Ribet, D. (2014). Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proceedings of the National Academy of Sciences of the United States of America, 111, 12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, J. K. , Andrews, N. W. , & Simon, S. M. (2002). Membrane proximal lysosomes are the major vesicles responsible for calcium‐dependent exocytosis in nonsecretory cells. The Journal of Cell Biology, 159, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathariou, S. , Metz, P. , Hof, H. , & Goebel, W. (1987). Tn916‐induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes . Journal of Bacteriology, 169, 1291–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, C. L. , Smith, D. J. , Lyras, D. , Chakravorty, A. , & Rodd, J. I. (2009). Programmed cellular necrosis mediated by the pore‐forming alpha‐toxin from Clostridium septicum . PLoS Pathogens, 5, e1000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschke, H. , Wiederanders, B. , Bromme, D. , & Rinne, A. (1989). Cathepsin S from bovine spleen. Purification, distribution, intracellular localization and action on proteins. Biochemical Journal, 264, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laulagnier, K. , Schieber, N. L. , Maritzen, T. , Haucke, V. , Parton, R. G. , & Gruenberg, J. (2011). Role of AP1 and Gadkin in the traffic of secretory endo‐lysosomes. Molecular Biology of the Cell, 22, 2068–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton, A. , Lakisic, G. , Job, V. , Fritsch, L. , Tham, T. N. , Camejo, A. , … Bierne, H. (2011). A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science, 331, 1319–1321. [DOI] [PubMed] [Google Scholar]
- Lebreton, A. , Stavru, F. , & Cossart, P. (2015). Organelle targeting during bacterial infection: Insights from Listeria . Trends in Cell Biology, 25, 330–338. [DOI] [PubMed] [Google Scholar]
- Lee, A. Y. , Gulnik, S. V. , & Erickson, J. W. (1998). Conformational switching in an aspartic proteinase. Nature Structural Biology, 5, 866–871. [DOI] [PubMed] [Google Scholar]
- Luzio, J. P. , Hackmann, Y. , Dieckmann, N. M. , & Griffiths, G. M. (2014). The biogenesis of lysosomes and lysosome‐related organelles. Cold Spring Harbor Perspectives in Biology, 6, a016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda, S. , Okada, N. , Kodama, T. , Honda, T. , & Iida, T. (2012). A cytotoxic type III secretion effector of Vibrio parahaemolyticus targets vacuolar H + −ATPase subunit c and ruptures host cell lysosomes. PLoS Pathogens, 8, e1002803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meixenberger, K. , Pache, F. , Eitel, J. , Schmeck, B. , Hippenstiel, S. , Slevogt, H. , … Opitz, B. (2010). Listeria monocytogenes‐infected human peripheral blood mononuclear cells produce IL‐1beta, depending on listeriolysin O and NLRP3. Journal of Immunology, 184, 922–930. [DOI] [PubMed] [Google Scholar]
- Peraro, M. D. , & van der Goot, F. G. (2016). Pore‐forming toxins: Ancient, but never really out of fashion. Nature Reviews. Microbiology, 14, 77–92. [DOI] [PubMed] [Google Scholar]
- Pillich, H. , Loose, M. , Zimmer, K. P. , & Chakraborty, T. (2012). Activation of the unfolded protein response by Listeria monocytogenes . Cellular Microbiology, 14, 949–964. [DOI] [PubMed] [Google Scholar]
- Pizarro‐Cerda, J. , Kuhbacher, A. , & Cossart, P. (2012). Entry of Listeria monocytogenes in mammalian epithelial cells: An updated view. Cold Spring Harbor Perspectives in Medicine, 2, pii: a010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy, D. A. , Jacks, P. S. , & Hinrichs, D. J. (1988). Role of hemolysin for the intracellular growth of Listeria monocytogenes . The Journal of Experimental Medicine, 167, 1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt, M. R. , Sekedat, M. D. , Chiang, K. P. , & Muir, T. W. (2009). Direct measurement of cathepsin B activity in the cytosol of apoptotic cells by an activity‐based probe. Chemistry & Biology, 16, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince, L. R. , Bianchi, S. M. , Vaughan, K. M. , Bewley, M. A. , Marriott, H. M. , Walmsley, S. R. , … Whyte, M. K. (2008). Subversion of a lysosomal pathway regulating neutrophil apoptosis by a major bacterial toxin, pyocyanin. Journal of Immunology, 180, 3502–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, A. , Caler, E. V. , & Andrews, N. W. (2001). Plasma membrane repair is mediated by Ca2 +‐regulated exocytosis of lysosomes. Cell, 106, 157–169. [DOI] [PubMed] [Google Scholar]
- Repnik, U. , Hafner Cesen, M. , & Turk, B. (2014). Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion, 19(Pt A), 49–57. [DOI] [PubMed] [Google Scholar]
- Repnik, U. , Stoka, V. , Turk, V. , & Turk, B. (2012). Lysosomes and lysosomal cathepsins in cell death. Biochimica et Biophysica Acta, 1824, 22–33. [DOI] [PubMed] [Google Scholar]
- Ribet, D. , Hamon, M. , Gouin, E. , Nahori, M. A. , Impens, F. , Neyret‐Kahn, H. , … Cossart, P. (2010). Listeria monocytogenes Impairs SUMOylation for efficient infection. Nature, 464, 1192–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, H. W. , Callery, M. P. , Deck, B. , & Unanue, E. R. (1996). Listeria monocytogenes Induces apoptosis of infected hepatocytes. Journal of Immunology, 156, 679–684. [PubMed] [Google Scholar]
- Schluter, D. , Domann, E. , Buck, C. , Hain, T. , Hof, H. , Chakraborty, T. , & Deckert‐Schluter, M. (1998). Phosphatidylcholine‐specific phospholipase C from Listeria monocytogenes is an important virulence factor in murine cerebral listeriosis. Infection and Immunity, 66, 5930–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre, C. , Fraldi, A. , Medina, D. L. , & Ballabio, A. (2013). Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nature Reviews. Molecular Cell Biology, 14, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy, L. M. , Hoppe, A. D. , Christensen, K. A. , & Swanson, J. A. (2006). Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cellular Microbiology, 8, 781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavru, F. , Bouillaud, F. , Sartori, A. , Ricquier, D. , & Cossart, P. (2011). Listeria monocytogenes Transiently alters mitochondrial dynamics during infection. Proceedings of the National Academy of Sciences of the United States of America, 108, 3612–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavru, F. , Palmer, A. E. , Wang, C. , Youle, R. J. , & Cossart, P. (2013). Atypical mitochondrial fission upon bacterial infection. Proceedings of the National Academy of Sciences of the United States of America, 110, 16003–16008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoorvogel, W. (1998). Analysis of the endocytic system by using horseradish peroxidase. Trends in Cell Biology, 8, 503–505. [DOI] [PubMed] [Google Scholar]
- Tam, C. , Idone, V. , Devlin, C. , Fernandes, M. C. , Flannery, A. , He, X. , … Andrews, N. W. (2010). Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. The Journal of Cell Biology, 189, 1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli, I. , Lembo‐Fazio, L. , Nigro, G. , Carneiro, L. A. , Ferraro, E. , Rossi, G. , … Bernardini, M. L. (2008). Intracellular bacteriolysis triggers a massive apoptotic cell death in Shigella‐infected epithelial cells. Microbes and Infection, 10, 1114–1123. [DOI] [PubMed] [Google Scholar]
- Turk, B. , Bieth, J. G. , Bjork, I. , Dolenc, I. , Turk, D. , Cimerman, N. , … Turk, V. (1995). Regulation of the activity of lysosomal cysteine proteinases by pH‐induced inactivation and/or endogenous protein inhibitors, cystatins. Biological Chemistry Hoppe‐Seyler, 376, 225–230. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe, T. , Vanlangenakker, N. , Parthoens, E. , Deckers, W. , Devos, M. , Festjens, N. , … Vandenabeele, P. (2010). Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death and Differentiation, 17, 922–930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Supporting info item
Figure S2. Supporting info item