

Abstract
Fast relaxation of cross-bridge generated force in the myocardium facilitates efficient diastolic function. Recently published research studying mechanisms that modulate the relaxation rate has focused on molecular factors. Mechanical factors have received less attention since the 1980s when seminal work established the theory that reducing afterload accelerates the relaxation rate. Clinical trials using afterload reducing drugs, partially based on this theory, have thus far failed to improve outcomes for patients with diastolic dysfunction. Therefore, we reevaluated the protocols that suggest reducing afterload accelerates the relaxation rate and identified that myocardial relengthening was a potential confounding factor. We hypothesized that the speed of myocardial relengthening at end systole (end systolic strain rate), and not afterload, modulates relaxation rate and tested this hypothesis using electrically-stimulated trabeculae from mice, rats, and humans. We used load-clamp techniques to vary afterload and end systolic strain rate independently. Our data show that the rate of relaxation increases monotonically with end systolic strain rate but is not altered by afterload. Computer simulations mimic this behavior and suggest that fast relengthening quickens relaxation by accelerating the detachment of cross-bridges. The relationship between relaxation rate and strain rate is novel and upends the prevailing theory that afterload modifies relaxation. In conclusion, myocardial relaxation is mechanically modified by the rate of stretch at end systole. The rate of myocardial relengthening at end systole may be a new diagnostic indicator or target for treatment of diastolic dysfunction.
Keywords: Diastole, Relaxation, Strain Rate, Cross-bridge, Afterload, Myocardium
INTRODUCTION
Rapid relaxation after ejection is essential for efficient filling of the left ventricle of the heart [1]. If the heart relaxes too slowly, there is not enough time for the heart to fill and cardiac function is compromised. Clinically, slow relaxation is an independent diagnostic criterion for Heart Failure with preserved Ejection Fraction (HFpEF) [2]. Improved understanding of the mechanisms that influence the rate of relaxation could lead to better treatments for diastolic dysfunction and HFpEF.
Cardiac relaxation ultimately reflects the reduction in the number of myosin heads that are bound to actin. This is a very complex process, and several molecular-level mechanisms are known to influence the rate of relaxation [3]. For example, myocardial relaxation can be quickened by increasing SERCA expression [4] or slowed by decreasing SERCA activity [5]. Increasing SERCA expression reduces the intracellular Ca2+ concentration during the latter stages of a twitch contraction, which in turn decreases the number of binding sites on actin to which myosin heads can attach.
Mechanical factors that influence relaxation have received less attention since the 1980s. Seminal work by D. L. Brutsaert and others used isolated intact trabeculae and working hearts to establish the theory that reducing afterload accelerates the myocardial relaxation rate [6–9]. In part due to the high prevalence of hypertension in patients with Heart Failure with preserved Ejection Fraction, clinical trials attempted to treat such patients with anti-hypertensive therapies such as inhibition of the renin-angiotensin-aldosterone system [10]. However, reducing afterload did not improve outcomes in these patients.
We were intrigued by these results and recent reports of reduced myocardial strain rates in HFpEF patients [11]. Therefore, we re-evaluated the original protocols used to suggest that afterload modifies the relaxation rate [6, 7, 9, 10]. We noted that both afterload and the myocardial strain rate at end systole (the speed of muscle relengthening) are simultaneously adjusted when using the conditions of the original protocol. However, the simultaneous change in afterload and strain rate suggests that strain rate may have been a confounding factor in the interpretation of the seminal experiments. The authors concluded that afterload was the mechanical modifier of relaxation, even though they noted that relengthening is necessary to modify relaxation [6, 9, 12].
This manuscript describes new experiments that we developed to isolate the role of myocardial strain rate from afterload. Specifically, we independently modified afterload and end-systolic strain-rate in experiments using electrically simulated trabeculae from mice, rats, and humans. We also used computer simulations to investigate the relationship between fast stretch and cross-bridge detachment. Our data shows a novel relationship between relaxation rate and end systolic strain rate and the likely molecular mechanism is enhanced cross-bridge detachment.
METHODS
ISOLATION OF RODENT VENTRICULAR TRABECULA
Female Sprague-Dawley or wild-type (mixed Sprague-Dawley/F344/Brown Norway) rats [13] and male C57BL/6 mice were used in this study. Animal use was approved by the Institutional Animal Use and Care Committees of the University of Kentucky and Wayne State University.
Each animal was anesthetized via an IP injection of sodium pentobarbital (50 mg kg−1) or inhalation of 3% isoflurane and heparinized via an IP administration of sodium heparin (1000 U kg−1). The animal was subsequently euthanized by exsanguination, and its heart was rapidly excised and rinsed in a cold (4°C), oxygenated (>10 ppm) perfusion solution (in mM: 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 1.2 MgSO4, 12 NaHCO3, 10 KHCO3, 10 2-[4-2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 30 Taurine, 5.5 glucose, 10 2,3-butanedione monoxine (BDM)). Additional perfusion solution was then flushed through the heart via an aortic cannula to remove any remaining blood. The heart was then placed in a dissecting dish that was coated with Sylgard (Dow Corning, Midland MI) and filled with cold perfusion solution. After opening the ventricles, the endocardial surfaces were inspected for free-standing trabeculae, including cylindrical papillary muscles. Potential preparations were removed from the heart by cutting a cuboid section of ventricular wall at each end of the trabecula. Contact with the trabecula itself was minimized to reduce the probability of induced damage.
ISOLATION OF HUMAN VENTRICULAR TRABECULA
One set of experiments was performed using a trabecula isolated from the right ventricle, between the mid-wall and apex, of an organ donor (60 year old female). The organ was not transplantable due to the donor’s history of left coronary artery disease. This experiment was approved by the Institutional Review Board of the University of Kentucky.
During the organ procurement, the heart was perfused with cardioplegia solution. The ventricles were subsequently excised and provided to the research team submerged in cardioplegia solution. A free-standing trabecula was located on the endocardial surface of the right ventricular free wall and carefully isolated from the organ.
INTACT TRABECULA MECHANICS
Each trabecula was transferred from the dissecting dish to the experimental chamber inside a bulb pipette using perfusion solution to maintain hydration. This prevented the trabecula from dehydrating during the transfer procedure. The experimental chamber had a volume of 350 μL and was continuously perfused with oxygenated Tyrode’s Solution (in mM: 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose) at 25°C or in one case increased to 37°C. One end of the trabecula was hooked to a high-speed length motor (Model 312C or 315C, Aurora Scientific, Aurora, Ontario, Canada) while the other end was secured to a force transducer (Model 403, Aurora Scientific). The trabecula was paced at 0.5 Hz using bipolar excitation approximately 1.2 times the threshold voltage and stretched to L0, the length where the maximal force was developed during isometric contractions. L0 and the cross sectional area were measured by video microscopy. The cross-sectional area was calculated by averaging the diameter of the trabecula at 4 positions along its length and assuming a cylindrical geometry. It was allowed to equilibrate for approximately 1 hr before data acquisition began.
Experiments were performed using SLControl software [14] and a newly written real-time control algorithm. The trabecula was held isometric except for the experimental load-clamped twitches that were performed at least 4 seconds apart to prevent history-dependent effects [15]. During a load-clamp trial, the trabecula isometrically contracted until force reached a predefined set-point (the afterload). Force was then isotonically maintained at the chosen afterload by adjusting the motor position to shorten and then relengthen the muscle in real-time using the control algorithm. If the load-clamp was stopped and the muscle was held isometric before the minimum length was achieved, additional systolic force was generated before the muscle could relax (Supplemental Figure S1). To focus on the mechanical behaviors of relaxation, the load-clamp was terminated and the muscle allowed to relax isometrically after one of three events. These events were: 1) the trabecula relengthened back to its original length, 2) the trabecula began to relengthen (i.e, the trabecula was held at the minimum attained length), or 3) the trabecula relengthened by a pre-set amount. Different termination criteria were used in successive trials to investigate different mechanical behaviors. Once the load-clamp was terminated, the trabecula was allowed to relax under isometric conditions at that length. If necessary, the trabecula was returned to its original length after the trial was complete. Load-clamps using different termination conditions were repeated at various afterloads (typically between 25–75% of the maximum isotonic force). Experiments were stopped if the developed isometric force decreased by >20% from the equilibrated maximal force at L0, which typically occurred >6 hours after the muscle was equilibrated.
Data were analyzed offline using custom scripts written in MATLAB (The MathWorks, Natick, MA). Force records were smoothed using a Savitzky-Golay filter and normalized to the measured cross-sectional area. Data were aligned using either the stimulus pulse or a threshold force when the stimulus was not available for technical reasons. Changes in muscle strain were calculated from the muscle length and the position of the motor. No corrections were made to allow for series compliance in the attachments to the experimental apparatus. For load-clamp twitches, the onset of relaxation was defined as the time-point at which the motor stopped moving. The relaxation rate was determined using the Glantz Method calculated in the pressure-phase plane [16, 17]. The rate is defined as 1/tau, where tau is the typical time constant of isovolumic relaxation.
MATHEMATICAL MODELING
Twitch contractions were simulated using a mathematical model consisting of a single half-sarcomere connected in series to an elastic spring. The half-sarcomere was represented by a population of cross-bridges cycling through a 2-state scheme and a parallel elastic component that produced the system’s passive resting tension. Strain dependent myosin kinetics were included in the model. The series elastic spring was non-linear and mimicked the compliance in the trabecula’s attachments to the experimental apparatus. All calculations were performed using freely-available MyoSim software [18] that can be downloaded from http://www.myosim.org. The data files necessary to reproduce these simulations are included as Supplementary Material.
The half-sarcomere was activated in every simulation by an identical idealized Ca2+ transient of the form
where as described by Rice et al [19].τ1=0.029 s and τ2=0.17 s were chosen to match calcium transient parameters obtained from rat cardiomyocytes isolated from age and gender matched rats [20].
A test suite of 5 representative force records was selected from the experimental data. These records correspond to: an isometric twitch and 4 load-clamped twitches (25% or 50% afterload, with zero or full relengthening). Multidimensional optimization was then used to deduce model parameters that produced the best attainable fit to the experimental data.
The resulting model was then used to simulate the mechanical behavior during 30 different combinations of afterload and relengthening. Twitches with relaxation rates less than 100 s−1 were subsequently analyzed using the same methods as described above for the experimental data.
RESULTS
LOAD-CLAMP EXPERIMENTS
Figure 1 shows the mechanical behavior of a single rat trabecula during isometric twitches and different load-clamp conditions. The left panels show a single isometric trial and 5 load-clamp trials in which the trabecula shortened and was then re-stretched to its starting length (L0) before relaxing isometrically. The theory that reduced afterload leads to faster relaxation is based on this protocol [6, 21]. The trabecula relaxed faster after it contracted against a low afterload. However, the magnitude and speed of relengthening also varied with afterload.
Figure 1.
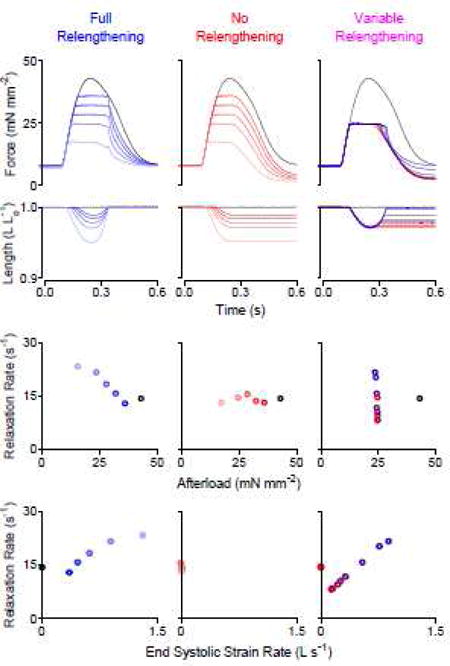
Load-clamp protocols indicate that relaxation rate depends on the end systolic strain rate, not afterload. Top: Force and length versus time traces; Middle: Relationship between relaxation rate and afterload; Bottom: Relationship between relaxation rate and end systolic strain rate. Left (blue): overlay of 6 twitches that were load-clamped at various afterloads; the muscle was relengthened back to its original length before being allowed to relax. Center (red): overlay of 6 twitches; the muscle relaxed while being held at the minimum attained length. Right: overlay of 9 twitches that were load-clamped at ~50% of the peak isometric force; the load-clamp was stopped with variable amounts of relengthening before being allowed to relax. Muscle relengthening was necessary to obtain an inverse relationship between relaxation rate and afterload. Relengthening alone (without modifying afterload) was sufficient to modify the relaxation rate. All data in this figure measured from the same rat trabecula.
The middle panels show additional trials in which the load-clamp was terminated as soon as the control algorithm indicated that the trabecula needed to relengthen. The trabecula therefore relaxed isometrically at the minimum length that it attained while contracting against the set afterload. The bottom panel shows that the rate of relaxation was not affected by afterload under these conditions, which suggests that relengthening is necessary to modify relaxation rate.
The right-hand panels show trials where the trabecula contracted against a fixed afterload but was allowed to re-stretch by different amounts before relaxing. The rate of relaxation increased from 8.1 to 23.6 s−1 as the re-stretch varied from zero to full relengthening. These data suggest that relengthening itself is sufficient to modify relaxation rate regardless of afterload.
Figure 2 shows twitch force and muscle length data from mouse, rat, and human trabeculae and Supplemental Figure S2 shows collated data from 6 additional trabeculae, each from individual rats. Supplemental Figure S3 shows that relaxation rate is dependent on end systolic strain rate at physiologic temperatures. These data show that the relaxation rate is not well correlated with afterload, but relaxation rate increased monotonically with end systolic strain rate. (We use the term end systolic strain rate because the load-clamp mimics ejection: e.g. the end of the clamp is similar to aortic valve closure at end systole.)
Figure 2.
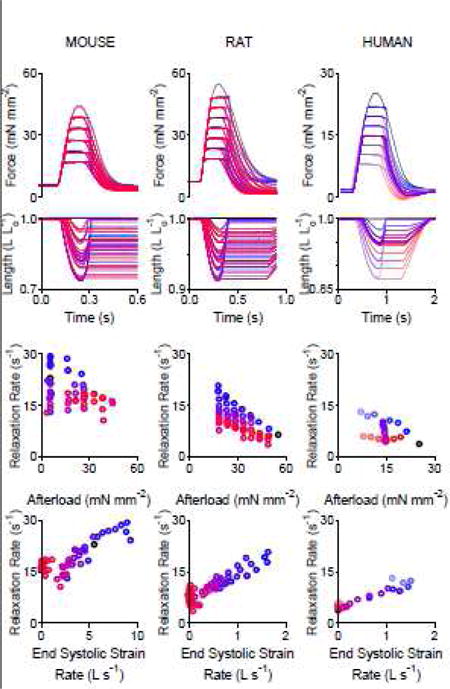
Relaxation rate is dependent on the end systolic strain rate in mouse, rat, and human myocardial trabeculae. Top: Force and length versus time traces. Bottom: Relationship between relaxation rate and end systolic strain rate. Each column shows multiple afterload-clamped twitches overlaid from a single mouse trabecula (53 twitches), rat trabecula (62 twitches), and human (25 twitches) trabecula. Six additional rat trabeculae shown in Supplementary Figure S2.
MATHEMATICAL MODELING
We investigated the molecular mechanisms underlying the relationship between end systolic strain rate and relaxation rate by simulating load-clamped twitch contractions using MyoSim software. We started this process by fitting a two-state cross-bridge model to a representative set of 5 experimental records (isometric twitch plus records with full and no relengthening at two different afterloads) (Supplemental Fig. S4).
The model was then used to predict the mechanical response of a trabecula to 30 different combinations of afterload and relengthening. These simulations exhibited similar trends to the experimental data (Fig. 3), specifically: (1) The rate of myocardial relaxation scaled inversely with afterload when the muscle relengthened fully before relaxation. (2) Relaxation rate was essentially constant when the muscle did not relengthen. (3) The rate of relaxation increased monotonically with the end-systolic strain rate.
Figure 3.
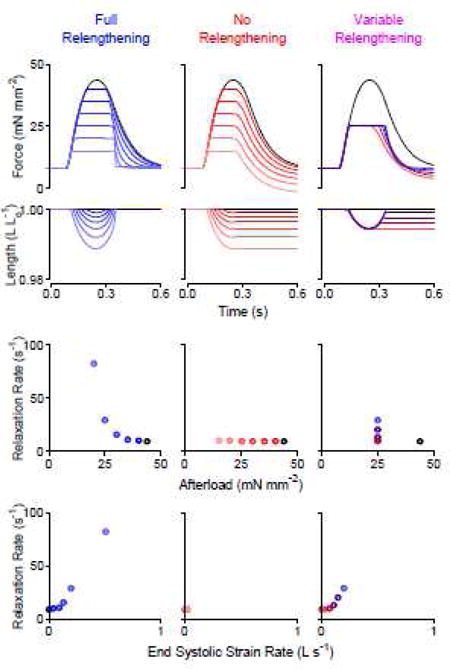
Model simulation mimics load-clamp experiments from a rat cardiac trabecula. Top: Mathematical modeling simulated force and length versus time traces; Middle: Relationship between relaxation rate and afterload, and bottom: Relationship between relaxation rate and end systolic strain rate observed in simulated data. These data show that relaxation rate is 1) only dependent on afterload only when the muscle relengthens and 2) dependent on the end systolic strain rate.
In these simulations, the magnitude of myocardial relengthening (strain) was related to the speed of relengthening (strain rate). Both parameters predicted the rate of relaxation but the relationship between relaxation rate and strain rate was more linear (Supplemental Fig. S5). These data suggest that strain rate, not strain, is a strong predictor of the relaxation rate.
These simulations also suggest that ~6.8% of myosin heads are bound to actin at the peak of an isometric twitch (Fig. 4). If the trabecula was load-clamped at half the peak isometric force, the rapid shortening during the initial stages of the clamp lowered the proportion of bound cross-bridges to ~4.5%. If the trabecula relaxed at the minimum length after a load-clamp, the cross-bridge detachment rate was similar to the purely isometric twitch (10.8 s−1 vs. 10.2 s−1), leading to a relatively slow rate of force decline (8.4 s−1) that is similar to that of the isometric contraction (6.8 s−1). In contrast, if the load-clamp was maintained until the trabecula had retained its original length, the quick re-stretch detached the bound cross-bridges up to ~30.0 s−1 and accelerated the rate of force decline dramatically (23.4 s−1). The monotonic relationship between relaxation rate and end systolic strain rate (Fig. 4C) is mimicked by the relationship between the rate of decline in bound cross-bridges and the end systolic strain rate (Fig. 4D).
Figure 4.
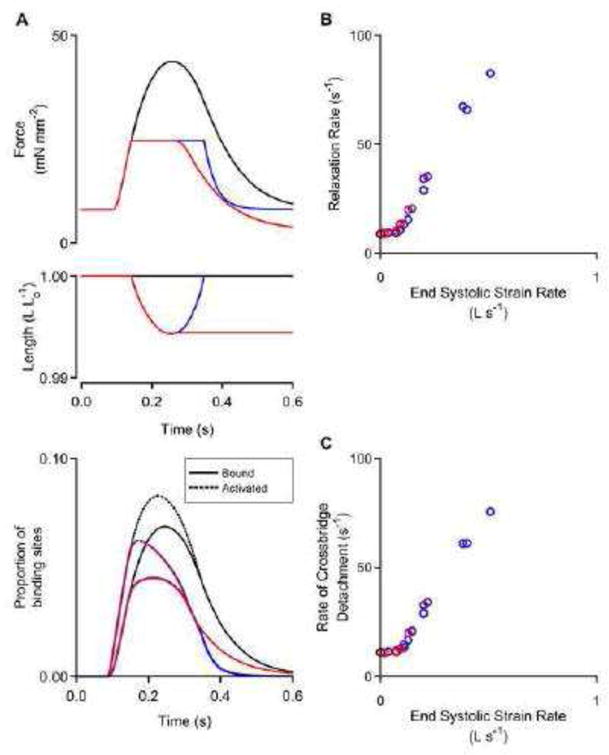
Mathematical modeling reveals molecular mechanisms. A) Model predicted force, length, and acto-myosin binding site status as a function of time. Black: isometric twitch. Red: load-clamped twitch held at minimum length. Blue: load-clamp twitch allowed to fully relengthen before relaxation. B) Relaxation rate versus end systolic strain rate and C) cross-bridge detachment rate versus end systolic strain rate for multiple simulated twitches. This modeling data set shows that cross-bridge detachment rates are slow unless a fast stretch is applied before relaxation.
DISCUSSION
Our data show that in cardiac trabeculae from mice, rats, and humans, the rate of relaxation scales with the strain rate at end systole but is not independently modulated by afterload. This result contrasts with the standard, clinically-accepted theory that afterload is the modifying mechanism [6, 9, 10]. Computer simulations reproduce this behavior and suggest that fast relengthening at end systole quickens relaxation by accelerating the detachment of bound cross-bridges.
MECHANICAL CONTROL OF RELAXATION
Reduced afterload was reported to accelerate myocardial relaxation at least as early as 1968 [22]. To our knowledge, the ex vivo studies that support this theory [6, 7, 21, 23] used a protocol that included relengthening of the muscle, similar to the one shown in Figure 1 (left). Superficially, these data show that trabeculae relax faster if they have previously contracted against a low afterload. However, the end systolic strain rate produced by relengthening increased as afterload was reduced.
Since strain rate could be a potential confounding factor we developed new protocols that allowed us to vary afterload and strain-rate independently. These new experiments showed that the rate of relaxation did not depend on afterload if the muscle was prevented from relengthening (Fig 1, center). Of note, Brutsaert et al coined the term “relaxation loading” to describe the relengthening period and concluded that the lengthening “allows load-dependent relaxation to become manifest” [12, 23]. This statement and our data suggest that relengthening is necessary to mechanically modify the relaxation rate. Moreover, the rate of relaxation increased monotonically with end-systolic strain rate for different combinations of afterload and relengthening (Fig 1, right). These data suggest that relengthening is sufficient to modify the relaxation rate, regardless of afterload.
Both our experimental data and computational modeling data suggests that a tight relationship between strain and strain rate exists during the load-clamp. However, small strains are insufficient to modify relaxation rate, while relaxation appears to be quite sensitive to even small strain rates (Supplemental Fig. S5). The strong monotonic relationship between relaxation rate and strain rate has not been previously described. In summary, relengthening is not only necessary but it is sufficient to mechanically modify the relaxation rate, which is dependent on the strain rate.
MOLECULAR CONTROL OF RELAXATION
Myocardial relaxation occurs as the number of bound cross-bridges falls. It is known that the population of bound cross-bridges is affected by changes in protein isoforms and post-translational modifications that modify calcium reuptake, thin filament deactivation, and/or cross-bridge detachment kinetics [3, 24].
Our mathematical model corroborates experimental work that suggests changes in strain can also play a critical role in determining how quickly cross-bridges detach [25]. Calcium activated skinned myofibril (and tetanically stimulated intact skeletal muscle) experiments exhibit a biphasic relaxation after calcium is removed [26–28]. Muscle force first relaxes in a slow linear phase that suddenly transitions to a fast decay, which is thought to be a strain-induced rapid detachment of cross-bridges [29, 30]. Importantly, externally inducing a strain by stretching the preparation soon after activating calcium is removed will accelerate the rapid detachment of cross-bridges and initiate the fast phase of relaxation [27]. In intact cardiac muscle, Janssen and colleagues showed that oscillating stretches during the late systolic phase reduced tension, with higher frequencies generating a greater reduction [31].
The transition between slow and fast relaxation in a myofibril coincides with a sudden yielding (relengthening) of a single half sarcomere [32] and chaotic intra-fiber movement [28, 33] in the otherwise isometric preparation. Brutsaert noted that non-uniformity in sarcomere lengths can enhance relaxation [6, 9]. This effect, along with potentially damaged sarcomeres [34] and elastic chordae near the fiber ends are simplified in our model as a series elastic element that generates series compliance. A recent study showed that such a series elastic element is required to model the experimentally measured force decay pattern [35]. Regardless of utilizing individual sarcomeres or a series elastic element, the rapid decline in force is related to rapid detachment of cross-bridges upon a change in strain within the myofibril or myofiber.
The simulations presented in this work suggest that similar mechanisms also influence the relaxation of electrically stimulated trabeculae. As shown in Fig 4A, the rapid shortening at the onset of the load-clamp decreased the proportion of cross-bridges that were bound between the filaments [24]. This decrease occurred because the interfilamentary movement displaced bound heads from their equilibrium positions, increasing their mean detachment rate. However, as long as the thin filament activation level remained high, many of the heads that detached were quickly replaced by newly attaching cross-bridges [12, 24]. The high level of activation also allowed the proportion of bound heads to remain relatively stable throughout the shortening phase of the load-clamp. (The high level of thin filament activation also explains why additional force is developed if a load-clamp is stopped prior to reaching minimum length (Supplementary Figure S1).)
As the muscle deactivated and the number of available binding sites declined, the behavior in the simulations became more complex. If the muscle relaxed isometrically at its shortest attainable length, bound heads remained close to their equilibrium positions and relaxation was relatively slow. Conversely, rapid re-stretch displaced cross-bridges and caused them to detach from the thin filament. The newly vacated binding sites on actin quickly deactivated because the Ca2+ concentration had declined. This binding site closure prevented detached heads from being replaced as they had during the activation phase. This induced further strain in each remaining myosin head and initiated a positive feedback loop that quickly detached additional bound heads. Brutsaert had previously suggested that fast lengthening would induce “back-rotation and detachment of force generating cross bridges” [9]. Our simulations confirm that mechanical control of relaxation is induced by detachment of cross-bridges, likely due to strain-dependent myosin kinetics. Furthermore, our simulations represent the first evidence that end systolic strain rate modifies the cross-bridge detachment rate in the same manner it modifies the relaxation rate (Fig. 4B,C).
IN VIVO PHYSIOLOGY AND RELENGTHENING
One may assume that relengthening does not occur during systole because the ventricular volume does not increase before aortic valve closure. However, myocardial strain is not identical to volume. In healthy hearts, a small amount relengthening or untwist is observed at the end of systole using echocardiography and magnetic resonance imaging techniques [36–40]. The theory that reduced afterload accelerates relaxation rate was supported by data from intact heart and in vivo studies where myocardial strain rate was not measured [7]. Figure 5 shows the effect of a modest but quick relengthening, which mimics the in vivo condition [38], in a trabecula at two different afterloads. These data suggest that a brief relengthening before aortic valve closure explains why reducing the afterload increased the relaxation rate in intact hearts and in vivo. Although further work is required to confirm that end systolic relengthening accelerates relaxation rate in vivo, it is interesting to note strain rates are reduced [11] and end systolic relengthening is lost [41] in patients with HFpEF and subjects with hypertension or hypertrophy, who typically exhibit slow isovolumic relaxation.
Figure 5.
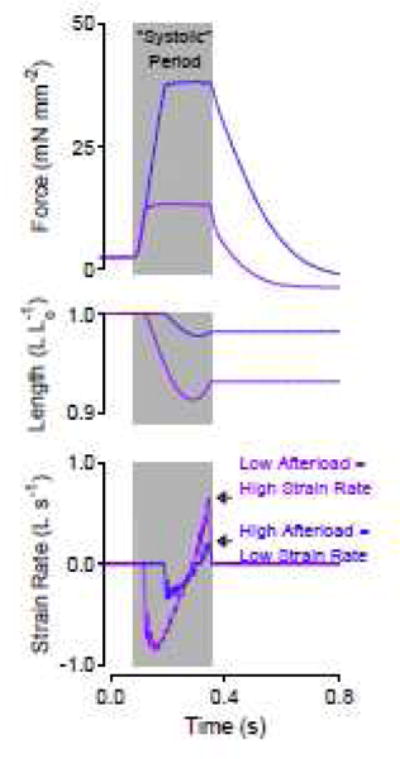
A short, quick relengthening explains why relaxation rate can be faster at reduced afterloads. Force, length, and strain rate versus time for two twitches load-clamped at different afterloads but allowed to relengthen for equal durations. Afterload may have been mistaken for the mechanical factor that modified relaxation rate in intact hearts because strain and strain rate were not measured.
Our in vitro system used a servo motor to drive the relengthening, but internal forces must relengthen the myocardium in vivo. It is already hypothesized that elastic elements such as titin and collagen help drive the myocardium to lengthen during filling [42, 43]. These molecular elastic elements are likely to have stored strain during ejection and shortening, which provides a driving force for the myocardium to relengthen before aortic valve closure in normal hearts.
Furthermore, the molecular mechanisms described here may also provide insight into why slow isovolumic relaxation has been correlated with impaired relaxation during early filling [44–46]. In healthy myocardium with end systolic relengthening, cross-bridges are more rapidly detached, minimizing their influence on filling. In patients with HFpEF whose myocardial strain rate is reduced [11], a larger population of bound cross-bridges needs to be detached during early filling.
LIMITATIONS
Like the previous work of Brutsaert and others [6, 7, 9], this work also utilized muscle fiber length control instead of sarcomere length control, which is difficult in muscle fibers. As noted above, our model includes non-uniform sarcomere lengths as part of a lumped series elastic element, whose compliance is dictated by damaged sarcomeres, averaging of multiple sarcomeres, and/or elastic chordae attached to the ends of the muscle. Experiment [27], recent computational models [35], and our current data suggests that local changes in sarcomere lengths are necessary to modify the relaxation rate. This is reflected in sarcomeres moving against the series elastic element or an overall change in muscle length.
Detailed evaluation of sarcomere length is of interest because cross-bridge kinetics appear to differ when sarcomere length is controlled instead of muscle length [34]. However, sarcomere length control induces muscle length changes that are not physiologic. A study using sarcomere length control instead of muscle length control suggested that minimum sarcomere length, not afterload, modified the relaxation rate [47]. While they speculated that the molecular mechanisms were similar (cross-bridge detachment), maintaining sarcomere length in the center of their preparation required muscle lengthening during the ejection phase and muscle shortening during relaxation. This is inconsistent with normal in vivo myocardial strain, where fibers typically shorten during ejection and relengthen during isovolumic relaxation [37]. Since muscle length, not sarcomere length, is most translatable to clinical cardiac imaging showing reduced strain rates in patients with diastolic dysfunction [11, 37–39, 41], we believe muscle length is the most appropriate index for this study.
While the general time course of our simulation data mimics the experimental data, the model over-estimates the relaxation rate. This may be due to the mathematical model, which represents the muscle system as a single half-sarcomere connected in series with a spring. Such a model omits a number of factors, such as inter-cell and inter-half-sarcomere heterogeneity. However, this simplification seems to provide useful data and mimics the function of our trabeculae, as it has previously for myofibrils [35].
CONCLUSION
This study is the first to establish that fast stretch (end systolic strain rate), not reduced afterload, is both necessary and sufficient to mechanically accelerate the myocardial relaxation rate. Rapid relengthening leads to enhanced cross-bridge detachment, which relates the mechanical and molecular mechanisms. The relationship between end systolic strain rate and relaxation rate may be important to clinical treatment or diagnosis of diastolic diseases such as HFpEF.
Supplementary Material
HIGHLIGHTS.
Intact cardiac trabeculae were used to study cardiac relaxation.
Relaxation rate is not directly modified by afterload.
Relaxation rate is directly related to fast myocardial relengthening at end systole.
Relengthening detaches myosin-actin cross-bridges.
Acknowledgments
SOURCES OF FUNDING
Supported by a Scientist Development Grant from the American Heart Association (14SDG20100063) to CSC and the National Institutes of Health R01 HL090749 to KSC and CTSA UL1TR000117.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES/CONFLICTS OF INTEREST
None
References
- 1.Borlaug BA, Jaber WA, Ommen SR, Lam CS, Redfield MM, Nishimura RA. Diastolic relaxation and compliance reserve during dynamic exercise in heart failure with preserved ejection fraction. Heart. 2011;97:964–9. doi: 10.1136/hrt.2010.212787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paulus WJ, Tschope C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, et al. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J. 2007;28:2539–50. doi: 10.1093/eurheartj/ehm037. [DOI] [PubMed] [Google Scholar]
- 3.Biesiadecki BJ, Davis JP, Ziolo MT, Janssen PM. Tri-modal regulation of cardiac muscle relaxation; intracellular calcium decline, thin filament deactivation, and cross-bridge cycling kinetics. Biophys Rev. 2014;6:273–89. doi: 10.1007/s12551-014-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, et al. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997;100:380–9. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babu GJ, Bhupathy P, Petrashevskaya NN, Wang H, Raman S, Wheeler D, et al. Targeted overexpression of sarcolipin in the mouse heart decreases sarcoplasmic reticulum calcium transport and cardiac contractility. J Biol Chem. 2006;281:3972–9. doi: 10.1074/jbc.M508998200. [DOI] [PubMed] [Google Scholar]
- 6.Brutsaert DL, Rademakers FE, Sys SU. Triple control of relaxation: implications in cardiac disease. Circulation. 1984;69:190–6. doi: 10.1161/01.cir.69.1.190. [DOI] [PubMed] [Google Scholar]
- 7.Leite-Moreira AF, Correia-Pinto J, Gillebert TC. Afterload induced changes in myocardial relaxation: a mechanism for diastolic dysfunction. Cardiovasc Res. 1999;43:344–53. doi: 10.1016/s0008-6363(99)00099-1. [DOI] [PubMed] [Google Scholar]
- 8.Leite-Moreira AF, Gillebert TC. Nonuniform course of left ventricular pressure fall and its regulation by load and contractile state. Circulation. 1994;90:2481–91. doi: 10.1161/01.cir.90.5.2481. [DOI] [PubMed] [Google Scholar]
- 9.Brutsaert DL, Sys SU. Relaxation and diastole of the heart. Physiol Rev. 1989;69:1228–315. doi: 10.1152/physrev.1989.69.4.1228. [DOI] [PubMed] [Google Scholar]
- 10.Andersen MJ, Borlaug BA. Heart failure with preserved ejection fraction: current understandings and challenges. Curr Cardiol Rep. 2014;16:501. doi: 10.1007/s11886-014-0501-8. [DOI] [PubMed] [Google Scholar]
- 11.Smith SP, Secomb TW, Hong BD, Moulton MJ. Time-Dependent Regional Myocardial Strains in Patients with Heart Failure with a Preserved Ejection Fraction. Biomed Res Int. 2016;2016:8957307. doi: 10.1155/2016/8957307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brutsaert DL, Housmans PR, Goethals MA. Dual control of relaxation. Its role in the ventricular function in the mammalian heart. Circ Res. 1980;47:637–52. doi: 10.1161/01.res.47.5.637. [DOI] [PubMed] [Google Scholar]
- 13.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766–73. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell KS, Moss RL. SLControl: PC-based data acquisition and analysis for muscle mechanics. Am J Physiol Heart Circ Physiol. 2003;285:H2857–64. doi: 10.1152/ajpheart.00295.2003. [DOI] [PubMed] [Google Scholar]
- 15.Xu Y, Monasky MM, Hiranandani N, Haizlip KM, Billman GE, Janssen PM. Effect of twitch interval duration on the contractile function of subsequent twitches in isolated rat, rabbit, and dog myocardium under physiological conditions. J Appl Physiol (1985) 2011;111:1159–67. doi: 10.1152/japplphysiol.01170.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raff GL, Glantz SA. Volume loading slows left ventricular isovolumic relaxation rate. Evidence of load-dependent relaxation in the intact dog heart. Circ Res. 1981;48:813–24. doi: 10.1161/01.res.48.6.813. [DOI] [PubMed] [Google Scholar]
- 17.Chung CS, Kovacs SJ. Physical determinants of left ventricular isovolumic pressure decline: model prediction with in vivo validation. Am J Physiol Heart Circ Physiol. 2008;294:H1589–96. doi: 10.1152/ajpheart.00990.2007. [DOI] [PubMed] [Google Scholar]
- 18.Campbell KS. Dynamic coupling of regulated binding sites and cycling myosin heads in striated muscle. J Gen Physiol. 2014;143:387–99. doi: 10.1085/jgp.201311078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rice JJ, Wang F, Bers DM, de Tombe PP. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys J. 2008;95:2368–90. doi: 10.1529/biophysj.107.119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung CS, Campbell KS. Temperature and transmural region influence functional measurements in unloaded left ventricular cardiomyocytes. Physiol Rep. 2013;1:e00158. doi: 10.1002/phy2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brutsaert DL, de Clerck NM, Goethals MA, Housmans PR. Relaxation of ventricular cardiac muscle. J Physiol. 1978;283:469–80. doi: 10.1113/jphysiol.1978.sp012513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noble MI. The contribution of blood momentum to left ventricular ejection in the dog. Circ Res. 1968;23:663–70. doi: 10.1161/01.res.23.5.663. [DOI] [PubMed] [Google Scholar]
- 23.Gillebert TC, Sys SU. Physiologic control of relaxation in isolated cardiac muscle and intact left ventricle. In: Gaasch WH, Lewinter MM, editors. Left Ventricular Diastolic Dysfunction and Heart Failure: Lea & Febiger. pp. 1994pp. 25–44. [Google Scholar]
- 24.Hunter WC. Role of myofilaments and calcium handling in left ventricular relaxation. Cardiol Clin. 2000;18:443–57. doi: 10.1016/s0733-8651(05)70155-7. [DOI] [PubMed] [Google Scholar]
- 25.Yadid M, Landesberg A. Stretch increases the force by decreasing cross-bridge weakening rate in the rat cardiac trabeculae. J Mol Cell Cardiol. 2010;49:962–71. doi: 10.1016/j.yjmcc.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Huxley AF, Simmons RM. Rapid ‘give’ and the tension ‘shoulder’ in the relaxation of frog muscle fibres. J Physiol. 1970;210:32P–3P. [PubMed] [Google Scholar]
- 27.Tesi C, Piroddi N, Colomo F, Poggesi C. Relaxation kinetics following sudden Ca(2+) reduction in single myofibrils from skeletal muscle. Biophys J. 2002;83:2142–51. doi: 10.1016/S0006-3495(02)73974-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brunello E, Fusi L, Reconditi M, Linari M, Bianco P, Panine P, et al. Structural changes in myosin motors and filaments during relaxation of skeletal muscle. J Physiol. 2009;587:4509–21. doi: 10.1113/jphysiol.2009.176222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stehle R, Solzin J, Iorga B, Poggesi C. Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflugers Arch. 2009;458:337–57. doi: 10.1007/s00424-008-0630-2. [DOI] [PubMed] [Google Scholar]
- 30.Poggesi C, Tesi C, Stehle R. Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Arch. 2005;449:505–17. doi: 10.1007/s00424-004-1363-5. [DOI] [PubMed] [Google Scholar]
- 31.Janssen PM, Honda H, Koiwa Y, Shirato K. The effect of diastolic vibration on the relaxation of rat papillary muscle. Cardiovasc Res. 1996;32:344–50. doi: 10.1016/0008-6363(96)00094-6. [DOI] [PubMed] [Google Scholar]
- 32.Stehle R, Kruger M, Scherer P, Brixius K, Schwinger RH, Pfitzer G. Isometric force kinetics upon rapid activation and relaxation of mouse, guinea pig and human heart muscle studied on the subcellular myofibrillar level. Basic Res Cardiol. 2002;97(Suppl 1):I127–35. doi: 10.1007/s003950200041. [DOI] [PubMed] [Google Scholar]
- 33.Telley IA, Denoth J, Stussi E, Pfitzer G, Stehle R. Half-sarcomere dynamics in myofibrils during activation and relaxation studied by tracking fluorescent markers. Biophys J. 2006;90:514–30. doi: 10.1529/biophysj.105.070334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Tombe PP, ter Keurs HE. Cardiac muscle mechanics: Sarcomere length matters. J Mol Cell Cardiol. 2016;91:148–50. doi: 10.1016/j.yjmcc.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell KS. Compliance Accelerates Relaxation in Muscle by Allowing Myosin Heads to Move Relative to Actin. Biophys J. 2016;110:661–8. doi: 10.1016/j.bpj.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burns AT, La Gerche A, Prior DL, Macisaac AI. Left ventricular untwisting is an important determinant of early diastolic function. JACC Cardiovasc Imaging. 2009;2:709–16. doi: 10.1016/j.jcmg.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 37.Notomi Y, Popovic ZB, Yamada H, Wallick DW, Martin MG, Oryszak SJ, et al. Ventricular untwisting: a temporal link between left ventricular relaxation and suction. Am J Physiol Heart Circ Physiol. 2008;294:H505–13. doi: 10.1152/ajpheart.00975.2007. [DOI] [PubMed] [Google Scholar]
- 38.Rosen BD, Gerber BL, Edvardsen T, Castillo E, Amado LC, Nasir K, et al. Late systolic onset of regional LV relaxation demonstrated in three-dimensional space by MRI tissue tagging. Am J Physiol Heart Circ Physiol. 2004;287:H1740–6. doi: 10.1152/ajpheart.00080.2004. [DOI] [PubMed] [Google Scholar]
- 39.Weiner RB, Weyman AE, Khan AM, Reingold JS, Chen-Tournoux AA, Scherrer-Crosbie M, et al. Preload dependency of left ventricular torsion: the impact of normal saline infusion. Circ Cardiovasc Imaging. 2010;3:672–8. doi: 10.1161/CIRCIMAGING.109.932921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haugaa KH, Amlie JP, Berge KE, Leren TP, Smiseth OA, Edvardsen T. Transmural differences in myocardial contraction in long-QT syndrome: mechanical consequences of ion channel dysfunction. Circulation. 2010;122:1355–63. doi: 10.1161/CIRCULATIONAHA.110.960377. [DOI] [PubMed] [Google Scholar]
- 41.Saito M, Okayama H, Yoshii T, Hiasa G, Sumimoto T, Inaba S, et al. The differences in left ventricular torsional behavior between patients with hypertrophic cardiomyopathy and hypertensive heart disease. Int J Cardiol. 2011;150:301–6. doi: 10.1016/j.ijcard.2010.04.040. [DOI] [PubMed] [Google Scholar]
- 42.Helmes M, Lim CC, Liao R, Bharti A, Cui L, Sawyer DB. Titin determines the Frank-Starling relation in early diastole. J Gen Physiol. 2003;121:97–110. doi: 10.1085/jgp.20028652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson TF, Factor SM, Sonnenblick EH. The heart as a suction pump. Sci Am. 1986;254:84–91. doi: 10.1038/scientificamerican0686-84. [DOI] [PubMed] [Google Scholar]
- 44.Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure–abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med. 2004;350:1953–9. doi: 10.1056/NEJMoa032566. [DOI] [PubMed] [Google Scholar]
- 45.Borlaug BA, Kass DA. Invasive hemodynamic assessment in heart failure. Cardiol Clin. 2011;29:269–80. doi: 10.1016/j.ccl.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Chung CS, Ajo DM, Kovacs SJ. Isovolumic pressure-to-early rapid filling decay rate relation: model-based derivation and validation via simultaneous catheterization echocardiography. J Appl Physiol (1985) 2006;100:528–34. doi: 10.1152/japplphysiol.00617.2005. [DOI] [PubMed] [Google Scholar]
- 47.De Tombe PP, Little WC. Inotropic effects of ejection are myocardial properties. Am J Physiol. 1994;266:H1202–13. doi: 10.1152/ajpheart.1994.266.3.H1202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.