

Abstract
Objective:
The objective was to evaluate the safety and efficacy of TV-45070 ointment, as a treatment for postherpetic neuralgia, and to explore the response in patients with the Nav1.7 R1150W gain-of-function polymorphism.
Materials and Methods:
This was a randomized, placebo-controlled, 2-period, 2-treatment crossover trial. Patients with postherpetic neuralgia with moderate or greater pain received TV-45070 and placebo ointments, each applied twice daily for 3 weeks. The primary efficacy measure was the difference in change in mean daily pain score from baseline compared with the last week of placebo and active treatment. Secondary endpoints included responder rate analyses and a further exploratory analysis of response in carriers of the Nav1.7 R1150W polymorphism was conducted.
Results:
Seventy patients were enrolled and 54 completed the study. TV-45070 was safe and well tolerated. No statistical difference was observed between treatments for the primary endpoint. However, the proportion of patients with ≥50% reduction in mean pain scores at week 3 was greater on TV-45070 than on placebo (26.8% vs. 10.7%, P=0.0039). Similarly, a greater proportion of patients on TV-45070 had a ≥30% reduction in mean pain scores at week 3 (39.3% on TV-45070 vs. 23.2% on placebo, P=0.0784). Of note, 63% of patients with the R1150W polymorphism versus 35% of wild-type carriers had a ≥30% reduction in mean pain score on TV-45070 at week 3 (no inferential analysis performed).
Conclusions:
The 50% responder analysis suggests a subpopulation may exist with a more marked analgesic response to TV-45070.The trend toward a larger proportion of responders within Nav1.7 R1150W carriers warrants further investigation.
Key Words: postherpetic neuralgia, neuropathic pain, R1150W, analgesic, Nav1.7
It is well recognized that existing analgesic drugs provide clinical benefit only for a proportion of patients.1 This suggests that subpopulations of patients with pain might exist where response to a given pharmacological intervention might be mechanism based. With an ever improving understanding of the molecular mechanisms underlying pain signaling in humans, studying mechanism-based pharmacodynamic response to analgesics may help to better explain responder subsets in clinical trials and may also aid in developing more targeted therapeutic approaches.
The sodium channel Nav1.7 is a target that appears to play an essential role in human pain sensing.2 Congenital indifference to pain, a rare inherited disorder of human analgesia is caused by deficiency of Nav1.7.3,4 Congenital indifference to pain patients and mouse knockouts of Nav1.7 are both analgesic to neuropathic and nociceptive stimuli.3–6 Gain-of-function mutations increasing the activity of Nav1.7 underlie paroxysmal extreme pain disorder and a subset of patients with inherited erythromelalgia (IEM), both disorders of spontaneous or easily evoked often severe pain.7,8 In addition, a common variant in Nav1.7, where an arginine is replaced with a tryptophan at position 1150 (R1150W) in the protein sequence is present in 15% to 30% of the population and is also capable of causing increased activity of Nav1.7. This Nav1.7 variant has been reported to be associated with increased sensitivity to pain.9 The validation for the Nav1.7 target provided by multiple human genetic studies, lends support for inhibition of this target as a strategy for analgesic development.
TV-45070 (formally XEN402) is a novel potent inhibitor of the sodium channel Nav1.7 and other Nav channels expressed in the peripheral nervous system.10 We have previously reported evidence of analgesic activity for the active ingredient of TV-45070 when administered orally in patients with IEM.11 Each of these IEM patients had inherited a causal gain-of-function mutation in Nav1.7 suggesting the analgesic effect of TV-45070 was by its inhibition of Nav1.7. To maximize inhibition of Navs locally in skin and subcutaneous tissue and minimize systemic adverse events, topical TV-45070 was developed to depot in the skin and underlying tissue while maintaining low plasma concentrations.
The current proof-of-concept study was designed to evaluate the safety and efficacy of topically applied TV-45070 as a treatment for postherpetic neuralgia (PHN). In addition, we explored the therapeutic response to TV-45070 among carriers of the common R1150W variant.
MATERIALS AND METHODS
Study Design
This randomized, double blind, placebo-controlled, 2-period, 2-treatment crossover trial evaluated the safety and efficacy of 21 days of twice daily (bid) treatment with TV-45070 ointment in PHN patients. The study was conducted between August, 2010 and March, 2011 at 24 sites in the United States. The study included 2, 1-week patient-blind placebo run-in periods (before each treatment period), and 2, 3-week double-blind treatment periods (Fig. 1). A 1-week washout separated treatment period 1 (TP1) and the second placebo run-in period, effectively separating each randomized treatment period by 2 weeks.
FIGURE 1.
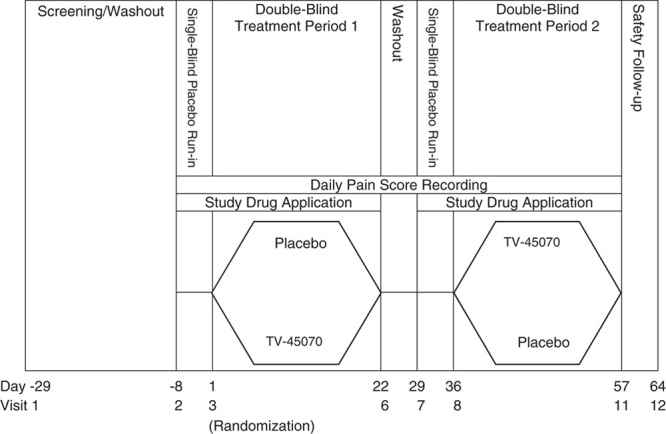
Study design. Screening/washout lasted up to 3 weeks (day-29 to day-8). The first single-blind, placebo run-in period was 1 week in duration (day-7 to day-1). At randomization (visit 3), patients were randomized in a crossover design to 1 of 2 treatment sequences: TV-45070/placebo or placebo/TV-45070. The duration of TP1 was 3 weeks. During the 1-week between-treatment washout period (between visits 6 and 7), study medication was not applied, but patients continued to record pain scores using the interactive voice response system. The second single-blind, placebo run-in period was 1 week in duration (day-29 to day-35). In TP2, patients received the medication that was not received during TP1. The duration of TP2 was 3 weeks. One week after the end of TP2, patients returned for a follow-up visit (visit 12). TP1 indicates treatment period 1; TP2, treatment period 2.
Patients with mean daily pain scores of ≥4 (using an 11-point Numerical Rating Scale) for at least 4 days during the initial placebo 7 day run-in period were randomized in a 1:1 ratio through an interactive voice response system to 1 of 2 treatment sequences (TV-45070/placebo or placebo/TV-45070).
Throughout the study (including washout), patients recorded pain scores 4 times each day (upon waking, 12 noon±1 h, 4 PM±1 h, and 8 PM±1 h) using an 11-point Numerical Rating Scale, and reported them through telephone to the interactive voice response system.
After treatment period 1 (TP1) and the 1-week washout, patients entered a second patient-blinded, placebo run-in period. Unlike TP1, patients were not required to report a minimum mean daily pain score before entering treatment period 2.
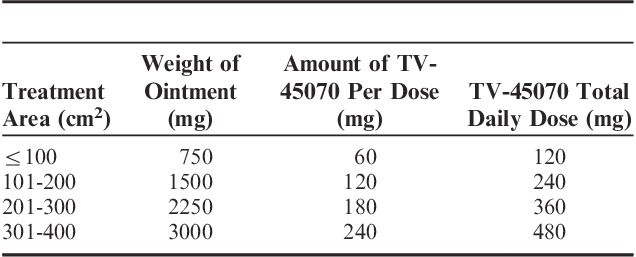
All patients applied ∼7.5 μL of ointment per cm2 of affected skin, covering the entire painful area (up to a maximum of 400 cm2). The actual dose applied varied according to the size of the treatment area (determined by the investigator). Patients were given 1 of 4 area-specific dosing cards, indicating the amount of ointment to be applied (60, 120, 180, or 240 mg of TV-45070 bid, Table 1). Placebo was a vehicle-only ointment identical in appearance to TV-45070 ointment. Patients were allowed to rescue with acetaminophen (up to 4000 mg/d) and recorded any use in a diary.
TABLE 1.
Doses Administered by Size of Treatment Area
Standard Protocol Approvals, Registrations, and Patient Consents
A central independent review board approved the study protocol and amendments. All participants provided written informed consent. The study was performed in accordance with the Declaration of Helsinki and International Conference on Harmonization Guideline on Good Clinical Practice and is registered with ClinicalTrials.gov (NCT01195636).
Study Population
Males and females of nonchildbearing potential, aged 18 to 80 years, with a clinical diagnosis of PHN, persistent pain for >6 months from the appearance of herpes zoster rash and intact skin over the treatment area were eligible for inclusion.
Exclusion criteria were patients with systemic disease that would have put them at an additional risk or limited their ability to participate, PHN on the face or in proximity to mucous membranes, creatinine clearance <30 mL/min, history of serious mental or psychiatric illness, known sensitivity to topical products, active herpes zoster lesions or dermatitis, other severe or chronic pain conditions, or patients who had participated in >1 topical pain study and/or >3 PHN studies.
Patients were also excluded if they had received a local anesthetic in 2 weeks before randomization, nerve blocks within 30 days of randomization, Qutenza or other capsaicin preparations in the 90 days before screening, or if they used opioids, local prescription or nonprescription therapy (lidocaine patch, transcutaneous electrical nerve stimulation, etc.) and were unable to washout for the study.
Patients were permitted to remain on stable doses of up to 2 of the following: gabapentin, pregabalin, duloxetine, or amitriptyline. Stable doses were defined as remaining unchanged for the 4 weeks before the start of the first placebo run-in period, with no planned doses changes during the study. Patients were to be excluded if doses were changed during the study. This did not occur for any patients.
Assessments
Safety assessments included adverse event reporting, laboratory measurements (serum chemistry, hematology, and urinalysis), 12-lead electrocardiograms, vital signs, and physical examinations (including assessments for local irritation and numbness were performed at regular clinic visits. Pharmacokinetic plasma samples were collected at each weekly visit to determine the extent of systemic exposure of TV-45070 using a validated bioanalytical method.
Efficacy assessments included patient reported pain intensity (4 times per day) and daily sleep interference scores (DSIS, an 11-point Numerical Rating Scale where 0=pain does not interfere with sleep and 10=completely interferes). The 4 daily pain intensity scores were combined to generate a mean daily pain score. The Neuropathic Pain Symptom Inventory (NPSI, a 12-point questionnaire to evaluate different neuropathic pain symptoms) and patient global impression of change (PGIC) were also assessed at clinic visits. As an exploratory endpoint, for patients who consented to genetic testing, a DNA sample was collected and genotyped for Nav1.7 R1150W carrier status.
Statistical Analyses
The sample size was estimated to provide 80% power to detect a 1.0-point difference in mean pain intensity. The calculation assumed a significance level of 0.05 and a within-patient SD of 2.25. The treatment effect and SD were selected based on review of other similar PHN studies.12–14 An interim analysis was performed after 21 patients completed the study to determine if the estimated SD was appropriate and if the sample size required adjustment. As a result, the significance level for the final analysis of the primary variable was adjusted to α=0.049. No changes were made to the sample size, or any other analyses.
Safety variables including pharmacokinetic exposure measures were summarized using descriptive statistics for continuous variables and frequency and percentage for categorical variables. No formal hypothesis testing was performed to compare safety parameters.
The primary efficacy endpoint was the difference in mean change from baseline in mean daily pain score between the last week (week 3) of TV-45070 treatment and the last week of placebo treatment. Baseline for each treatment period was the average of the 7 mean daily pain measurements in the preceding patient-blind placebo run-in period. The mean daily pain scores for weeks 1, 2, and 3 of each treatment period were similarly defined. If the third week in a treatment period had <4 days of recordings, the last observation carried forward (LOCF) algorithm was used (ie, the average of the last 4 d with at least 1 pain score). The hypothesis that the mean change in mean daily pain scores from baseline would differ between TV-45070 and placebo was tested using a mixed effects analysis of covariance model, with period and sequence as fixed effects, patient within treatment sequence as a random effect, and the baseline pain score as a covariate. Secondary efficacy analyses included:
Number of patients with ≥30% and ≥50% improvement in mean pain score from baseline.
Changes in mean daily pain scores from baseline to each week of treatment and over the entire treatment period.
Number of patients with a mean improvement from baseline of 1, 2, and 3 or more points for each week of treatment and overall.
Number of patients using rescue medication per treatment.
Changes in DSIS, NPSI, and PGIC scores from baseline to the end of treatment.
Treatment groups were compared using McNemar test when appropriate. Continuous variables were summarized using similar methods as for the primary efficacy variable. For categorical variables the numbers and percentages of patients in each treatment group in each category were summarized.
Exploratory analyses included examining response to treatment in the subgroup of patients who were carriers of the Nav1.7 R1150W polymorphism.
RESULTS
Patient Demographics and Disease Characteristics
In total, 129 patients were screened and 109 (84.5%) entered the first placebo run-in period, 70 patients were randomized: 35 (50.0%) to each treatment sequence. Fifty-four (77.1%) patients completed the study and 16 (22.9%) discontinued (Fig. 2). Reasons for discontinuation included adverse event (8 [11.4%] patients), withdrawal of consent (6 [8.6%] patients), protocol violation (1 [1.4%] patient), and lost to follow-up (1 [1.4%] patient).
FIGURE 2.
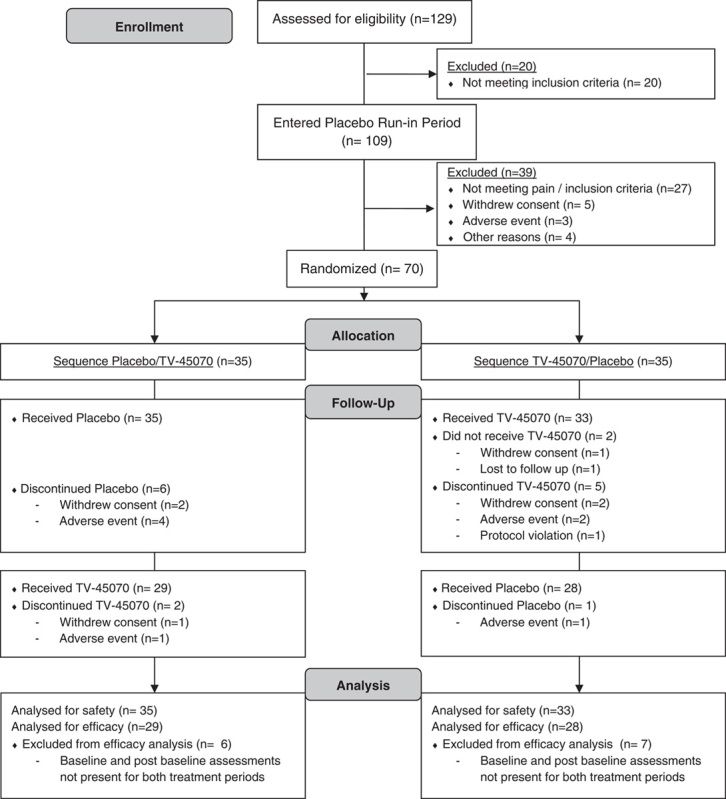
Consort flow diagram.
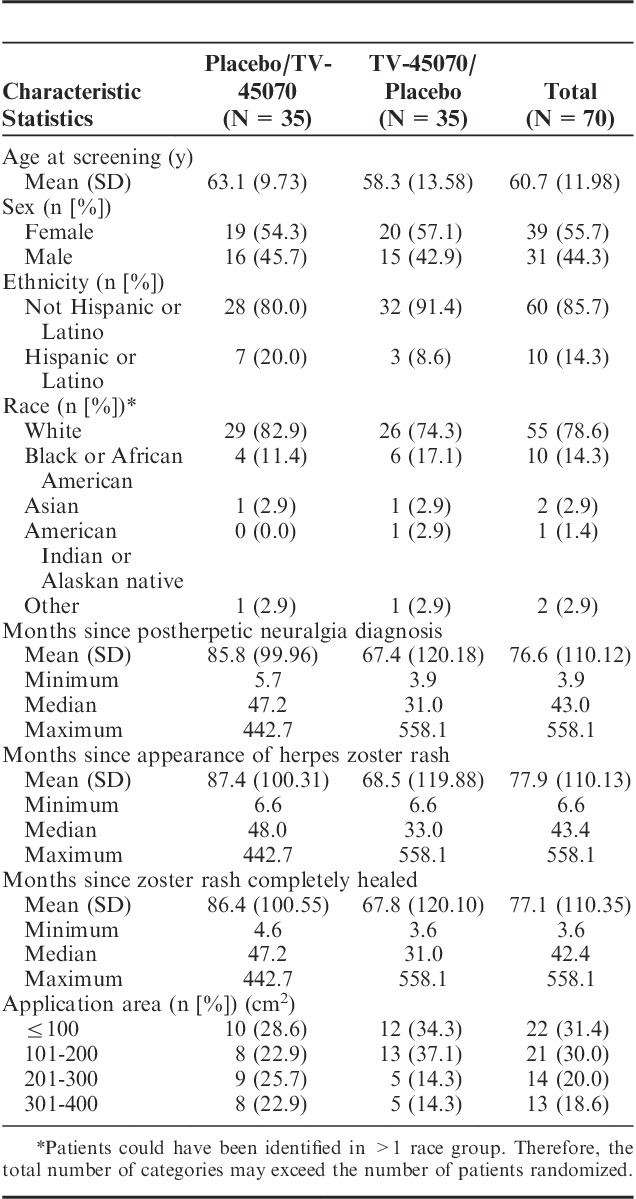
Patient demographics and disease characteristics are provided in Table 2. The TV-45070/placebo sequence had a slightly lower mean age than the placebo/TV-45070 sequence (58.3 vs. 63.1 y). The treatment sequences were otherwise comparable with respect to demographic characteristics. Overall, the mean number of months since herpes zoster rash healing was 77.1 months (range, 3.6 to 558.1 mo). The majority of patients (80.0%) had not previously participated in PHN trials. Patients were relatively evenly distributed between the 4 categories of treatment area; 61.4% of patients were allocated to the 2 smaller areas of application (ie, <201 cm2). Seventeen (24.3%) patients remained on one or more of the permitted PHN medications during the study, (15 [21.4%] patients remained on gabapentin and 2 [2.9%] on pregabalin). Thirteen (76.5%) of these patients reported receiving “a little” or “some” pain relief from these medications when asked at screening.
TABLE 2.
Patient Demographics and Disease Characteristics by Treatment Sequence for All Randomized Patients
Safety
The safety population (N=68) comprised patients who received at least 1 application of study medication. There were no deaths. There were 2 serious adverse events (SAEs), neither considered related to study medication: an SAE of tooth abscess occurred during placebo treatment, and an SAE of worsening of coronary artery disease (CAD) occurred on the first day of TV-45070 treatment. This cardiac event occurred in a patient with well-documented CAD (including angina and a prior myocardial infarction). The patient had previously undergone stenting and cardiac catheterization. Angiography performed at the time of the SAE revealed coronary artery occlusion that was managed with angioplasty and stent placement to the left anterior descending coronary artery. The patient was withdrawn from the study.
There were no clinically meaningful changes in blood chemistry, hemodynamic parameters, electrocardiograms, vital signs, or in physical examination findings.
Treatment emergent adverse events (TEAEs) are summarized in Table 3. The incidence of TEAEs was similar between TV-45070 and placebo (53.2% and 50.8%, respectively). However, study medication-related TEAEs were more frequent on placebo than TV-45070 (30.2% vs. 17.7%). Generally, application site related TEAEs were more common on placebo treatment, for example, application site pain and pruritus (Table 3). In addition, a very low incidence of dizziness was reported and no somnolence was observed (Table 3).
TABLE 3.
Summary and Incidence of the Most Frequently Reported Treatment Emergent Adverse Events—Safety Population
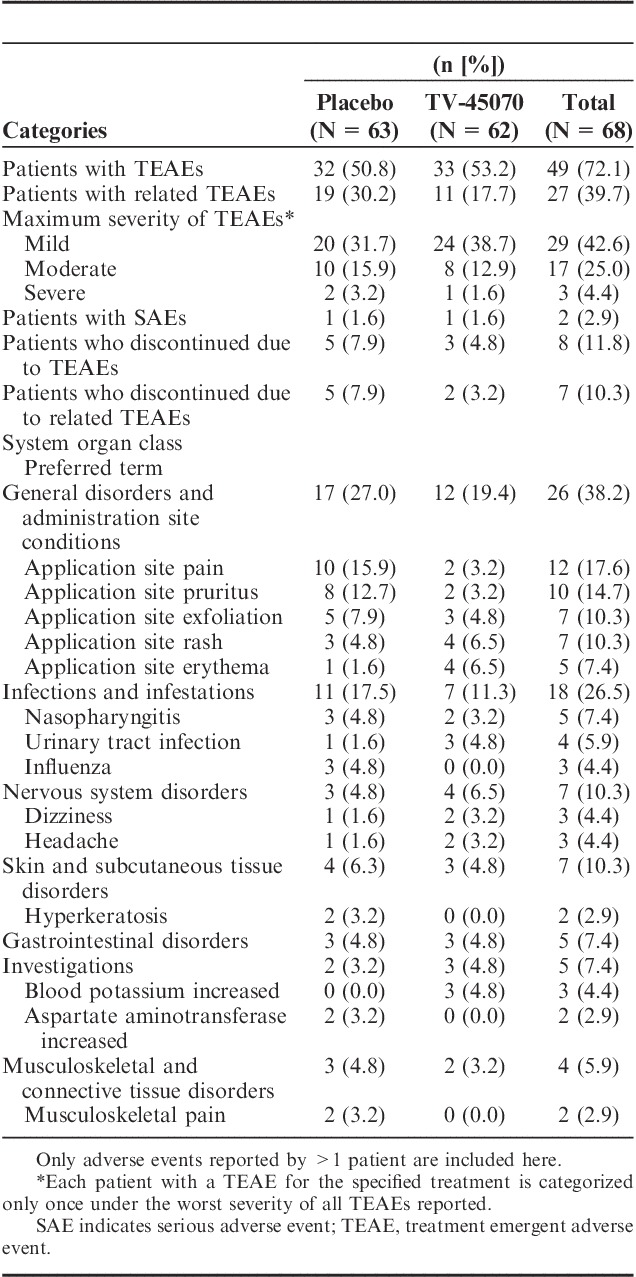
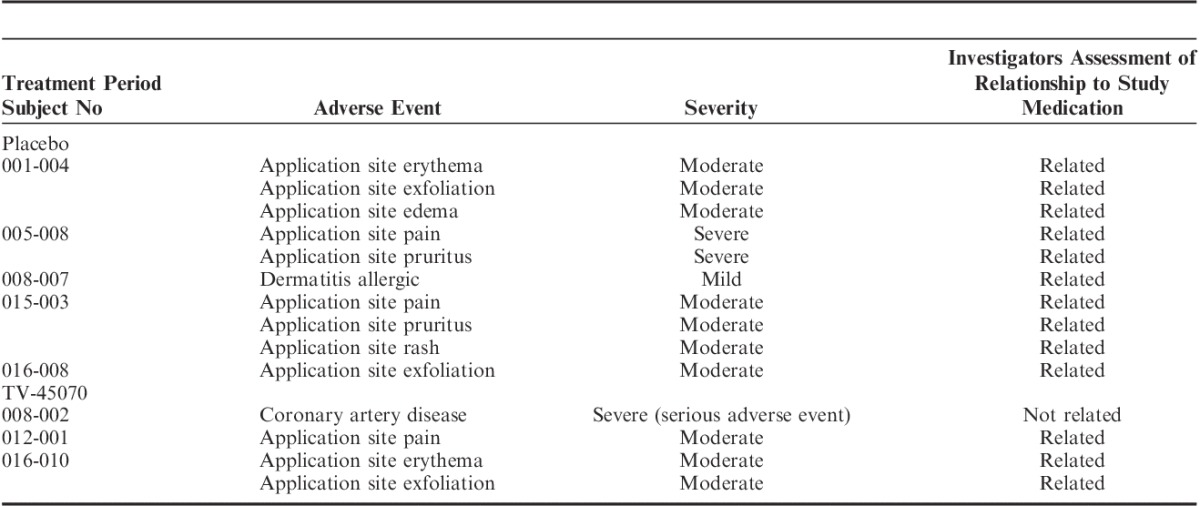
Eight patients discontinued because of a TEAE. For 7 of these 8 patients, the events leading to discontinuation were local skin reactions (5 during placebo and 2 during TV-45070; Table 4) and the other discontinuation was the patient with the worsening CAD.
TABLE 4.
Adverse Events Leading to Discontinuation
Plasma Drug Concentration
Overall, systemic exposure was low, ranging from <0.1 ng/mL (lower limit of quantitation) to 13.7 ng/mL. The mean maximum plasma concentration (Cmax) was comparable between treatment sequences: the mean Cmax for the TV-45070/placebo sequence was 2.1 ng/mL and the mean Cmax for the placebo/TV-45070 treatment sequence was 2.2 ng/mL. Similar findings of low plasma levels have been observed with application of topical TV-45070 in a prior human phase 1 study (unpublished) which also showed high skin concentration (mean concentration 12,000 ng/g).
Efficacy
The efficacy evaluable population (N=57) comprised all patients with baseline and postbaseline efficacy data in both treatment periods. This population was used for all planned efficacy analyses.
The primary efficacy analysis did not show a difference between treatment groups. The least squares (LS) mean change in mean daily pain score from baseline to week 3 with LOCF was −0.97 with placebo and −0.94 with TV-45070 (difference in change from baseline LS means between treatments=0.03, 95% confidence interval, −0.38, 0.43; P=0.8885).
Analysis of the change in mean daily pain scores by each week of treatment did not result in significant differences between TV-45070 and placebo and there were no significant differences in the numbers or percentages of patients with a mean improvement from baseline of ≥1, ≥2, and ≥3 points for each week of treatment. However, a significant difference in responder rates during TV-45070 treatment was observed. More patients achieved ≥50% improvement with TV-45070 (n=15, 26.8%) than with placebo (n=6, 10.7%) at week 3 with LOCF (P=0.0039). A similar trend was observed in the proportion of patients achieving ≥30% improvement at week 3 with LOCF, with 22 (39.3%) patients achieving ≥30% improvement on TV-45070 versus 13 (23.2%) patients on placebo (P=0.0784). Over the entire 3-week treatment period, the difference between treatments in the proportion of patients achieving ≥30% improvement was statistically significant (P=0.0213), with 19 (33.9%) patients achieving ≥30% improvement on TV-45070 versus 9 (16.1%) patients on placebo (Table 5). The entire distribution of percent change from baseline scores is illustrated in Figure 3. The separation between treatments increased over time—a week-to-week improvement in the percentage of responders on TV-45070 versus placebo was observed (Table 5).
TABLE 5.
Proportion of Patients Achieving at Least 30% or 50% Pain Improvements—Efficacy Evaluable Population
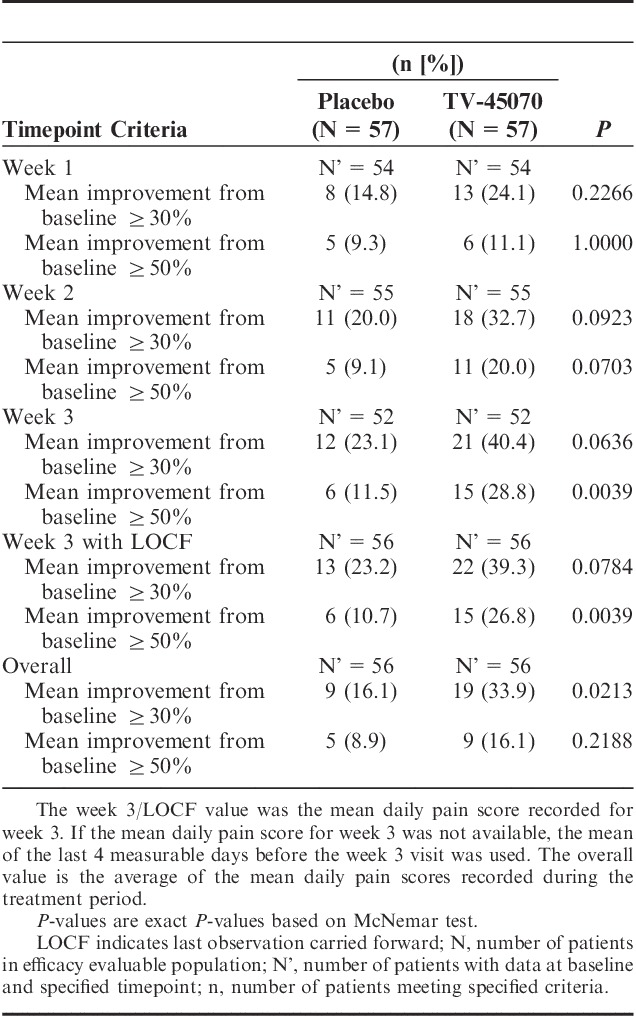
FIGURE 3.
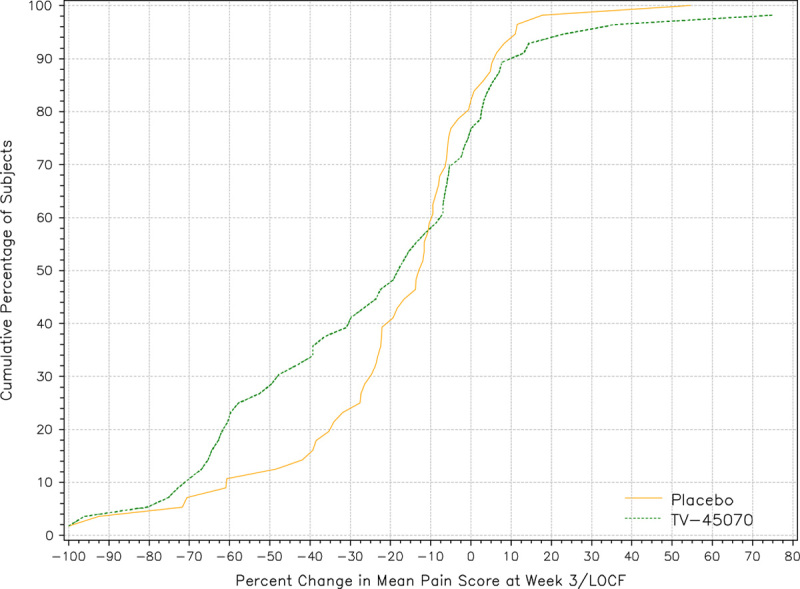
Cumulative percentage of patients versus percent change in mean daily pain score at week 3/LOCF. The figure displays the cumulative percentage of patients versus percent change in mean daily pain score at week 3 with LOCF for the efficacy evaluable population. There is a noticeable separation between the 2 curves, indicating that more patients experienced a 10% to 80% reduction in mean daily pain scores at the end of TV-45070 treatment compared with placebo treatment. For example, approximately 40% of patients reported a 30% or greater reduction in pain when on TV-45070 treatment, compared with just 24% of patients during placebo treatment. LOCF indicates last observation carried forward.
Overall, the mean total number of rescue medication pills taken was slightly lower with TV-45070 treatment compared with placebo treatment (24.8 vs. 29.2 pills, not statistically significant).
Analyses of DSIS, NPSI, and PGIC did not demonstrate any significant differences between treatments. The LS mean change in DSIS score from baseline to week 3 with LOCF was −0.84 on placebo and −0.81 on TV-45070. The treatment difference was not statistically significant (P=0.8801).
However, a statistically significant increase, favoring TV-45070, in the proportion of patients with ≥30% and ≥50% improvement in sleep interference scores in week 3 was observed (P<0.05). The mean change in NPSI score from baseline was −6.1 on placebo and −6.7 on TV-45070.
For PGIC, patients reported their overall status during each treatment period as follows: “very much improved” (14.3% TV-45070: 8.9% placebo), “much improved” (28.6% TV-45070: 28.6% placebo), “minimally improved” (17.9% TV-45070: 33.9% placebo), “no change” (25.0% TV-45070: 21.4% placebo), “minimally worse” (8.9% TV-45070: 5.4% placebo), “much worse” (5.4% TV-45070: 1.8% placebo), or “very much worse” (0% TV-45070: 0% placebo).
One randomized patient discontinued due to lack of efficacy (in treatment period 2 while on placebo treatment due to worsening of pain at the site of PHN).
Responder Rates by Nav1.7 R1150W Genotype
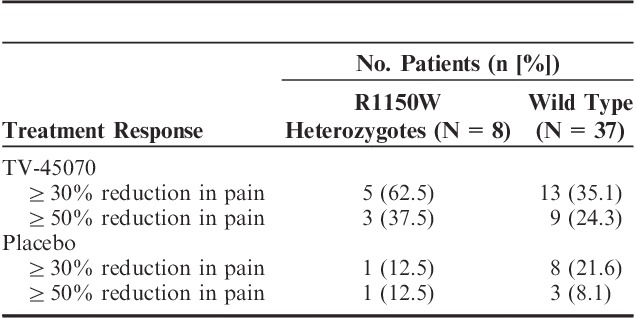
Fifty-two patients provided a DNA sample for genetic analysis. Forty-five of these patients were included in the efficacy evaluable population and contributed data to the exploratory R1150W subgroup analyses. Thirty-seven patients did not have the R1140W genotype, whereas 8 were heterozygous carriers of the Nav1.7 R1150W polymorphism. Given the small number of patients contributing data, no inferential analyses were performed and results were summarized.
Sixty-three percent of R1150W carriers achieved ≥30% reduction in pain scores on TV-45070 treatment compared with 35% of wild-type patients, and 38% of R1150W carriers achieved ≥50% reduction in pain scores compared with 24% of wild-type patients (Table 6). In addition, the R1150W carriers showed a greater reduction in mean pain after 3 weeks of TV-45070 treatment compared with their response to placebo treatment (mean treatment difference=0.78 points).
TABLE 6.
R1150W Mutation Status and Response to TV-45070
DISCUSSION
We conducted a proof-of-concept trial in 70 patients with PHN using TV-45070, a topical novel potent inhibitor of Nav1.7 and additional Navs.10 Overall, TV-45070 was safe and well tolerated and unlike many commonly used orally active medications used to treat neuropathic pain, topically applied TV-45070 demonstrated an absence of drug-related central nervous system (CNS) TEAEs. This was an expected property of the product due to the low plasma concentrations (mean Cmax was ∼2.1 ng/mL with a maximal individual Cmax of ∼14 ng/mL) relative to plasma levels previously shown to mediate CNS effects (data not shown). The most common adverse events were local skin reactions where application site pain and pruritus were observed more frequently on placebo than TV-45070 treatment, suggesting that the TV-45070 API itself does not primarily cause dermal irritation. The local skin reactions were mostly mild (66%), with occasional moderate events (28%). These resolved spontaneously without intervention. Eight patients terminated because of an adverse event (5 during placebo treatment and 3 during TV-45070 treatment). Neither of the 2 SAEs reported in this trial were considered related to study treatment.
The PHN population studied was consistent with the typical demographic profile for other reported PHN trials with one major exception; the mean duration since diagnosis was substantially higher (76.6 mo) than reported in other PHN studies (∼30 mo).12,13,15 Although some studies have been able to demonstrate efficacy in PHN patients who have failed prior therapies, in this study the longer duration of disease suggests a more refractory population that may have compromised the ability to demonstrate an overall treatment difference in this study.
Nevertheless, despite a potentially more refractory study population, while the primary efficacy analysis did not show differences between the treatment arms, statistical significance was observed for responder rate analyses. A statistically higher percentage of patients had a ≥50% improvement in pain during TV-45070 treatment compared with placebo and a trend to ≥30% improvement in pain during TV-45070 treatment compared with placebo was observed.
This positive responder rate observation suggests that a subpopulation of PHN patients exists that is more likely to have an analgesic effect and, further suggests the possibility exists that this response to topical TV-45070 could be mechanism based. Although the sample size is very small and definitive conclusions cannot be drawn, PHN patients who were carriers of the common gain-of-function R1150W polymorphism appeared more likely to experience a more robust response to TV-45070. An increase in the proportion of responders for both ≥30% and ≥50% improvement in pain was observed for heterozygous carriers compared with wild type. Interestingly, the placebo response was reduced in the R1150W carriers, further widening the effect size seen in this carrier subpopulation.
Although the Nav1.7 R1150W polymorphism has been associated with an increase in pain perception for a variety of painful disorders including both nociceptive and neuropathic pain as well as experimentally induced heat pain (mean increase in Visual Analog Scale of 2.0 for carriers vs. wild type),9 these data have not yet been replicated.16 Using in vitro electrophysiological testing in transfected dorsal root ganglion cells, the R1150W variant channel has previously been shown to demonstrate gain-of-function and hyperactivity.17 We postulate that enhanced pain perception mediated through an increased activity of the R1150W polymorphism may render patients more susceptible to pharmacological intervention by Nav1.7 inhibitors such as TV-45070 compared with their wild-type counterparts. These preliminary findings reported herein, when combined with the previously reported efficacy of the active ingredient of TV-45070 in IEM patients where the mutations in the SCN9A gene are known to cause a gain-of-function of Nav1.7 channel, support the continued evaluation of the R1150W polymorphism in larger clinical studies in common disorders of pain.
Human genetics has uncovered the vital importance of Nav1.7 in pain perception illuminating a critical mechanism for analgesic development.3,4,18 Here, we show that topically applied TV-45070, which has potent Nav1.7 inhibition and is currently in clinical development, was safe, well tolerated, and devoid of drug-related CNS or cardiac toxicity. Although a mean difference between treatment groups was not detected, significant improvements were demonstrated with TV-45070 in a subset of PHN patients. Furthermore, based on a small analysis subpopulation, heterozygous carriers of the R1150W polymorphism appeared more likely to report a clinically meaningful response than their wild-type counterparts. These results support the potential clinical utility of pharmacological inhibition of Nav1.7 for the treatment of peripheral neuropathic pain. Given the favorable safety profile coupled with demonstrated efficacy in this PHN study, these data warrant further development of topical TV-45070 in larger, longer, parallel-design studies of patients with PHN or other peripheral neuropathic pain conditions.
ACKNOWLEDGMENTS
The authors would like to thank the following co-investigators and all of the site teams and Medpace Inc. personnel involved in conducting this study: Maxwell L. Axler, MD (Clinical Trial Network, Texas); Lisa M. Cohen, DO (Suncoast Clinical Research, Inc., Florida); Barry J. Cutler, MD (Neurology Clinical Research, Inc., Florida); Mark A. Fisher, MD (Lynn Health Science Institute, Oklahoma); James E. Greenwald, MD, PhD (Medex Healthcare Research, Inc., Missouri); Robert M. Griffin Jr., DO (Clinical Research Center of Reading, LLP, Pennsylvania); Daniel M. Gruener, MD (CRI Worldwide, LLC, Pennsylvania); John D. Hudson, MD (FutureSearch Clinical Trials of Neurology, Texas); Thomas Hurd, MD (Drug Studies America, Georgia); Yekaterina Khronusova, MD (Advanced Biomedical Research of America, Nevada); Paula J. Lane, MD (Lovelace Scientific Resources, Inc., New Mexico); W. Alvin McElveen, MD (Bradenton Research Center, Inc., Florida); Sathish R. Modugu, MD (Drug Trials America, New York); John P. Nardandrea Jr., MD (Renstar Medical Research, Florida); Marshall L. Nash, MD FAHA CPI (Dekalb Neurology Associates, LLC, Georgia); John F. Peppin, DO FACP (The Pain Treatment Center of the Bluegrass, Kentucky); Edward B. Portnoy, MD (Westlake Medical Research, California); Jeffrey A. Potts, MD (Great Lakes Research Group, Inc., Michigan); Ronald M. Pucillo, MD (Breco Research, Ltd, Texas); Marina Raikhel, MD FAAFP (Torrance Clinical Research, California); Gladstone A. Sellers, MD (Mount Vernon Clinical Research, LLC, Georgia); Mary L. Stedman, MD (Stedman Clinical Trials, LLC, Florida); Ashley Tunkle, MD (Anchor Research Center, Florida); Mark C. Wood, MD (Clopton Clinic, Arizona).
Footnotes
This was an industry-sponsored study (Xenon Pharmaceuticals Inc.). Several authors are current or former employees of Xenon Pharmaceuticals Inc. (N.P., R.N., J.N., K.J.W.P., R.P.S., S.N.P., Y.P.G.).
REFERENCES
- 1.Dworkin RH, Turk DC. Accelerating the development of improved analgesic treatments: the ACTION public-private partnership. Pain Med. 2011;12(suppl 3):S109–S117. [DOI] [PubMed] [Google Scholar]
- 2.Dib-Hajj SD, Yang Y, Black JA, et al. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62. [DOI] [PubMed] [Google Scholar]
- 3.Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldberg YP, MacFarlane J, MacDonald ML, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71:311–319. [DOI] [PubMed] [Google Scholar]
- 5.Gingras J, Smith S, Matson DJ, et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One. 2014;9:e105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minett MS, Nassar MA, Clark AK, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. 2012;3:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41:171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fertleman CR, Baker MD, Parker KA, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. [DOI] [PubMed] [Google Scholar]
- 9.Reimann F, Cox JJ, Belfer I, et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc Natl Acad Sci U S A. 2010;107:5148–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg YP, Cohen CJ, Namdari R, et al. Letter to the editor. Pain. 2014;155:837–838. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg YP, Price N, Namdari R, et al. Treatment of Na(v)1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain. 2012;153:80–85. [DOI] [PubMed] [Google Scholar]
- 12.Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic neuralgia. Neurology. 1998;50:1837–1841. [DOI] [PubMed] [Google Scholar]
- 13.Rowbotham M, Harden N, Stacey B, et al. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA. 1998;280:1837–1842. [DOI] [PubMed] [Google Scholar]
- 14.Wu CL, Agarwal S, Tella PK, et al. Morphine versus mexiletine for treatment of postamputation pain: a randomized, placebo-controlled, crossover trial. Anesthesiology. 2008;109:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dworkin RH, Corbin AE, Young JP, Jr, et al. Pregabalin for the treatment of postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2003;60:1274–1283. [DOI] [PubMed] [Google Scholar]
- 16.Valdes AM, Arden NK, Vaughn FL, et al. Role of the Nav1.7 R1150W amino acid change in susceptibility to symptomatic knee osteoarthritis and multiple regional pain. Arthritis Care Res (Hoboken). 2011;63:440–444. [DOI] [PubMed] [Google Scholar]
- 17.Estacion M, Harty TP, Choi JS, et al. A sodium channel gene SCN9A polymorphism that increases nociceptor excitability. Ann Neurol. 2009;66:862–866. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg YP, Pimstone SN, Namdari R, et al. Human Mendelian pain disorders: a key to discovery and validation of novel analgesics. Clin Genet. 2012;82:367–373. [DOI] [PubMed] [Google Scholar]