

Abstract
Background and aims
Mutations in the 5′-nucleotidase ecto (NT5E) gene that encodes CD73, a nucleotidase that converts AMP to adenosine, are linked to arterial calcification. However, the role of purinergic receptor signaling in the pathology of intimal calcification is not well understood. In this study, we examined whether extracellular nucleotides acting via P2Y2 receptor (P2Y2R) modulate arterial intimal calcification, a condition highly correlated with cardiovascular morbidity.
Methods
Apolipoprotein E, P2Y2R double knockout mice (ApoE−/−P2Y2R−/−) were used to determine the effect of P2Y2R deficiency on vascular calcification in vivo. Vascular smooth muscle cells (VSMC) isolated from P2Y2R−/− mice grown in high phosphate medium were used to assess the role of P2Y2R in the conversion of VSMC into osteoblasts. Luciferase-reporter assays were used to assess the effect of P2Y2R on the transcriptional activity of Runx2.
Results
P2Y2R deficiency in ApoE−/− mice caused extensive intimal calcification despite a significant reduction in atherosclerosis and macrophage plaque content. The ectoenzyme apyrase that degrades nucleoside di- and triphosphates accelerated high phosphate-induced calcium deposition in cultured VSMC. Expression of P2Y2R inhibits calcification in vitro inhibited the osteoblastic trans-differentiation of VSMC. Mechanistically, expression of P2Y2R inhibited Runx2 transcriptional activation of an osteocalcin promoter driven luciferase reporter gene.
Conclusions
This study reveals a role for vascular P2Y2R as an inhibitor of arterial intimal calcification and provides a new mechanistic insight into the regulation of the osteoblastic trans-differentiation of SMC through P2Y2R-mediated Runx2 antagonism. Given that calcification of atherosclerotic lesions is a significant clinical problem, activating P2Y2R may be an effective therapeutic approach for treatment or prevention of vascular calcification.
Keywords: ATP, Atherosclerosis, Nucleotide receptor, Vascular calcification, Inflammation
Introduction
Vascular calcification is a hallmark of many diseases including atherosclerosis, type 2 diabetes, end-stage renal disease, and is highly correlated with cardiovascular morbidity (1–2). Arterial medial calcification occurs in the absence of inflammation, and is associated with chronic kidney disease (CDK) in adults (3–5). Medial calcification causes vessel stiffening, elevated pulse-wave velocity, and increased left ventricular (LV) load with hypertrophy (6–9). In contrast, intimal calcification occurs in the context of atherosclerosis and is associated with plaque rupture and myocardial infarction (10–12). Vascular calcification is an active and regulated process reminiscent of physiological bone formation (12). VSMC are the main cell type involved in arterial calcification, and can trans-differentiate into an osteoblast-like cell (13–14). This phenotypic transition is characterized by the expression of alkaline phosphatase, type I collagen, osteocalcin and the formation of mineralized bone-like structures (15).
Runt-related transcription factor (Runx) 2 is a key regulator of osteoblast differentiation (16). Runx2-null mice are devoid of osteoblasts and lack the ability to form bone (17–18). Runx2 mediates osteoblast differentiation as well as mineralization in immature mesenchymal cells and osteoblastic cells in vitro (19–20). Runx2 is expressed in calcified human atherosclerotic lesions as well as in calcifying mouse aortic SMC (21–23). Expression of Runx2 alone does not induce VSMC calcification in vitro (24), however, Runx2 deficiency in VSMC has been shown to inhibit vascular calcification in vivo (25). Given the pivotal role of Runx2 in SMC calcification, identifying factors that regulate this transcription factor may lead to new medical therapies to prevent or treat cardiovascular calcification.
A study of idiopathic infantile arterial calcification first led to the hypothesis that the purinergic signaling may play a role in the pathogenesis of vascular calcification (26). This autosomal recessive disease is caused by a mutation in the ectonucleotide pyrophosphatase-phosphodiesterase 1 gene (ENPP1) that catalyzes the hydrolysis of pyrophosphate and phosphodiester bonds in nucleotide triphosphates and oligonucleotides, respectively, to generate nucleoside 5′-monophosphates. In addition, homozygous nonsense mutations in the 5′-nucleotidase ecto (NT5E) gene that encodes CD73, a nucleotidase that converts AMP to adenosine, have been linked to symptomatic arterial and joint calcifications (27). Together, these observations suggest a role for the AMP/adenosine metabolic pathway in inhibiting ectopic tissue calcification. Indeed, increased cAMP signaling synergizes with elevated extracellular inorganic phosphate (Pi) to induce calcification of vascular SMC (28). However, the role of purinergic receptor signaling in the pathogenesis of vascular calcification, particularly in the setting of atherosclerosis, is not understood.
The P2Y2 is expressed in vascular cells and its activation mediates inflammatory responses that contribute to intimal hyperplasia in response to injury (29–31). We recently showed that P2Y2R deletion prevents fatty-streaks formation in ApoE-null mice (32). In the present study, we examined the effects of P2Y2R deletion on arterial intimal calcification during atherosclerosis. We utilized the ApoE knockout mouse model of atherosclerosis which develops spontaneous atherosclerosis without dietary manipulations (33). By crossing ApoE knockout mice with P2Y2R knockout mice, we show that although loss of P2Y2R attenuates atherosclerotic plaque development, it leads to extensive calcification of the atherosclerotic lesions. In vitro, high Pi induced-calcium deposition in VSMC and expression of stage-specific osteoblast markers were exacerbated in the absence of P2Y2R. Finally, we demonstrated that P2Y2R prevents the osteoblastic trans-differentiation of VSMC by inhibiting Runx2 transcriptional activity. Our findings reveal a novel role of P2Y2R in the control of vascular calcification. Therefore, targeting P2Y2R signals in VSMC may represent a novel strategy for therapies targeting vascular calcification.
Materials and methods
Animals
Animal protocols were approved by the Animal Care and Use Committee of Indiana University and the procedures followed were in accordance with institutional guidelines. C57BL/6J, ApoE−/− and P2Y2R−/− mice were purchased from Jackson laboratory. P2Y2R−/− mice were bred to the ApoE−/− background to generate ApoE−/−/P2Y2R−/− mice. All of these mice are on a C57BL/6J background. All animals were fed a standard chow diet. Only males were used in experimental groups.
Analysis of atherosclerotic lesions in the aortic root
Mouse hearts were perfused, fixed in 4% paraformaldehyde and embedded in paraffin. Five-micrometer sections at 50-μm intervals were stained with Masson’s trichrome for lesion area measurement and morphometric analysis as described in our previous studies (32). Apoptosis was assessed by TUNEL (Tdt-mediated dUTP nick end labeling) method using the TACS 2 TdT in situ apoptosis detection kit (Trevigen). For the quantification of plaque necrosis, aortic root lesions were stained with hematoxylin/eosin. Necrotic areas were identified as large non-stained areas with a 3,000-μm2 threshold, thus excluding very small clear areas, and quantified as previously described (34).
In situ detection of calcification
Intimal calcification was detected by performing Von Kossa staining on tissue sections. Von Kossa-stained sections were then counterstained for hematoxylin and eosin. Von Kossa reactivity was quantified and expressed as a percentage of Von Kossa positive area divided by total lesion area.
Tissue and cell quantification of calcium
Aortic arch specimens from mice were lyophilized to a constant weight. The calcium extracted from lyophilized tissue with 0.6 N HCl at 37°C for 48 hours was measured by a colorimetric assay using the o-cresolphsthalein complexone kit (calcium diagnostic kit; Sigma-Aldrich). The amount of calcium was normalized to the tissue dry weight. Calcium content in cultured VSMC was determined using a similar procedure as described above, and expressed as micrograms of calcium per milligram of cellular protein. Protein content was measured using the bicinchoninic acid assay protein assay kit (Thermo Scientific).
Immunohistochemistry
Immunohistochemical staining was performed by the labeled streptavidin biotin method. Monoclonal anti-smooth muscle α-actin antibody (clone 1A4, Sigma, 1/1000 dilution) was used to detect smooth muscle cells. Macrophages were identified by immunostaining with a rat anti-mouse Mac-3 monoclonal antibody (M3/84, 550292, BD PharMingen, San Diego, CA; 1/100 dilution).
VSMC calcification assay
Aortic samples were collected from C57BL/6 and P2Y2R−/− mice and VSMC were isolated using the explant technique as described in our previous studies (34). SMC were seeded in growth medium at a density of 1.5 × 104/cm2 in multi-well plates. Cells were grown to confluence and incubated with calcification medium containing NaH2PO4 and Na2HPO4 at pH 7, to a final concentration of 3 mM phosphate. VSMC were incubated for up to 14 days in 95% air/5% CO2 and medium was changed every four days. Retroviral transduction of DNA construct into VSMC was performed as described in our previous studies (35).
Alizarin Red S staining
Cells were incubated in 10%-PFA for 10 minutes then rinsed with distilled water and dehydrated to 70% alcohol. Cells were stained with 2% Alizarin Red S solution for 5 minutes then blotted dry. The slides were then dehydrated in acetone and 1:1 acetone-xylene solution, cleared in xylene, and mounted using a synthetic mounting medium.
Luciferase reporter assays
Cells were transfected with firefly luciferase reporter 6×OSE2-Luc plus Renilla luciferase reporter pRL-TK. At 48 hours after transfection, cells were harvested and firefly and Renilla luciferase activities in cell lysates were measured using a dual luciferase kit (Promega). Renilla luciferase activities in cells were used as internal control.
Gene expression analysis
Quantitative polymerase chain reaction (qPCR) was performed as previously described (36). Total RNA was extracted from VSMC lysed in TRIzol reagent (Invitrogen), using an RNeasy mini kit and treated with DNase I (Qiagen). High quality RNA was extracted (0.5 μg/sample was used for synthesis of single-strand cDNA with Superscript II (Invitrogen). Syber green based real time PCR was conducted with 45 ng of cDNA using an ABI Prism 7900HT PCR machine (Applied Biosystems). The following primers (Integrated DNA Technologies) were used for Runx2: forward 5′-ACC ATA ACA GTC TTC ACA AAT CCT-3′, and reverse: 5′-CAG GCG ATC AGA GAA CAA ACT A-3′. The primers for osteocalcin were as follows: forward 5′-CTGACCTCACAGATGCCAAG-3′, and reverse: 5′-GTAGCGCCGGAGTCTGTTC-3′. GAPDH primers were used as internal control. The forward GAPDH primer was: 5′-GGT GGC AGA GGC CTT TG-3′. The reverse GAPDH primer was: 5′-TGC CCA TTT AGC ATC TCC TT-3′.
Statistical analysis
Data are expressed as means ± SEM. Differences in data between groups were compared with Student’s paired 2-tailed t test or 1-way ANOVA where appropriate. A p value less than 0.05 was considered statistically significant.
Results
P2Y2 receptor deficiency promotes calcification of atherosclerotic lesions
We assessed the effects of P2Y2R deletion on arterial calcification in ApoE−/− mice maintained on a standard mouse chow diet for 30 weeks. Strikingly, despite a significant decrease in atherosclerotic lesion area compared to ApoE−/− mice (p<0.01; Fig. 1A and B), ApoE−/−/P2Y2R−/− mice (15 out of 15 mice) exhibited extensive intimal calcification covering more than 10% of the plaque area (p<0.001; Fig. 1C–E). Micro-calcification in the form of spotty or granular calcium deposits was also observed in ApoE−/−/P2Y2R−/− mice (Fig. 1C). In contrast, large areas of calcification were absent from ApoE−/− lesions (Fig. 1C), and only 1 out of 15 ApoE−/− mice exhibited very few granular calcium deposits. Notably, no calcification was observed in the media layer in either genotype. Fig. 1C shows a typical lesion in the aortic root, with extensive calcification in close proximity of the endothelium. The amount of extractable calcium that was deposited in the aortic root in P2Y2R−/−/ApoE−/− mice was 10-fold greater compared to that of ApoE−/− mice (Fig. 1D). The calcified area occupied about 13% of the total lesion area (Fig. 1E). Vascular calcification observed in P2Y2R−/−/ApoE−/− mice was not due to an increase in plasma cholesterol since P2Y2R-deficiency did not alter total plasma cholesterol in ApoE−/− mice (Table 1). These data demonstrate that absence of P2Y2R result in early calcification of atherosclerotic lesions in ApoE−/− mice.
Fig. 1. P2Y2 receptor deficiency reduces atherosclerosis and increases calcification of atherosclerotic lesions.

(A) Light micrographs of representative Masson’s trichrome stained aortic root lesions from ApoE−/− and ApoE−/−/P2Y2R−/− mice, showing atherosclerotic lesion (area between yellow and red lines) as well as collagen and muscle fibers in the lesions. (B) Quantitative analysis of lesions in the aortic sinus. Data represent the mean ± SEM lesion area for 5 consecutive sections in each of the 12 mice examined for each genotype. (*p<0.001). Scale represents 100 μm. Arrows indicate lesions. (C) Representative images of sections stained with Von Kossa to detect calcification. Areas in insert have been magnified. Arrows indicate spotty calcium deposits. Scale bar = 100μm. (D) Quantification of the sinus aortic calcium content expressed as mmol/g of dried weight and (E) calcification presented as percentage of Von Kossa-positively stained areas in the total aortic lesion. Quantification was done using 5 cross sections in each of the 12 ApoE−/− mice and 12 ApoE−/−/P2Y2R−/− mice. Bar values are means ± SEM, *p<0.001.
Table 1.
Body weight and plasma lipid analysis in standard chow-fed ApoE−/− and ApoE−/−/P2Y2R−/− mice.
| Week 30 (n=11) | ApoE−/− | ApoE−/−/P2Y2R−/− |
|---|---|---|
| Body weight (g) | 37.2.5 ± 2 | 36 ± 1.8 |
| Total cholesterol (mg/dl) | 459 ± 35 | 452 ± 29 |
| Triglycerides (md/dl) | 74 ± 9 | 76 ± 7 |
P2Y2R deficiency reduces plaque cellularity
P2Y2R deficiency led to other differences in intimal plaque composition as well. The increased intimal calcification attributable to the loss of the P2Y2R gene correlated with a sharp reduction in the total plaque area occupied by Mac-3 positive macrophages (p<0.001, Fig. 2A) and a significant decrease in SMC content (p<0.001; Fig. 2B). Consequently, plaque cellularity (Fig. 3A) was decreased in lesions observed in P2Y2R−/−/ApoE−/− mice. Analysis of lesions for acellular/necrotic areas revealed a significant increase in plaque necrosis in the ApoE−/−/P2Y2R−/− lesions (p<0.001; Fig. 3B). We next examined if the increased necrosis in ApoE−/−/P2Y2R−/− lesions was due to an increase in cell apoptosis. As shown in Fig. 3C, apoptosis as measured by TUNEL-positive cells was 40% (p<0.001) higher in the ApoE−/−/P2Y2R−/− lesions. The formation of plaques with large necrotic/apoptotic cores is followed by conversion to highly fibrotic nodules, and many of the cells within the nodules express markers of chondrocytes and osteoblasts (37). Notably, cells within the calcified areas in the ApoE−/−/P2Y2R−/− lesions exhibited chondrocyte appearance and features such as abundant collagen fibers and proteoglycans reacting with the hematoxylin counterstaining (Fig. 3D),
Fig. 2. P2Y2 receptor modulates cellular composition and cellularity of atherosclerotic lesions.

(A and B) Representative images of immunohistological staining of atherosclerotic lesions in the aortic sinus. Adjacent sections were stained with (A) Mac-3 or (B) smooth muscle α-actin antibodies, respectively. The percentage of Mac-3 or smooth muscle α-actin-positive areas in A and B was calculated by dividing the positively stained areas by the total cross-sectional area of the lesion. Bar values are means ± SEM. Five cross sections were evaluated in 12 mice for each genotype. ***p<0.001. Arrows indicate the localization of the staining. Scale bar represents 100 μm.
Fig. 3. Reduction of plaque cellularity in ApoE−/−/P2Y2R−/− mice.

(A) Total nuclei were counted in each hematoxylin eosin-stained cross sections (n=5) from 12 mice in each group and normalized to the area lesion. **p<0.01. Scale bar represents 100 μm. (B) Evaluation of plaque necrosis. Representative images of aortic root sections (n=5) from 12 mice of each group genotype were stained with hematoxylin and eosin, and plaque necrosis was quantified. Necrotic areas are indicated with an asterisk. ***p<0.001. Scale bar represents 100 μm. (C) TUNEL-positive nuclei were quantified on lesions from 12 ApoE−/− and 13 ApoE−/−/P2Y2R−/− mice.***p<0.001. (D) High magnification of aortic root cross section from ApoE−/−/P2Y2R−/− mice showing chondrocyte-like cells (asterisk) within large areas of calcification. Scale bar represents 25 μm.
Expression of P2Y2 receptor inhibits VSMC calcification in vitro
As described previously by others (38), we confirmed that high Pi induces a significant increase in VSMC calcium deposition, evident after 14 days in culture as compared to cells cultured in control medium (Fig. 4A). Notably, adding an enzyme that degrades nucleoside di- and triphosphates (potato apyrase grade III, 0.1 unit/ml) greatly accelerated SMC calcification such that high levels of calcium content were measurable by 7 days of culture (Fig. 4B), suggesting that nucleotides released by VSMC inhibit calcification. P2Y2R deficiency in VSMC significantly increased high Pi-induced calcification as demonstrated by a sharp increase in the amount of incorporated calcium (p<0.001) compared to VSMC from WT mice (Fig. 5A; p<0.001) after 7 days of culture. Interestingly, the extent of calcification in P2Y2−/− SMC was similar to cultures lacking nucleoside di- and triphosphates (Fig. 5A compared to Fig. 4B).
Fig. 4. P2Y2R deficiency in VSMC enhances high phosphate-induced calcification in VSMC.

(A) Wild-type (WT) and P2Y2R−/− VSMC were incubated in high phosphate (High Pi) or control medium (Low Pi) for 7 or 14 days and stained with alizarin red. Calcium deposition was quantified. Results are presented as mean ± SEM. *p<0. 05; ***p < 0.001; n=4 independent experiments. (B) VSMC were cultured in high or low phosphate medium in the absence or presence of apyrase. Calcium deposition was quantified as in (A). Data represent mean ± SEM of 5 independent experiments. NS= Not significant; ***p < 0.001. Scale bar =100 μm.
Fig. 5. Expression of P2Y2 receptor prevents smooth muscle cell calcification in vitro.

(A) Wild-type (WT) and P2Y2R−/− VSMC were incubated in high phosphate (High Pi) or control medium (Low Pi) for 7 days and stained with alizarin red. Scale bar =100μm. (B) Calcium deposition in VSMC was quantified as in Fig. 4. Fold changes in the mRNA expression of osteogenic markers Runx2 and osteocalcin relative to that seen in WT cells are shown in bar graphs. Results are presented as mean ± SEM. ***p < 0.001; n=4 independent experiments. Transduction of WT P2Y2R into P2Y2R−/− inhibits (C) calcium deposition as well as (D) relative mRNA levels for Runx2 and osteocalcin. ***p < 0.001; n=3 independent experiments; NS= not significant.
P2Y2 receptor inhibits the osteogenic trans-differentiation of VSMC
Calcifying VSMC adopt an osteoblast-like phenotype, including expression of osteoblast markers (25). Therefore, we measured the expression of the stage-specific osteoblast markers Runx2 and osteocalcin in VSMC from WT or P2Y2R−/− mice cultured in high Pi medium (Fig. 5B). Both osteoblast markers were expressed at significantly higher levels in P2Y2R−/− VSMC, suggesting that the absence of P2Y2R sensitizes them to calcification promoting conditions. Conversely, rescue of P2Y2R expression in knockout VSMC (via retroviral transduction of full-length WT mouse P2Y2R into P2Y2R−/− cells) decreased osteoblast-specific gene expression and total calcium content to levels comparable with WT (Fig. 5C and D). These results demonstrate that P2Y2R in VSMC is required to inhibit osteogenic trans-differentiation.
P2Y2 receptor represses Runx2 transcriptional activity
As Runx2 is known to be required for VSMC calcification in vitro and in vivo (25, 39) and its expression was increased in P2Y2R−/− VSMC, we examined whether expression of P2Y2R inhibits the transcriptional activity of Runx2. Overexpression of Runx2 increased the activity of an OC promoter driven luciferase reporter gene (40) in P2Y2R-deficient VSMC (Fig. 6). However, this Runx2-induced OC promoter activity was significantly attenuated in P2Y2R-deficient VSMC transduced with WT P2Y2R (Fig. 6). These data clearly indicate that P2Y2R regulates the osteogenic trans-differentiation of VSMC through Runx2 antagonism.
Fig. 6. P2Y2R inhibits Runx2-activation of the osteocalcin promoter.
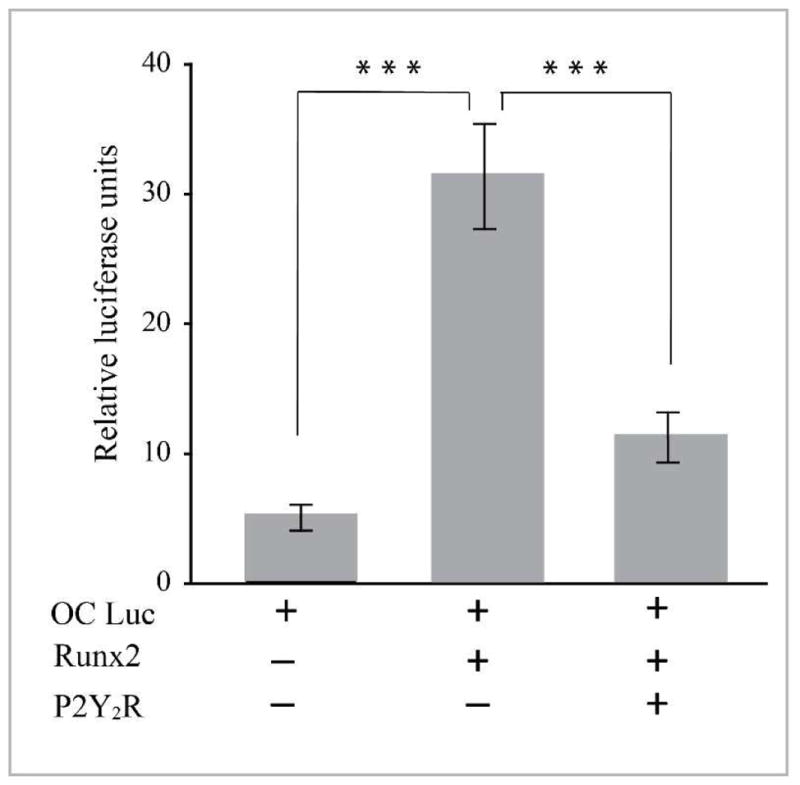
P2Y2R-null VSMC were transfected with an osteocalcin promoter luciferase reporter gene, an internal control pRL-TK plasmid, a Runx2 expression plasmid, and a full length P2Y2R expression plasmid as indicated. Lysates were harvested 48 h after transfection and were normalized to the expression of the pRL-TK plasmid. Results are presented as the mean ± SD of triplicates of cells and are representative of three independent experiments. ***p<0.001.
Discussion
The main finding of this study supports a dual role for P2Y2 receptor as a mediator of atherosclerosis but an inhibitor of arterial intimal calcification. P2Y2R deficiency leads to reduced atherosclerotic lesion sizes, increased plaque necrosis with extensive calcification. Furthermore, loss of P2Y2R accelerates in vitro calcification and the osteoblastic trans-differentiation of VSMC. Mechanistically, P2Y2R acts as a negative regulator of Runx2 transcriptional activity.
Intimal calcification is apparent in normal chow fed ApoE−/− mice at around 45 weeks of age, where small deposits of hydroxyapatite are observed (37). Surprisingly, calcium deposits are apparent in ApoE−/−/P2Y2R−/− mice at high frequency, as early as 30 weeks of age, indicating that absence P2Y2R accelerates intimal calcification. The calcified lesions were associated with decrease cellularity that is associated with a significant reduction in both macrophage and VSMC content of the plaques. The decrease in lesion cellularity observed in ApoE−/−/P2Y2R−/− mice was in part due to increased cell apoptosis.
Several factors may account for the increased calcification of the lesions in the ApoE−/− mice in the absence of P2Y2R. Increased circulating LDL and decreased HDL have been shown to increase plaque burden and coronary artery calcification (42). However, since P2Y2R deficiency does not alter plasma lipid content in the ApoE−/− mice (Table 1), this mechanism likely does not account for the increased calcification in these mice. The observation of bone-like regions within the lesions in P2Y2R−/−/ApoE−/− mice suggests that lack of P2Y2R stimulates an active process similar to bone formation in the lesions.
Depending on the type of calcium deposition, intimal calcification may either promote the formation of rupture-prone lesions or stabilize the plaque (43). Our data indicate that P2Y2R-deficiency drives the formation of both spotty calcium deposits (micro-calcification) and large calcified areas (macro-calcification). Micro-calcification which induces further inflammation is associated with unstable plaques while macro-calcification favors plaque stabilization (43). It is possible that the large areas of calcium deposits in lesions from ApoE−/−/P2Y2R−/− mice represent an adaptive mechanism to counter the initial detrimental effects caused by small granular calcium deposits. The increased stability associated with large calcium deposits would be consistent with increased plaque stability resulting from the significantly reduced macrophage content in lesions from ApoE−/−/P2Y2R−/− mice. This reduction in plaque macrophage content was also correlated with the presence of chondrocyte-like cells that are often seen in stabilized plaques (37).
Osteogenic trans-differentiation of VSMC is known to contribute directly to the pathogenesis of atherosclerotic calcification (25). Our data show that extracellular nucleotides act as inhibitors of VSMC calcification in vitro. Indeed, the enzyme potato apyrase that degrades nucleoside di- and triphosphates accelerated VSMC calcification under an osteogenic environment. The role of nucleotides in vascular calcification has been largely attributed to inorganic pyrophosphate. Genetic studies have shown the important roles for inorganic pyrophosphate (PPi) as inhibitors of vascular mineralization. Deficiency in the mineralization inhibitor PPi, that arises from genetic disruption of the murine ectoezyme NPP1 (ectonucleotide pyrophosphatase/phosphodiesterase I), causes arterial calcification (26). In addition, PPi deficiency due to NPP1 loss-of-function mutations is associated with a rare congenital disorder characterized by generalized arterial calcification in infants (27). The present study identifies an alternative and robust pathway by which extracellular nucleotides acting through P2Y2R inhibit VSMC calcification. Using loss-of-function and gain-of-function experiments, we unequivocally established that P2Y2R is a powerful inhibitor of VSMC calcification. Indeed, we demonstrated that loss of P2Y2R accelerated high phosphate-induced calcium deposition in VSMC. Furthermore, retroviral transduction of full length wild-type P2Y2R into P2Y2R−/− VSMC reduced calcium deposition to levels comparable with those observed in VSMC from WT mice.
Mechanistically, our studies demonstrate that P2Y2R mediated inhibition of VSMC calcification at least in part through repression of Runx2 transcriptional activity. Runx proteins are targeted to gene regulatory micro-environments within the nucleus, via a specific sub-nuclear targeting signal (44). However, the mechanisms that shuttle Runx2 between the cytoplasm and the nucleus are poorly understood. It was reported that when endogenous Runx2 associates with stabilized microtubules it is held in the cytoplasm and that the Runx2 amino terminus mediates the microtubule association through the alpha/beta tubulin subunits (45) Thus, it is possible that P2Y2R-mediated microtubule/actin organization regulates Runx2 trafficking between the cytoplasm and the nucleus, a process that directly affects Runx2 transcriptional activity.
In conclusion, this study reveals a previously unknown function of P2Y2R in vascular calcification, which is distinct from deficits in the mineralization inhibitor inorganic pyrophosphate. Our findings provide a new mechanistic insight into the regulation of the osteoblastic trans-differentiation of VSMC by a nucleotide receptor, through Runx2 antagonism. Given that calcification of atherosclerotic lesions is a highly regulated pathological process potentially subject to intervention, P2Y2R may be targeted for the treatment or prevention of vascular calcification.
Highlights.
P2Y2 receptor deficiency in ApoE−/− mice reduces atherosclerosis
Loss of P2Y2 receptor accelerates arterial intimal calcification in ApoE−/− mice
Expression of P2Y2 receptor inhibits high phosphate-induced smooth muscle cell calcification
P2Y2 receptor negatively regulates Runx2 transcriptional activity in smooth muscle cells.
Acknowledgments
Financial support
This work was supported by a grant from the National Institutes of Health (1R01HL112883) to Cheikh I. Seye.
Abbreviations
- CDK
chronic kidney disease
- ENPP1
ectonucleotide pyrophosphatase-phosphodiesterase 1
- P2Y2R
P2Y2 nucleotide receptor
- Runx2
Runt related transcription factor-2
- TRAP
tartrate-resistant acid phosphatase
- VCAM-1
vascular cell adhesion molecule-1
- SMC
smooth muscle cells
Footnotes
Conflict of interest
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Block GA. Prevalence and clinical consequences of elevated Ca x P product in hemodialysis patients. Clin Nephrol. 2000;54:318–324. [PubMed] [Google Scholar]
- 2.Zhu D, Mackenzie NCW, Farquharson C, et al. Mechanisms and clinical consequences of vascular calcification. Front Endocrinol. 2012;3:95. doi: 10.3389/fendo.2012.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, Hiorns M, Donald AE, Deanfield J, Rees L, Shanahan CM. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–1757. doi: 10.1161/CIRCULATIONAHA.108.783738. [DOI] [PubMed] [Google Scholar]
- 4.Goodman WG, London G, Amann K, Block GA, Giachelli C, Hruska KA, Ketteler M, Levin A, Massy Z, McCarron DA, Raggi P, Shanahan CM, Yorioka N. Vascular calcification in chronic kidney disease. American journal of kidney diseases. 2004;43:572–579. doi: 10.1053/j.ajkd.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Moorthi RN, Moe SM. CKD-mineral and bone disorder: core curriculum. Am J KidneyDis. 2011;58:1022–1036. doi: 10.1053/j.ajkd.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerin AP, Pannier B, Metivier F, Marchais SJ, London GM. Assessment and significance of arterial stiffness in patients with chronic kidney disease. Curr Opin Nephrol Hypertens. 2008;17:635–641. doi: 10.1097/mnh.0b013e32830dcd5c. [DOI] [PubMed] [Google Scholar]
- 7.London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731–1740. doi: 10.1093/ndt/gfg414. [DOI] [PubMed] [Google Scholar]
- 8.Kelly RP, Tunin R, Kass DA. Effect of reduced aortic compliance on cardiac efficiency and contractile function of in situ canine left ventricle. Circ Res. 1992;71:490–502. doi: 10.1161/01.res.71.3.490. [DOI] [PubMed] [Google Scholar]
- 9.Niederhoffer N, Lartaud-Idjouadiene I, Giummelly P, Duvivier C, Peslin R, Atkinson J. Calcification of medial elastic fibers and aortic elasticity. Hypertension. 1997;29:999–1006. doi: 10.1161/01.hyp.29.4.999. [DOI] [PubMed] [Google Scholar]
- 10.Beadenkopf WG, Daoud AS, Love BM. Calcification in the coronary arteries and its relationship to arteriosclerosis and myocardial infarction. Am J Roentgenol Radium Ther Nucl Med. 1964;92:865–887. [PubMed] [Google Scholar]
- 11.Kelly-Arnold A, Maldonado N, Laudier D, Aikawa E, Cardoso L, Weinbaum S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci. 2013;110:10741–10746. doi: 10.1073/pnas.1308814110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehara S, Kobayashi Y, Yoshiyama M, Shimada K, Shimada Y, Fukuda D, Nakamura Y, Yamashita H, Yamagishi H, Takeuchi K, Naruko T, Haze K, Becker AE, Yoshikawa J, Ueda M. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation. 2004;110:3424–3429. doi: 10.1161/01.CIR.0000148131.41425.E9. [DOI] [PubMed] [Google Scholar]
- 13.Shroff RC, McNair R, Figg N, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–1757. doi: 10.1161/CIRCULATIONAHA.108.783738. [DOI] [PubMed] [Google Scholar]
- 14.Zhu D, Mackenzie NC, Millan JL, et al. The appearance and modulation of osteocyte marker expression during calcification of vascular smooth muscle cells. PloS ONE. 2011;6:e19595. doi: 10.1371/journal.pone.0019595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collin-Osdoby P. Circ Res. 2004;95:1046–1057. doi: 10.1161/01.RES.0000149165.99974.12. [DOI] [PubMed] [Google Scholar]
- 16.Schroeder TM, Jensen ED, Westendorf JJ. Runx2: a master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res C Embryo Today. 2005;75:213–225. doi: 10.1002/bdrc.20043. [DOI] [PubMed] [Google Scholar]
- 17.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 18.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 19.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: atranscriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 20.Lian JB, Javed A, Zaidi SK, Lengner C, Montecino M, van Wijnen AJ, Stein JL, Stein GS. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit Rev Eukaryot Gene Expr. 2004;14:1–41. [PubMed] [Google Scholar]
- 21.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality Molecular Imaging Identifies Proteolytic and Osteogenic Activities in Early Aortic Valve Disease. Circulation. 2007;115:377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 22.Engelse MA, Neele JM, Bronckers AL, Pannekoek H, de Vries CJ. Vascular calcification: expression patterns of the osteoblast-specific gene core binding factor α-1 and the protective factor matrix gla protein in human atherogenesis. Cardiovasc Res. 2001;52:281–289. doi: 10.1016/s0008-6363(01)00375-3. [DOI] [PubMed] [Google Scholar]
- 23.Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/Chondrocytic Transcription Factors and Their Target Genes Exhibit Distinct Patterns of Expression in Human Arterial Calcification. Arterioscler Thromb Vasc Biol. 2003;23:489–494. doi: 10.1161/01.ATV.0000059406.92165.31. [DOI] [PubMed] [Google Scholar]
- 24.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111(5):543–52. doi: 10.1161/CIRCRESAHA.112.267237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–81. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 27.St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, Siegenthaler MP, Arduino C, Mancini C, Stanescu HC, Zdebik AA, Chaganti RK, Nussbaum RL, Kleta R, Gahl WA, Boehm M. NT5E Mutations and Arterial Calcifications. N Engl J Med. 2011;364(5):432–442. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prosdocimo DA, Wyler SC, Romani AM, O’Neill WC, Dubyak GA. Regulation of vascular smooth muscle cell calcification by extracellular pyrophosphate homeostasis: synergistic modulation by cyclic AMP and hyperphosphatemia. Am J Physiol Cell Physiol. 2010;298(3):C702–C713. doi: 10.1152/ajpcell.00419.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seye CI, Yu N, Jain R, Kong Q, Minor T, Newton J, Erb L, Gonzalez FA, Weisman GA. The P2Y2 nucleotide receptor mediates UTP-induced vascular cell adhesion molecule-1 expression in coronary artery endothelial cells. J Biol Chem. 2003;278:24960–24965. doi: 10.1074/jbc.M301439200. [DOI] [PubMed] [Google Scholar]
- 30.Seye CI, Yu N, Gonzalez FA, Erb L, Weisman GA. The P2Y2 Nucleotide Receptor Mediates Vascular Cell Adhesion Molecule-1 Expression through Interaction with VEGF Receptor-2 (KDR/Flk-1) J Biol Chem. 2004;279:35679–35686. doi: 10.1074/jbc.M401799200. [DOI] [PubMed] [Google Scholar]
- 31.Seye CI, Kong Q, Erb L, Garrad RC, Krugh B, Wang M, Turner JT, Sturek M, Gonzalez FA, Weisman GA. Functional P2Y2 nucleotide receptors mediate uridine 5′-triphosphate-induced intimal hyperplasia in collared rabbit carotid arteries. Circulation. 2002;106:2720–2726. doi: 10.1161/01.cir.0000038111.00518.35. [DOI] [PubMed] [Google Scholar]
- 32.Qian Shaomin, Hoggatt April, Jones-Hall Yava L, Ware Carl F, Herring Paul, Seye Cheikh I. Deletion of P2Y2 Receptor Reveals a Role for Lymphotoxin-α in Fatty Streak Formation. Vascul Pharmacol. 2016;85:11–20. doi: 10.1016/j.vph.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Vlijmen BJ, et al. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. 1994;93:1403–10. doi: 10.1172/JCI117117. [DOI] [PMC free article] [PubMed] [Google Scholar]; Westenfeld R, Schäfer C, Smeets R, Brandenburg VM, Floege J, Ketteler M, Jahnen-Dechent W. Fetuin-A (AHSG) prevents extraosseous calcification induced by uraemia and phosphate challenge in mice. Nephrol Dial Transplant. 2007;22(6):1537–46. doi: 10.1093/ndt/gfm094. [DOI] [PubMed] [Google Scholar]
- 34.Feng B, Zhang D, Kuriakose G, Devlin CM, Kockx M, Tabas I, Niemann-Pick C. Heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:10423–10428. doi: 10.1073/pnas.1732494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu N, Erb L, Shivaji R, Weisman GA, Seye CI. Binding of the P2Y2 nucleotide receptor to filamin A regulates migration of vascular smooth muscle cells. Circ Res. 2008;102:581–588. doi: 10.1161/CIRCRESAHA.107.162271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seye CI, Agca Y, Agca C, Derbigny W. P2Y2 receptor-mediated lymphotoxin-α (LTA) secretion regulates intercellular cell adhesion molecule (ICAM)-1 expression in vascular smooth muscle cells. J Biol Chem. 2012;287:10535–10543. doi: 10.1074/jbc.M111.313189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rattazzi M, Bennett BJ, Bea F, Kirk EA, Ricks JL, Speer M, Schwartz SM, Giachelli CM, Rosenfeld ME. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arterioscler Thromb Vasc Biol. 2005;25:1420–1425. doi: 10.1161/01.ATV.0000166600.58468.1b. [DOI] [PubMed] [Google Scholar]
- 38.Speer MY, Chien YC, Quan M, Yang HY, Vali H, McKee MD, Giachelli CM. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc Res. 2005;66(2):324–33. doi: 10.1016/j.cardiores.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 39.Lin ME, Chen T, Leaf EM, Speer MY, Giachelli CM. Runx2 Expression in Smooth Muscle Cells Is Required for Arterial Medial Calcification in Mice. Am J Pathol. 2015;185(7):1958–1969. doi: 10.1016/j.ajpath.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, Jensen ED, Westendorf JJ, Cheng L, Huang H. FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res. 2011;71(9):3257–3267. doi: 10.1158/0008-5472.CAN-10-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find me signal to promote phagocytic clearanc. Nature. 2009;461(7261):282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Achenbach S, Ropers D, Pohle K, Leber A, Thilo C, Knez A, Menendez T, Maeffert R, Kusus M, Regenfus M, Bickel A, Haberl R, Steinbeck G, Moshage W, Daniel WG. Influence of lipid-lowering therapy on the progression of coronary artery calcification: a prospective evaluation. Circulation. 2002;106(9):1077–1082. doi: 10.1161/01.cir.0000027567.49283.ff. [DOI] [PubMed] [Google Scholar]
- 43.Pugliese G, Iacobini C, Blasetti Fantauzzi C, Menini S. The dark and bright side of atherosclerotic calcification. Atherosclerosis. 2015;238(2):220–30. doi: 10.1016/j.atherosclerosis.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 44.Zaidi Sayyed K, Young Daniel W, Choi Je-Yong, Pratap Jitesh, Javed Amjad, Montecino Martin, Stein Janet L, van Wijnen Andre J, Lian Jane B, Stein Gary S. The dynamic organization of gene-regulatory machinery in nuclear microenvironments. EMBO Rep. 2005;6(2):128–133. doi: 10.1038/sj.embor.7400337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Addario M, Arora PD, Ellen RP, McCulloch CA. Regulation of tension-induced mechanotranscriptional signals by the microtubule network in fibroblasts. J Biol Chem. 2003;278(52):53090–53097. doi: 10.1074/jbc.M309027200. [DOI] [PubMed] [Google Scholar]
- 46.Pockwinse SM, Rajgopal A, Young DW, Mujeeb KA, Nickerson J, Javed A, Redick S, Lian JB, van Wijnen AJ, Stein JL, Stein GS, Doxsey SJ. Microtubule-dependent nuclear-cytoplasmic shuttling of Runx2. J Cell Physiol. 2006;206(2):354–362. doi: 10.1002/jcp.20469. [DOI] [PubMed] [Google Scholar]