

Abstract
Vitamin D has so far not fulfilled its early promise as an antineoplastic agent, in spite of compelling in vitro data. With the aim of bringing vitamin D or its derivatives (VDDs) effectively to the clinic, we developed a two-pronged approach. First, by adding the plant-derived Carnosic Acid (CA) to a vitamin D2 derivative Doxercalciferol we increased its differentiation potency without increasing it hypercalcemic properties. Second, we added these two agents together to AML cells already treated with Cytarabine (AraC), the standard drug for the treatment of patients with AML. We now report that BRAF, a part of the MAPK signaling pathway, is required for the optimally increased cell death in this system and acts upstream of BIM, the regulator of the caspase cascade that leads to cell death by apoptosis. It is proposed that this therapeutic regimen should be tested in a clinical trial.
Keywords: Vitamin D, BRAF, BIM, Apoptosis, Cytarabine, AML
Graphical abstract
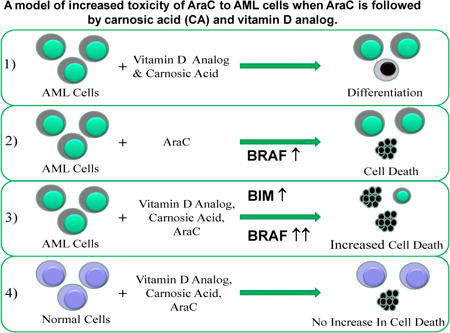
1. Introduction
The original enthusiasm for the use of vitamin D derivatives (VDDs) for the treatment of neoplastic diseases was greatly diminished by the publication of the Institute of an Medicine (IOM) report, which concluded that in contrast to the positive effect of vitamin D on bone health, there is no credible evidence of a similar effect on malignant cells [1]. This opinion was largely based on the published record indicating that although there is clear evidence of an “anti-cancer” effect of VDDs in preclinical studies (eg [2-5]), multiple attempts to demonstrate such an effect in the clinic were in almost all cases negative (eg. [6-10] also www.clinicaltrials.gov). The few publications with positive results were based on small numbers of patients, and there remains the concern that there are confounding factors in the interpretation of the results (eg [11-13]).
However, others in the field of vitamin D and cancer have suggested that the clinical trials performed so far were not performed in a manner that guarantees detection of positive effects of VDDs on cancer treatment (eg; [14-16]), and this suggests that future trials, well designed, are warranted. This seems to be particularly true regarding the use of VDDs in the treatment of Acute Myeloid Leukemia (AML). The prognosis for most AML patients is abysmal, but while it is fortunate that the disease has low incidence, this makes it difficult to accrue patients to large clinical trials. Thus, an understanding of the mechanistic basis of the actions of VDDs in the proposed trials seem essential in order to structure such trials to detect small differences between the results of therapeutic regimens offered to patients with AML, other than the subtype APL, where there has been significant success [17].
It is arguable that it will not be possible to use VDDs alone in the clinic, as the concentrations of these compounds, even the currently available vitamin D analogs with claimed low calcemic activity, are still hypercalcemic in vivo if used alone as anti-cancer treatment. Thus, numerous attempts have been made to combine VDDs such as Calcitriol (1,25D), or analogs, with either toxic or non-toxic compounds, but to date these combinations have not resulted in clinically demonstrated advances of the field [5, 14]. Recently, we presented in vitro studies of human AML blasts, which demonstrated a selective increase in cytotoxicity of Arabinocytosine (AraC, Cytarabine), the key therapeutic for AML [18, 19], when the exposure of the AML cells to AraC was followed by a combination of differentiating agents [20, 21]. The combination consisted of the vitamin D2 analog Doxercalciferol (D2) already approved for human administration, together with a plant-derived anti-oxidant Carnosic Acid (CA), currently used as a food flavoring agent [22], and previously reported to enhance1,25-dihydroxyvitamin D3-induced differentiation of AML cells [23]. Our studies were conducted using patient blasts ex vivo as well as in established culture, and the selectivity to malignant blasts was demonstrated by the finding that the addition of the D2/CA combination to normal bone marrow (NBM) cells treated with AraC did not kill more cells than the treatment with AraC alone [20].
While our initial studies of the mechanisms responsible for the effect of differentiating agents on cells with AraC-induced DNA damage showed that the Vitamin D Receptor (VDR), needed for the induction of monocytic differentiation by VDDs, and the BCL2 interacting mediator of apoptosis (BIM, BCL2L11) were required for the optimal enhancement of AraC cytotoxicity [20] it was not clear how BIM was upregulated in this “enhancement of cytotoxicity” model. Here we report that BRAF is a part of the signaling pathway from VDR to BIM, which then activates the caspase cascade.
2. Materials and Methods
Cell Lines and Chemicals
Two AML cell lines, HL60 (cultured from a patient with acute promyeloblastic leukemia [24], and U937 (monocytes from histiocytic lymphoma) [25], were cultured as previously described [21].
Arabinocytosine (AraC) and Doxercalciferol (1α-hydroxyvitamin D2; D2) were purchased from Sigma-Aldrich (St. Louis, MO). Carnosic acid (CA) was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY). BRAF inhibitors SB590885 and TAK632 were purchased from Selleck Inc., stated to inhibit both wild type and mutated BRAF. The antibodies used for Western blots were: BRAF, CRAF, BIM (sc-8625), VDR (sc-1008), and CRK-L, the V-Crk Avian Sarcoma Virus CT10 Oncogene Homolog-Like (sc-319), were obtained from Santa Cruz Biotechnology (Dallas, TX). Phospho-H2AX-Ser139 (#9718), phospho-BIM-Ser69 (#4581) and HRP-linked anti-rabbit (#7074) antibodies were from Cell Signaling Technologies (Danvers, MA). The siRNA transfection reagents were from Santa Cruz Biotechnology (Dallas, TX).
Isolation of mononuclear cells from peripheral blood or bone marrow samples and their culture
Peripheral blood specimens from patients with AML, and a bone marrow sample from a volunteer, were obtained with written consent according to an IRB approved protocol. Mononuclear cells were isolated from these samples by using Histopaque-1077 (Sigma-Aldrich) gradient centrifugation, as previously described [26]. Briefly, isolated mononuclear cells were divided into a group exposed for 72 h to 100 nM AraC or the vehicle (0.1% DMSO). The cells were washed with control medium after this 72 h pretreatment, and each of these groups was further divided for addition of D2 (100 nM), or CA (10 μM), or both, for 96 h. Cell viability was determined by Trypan blue (TB) exclusion using a Neubauer hemocytometer as described before [21], and apoptosis, and necrosis by Annexin V as described below.
Knockdown of VDR or BRAF expression
AML blasts or HL60 cells were transfected with 10 nM of VDR siRNA or BRAF siRNA or scrambled Control siRNA, using Endo-Porter delivery reagent from Gene Tools Inc (Philomath, OR) before exposure to other agents. The cells were allowed to recover in RPMI 1640 medium with 10% FCS for 24 h, and then were exposed to the indicated compounds for times indicated in the individual experiments. The reduction of target protein or mRNA by siVDR or siBRAF transfection in groups designed to show cell death enhancement changes was approximately 40%-50%. We transfected cells with siRNA (20nM) for 72 hr before adding other agents, then we replenished with half of the original amount of siRNA every 48 hrs throughout the whole experiment.
For inhibition of BRAF kinase activity, AML cells were pretreated with 100 nM AraC for 72 h, and then incubated with either of the BRAF inhibitors for 1 h before adding other agents. However, it is possible that knockdown of BRAF was incomplete under our conditions which were not toxic to the cells.
Flow cytometry analysis for Annexin V and propidium iodide staining
Cells were washed twice with 1×PBS, then re-suspended in the binding buffer, containing 0.14 M NaCl and 2.5 mM CaCl2, pH 7.5, and incubated with 50 μg/ml Annexin V-FITC (Kit from Sigma) and 20 μg/ml propidium iodide in 1× binding buffer at room temperature in the dark for 15 minutes, then immediately analyzed by flow cytometry (EPICS XL). Annexin V-positive/ PI-negative cells were considered as early apoptotic, cells both Annexin V and PI positive, as late apoptotic, and Annexin negative but PI positive as “necrotic”, likely a variety of caspase independent modes of cell death [27].
Quantitative Real Time PCR analysis
Total RNA was extracted by using Trizol (Invitrogen, CA) according to manufacturer's protocol, as described before [21]. The Human Apoptosis RT2 Profiler PCR Array profiles (PAHS-012Z, Sabiosciences,) which contains 84 key genes reported to be involved in programmed cell death, were performed according to the manufacturer's instructions. Briefly, Reverse Transcription was performed by using 1 ug of total RNA. cDNA template, which was then run on the PCR array plate in an ABI 7500 Real-Time PCR System. Data analysis was performed by using the web-based analysis tool (www.sabiosciences.com/pcrarraydataanalysis.php), which includes fold change and descriptive statistics. Quantitative RT-PCR was carried out using an ABI SYBR Green master kit (Applied Biosystems, Foster City, CA). Fold changes of mRNA levels in target gene relative to the RNA polymerase II (RPII) control were calculated by relative quantification analysis. Primers used for BRAF were upstream 5′AGGTAATGTACTTAGGGTGAA3′, downstream 5′TGTTAGAAACTTTTGGAGGAG BIM were: upstream 5′-AGTTCTGAGTGTGACCGAGAAGGT-3′, downstream 5′-TCCTGTCTTGTGGCTCTG TCTGTA-3′; For VDR: forward primer, 5′-CTTCAGGCGAAGCATGAAGC-3′; reverse primer, 5′-CCTTCATCATGCCGATGTCC-3′; for RP II, the internal control for RNA loading, upstream 5′-GCACCA CGTCCAATGACAT-3′, downstream 5′-GTGCGGCTGCTTCCATAA-3′. The quality of PCR products were monitored using post-PCR melting curve analysis.
Western blotting
Western blotting was performed using 50 μg of total cell extracts as previously described [28]. Each membrane was stripped and re-probed with CRK-L to serve as an internal loading control. The optical density (OD) of each band was quantitated using ImageQuant 5.0 program (Molecular Dynamics, Sunnyvale, CA). The OD signals on Westerns were always normalized to CRK-L control.
Statistical analysis
Each experiment was repeated at least 3 times. The results are presented as the mean ± SD. Statistical significance of the differences between mean values was performed by a 2-tailed Student's T-test. All computations were done with an IBM-compatible personal computer using Microsoft EXCEL program.
3. Results
3.1. Genes showing significant upregulation of expression in the phase of enhancement of AraC toxicity
We have reported that the SH3-only pro-apoptotic protein BIM is required for the optimal enhancement of cell death in AraC-treated cells followed by differentiation agents [20]. However, it seemed likely that other genes participate in this effect. We therefore conducted an Array study of the expression of eighty apoptosis-related genes during the enhancement phase of AraC cytotoxicity. Interestingly, in this phase, the Array showed that the expression of only two out of the eighty genes, BIM and BRAF, was upregulated significantly and consistently between the three AML cell types studied here (Table 1). The upregulation of BRAF was confirmed by quantitative RT-PCR in both HL60 and U937 cells (Fig 1), and we have previously confirmed BIM upregulation using this protocol at mRNA and protein levels in ex vivo blasts and cell lines [20]. Of note, the initial treatment with AraC alone, or D2/CA alone, also showed significantly increased BRAF mRNA expression (Fig 1).
Table 1. Genes showing significant upregulation in the enhancement phase of AraC toxicity.
| HL60 | Fold Change (comparing to control) | Difference | P value | |
|---|---|---|---|---|
| Symbol | HL60-AraC | HL60-AraC-D2/CA | Combo - AraC | Combo vs AraC |
| BCL2L11 (BIM) | 2.020 | 5.229 | 3.210 | 0.031 |
| BRAF | 0.749 | 3.189 | 2.440 | 0.028 |
| U937 | Fold Change (comparing to control) | Difference | p-values | |
| Symbol | U937-AraC | U937-AraC-D2/CA | Combo - AraC | Combo vs AraC |
| BRAF | 0.985 | 1.791 | 0.806 | 0.013 |
| BCL2L11 (BIM) | 1.661 | 2.415 | 0.754 | 0.044 |
| AML-Ex vivo | Fold Change (comparing to control) | p-values | ||
| Symbol | AML-ex vivo-AraC | AML-AraC-D2/CA | Combo - AraC | Combo vs AraC |
| BRAF | 1.22 | 2.94 | 1.728 | N/A |
| BCL2L11 (BIM) | 1.58 | 2.73 | 1.146 | N/A |
The genes are listed in the order of decreasing magnitude of the difference between AraC-D2/CA (Combo) and AraC alone. For details of the procedure see Materials and Methods Section. The genes shown here had the highest scores (the increase in expression when D2/CA added after AraC exposure) out of the 80 genes in the Array
Fig 1.
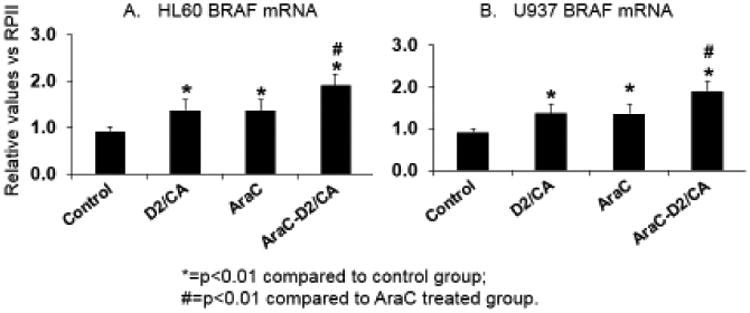
Expression of BRAF mRNA increases after treatment with components and the combination of cytotoxic and differentiation agents. A. HL60 cells. B. U937 cells. Note that in both cell lines the exposure for 96 hr to the combination of D2 (100 nM) and CA (10 uM) (labelled D2/CA in the figure), or AraC alone for 72 hr, or followed by 96 hr in normal medium (labeled AraC) or with D2+CA (labeled AraC-D2/CA),results in a significant increase in BRAF mRNA *=p<0.01 compared to control group; #=p<0.01 compared to AraC-only treated group; n= 3.
BRAF expression was increased also at protein level in the two cell lines (top Westerns in Fig 2, lanes 1-4). While in HL60 cells AraC alone showed an increase in BRAF protein level, this was further increased by the D2/CA differentiating agents, whereas in U937 cells the triple combination was needed to see a significant increase. In contrast to BRAF mRNA levels, the protein levels of BRAF were not increased by D2/CA alone, suggesting a different level of regulation of the BRAF gene by toxic (eg AraC) and non-toxic (eg D2/CA) agents.
Fig 2.
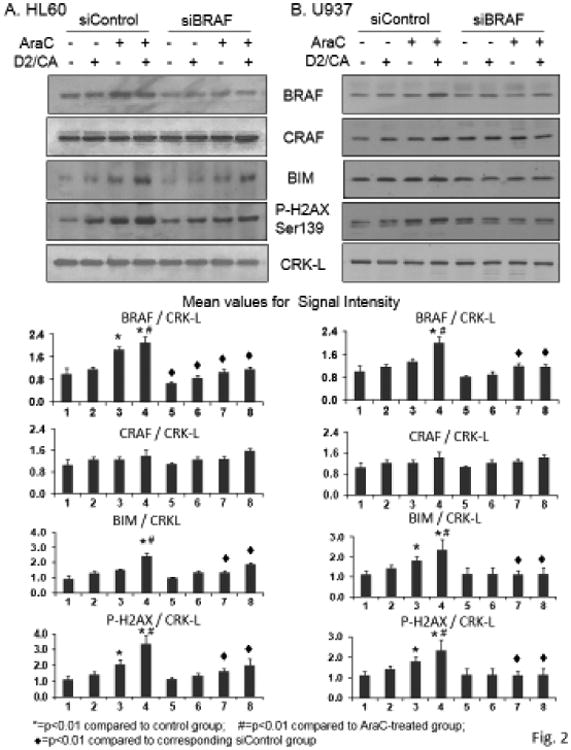
Silencing of BRAF expression selectively reduces BRAF protein expression as well as the phosphorylated H2AX and BIM protein levels. A. HL60 cells.The first four signals in each Western blot show that the incubation with a control silencing oligonucleotides, according to the protocol detailed in Fig 1, results in increased levels of BRAF but not CRAF protein when treated with AraC alone, or followed by D2+CA (lanes 3 and 4, respectively). In parallel, P-H2AX and BIM protein levels are also increased. In contrast, CRAF shows no upregulation by the treatment protocols used in this study, and the levels of CRAF proteins are unaffected by the exposure of the cells to siBRAF oligonucleotides. However, the marker of DNA damage P-H2AX and the pro-apoptotic BIM showed reduced levels following silencing of BRAF gene. CRK-L signals served as a loading control. B. U937 cells show similar effects to HL60 cells of BRAF dependence on AraC effects on P-H2AX increase, and its enhancement by D2/CA, as well as BIM phosphorylation. *=p<0.01 compared to control group; #=p<0.01 compared to AraC treated group; ◆=p<0.01 compared to corresponding siControl group, n= 3.
3.2. A silencing BRAF construct selectively reduces BRAF and BIM protein levelsand diminishes the enhancement of cell death by D2/CA
The specificity of the silencing construct for BRAF, as opposed for the related CRAF, was demonstrated in Fig 2 by the lack of effect of the siBRAF construct on CRAF protein expression (the second Western in Fig 2). To demonstrate that BRAF had a functional role in the enhancement phase of AraC cytotoxicity we silenced its expression, as shown in the subsequent lanes in this Western (Fig 2, lanes 5-8). The diminished levels of BIM, a regulator of apoptosis, after siBRAF exposure of the cells, suggested that in cells with DNA damage (P-H2AX Western in Fig 2) and in the presence of differentiation agents, BRAF has a pro-apoptotic function (Fig 2). This is in contrast to its well established pro-proliferation role in tumorigenesis. This was confirmed by determinations of cell viability by Trypan blue exclusion (TB) and by Annexin V staining (Table 2). Both methods showed that siBRAF reduced the enhancement of cell death when D2/CA followed the AraC treatment (compare groups 3 and 4 vs groups 7 and 8 in Table 2, A and B).
Table 2. Silencing BRAF expression reduces cell death enhancement induced by differentiation agent combination in HL60 and U937 cells.
| A | TB exclusion | P value | P value | Annexin V assay | |||
| HL60 cells | Cell death-Mean | vs CTL | vs AraC | Apoptosis | Necrosis | Total death | |
| 1. siControl-Control | 7.5% | 4.9% | 4.8% | 9.7% | |||
| 2. siControl-D2/CA | 12.8% | 0.009 | 5.8% | 6.4% | 12.2% | ||
| 3. siControl-AraC | 33.0% | 0.003 | 12.0% | 13.5% | 25.5% | ||
| 4. siControl-AraC-D2/CA | 47.3% | 0.001 | 0.001 | 15.0% | 18.9% | 33.9% | |
| 5. siBRAF-Control | 11.0% | 6.2% | 6.5% | 12.7% | |||
| 6. siBRAF-D2/CA | 12.5% | 0.339 | 5.9% | 6.5% | 12.3% | ||
| 7. siBRAF-AraC | 29.0% | 0.007 | 10.5% | 10.1% | 20.6% | ||
| 8. siBRAF-AraC-D2/CA | 39.0% | 0.003 | 0.009 | 12.4% | 14.1% | 26.4% | |
| B | TB exclusion | P value | P value | Annexin V assay | |||
| U937 cells | Cell death-Mean | vs CTL | vs AraC | Apoptosis | Necrosis | Total death | |
| 1. siControl-Control | 9.0% | 7.8% | 4.6% | 12.4% | |||
| 2. siControl-D2/CA | 10.3% | 0.691 | 10.1% | 6.0% | 16.1% | ||
| 3. siControl-AraC | 49.7% | 0.006 | 26.7% | 14.1% | 40.8% | ||
| 4. siControl-AraC-D2/CA | 58.3% | 0.009 | 0.049 | 36.6% | 25.6% | 62.2% | |
| 5. siBRAF-Control | 10.7% | 7.5% | 5.5% | 13.0% | |||
| 6. siBRAF-D2/CA | 11.3% | 0.529 | 8.8% | 7.0% | 15.8% | ||
| 7. siBRAF-AraC | 47.7% | 0.001 | 26.5% | 13.2% | 39.7% | ||
| 8. siBRAF-AraC-D2/CA | 54.0% | 0.004 | 0.092 | 33.2% | 21.1% | 54.3% | |
Experimental details are described in Materials and methods and the legend for Fig 1.
3.3. Inhibition of BRAF kinase activity reduces of the enhancement of AraC-induced cell death
To confirm the participation of BRAF in the enhancement of AML cell death, we inhibited its kinase activity using two pharmacological agents which potently inhibit BRAF, wild type or mutated, and considerably less effectively CRAF, but show little inhibition of other kinases [29-31]. Similar to siBRAF experiments, both BRAF inhibitors reduced the D2/CA-induced enhancement of cell death as determined by the TB and Annexin V methods (Table 3, A and B). This inhibition of BRAF kinase activity also reduced BIM protein levels, as well as the P-H2AX level (Fig 3), supporting the data on DNA damage and cell death reduction by siBRAF.
Table 3. The effects of inhibition of BRAF by pharmacological inhibitors on AraC-D2/CA enhancement of cell death in HL60 cells.
| A. TB exclusion | p value | p value | B. Annexin V assay | |||
|---|---|---|---|---|---|---|
| HL60 cells | Cell Death-Mean | vs AraC | vs AraC-D2/CA | Apoptosis | Necrosis | Total death |
| Control | 8.0% | 6.8% | 6.7% | 13.5% | ||
| AraC | 31.7% | 12.5% | 17.1% | 29.6% | ||
| AraC-D2/CA | 47.0% | 0.002 | 20.4% | 26.3% | 46.7% | |
| SB590885 (SB)- 1 nM | 8.8% | 7.2% | 7.7% | 14.9% | ||
| AraC-SB | 28.7% | 9.5% | 16.8% | 26.3% | ||
| AraC-SB-D2/CA | 41.5% | 0.017 | 0.006 | 16.7% | 18.9% | 35.6% |
| TAK632 (TAK)-10 nM | 8.0% | 7.4% | 9.1% | 16.5% | ||
| AraC-TAK | 27.0% | 11.7% | 13.6% | 25.3% | ||
| AraC-TAK-D2/CA | 38.5% | 0.002 | 0.008 | 14.5% | 18.5% | 33.0% |
Experimental details are described in Materials and methods and the in legend for Fig 3.
Fig 3.
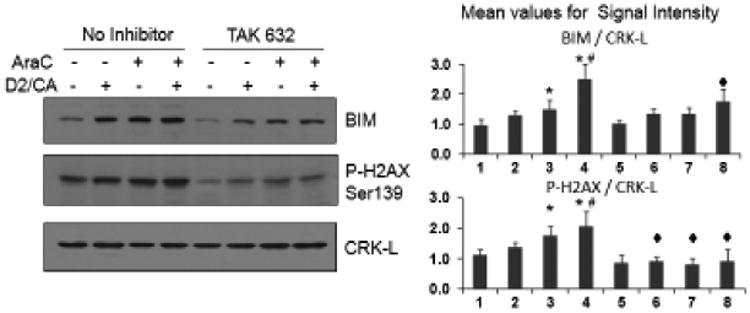
BRAF kinase activity is required for the induction of DNA damage by AraC and by AraC-D2/CA combination and contributes to the increased expression of BIM. The kinase activity of BRAF is inhibited by 96 hr treatment with the selective BRAF inhibitor TAK632 (10 nM). Note that P-H2AX levels are reduced below the basal levels, and BIM expression is reduced in the combination group. *=p<0.01 compared to control group; #=p<0.01 compared to AraC treated group; ◆=p<0.01 compared to corresponding siControl group, n= 3.
3.4. Vitamin D Receptor (VDR) is required for the optimal enhancement of BRAF expression by combination of AraC-D2/CA
To establish that the increased expression of BRAF is due to the differentiation-agent combination D2/CA, we reduced the expression of VDR, the transcription factor activated by VDDs, using siVDR constructs. As shown in Fig 4 (top Western) the siVDR construct markedly reduced VDR levels, and this was accompanied by the reduction of the upregulation of BRAF protein by AraC and D2/CA, as well as of the marker of DNA damage, P-H2AX. In this system total H2AX (T-H2AX) protein also increased in parallel. Thus, BRAF lies in the previously reported VDR→ BIM pathway, downstream from VDR (Fig 4), but upstream from BIM (Fig 2) and H2AX (Figs 2 and 3). However, there is a possibility that BRAF is only partially under the control of VDR in this system, as after our VDR knockdown BRAF still increases, though to a lesser extent, when exposed to D2/CA. Also, while it is possible that there a cell cycle effect of the combined treatment that might affect susceptibility to AraC, for example more or less cells in S phase, difficult to detect in this system, and remain to be investigated in future studies.
Fig 4.
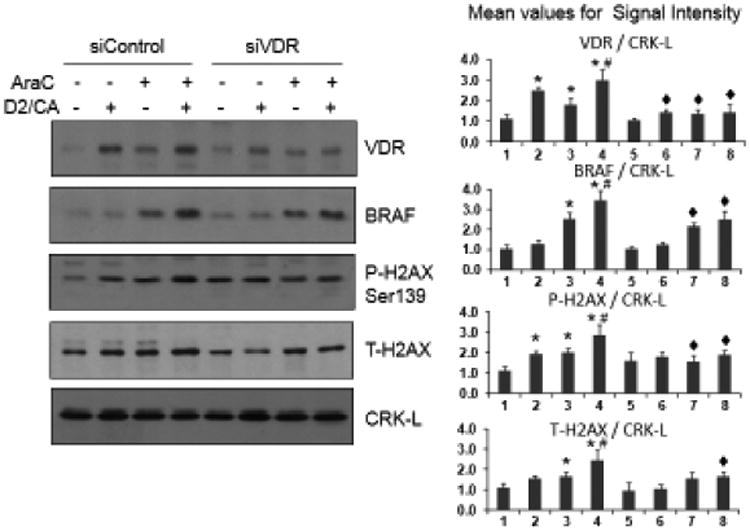
Reduced enhancement by D2/CA of the AraC effects on BRAF, P-H2AX and T-H2AX protein expression after partial silencing of VDR expression. HL60 cells were treated as detailed in Fig 1, and half of the cells were exposed to control oligonucleotides, while the remaining cells were treated with siVDR oligos (10 nM). The induction of VDR by the DNA damage-inducing agents were reduced approximately by 50%, along with somewhat smaller, but significant reductions in BRAF and H2AX proteins. *=p<0.01 compared to control group; #=p<0.01 compared to AraC treated group; ◆=p<0.01 compared to corresponding siControl group, n= 3.
3.5. AML patient samples ex vivo corroborate the findings with AML cell lines that BRAF is upregulated by AraC-D2/CA and contributes to AML cell death
Following informed consent in the context of IRB approved protocols, small leukemic blast samples were obtained from two patients with newly diagnosed AML, and one bone marrow (BM) sample from a healthy subject. These limited studies allowed confirmation of the generality of the results from cell lines that the expression of BRAF increases following AraC exposure, and that this is significantly increased by D2/CA treatment (Fig 5A). Further, siBRAF markedly reduced the expression of BIM protein (Fig 5B), and reduced the cell death differential between AraC alone and AraC-D2/CA (Figs 5, C and D). Notably, this differential, though modest, was significant (patient 1 (FAB M5, p= 0.012; patient 2 FAB M4, p=0.029) in the siControl groups, but lost statistical significance when siBRAF was transfected (Fig 5C, column “vs AraC”). Annexin V assay confirmed the overall result that BRAF is required for optimal cell death enhancement in freshly obtained AML blasts, though insufficient number of cells was available for replicate assays.
Fig 5.
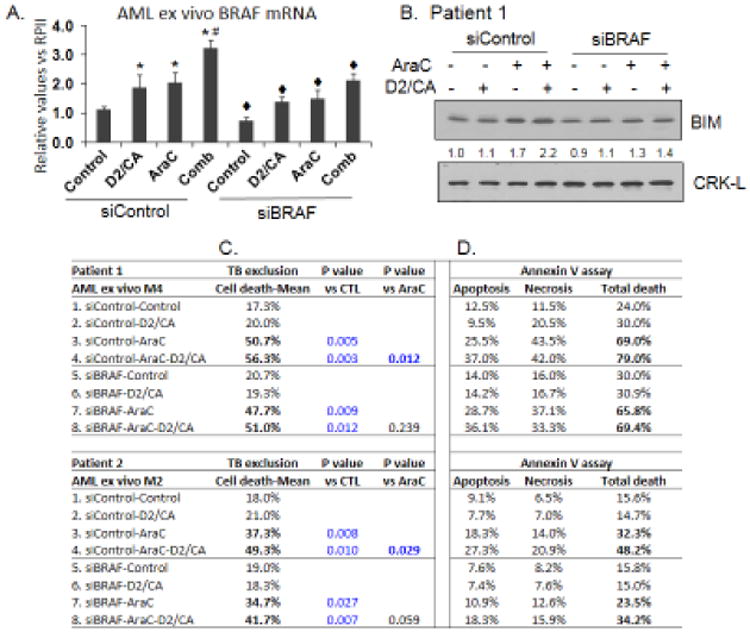
Reduced BRAF expression impedes BIM expression and cell death enhancement by D2/CA in patients AML blasts ex vivo. A. BRAF mRNA increases in the three treatment groups, but this is significantly reduced by siBRAF. Repeated RT-PCR determinations were performed on a sample taken from one patient. B. BIM protein increases when ex vivo cells are treated with AraC or with AraC-D2/CA, but this is inhibited by siBRAF. C. Cell death determinations by Trypan blue permeability, or D, by Annexin V flow cytometry, show that cell death difference (ie the enhancement of cytotoxicity) between treatment group 4 minus group 3, are reduced in both patients studied when BRAF expression is reduced by siBRAF (group 8 minus group 7). *=p<0.01 compared to control group; #=p<0.01 compared to AraC treated group; ◆=p<0.01 compared to corresponding siControl group, n= 3.
In the normal bone marrow (NBM) sample AraC also increased the expression of BRAF mRNA, but as expected, there was no significant increase following the addition of D2/CA, and inhibition of kinase activity of BRAF had no effect on BRAF expression (Table 4A). Consistent with the previously reported results that D2/CA have no effect on AraC-induced cell death in normal BM cells [20], and the current finding that BRAF is upstream of pro-apoptotic BIM but shows no change in the expression following the addition of D2/CA to AraC-treated cells, the inhibition of BRAF activity had no effect on the survival of normal BM cells (Table 4B).
Table 4. Effect of the inhibition of BRAF kinase activity on cell death and BRAF mRNA expression in mononuclear cells from a normal bone marrow (BM).
| A. BRAF mRNA (RT-qPCR) | B. TB exclusion | |||||
|---|---|---|---|---|---|---|
| BRAF | P value | P value | Cell death | P value | P value | |
| Normal BM | Mean | vs CTL | vs AraC | Mean | vs CTL | vs AraC |
| Control | 1.13 | 17% | ||||
| AraC | 2.44 | 0.010 | 47% | 0.005 | ||
| AraC-D2/CA | 2.49 | 0.005 | 0.665 | 49% | 0.002 | 0.160 |
| TAK632 (TAK) | 0.88 | 20% | ||||
| AraC-TAK | 2.20 | 0.048 | 48% | |||
| AraC-TAK-D2/CA | 2.38 | 0.033 | 0.053 | 47% | 0.401 | |
For details of the procedures see Materials and Methods Section.
Discussion
We demonstrate here, for the first time to our knowledge, that in contrast to any other mammalian cell type studied to date, in this system BRAF has pro-apoptotic activity for malignant hematopoietic cells with DNA damaged by AraC (Tables 2 and 3, Fig 5, C and D). In the in vitro experiments, in which exposure to D2/CA is only 4 days due the difficulty of maintaining ex vivo cells in culture, the reduction in AraC-induced cell death is quite small, but in the projected clinical situation the patients will be maintained on D2/CA for at least two months, so it is reasonable to expect that the effect will be more robust. The reduction of cell survival by BRAF in the context of exposure to differentiation inducers D2/CA is in stark contrast to many reports that BRAF has cell survival-enhancing activity, seen most dramatically when BRAF has mutations that drive neoplasms such as melanoma, colon cancer, and hairy cell leukemia [32-34]. However, in the protocol used in this study, in AML cells with DNA damage, BRAF -further upregulated by D2/CA combination-has an opposite effect- it promotes cell death. This emphatically illustrates that not only cell type, but also the cell's integrity, determines a biological effect of signals from the cell's environment.
Rather similar to our study, it was noted in a recent report that in primary human keratinocytes BRAF inhibitors can suppress apoptosis independently of MEK/ERK signaling. However, the authors proposed that this was not due to BRAF activity, but to an off-target inhibition of JNK signaling by BRAF inhibitors used in their study [35]. Although these BRAF inhibitors, Vemurafenib and Dabrafenib, are selective inhibitors of the oncogenic, ie mutant BRAF, the BRAF inhibitors used in our study can inhibit the mutated as well as wild type BRAF [29-31]. Thus, the possibility needs to be excluded that in the quoted study direct inhibition of BRAF resulted in the suppression of apoptosis in human keratinocytes. Of note, most likely there was DNA damage in those cells, as the cells were irradiated in their experiments, as in well as the cultured cutaneous squamous carcinoma cells (Ref 32, Fig 2 in that reference). This suggests the wider existence of BRAF-induced pro-apoptosis events when cellular DNA damaged.
Our studies on the anti-cancer actions of vitamin D have some carry over to the understanding of the mechanistic aspects of AML chemotherapy with AraC. For instance, we extend the previous observation that doxorubicin or etoposide-induced DNA damage can upregulate VDR expression in human non-small lung carcinoma H1299 and human osteosarcoma cell lines [36] to human AML cells treated with AraC (Fig 4). Also novel is our finding that the exposure to AraC alone upregulates BRAF expression (Fig 1 and Fig 2A), though it does not appear to be upregulated by VDR (Fig 4).
As yet there is no clear correlation that we have identified between the World Health Organization subtypes of AML and the intensity of the response to D2/CA therapy, but we are actively investigating which, if any, clinical features of AML correlate with responsiveness to this approach. However, it is important to realize that in every sample of AML patient blasts that we studied, or cell line investigated, there was some degree of cell death enhancement by D2/CA following AraC exposure of the blasts (19).
In summary, we report here an unusual pro-apoptotic, tumor-suppressor-like function for the frequently oncogenic BRAF, which may underpin the projected application of vitamin D-based regimen adjunct to the standard therapy of patients with AML.
Highlights.
The exposure of human AML cells to a sequential treatment using AraC followed by a Vitamin D analog/ plant anti-oxidant combination results in improved potential for effective therapeutic use of AraC, and is associated with an upregulated expression of BRAF.
Inhibition of BRAF expression or BRAF kinase activity inhibits BIM expression and reduces the potential therapeutic effect in this system.
The results obtained with AML cell lines are confirmed by ex vivo studies of AML blasts, and the selectivity of the increased cytotoxicity induced by Doxercalciferol (D2) / Carnosic Acid (CA) to malignant cells is shown by the lack of effect of D2/CA on BRAF expression or cell death in normal bone marrow cells.
Acknowledgments
We thank Dr E Gocek of Faculty of Biotechnology, the University of Wroclaw for helpful comments on the manuscript. The studies were supported by NIH grant R01-CA-044722-26 to GPS.
Footnotes
Disclosure: The authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross AC, et al., editors. Dietary Reference Intakes for Calcium and Vitamin D. Washington (DC): 2011. [PubMed] [Google Scholar]
- 2.Dalhoff K, Dancey J, Astrup L, Skovsgaard T, Hamberg KJ, Lofts FJ, Rosmorduc O, Erlinger S, Bach Hansen J, Steward WP, Skov T, Burcharth F, Evans TR. A phase II study of the vitamin D analogue Seocalcitol in patients with inoperable hepatocellular carcinoma. Br J Cancer. 2003;89:252–7. doi: 10.1038/sj.bjc.6601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu G, Wilding G, Staab MJ, Horvath D, Miller K, Dresen A, Alberti D, Arzoomanian R, Chappell R, Bailey HH. Phase II study of 1alpha-hydroxyvitamin D(2) in the treatment of advanced androgen-independent prostate cancer. Clin Cancer Res. 2003;9:4077–83. [PubMed] [Google Scholar]
- 4.Kim M, Mirandola L, Pandey A, Nguyen DD, Jenkins MR, Turcel M, Cobos E, Chiriva-Internati M. Application of vitamin D and derivatives in hematological malignancies. Cancer Lett. 2012;319:8–22. doi: 10.1016/j.canlet.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Harrison JS, Bershadskiy A. Clinical experience using vitamin d and analogs in the treatment of myelodysplasia and acute myeloid leukemia: a review of the literature. Leuk Res Treatment. 2012;2012:125814. doi: 10.1155/2012/125814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans TR, Colston KW, Lofts FJ, Cunningham D, Anthoney DA, Gogas H, de Bono JS, Hamberg KJ, Skov T, Mansi JL. A phase II trial of the vitamin D analogue Seocalcitol (EB1089) in patients with inoperable pancreatic cancer. Br J Cancer. 2002;86:680–5. doi: 10.1038/sj.bjc.6600162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheung FS, Lovicu FJ, Reichardt JK. Current progress in using vitamin D and its analogs for cancer prevention and treatment. Expert Rev Anticancer Ther. 2012;12:811–37. doi: 10.1586/era.12.53. [DOI] [PubMed] [Google Scholar]
- 8.Clark AS, Chen J, Kapoor S, Friedman C, Mies C, Esserman L, DeMichele A. Pretreatment vitamin D level and response to neoadjuvant chemotherapy in women with breast cancer on the I-SPY trial (CALGB 150007/150015/ACRIN6657) Cancer Med. 2014;3:693–701. doi: 10.1002/cam4.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta D, Trukova K, Popiel B, Lammersfeld C, Vashi PG. The association between pre-treatment serum 25-hydroxyvitamin D and survival in newly diagnosed stage IV prostate cancer. PLoS One. 2015;10:e0119690. doi: 10.1371/journal.pone.0119690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baron JA, Barry EL, Mott LA, Rees JR, Sandler RS, Snover DC, Bostick RM, Ivanova A, Cole BF, Ahnen DJ, Beck GJ, Bresalier RS, Burke CA, et al. A Trial of Calcium and Vitamin D for the Prevention of Colorectal Adenomas. N Engl J Med. 2015;373:1519–30. doi: 10.1056/NEJMoa1500409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrero D, Campa E, Dellacasa C, Campana S, Foli C, Boccadoro M. Differentiating agents + low-dose chemotherapy in the management of old/poor prognosis patients with acute myeloid leukemia or myelodysplastic syndrome. Haematologica. 2004;89:619–20. [PubMed] [Google Scholar]
- 12.Ferrero D, Crisa E, Marmont F, Audisio E, Frairia C, Giai V, Gatti T, Festuccia M, Bruno B, Riera L, Passera R, Boccadoro M. Survival improvement of poor-prognosis AML/MDS patients by maintenance treatment with low-dose chemotherapy and differentiating agents. Ann Hematol. 2014;93:1391–400. doi: 10.1007/s00277-014-2047-7. [DOI] [PubMed] [Google Scholar]
- 13.Slapak CA, Desforges JF, Fogaren T, Miller KB. Treatment of acute myeloid leukemia in the elderly with low-dose cytarabine, hydroxyurea, and calcitriol. Am J Hematol. 1992;41:178–83. doi: 10.1002/ajh.2830410307. [DOI] [PubMed] [Google Scholar]
- 14.Krishnan AV, Trump DL, Johnson CS, Feldman D. The role of vitamin D in cancer prevention and treatment. Endocrinol Metab Clin North Am. 2010;39:401–18. doi: 10.1016/j.ecl.2010.02.011. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnan AV, Trump DL, Johnson CS, Feldman D. The role of vitamin D in cancer prevention and treatment. Rheum Dis Clin North Am. 2012;38:161–78. doi: 10.1016/j.rdc.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo W, Johnson CS, Trump DL. Vitamin D Signaling Modulators in Cancer Therapy. Vitam Horm. 2016;100:433–72. doi: 10.1016/bs.vh.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, Shi JY, Zheng PZ, Yan H, Liu YF, Chen Y, Shen Y, Wu W, et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2004;101:5328–35. doi: 10.1073/pnas.0400053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–62. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 19.Momparler RL. A model for the chemotherapy of acute leukemia with 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1974;34:1775–87. [PubMed] [Google Scholar]
- 20.Harrison JS, Wang X, Studzinski GP. The role of VDR and BIM in potentiation of cytarabine-induced cell death in human AML blasts. Oncotarget. 2016 doi: 10.18632/oncotarget.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Harrison JS, Studzinski GP. Enhancement of arabinocytosine (AraC) toxicity to AML cells by a differentiation agent combination. J Steroid Biochem Mol Biol. 2015 doi: 10.1016/j.jsbmb.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birtic S, Dussort P, Pierre FX, Bily AC, Roller M. Carnosic acid. Phytochemistry. 2015;115:9–19. doi: 10.1016/j.phytochem.2014.12.026. [DOI] [PubMed] [Google Scholar]
- 23.Steiner M, Priel I, Giat J, Levy J, Sharoni Y, Danilenko M. Carnosic acid inhibits proliferation and augments differentiation of human leukemic cells induced by 1,25-dihydroxyvitamin D3 and retinoic acid. Nutr Cancer. 2001;41:135–44. doi: 10.1080/01635581.2001.9680624. [DOI] [PubMed] [Google Scholar]
- 24.Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F, Gallo R. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood. 1979;54:713–33. [PubMed] [Google Scholar]
- 25.Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17:565–77. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Harrison JS, Uskokovic M, Danilenko M, Studzinski GP. Silibinin can induce differentiation as well as enhance vitamin D3-induced differentiation of human AML cells ex vivo and regulates the levels of differentiation-related transcription factors. Hematol Oncol. 2010;28:124–32. doi: 10.1002/hon.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danilenko M, Wang X, Studzinski GP. Carnosic acid and promotion of monocytic differentiation of HL60-G cells initiated by other agents. J Natl Cancer Inst. 2001;93:1224–33. doi: 10.1093/jnci/93.16.1224. [DOI] [PubMed] [Google Scholar]
- 29.Takle AK, Brown MJ, Davies S, Dean DK, Francis G, Gaiba A, Hird AW, King FD, Lovell PJ, Naylor A, Reith AD, Steadman JG, Wilson DM. The identification of potent and selective imidazole-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett. 2006;16:378–81. doi: 10.1016/j.bmcl.2005.09.072. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013;73:7043–55. doi: 10.1158/0008-5472.CAN-13-1825. [DOI] [PubMed] [Google Scholar]
- 31.Okaniwa M, Hirose M, Arita T, Yabuki M, Nakamura A, Takagi T, Kawamoto T, Uchiyama N, Sumita A, Tsutsumi S, Tottori T, Inui Y, Sang BC, et al. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C-7-substituted 1,3-benzothiazole derivatives. J Med Chem. 2013;56:6478–94. doi: 10.1021/jm400778d. [DOI] [PubMed] [Google Scholar]
- 32.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 33.Oliveira C, Pinto M, Duval A, Brennetot C, Domingo E, Espin E, Armengol M, Yamamoto H, Hamelin R, Seruca R, Schwartz S., Jr BRAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogene. 2003;22:9192–6. doi: 10.1038/sj.onc.1207061. [DOI] [PubMed] [Google Scholar]
- 34.Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci R, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–15. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vin H, Ojeda SS, Ching G, Leung ML, Chitsazzadeh V, Dwyer DW, Adelmann CH, Restrepo M, Richards KN, Stewart LR, Du L, Ferguson SB, Chakravarti D, et al. BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. Elife. 2013;2:e00969. doi: 10.7554/eLife.00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kommagani R, Payal V, Kadakia MP. Differential regulation of vitamin D receptor (VDR) by the p53 Family: p73-dependent induction of VDR upon DNA damage. J Biol Chem. 2007;282:29847–54. doi: 10.1074/jbc.M703641200. [DOI] [PMC free article] [PubMed] [Google Scholar]