

Abstract
Coproheme decarboxylase catalyzes two sequential oxidative decarboxylations with H2O2 as the oxidant, coproheme III as substrate and cofactor, and heme b as the product. Each reaction breaks a C-C bond and results in net loss of hydride, via steps that are not clear. Solution and solid-state structural characterization of the protein in complex with a substrate analog revealed a highly unconventional H2O2-activating distal environment with the reactive propionic acids (2 and 4) on the opposite side of the porphyrin plane. This suggested that, in contrast to direct C-H bond cleavage catalyzed by a high-valent iron intermediate, the coproheme oxidations must occur through mediating amino acid residues. A tyrosine that hydrogen bonds to propionate 2 in a position analogous to the substrate in ascorbate peroxidase is essential for both decarboxylations, while a lysine that salt bridges to propionate 4 is required solely for the second. A mechanism is proposed in which propionate 2 relays an oxidizing equivalent from a coproheme compound I intermediate to the reactive deprotonated tyrosine, forming Tyr■. This residue then abstracts a net hydrogen atom (H■) from propionate 2, followed by migration of the unpaired propionyl electron to the coproheme iron to yield the ferric harderoheme and CO2 products. A similar pathway is proposed for decarboxylation of propionate 4, but with a lysine residue as an essential proton shuttle. The proposed reaction suggests an extended relay of heme-mediated e−/H+ transfers and a novel route for the conversion of carboxylic acids to alkenes.
Keywords: heme b, propionate, tyrosyl, decarboxylation
Graphical abstract
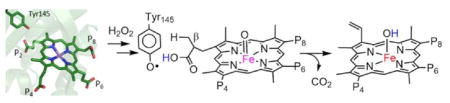
INTRODUCTION
Activation of O2 or H2O2 by heme often leads to the generation of high-valent iron-oxo (ferryl) intermediates.1 In reactions catalyzed by heme peroxidases, donation of two electrons from the Fe(III)-porphyrin complex results in heterolytic cleavage of the HO-OH bond. The resulting compound I intermediate carries one oxidizing equivalent on the iron and the other in the porphyrin π system, and is consequently described as Fe(IV)=O coupled to a porphyrin radical cation (por■+):2, 3
| (1) |
How this central intermediate forms and how it oxidizes compounds, sometimes over long distances, are longstanding questions in catalysis research. In reactions catalyzed by many cytochrome P450s and peroxidases,4 1 e− + 1 H+ are transferred from a substrate to compound I. Such reactions, known as proton coupled electron transfers,5 can occur in a single concerted proton/electron transfer step. Alternatively, as in the catalytic model for P450s,3 the proton may migrate to the ferryl oxygen while the electron enters an orbital that is porphyrin-based. The overall reaction is described as hydrogen atom transfer.6
The proton and electron may have distinct routes to their separate destinations, though in only few cases have these been rigorously described. In ascorbate peroxidase (APX), the ascorbate substrate binds near and donates a hydrogen bond (H-bond) to one of the heme propionates (Fig. S1).7, 8 Hydrogen atom transfer from ascorbate, in both APX and a synthetic heme complex designed to mimic it,9 occurs with the propionate acting both as a base toward the proton and as the conduit for the electron to por■+. The proton is shuttled to an active site arginine and water molecules before finally arriving at the ferryl oxygen,10 and the resulting Fe(IV)=O + H+ or Fe(IV)-OH complex is known as compound II.11 Transfer of a hydrogen atom from a second substrate leads back to the ferric heme and a molecule of water. Representing the substrate as SH gives the general reaction scheme:
| (2) |
| (3) |
The propionate in APX (Fig. S1) allows the heme to transmit oxidizing equivalents, from the site where the heme iron converts H2O2 to its catalytically activated compound I form, to the substrate binding site. In some peroxidases, including cytochrome c peroxidase (CCP) and lignin peroxidase (LnP),12–14 the sites of heme-Fe(III)/H2O2 reaction and substrate oxidation may be quite spatially remote. Redox-active amino acid side chains15–17 in addition to the propionates18 act as internal mediators within these enzymes. These supply the means to move oxidizing equivalents away from por■+ and to the site where substrates bind, greatly extending the catalytic reach of heme as an oxidation catalyst.
Here, we describe the combined structural and solution-state characterization of a H2O2-activating heme protein in which the iron and the pair of organic substrates have an unusual spatial and electronic relationship. The enzyme uses H2O2 as an oxidant in the successive decarboxylations of propionates 2 and 4 of iron coproporphyrin III (coproheme), which serves as both substrate and cofactor in the reactions (Scheme 1). Use of H2O2 as the biological oxidant in a decarboxylation or indeed any C-C bond cleavage is a highly uncommon feature of this enzyme,19 which is encoded by the hemQ gene and which is responsible for the last step in heme biosynthesis in many gram positive bacteria.20 The 1.8 A resolution structure of the protein in complex with its Mn(III)-containing substrate analog clearly shows the two reactive propionates residing below the coproheme plane, in a position where they cannot directly make contact with a ferryl intermediate. Instead, the structure suggests and the solution data support an electron/proton relay involving the heme, its propionates, and reactive amino acid side chains, leading to net transfers of an electron and a hydrogen atom from the reactive propionates themselves. The structurally-inferred, experimentally supported reaction mechanism is unprecedented in biology. As such, it suggests possible new avenues for extending the catalytic repertoire of synthetic heme complexes.
Scheme 1. Reaction catalyzed by coproheme decarboxylase†.
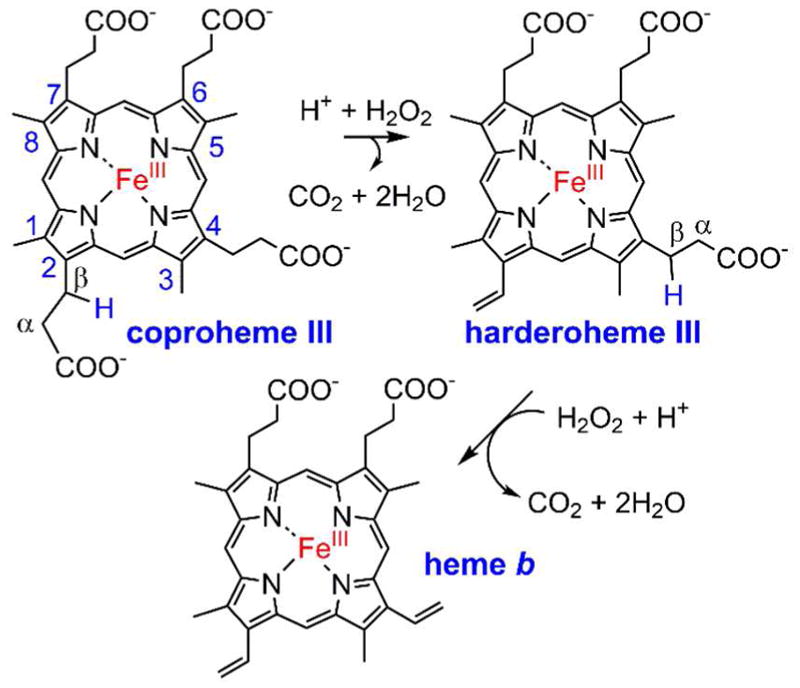
†Net loss of 2e− + H+ from the Cα-Cβ bond of each propionate occurs along with decarboxylation. Involvement of Fe(III), H2O2, and harderoheme III in the reaction scheme was previously verified.21, 22 The porphyrin species are drawn in their expected protonation states at pH 7.
EXPERIMENTAL PROCEDURES
Expression, purification, and ligand binding to WT and mutant coproheme decarboxylases
The sequence of the protein encoded by the hemQ from Geobacillus stearothermophilus (Gs, NCBI accession WP_053414189, PDB ID: 1T0T)23 was used to design a codon-optimized synthetic hemQ gene for expression in untagged form in Escherichia coli. The gene was inserted into a pET14(a) plasmid between the Nde1 and Xho1 restriction sites and used to transform competent E. coli Tuner (DE3) cells (Novagen). A similar construct for expressing the untagged, homologous protein from Staphylococcus aureus (Sa) was previously described.21 A QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) was used to generate site-directed mutants of Sa decarboxylase using the manufacturer’s instructions and primers in Table S1. For residues closer to the protein interior, substitutions were made from Tyr/Ser, or Trp/Phe- in order to retain the hydrophobic/hydrophilic character of the WT residue while dramatically changing its size or chemical functionality. Residues at the solvent interface were substituted by Ala.
Proteins were overexpressed and isolated at >95% purity by reverse-anion exchange and gel filtration chromatography as previously described.21 Concentrated proteins (Amicon, 10,000 MWCO centrifuge filters) were incubated in the dark with gentle stirring at 4 °C for 24 h with either the substrate ferric coproheme III or the substrate analog Mn(III) coproporphyrinate III (coproporphyrinate III = tetrakis-2,4,6,7-propionate-protoporphyrin, Frontier Scientific) in a 1:1 subunit: coproheme ratio. Unbound coproheme was removed and the protein-ligand complexes further purified on an S-200 Sephacryl gel filtration column (0.4 ml/min). Fractions were collected using an AKTA purification system and then screened via UV/Vis spectroscopy (Cary50) for coproheme content. Fractions with Rz values ≥0.8 (Rz = absorbance So-ret/absorbance 280nm) were pooled. The Bradford and pyridine hemochrome assays for protein and released coproheme, respectively, were used to determine the coproheme occupancy in the purified complexes. Extinction coefficients determined for the Gs coproheme decarboxylase complexes were εSoret = 110.6 mM−1 cm−1 (Fe-coproheme)21 and εSoret = 69.7 ± 6.6 mM−1 cm−1 (Mn-coproheme) (Fig. S2).
Structure Determination
The Gs decarboxylase in complex with Mn(III)coproheme was crystallized by hanging drop vapor diffusion at 20 °C. Drops were assembled by mixing the complex (7 mg/mL in 10 mM HEPES-NaOH pH 7.5) with an equal volume of reservoir solution (0.1 M citrate pH 5.0–5.5, 15–25 % (w/v) polyethylene glycol 3350). Bright red crystals of the complex formed after approximately 2 weeks. Crystals were briefly soaked in reservoir solution supplemented with 15% (w/v) glycerol as a cryoprotectant prior to freezing in liquid N2. An X-ray diffraction dataset to 1.8 A was measured at Stanford Synchrotron Radiation Lightsource beamline 14-1 and processed with autoxds.24 The structure was determined by molecular replacement using Phaser25 in the CCP4 program suite26. Phaser was supplied with the non-substrate bound structure as a search model (PDB ID 1T0T). Model building and refinement and was done using Coot26 and Phenix.27 Electron density maps of the Phaser solution showed the presence of a ligand adjacent to His172 (Sa sequence numbering, Fig. S3); Mn-coproheme (designated as 76R in the PDB file) was modeled into this electron density. Iterative rounds of model building with Coot and refinement with Phenix24 yielded the final model (PDB ID 5T2K) with Rfree = 17.6 % and Rwork = 15.4 %. The following residues are not included in the final model due to weak electron density: residues 115–123, 114–117, 112–117, 114–119 and 115–123 (all Gs numbering) in subunits A, B C, D and E, respectively. Molecular graphics were created with PyMOL (www.pymol.org).
Determining protein-heme dissociation constants
Values of KD for equilibrium binding of decarboxylases/hemes were determined by fluorescence quenching (Cary). Tryptophan fluorescence (5 μM subunit) was excited at 295 nm and emission monitored at 340 nm. Quenched emission at 340 nm was plotted versus the concentration of added heme ligand. Plots were fitted with the Langmuir-Hill equation to determine the KD.
| (4) |
θ is the fraction of ligand-binding sites occupied by the ligand, [L] is the ligand concentration, and n is the Hill coefficient describing cooperativity.
Reactions of decarboxylase-coproheme complexes monitored following titrimetric addition of oxidants
Complexes of decarboxylase with Mn- or Fe-coproheme (pH 7.4, 20 °C) were mixed with aliquots of H2O2 or peracetic acid from 1–50 mM stocks. The progress of reactions was monitored by UV/vis spectroscopy (Cary50, samples 10 μL in heme species). Reaction products were analyzed by high performance liquid chromatography (HPLC) and HPLC coupled to electrospray ionization mass spectrometry (HPLC-ESI-MS) (samples 40 μM in the heme species). MS was used to verify the identities of all reactants and products via their exact masses. Fe-bound coproheme, harderoheme (isomers III or IV), and heme b, or Mn-bound coproheme/heme b were separated and quantified via their integrated HPLC peak intensities referenced to standard curves:
HPLC: 20 μL samples were injected onto a Hypersil Gold PFP 5μm column (150 mm × 4.6 mm, Thermo Fisher) attached to an Agillent1100 series instrument. Solvent A was H2O with 0.1% trifluoroacetic acid (TFA) and Solvent B was acetonitrile (ACN) with 0.1% TFA. Samples were run at a flow rate of 2 mL/min starting with isocratic 70% A:30% B for 3 min, followed by a linear gradient transitioning from 70% A to 5% A over 12 min. This was followed by isocratic 5% A for 5 min and a final isocratic run of 30 % A for 3 min. UV/vis detection was at 411 nm. Standard curves ranging from 0 – 500 pmol of the tetrapyrroles versus peak areas were constructed. HPLC peak areas were determined in triplicate and error bars are reported as ± 1 standard deviation.
HPLC-ESI-MS: The chromatography conditions described above were used with an Agilent PLRP-S PSDVB column (PL1312-1300) and 1290 HPLC system. Mass spectra were obtained on an Agilent 6538 quadrupole time of flight (QTOF) instrument with ESI source (drying gas 8.0 L/min, drying gas heat at 350°C, nebulizer 55 psi, capillary voltage 3500 V, capillary exit 100 V). Spectra were collected in negative mode from 50 to 1700 m/z at a rate of 2 Hz.
Steady state kinetics of catalase reactions
The conversion of H2O2 to O2 and water by various decarboxylase-metalloporphyrin complexes was monitored continuously over time via polarography using a Clark O2 electrode (YSI). The electrode was equilibrated to 20 °C for 1 h using a circulating water bath and calibrated to the concentration of O2 in air-saturated distilled/deionized water. Reactions were carried out in 2 mL volumes (50 mM potassium phosphate buffer, pH 7.4) with 4 μM of enzyme-porphyrin complex and initiated by addition of 10–100 mM H2O2 stocks via airtight syringe (5–20 μL). Linear initial rates (vi) were fit by least-squares regression to the first 5–10% of the progress of reaction curves (Kaleidagraph). Plots of vi versus H2O2 concentration were fit to the Michaelis Menton equation:
| (5) |
Here, E is the concentration of enzyme-metalloporphyrin complex. Unreacted H2O2 was quantified at the end of reactions using horseradish peroxidase (HRP) and a colorimetric substrate (2,2′-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt, ABTS). 0.5U/mL HRP and 500 μM ABTS (final concentrations) were added to the samples and incubated (25 °C) for 1hr. The absorbance at 738 nm was recorded and compared with a standard curve generated over 5–100 μM H2O2].
Resonance Raman (rR) spectroscopy
Ferric samples for rR experiments were prepared at 25 to 60 μM coproheme-decarboxylase complex in 100 mM sodium phosphate at pH 7.5 Using Schlenk techniques, ferrous complexes were prepared anaerobically at 20 °C from ferric enzymes by reduction with an excess of buffered sodium dithionite. The CO complexes were prepared by flushing the ferrous enzymes with natural abundance CO or 13CO (99% 13C).
The rR spectra were recorded using the 135° backscattering geometry and f1 collection using a previously described spectrometer.21 Either the 406.7-nm or the 413.1-nm emission line from a Kr+ laser was used for Raman excitation, and the laser beam was focused to a line on a spinning 5 mm NMR tube. External spectral calibration standards for the rR spectra were toluene, acetone, methylene bromide, and d6-dimethylsulfoxide. Laser power for ferric samples ranged from 4 to 12 mW; ferrous CO complexes required laser irradiation powers of 1 to 3 mW to prevent CO photolysis. To check the integrity of the rR samples, their UV-visible spectra were checked before and after irradiation in the laser beam. Normal mode assignments were made by analogy to other heme proteins. Peak fitting analyses of the rR spectra were based on the minimum number of peaks required to achieve statistically valid fits of the spectra. Band shapes were assumed to be Gaussian and were well modeled by Gaussian line-shape functions.
RESULTS
Co-crystals of the decarboxylase with Mn-coproheme diffracted to 1.8 Å resolution
Our ongoing biochemical characterization of the coproheme decarboxylase and its genetic knock out has focused on S. aureus due to the importance of pathogenic strains of that species.21, 28 Robotic screening of thousands of crystallization conditions failed to generate diffraction-quality crystals of the enzyme from Sa, either alone or in complex with metalloporphyrins. However, the substrate-free structure of a coproheme decarboxylase from another member of its phylum, Geobacillus stearothermophilus, was available through a structural genomics consortium (PDB ID: 1T0T).23 The Gs and Sa proteins have 59% sequence identity (Fig. S3). UV-vis and rR spectra (Fig. S4–S5) and reactivity (Fig. S5) for the two are similar, though the Gs enzyme appears to require more H2O2 to convert bound coproheme or harderoheme to heme b, and the heme b complex with the protein is less stable (reflected in its higher KD, Table S2). The lower protein/heme b affinity for the Gs decarboxylase may be responsible for the increased susceptibility of the heme b to degradation by H2O2, apparent in the H2O2 titration data shown in Fig. S5. Given the overall similarity between the Sa and Gs proteins, we therefore attempted to generate crystals with Fe(III)coproheme using the Gs decarboxylase, which was likewise unsuccessful. The enzyme complex with the substrate analog Mn(III)coproheme, however, yielded large crystals that diffracted to 1.8 A resolution (Table S3). In solution, neither the Gs nor the Sa Mn(III)coproheme-decarboxylase complex reacted with H2O2 (up to 50 eq). However, the Mn(III) complexes reacted with peracetic acid (PAA), yielding the expected Mn-substituted harderoheme intermediate and heme b products (Fig. S6).22 These results suggest that the Mn(III)coproheme binds in the catalytic orientation but is incapable of activating H2O2 and therefore is stable as a solid state complex in the presence of O2 and ionizing X-ray radiation, conditions under which H2O2 may be spontaneously produced.
Structural changes associated with substrate binding to coproheme decarboxylase are confined to the C-terminus
Like other members of its family (Pfam 06778, which includes both chlorite dismutase [Cld] and coproheme decarboxylase enzymes),29 the crystal structure of the Gs coproheme decarboxylase is a homopentamer where each subunit consists of two ferrodoxin-like α/β domains connected by a pseudo-twofold axis (Fig. 1A). The same domain is shared by both non-canonical IsdG-type heme oxygenases and heme-binding DyP-type peroxidases, as previously noted.28 The superimposed structures of the ligand-free (PDB ID 1T0T)23 and -bound (5T2K) forms of subunit A are shown in Fig. 1B (Cα RMSD = 0.47 A). The peptide backbone and side chains mostly overlay, with the structural changes accompanying ligand binding confined to the C-terminal region housing the Mn-coproheme. In particular, residues 111–121 form part of a loop on the solvent-exposed exterior of the ligand-free homopentamer, where they might constitute the site of substrate entry/product egress. (Sa residue numbering is used except where noted.) The most dramatic rearrangements in the ligand-bound structure occur here (Fig. 1C). Though several of its side chains could not be modeled due to weak electron density in this region for the individual coproheme-bound subunits, the loop as a whole folds inwards toward the coproheme in the ligand-bound structure. This rearrangement brings the side chain of Asn112, which occupies a position inside the substrate-binding pocket, toward the protein exterior in the ligand-bound structure. Tyr113 and Arg131, by contrast, form new contacts with propionates 4 and 6, respectively, which could be significant for the oxidative decarboxylation mechanism (see below).
Figure 1. Superposition of the apo and ligand-bound forms of coproheme decarboxylase illustrate the effects of tetrapyrrole binding.
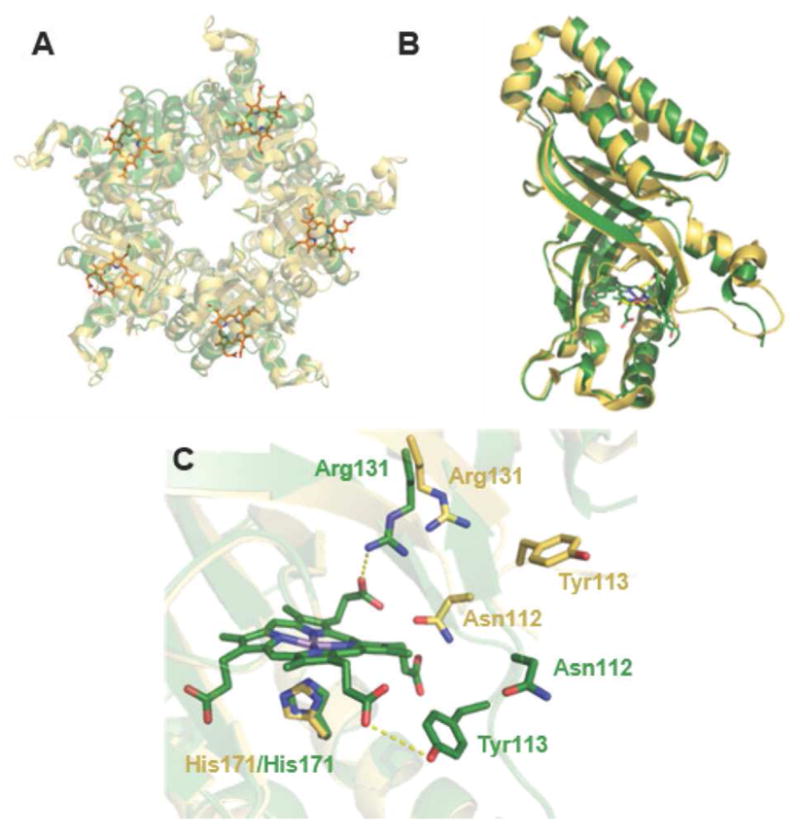
A. The Mn(III)-coproporphyrin-bound decarboxylase homopentamer is shown in green with the bound Mn(III)- coproporphyrin ligands as sticks (orange) (5T2K) and the ligand-free structure in yellow (1T0T). The view is down the pseudo-C5 axis with the coproheme-containing domains oriented toward the viewer. B. Superimposition of an individual subunit of coproheme decarboxylase in the apo (yellow) and ligand-bound (green) forms. Differences are localized around a flexible loop near residues 111–120 and the Mn(III)-coproporphyrin binding site. C. Close-up view of the Mn(III)-coproporphyrin binding site, illustrating prominent shifts in the positions of side chains for Asn112, Tyr113, and Arg131 in the apo- (yellow) and ligand-bound (green) forms.
Coproheme binding site structure: Implications for mechanism
The electron density map for Mn-coproheme bound to the active site (Fig. 2A) illustrates the unambiguous assignment of the atomic positions of the porphyrin and all of its side chains in the crystal structure. The porphyrin ring is slightly ruffled, occupying roughly the same position as the molecule of solvent in the ligand-free structure or the heme b which serves as a cofactor in Clds. However, the coproheme is rotated approximately 90° about the pseudo-C4 axis relative to the heme b in either Cld (Fig. S7) or energy-minimized predictions of the structure of the decarboxylase-coproheme complex.31 This orientation positions the two unreactive propionates (6 and 7, Scheme 1) pointing outward from the protein toward the solvent, where they occupy positions on the distal side of the heme plane (Fig. 2B). The two reactive propionates (2 and 4), by contrast, are rotated to the proximal side of the porphyrin plane and well beyond the reach of the open, distal coordination site.
Figure 2. The structure of the coproheme binding site in coproheme decarboxylase has important implications for the chemical mechanism.

A. View of the Mn-coproheme in subunit B with Fo-Fc omit electron density map contoured at 3σ. B. The tetrapyrrole is slightly ruffled, with the two reactive propionates (2 and 4) below its approximate plane. A close-up view of the His171 ligand in the (proximal) pocket beneath the substrate analog shows that it binds to the metal through the imidazole-Nε. The imidazole-Nδ forms an apparent hydrogen bond to a water molecule, which is itself hydrogen bonded to Ser223 and Asp220. C. The distal region above the coproheme plane is shown, highlighting the pair of unreactive propionates. The R131 side chain is part of a hydrogen bonding network involving propionate 6, Gln185, and a water molecule that occupies a position close to where the terminal oxygen of Fe-OOH might reside. The residue immediately over the coproheme plane is Ile187. The environment surrounding the reactive propionates 2 and 4 is shown in D and E, respectively.
On the other side of the plane (proximal pocket), histidine 172 coordinates the Mn of the coproheme (2.3 A) via the imidazole-Nε atom (Fig. 2B). The imidazole-Nδ is H-bonded to a water molecule, which is itself H-bonded to Ser222, Asp219, and the Met216 main chain carbonyl. Most H2O2-activating heme enzymes as well as Clds have an aspartic or glutamic acid as the hydrogen bonding donor in the analogous position,32–34 leading to some degree of negative charge in the proximal ligand. The negatively charged histidine supplies an electronic push that favors heterolytic cleavage of the O-O bond in the heme-Fe(III)-OOH that forms in trans. Water is a comparatively weak H-bonding partner; consistent with previously reported rR spectra,21 His172 is electronically neutral, leading to a more globin-like metal-His interaction.
The distal pocket (Fig. 2C) is likewise distinct from other H2O2-reactive heme proteins. Canonical peroxidases share a distal histidine with a relatively low pKa due to a nearby arginine.34 In its deprotonated form, the distal His acts as a base toward H2O2 (pKa = 11). Heterolytic bond cleavage in the resulting heme-Fe(III)-OOH complex is catalyzed by proton donation from the His-Arg pair, yielding Compound I and H2O. By contrast, the distal environment in the coproheme decarboxylase is relatively hydrophobic. Ile187 hovers over the center of the porphyrin plane. Gln185 resides at its side and in the same sequence/structural position occupied by the distal arginine from Clds (Fig. S8). This residue, which distinguishes the decarboxylase- and dismutase-encoding hemQ and cld genes, is important for the heme-dependent chlorite (ClO2−) decomposition mediated by Clds to yield Cl− and O2.35
At the edge of the distal pocket nearest the solvent interface, the Arg131 side chain sits at the center of a H-bonding network involving propionate 6, Gln185, and a water molecule (Fig 2C). This water occupies a position close to where the terminal oxygen of a coproheme-Fe(III)-OOH intermediate could reside. Though the solution pKa of the Arg side chain is high (12.5) and the propionate is low (3.2–3.5), the two together could in principle act as a base toward H2O2, an acid toward coproheme-Fe(III)-OOH, or both. An arginine side chain with a similar H-bond to a propionate has been proposed to act as a distal base in lieu of His-Arg in dye decoloring peroxidases.36 Prior work showed that the second order reaction between the decarboxylase-coproheme complex and H2O2, which leads to the ferric heme b complex, accelerates with pH (~3-fold, pKa = 7.4) and is solvent-isotope dependent (average k(H2O):k(D2O) = 2.2).22 Both results suggest the involvement of proton movement in the rate limiting step of the H2O2 reaction. However, pH has a much larger influence over the analogous, acid-base-catalyzed steps in heme peroxidases.34, 37 The roles of these residues and of pH therefore remain to be determined.
Reactive propionates 2 and 4 are each converted (net 2e−+1H+ lost per propionate) to a vinyl group and CO2 by a H2O2-mediated oxidation reaction (Scheme 1). Key residues surrounding each are shown in Figs. 2D and E. Propionate 2, which reacts first,21,22 forms H-bonds to the side chain hydroxyls of Ser223 (2.2 A) and Tyr145 (2.8 A). The former is part of a H-bonding network leading to the proximal His172. The latter is just 3.1 A away from the Cβ of the propionate (Scheme 1).
This was the observed site of net hydrogen atom removal catalyzed by coproporphyrinogen oxidase,38 an enzyme that carries out analogous oxidative decarboxylations on the metal-free, reduced analog of coproheme III. Removal of a hydrogen atom at this position leaves a carbon radical conjugated to, and consequently stabilized by, the tetrapyrrole. Additionally, propionate 2 is H-bonded to a water molecule which is in turn H-bonded to Arg218. Propionate 4 is directly H-bonded/salt-bridged to Lys149 (2.7 A), and indirectly to Trp156 and Tyr113 by a network of H-bonds including a bridging water molecule. Another H-bond network leads to Trp198 via the Oε1 atom of Glu109 and a second water molecule. The Nε of Trp198 has the closest through-space approach to the Cβ of propionate 4 (3.9 A). The occurrence of multiple residues with possible redox (Tyr, Trp) and proton-translocating activities suggests one or more may be involved in catalyzing the two oxidative decarboxylations.
The solution state Sa and Gs decarboxylase heme environments are similar to one another and distinct from chlorite dismutases. Resonance Raman spectra were measured for the Sa and Gs decarboxylases in complex with ferric coproheme and heme b (Fig. 3A). Comparison of the low frequency spectra of the coproheme complexes demonstrated similarities in their propionate bending (δ(CβCcCd), in this nomenclature Cc and Cd correspond to carbons labeled Cβ and Cα in Scheme 1 frequencies (Sa: 374 and 393 cm−1; Gs: 374 and 395 cm−1). Reaction of both coproheme-decarboxylase complexes with H2O2 (Fig. S3) yielded comparable rR spectral changes,21 including the appearance of bands corresponding to vinyl stretching and bending modes with very similar frequencies (1627 and 415–418 cm−1, respectively). The 374-cm−1 propionate bending frequency is unaffected by decarboxylation while the second propionate bending frequency decreases by 4 cm−1 in both decarboxylases. The similarities between the solution spectra and reactivities of the Gs and Sa enzyme-substrate complexes suggest it is reasonable to expect that their structures, especially the positioning of their reactive coproheme 2-and 4-propionate groups, are comparable.
Figure 3. Low frequency rR spectra report on the core and peripheral environment of hemes bound to decarboxylases.

A) The 406.7-nm excited rR spectra of ferric forms of a) coproheme: Sa decarboxylase, b) coproheme:Gs decarboxylase c) heme b:Sa decarboxylase, d) heme b:Gs decarboxylase, and e) DaCld(R183Q). Ferric samples were in 100 mM potassium phosphate at pH 7.4. B) Soret-excited rR spectra of CO complexes of a) coproheme:Sa decarboxylase, b) heme b:Sa decarboxylase, c) WT DaCld, and d) DaCld(R183Q). Both DaCld enzymes contain heme b. Spectra of CO samples in 100 mM potassium phosphate at pH 6.8 were acquired with 413.1 nm excitation and 2 mW power at the sample. Original spectra, black; fit spectra, red; component bands making up the fit spectra, gray.
Prior simulations of coproheme binding to the decarboxylase had the non-reactive propionates 6 and 7 in the same positions as the propionates of the heme b in the Cld structures, in contrast with the solid state structure shown here.31 Unique peripheral substituent environments of ferric heme b in decarboxylases and Clds are supported by their propionate and vinyl (δ(CβCaCb)) bending mode frequencies. The decarboxylase complexes exhibit δ(CβCcCd) and δ(CβCaCb) frequencies 8–11 cm−1 and 4–8 cm−1 lower, respectively, relative to those modes in WT DaCld and DaCld Arg183Gln mutant (Fig. 3A).35 The latter substitution exchanges the Cld-associated distal Arg with the distal Gln of the Sa decarboxylase, rendering their distal environments more similar. To further examine the solution state environment of the non-reactive propionates, the rR spectrum for the ferrous Sa decarboxylase-heme b complex with CO was measured and compared with counterpart spectra from WT DaCld and the DaCld(R183Q) (Fig. 3B). Comparison of the decarboxylase-CO and Cld-CO spectra clearly shows that the bands associated with propionate and vinyl bending, as well as the porphyrin ring, occur at different frequencies and with different band widths. The bending modes for propionates 6 and 7 of heme b:decarboxylase-CO are observed at 377 and 395 cm−1, in contrast to one broad band at 372 cm−1 in the DaCld-CO spectrum. The frequency of the vinyl bending mode for heme b:decarboxylase-CO is 10 cm−1 lower than that of WT DaCld (416 versus 426 cm−1). The DaCld R183Q mutation has little effect on these propionate frequencies, suggesting that the observed differences in the spectra are not due to differences in their distal environments. In addition to these differences in vinyl/propionate-associated frequencies, the ν8 core size marker band (M-N stretch) of the heme b:decarboxylase-CO occurs at 348 cm−1, and 8–9 cm−1 higher in frequency in the DaCld spectrum. Finally, a large band at 341 cm−1 that is tentatively assigned to the pyrrole tilting mode (γ6) in DaCld is absent from the decarboxylase spectrum. These differences collectively reveal distinct peripheral and core heme conformations for the decarboxylases and Clds.
Solution-state spectroscopy indicates retention of the weak Fe-His bond but diminishing distal interactions as the propionates are oxidized. The ferrous heme-CO complex is a sensitive probe of heme coordination and the electrostatic character of its binding site inside proteins.39–42 The CO complexes of the ferrous coproheme-, harderoheme-, and heme b-bound decarboxylase (Sa) were generated and their spectra analyzed for inclusion on a νFe−C/νC−O correlation plot (Fig. 4). Three isotope-sensitive (i.e. 12CO versus 13CO) bands were detected for each of the aforementioned hemes. Based on their frequencies and isotope shifts, they were assigned to the Fe-C and C-O stretching (νFe−C and νC−O) and FeCO bending (δFeCO) modes for CO-bound forms: coproheme, 513, 1941, and 582 cm−1; harderoheme, 502, 1955, and 579 cm−1; heme b, 498, 1958, and 577 cm−1, respectively (Fig. 4A). The same isotopically sensitive bands were observed for the heme b-CO, whether generated by binding ferric heme b to the enzyme or by the enzymatic activity of the decarboxylase.
Figure 4. rR characterization of ferrous CO complexes of the Sa decarboxylase report on their proximal ligands and distal environments.

A) Soret-excited rR spectra of the isotopomers of CO complexes of the decarboxylase bound to a) coproheme b) harderoheme III, and c) heme b at pH 6.8. Spectra were acquired with 413.1 nm excitation and 2 mW power at the sample. 12CO complex, black; 13CO complex, red; difference spectra 12CO−13CO, blue. B) Backbonding correlation plot of νFe-C versus νC-O for ferrous carbonyls of heme proteins shows the dependences of their positions on axial ligation and distal pocket properties. The black line correlates vFe-C with vC-O for six-coordinate Fe-CO adducts in which the sixth ligand is histidine (neutral imidazole). The gray dashed line represents six-coordinate Fe-CO adducts in which the sixth ligand is a thiolate or and imidazolate. Points used for these lines are tabulated in Table S1 of reference 43. Data for DaClds and NdClds are from references 43 and 44, respectively.
All of the CO complexes generated here fell on the inverse correlation line associated with charge-neutral proximal histidine ligands (Fig. 4B), consistent with the proximal pocket structure (Fig. 2B) and with retention of the proximal histidine as the substrate converts to product. However, the location along the histidine correlation line for the three decarboxylase complexes varied, consistent with a changing distal environment. Stronger interactions of the CO ligand with positively charged or H-bond donating residues on the distal side of the heme lead to greater π back bonding and, therefore, to points higher on the imidazole line. The decarboxylase complex with coproheme is highest on the line, in a position consistent with moderate H-bond donation to or electrostatic interaction with the bound CO and the distal pocket. The harderoheme complex is much lower on the correlation line and close to the position of heme b, which is lowest of all. The loss of Fe-C π bonding character and increase in the C-O π bonding character in the three CO complexes is consistent with the progressive loss of distal interaction as the substrate is oxidized.
The positions of these complexes on the line can be further interpreted in terms of distal structure using available data from Clds. Two conformers, described as closed and open forms, have been reported for the pH 6.5 and 9 crystal structures of DaCld as well as in rR spectra of DaCld−CO.45, 46 These have the distal Arg183 oriented toward and away from the open coordination position above the heme plane, respectively. The coproheme:decarboxylase-CO is comparable to the closed form of DaCld(Arg183Gln)-CO, suggesting similar distal environments. The closed form of WT DaCld-CO, with its distal Arg, is higher up on the correlation line; as one would predict, the distal Arg interacts with the bound CO in a stronger manner than the distal Gln in either coproheme: decarboxylase-CO or DaCld(Arg183Gln)-CO. The heme b:decarboxylase-CO falls at a position similar to the open form of DaCld-CO and the CO complex of the Cld from Candidatus Nitrospira defluvii.47 The degree of distal pocket interaction with the bound CO in these three complexes is similar and small.
Coproheme-bound CO interacts with the conserved distal Gln
The crystal structure suggests that Gln185 and/or Arg131 could make contact with the terminal oxygen of a ferric coproheme-OOH complex. To examine this hypothesis, rR spectra for the ferrous coproheme-CO complexes were used to assess the distal environment sampled by a bound diatomic ligand inside the Gln185Ala and Arg131Ala variant decarboxylases. The νFe−C and νC−O frequencies for the Gln185Ala-CO complex (498 and 1953 cm−1, respectively (Fig. S9) dramatically shift the Gln185Ala-CO point down the correlation line away from the WT coproheme complex and to a position close to the heme b-decarboxylase (Fig. 4B.). This indicates loss of distal interaction when Gln185 is absent. The Arg131Ala-CO complex falls between WT and Gln185Ala on the νFe−C/ νC−O correlation plot (Fig. 4B and S8) indicating that this mutation has a smaller effect on the distal pocket interaction than Gln185Ala. Its distal environment is comparable to those observed for mutants in which H-bonds/salt bridges to a propionate group have been disrupted (see below). This suggests that Arg131 plays a role at the periphery of the coproheme instead of in the distal pocket. Loss of the Arg charge could result in either a change in coproheme conformation because of loss of the interaction with propionate 6 or in an increase in solvent accessibility to the pocket. Either could be responsible for the characteristics of Arg131Ala-CO and its decreased activity with H2O2 described below. Consistent with these observations, the Gln185Ala but not the Arg131Ala exhibits impaired reactivity (described below).
Disruption of H-bonding to propionates 2 and 4 alters heme conformation
The presence of H-bonds or salt bridges to propionates 2 (Tyr145, Arg218) and 4 (Lys149) are predicted by the structure (Fig. 2). To test whether these apparent interactions modulate the electronic structure of the coproheme, the site-specific Tyr145Ser, Lys149Ala, and Arg218Ala mutants were generated. The rR spectra of these coproheme: mutant-CO complexes are compared in Fig. 4B and S8 and their νFe−C and νC−O frequencies given in Table S4. As judged by their position on the νFe−C/νC−O correlation plot (Fig. 4B) the mutant:coproheme-CO complexes, like WT coproheme-CO, retain the neutral His as the proximal ligand. The CO interactions with their distal environments, however, have been diminished. If the only role of the H-bonds/salt bridges to the reactive propionates is to position them for decarboxylation, loss of these interactions would not be expected to change exogenous ligand interactions with the distal pocket. This result therefore argues that the roles of the H-bond donors to propionates 2 and 4 are more nuanced than simply positioning their carboxylate groups and the changes in their reactivity could be due, in part, to changes in the distal pocket interactions and/or distortions of the coproheme plane that direct the reactivity of the high-valent intermediate toward oxidative decarboxylation of propionate 2 or 4.
Three bands have been tentatively assigned to propionate bending modes in the CO complexes of WT, Tyr145Ser, Arg218Ala and Lys149Ala coproheme:decarboxylases (Fig. S8B and Table S4). The Try145Ser mutant has a propionate bending band at 361 cm−1 that is 6 cm−1 lower than the comparable δ(CβCcCd) band in the WT CO complex; the other two δ(CβCcCd) frequencies are unaffected indicating that the band at 367 cm−1 in the WT CO spectrum is due to propionate-2. Loss of the Try145 H-bond to propionate 2 results in a significant alteration in its environment. In contrast, all three bands assigned to the propionate bending modes in Arg218Ala and Lys149Ala CO complexes exhibit decreases of 3 cm−1 in their frequencies of relative to WT. This indicates that disruption of the H-bond network between propionate 2 and Arg218 or the salt bridge between Lys149 and propionate 4 lead to more global changes in coproheme environment than the very localized effect of Tyr145 mutation. Interestingly, the ν8 band, a core size marker band that reports Fe-Npyrrole stretching, exhibits a decrease in frequency for all three mutants. This is consistent with increases in the porphyrin core size upon mutation of the amino acids that interact with the reactive propionates 2 and 4. These features suggest that the interaction of these residues with the coproheme are important in determining the degree to which the heme conformation deviates from planarity. Thus, stepwise changes in the heme planarity may contribute to the structural basis for regioselectivity in oxidizing propionates 2 and 4.
Tyr145 is essential for the decarboxylation of coproheme and harderoheme III; Lys149 contributes to decarboxylation of harderoheme III. Based on their proximity to the substrate and their catalytic roles in other enzymes, we hypothesized that several residues in Fig. 2 may have essential roles in catalysis. We made a series of mutations in the Sa enzyme at a total of eight key positions, including those described above (Table S2). All of the mutants bound coproheme with >80% occupancy, with KD values similar to WT and varying by less than an order of magnitude (Table S2, Fig. S10–11). The catalytic competence of each mutant-coproheme complex was examined to determine which residues were essential for turnover, specifically whether each generated the expected intermediate (harderoheme III) and product (heme b) and how much oxidant was required.
The WT enzyme required 5 eq of H2O2 to turn over 90% of the initially bound coproheme. Harderoheme III was the observed intermediate, maximizing after delivery of 2 eq H2O2 (Fig. S5) and fully converting to heme b with a total of 10–12 eq H2O2. Addition of further oxidant has been observed to bring about the complete degradation of the heme b chromophore.21
The most dramatic deviations from WT behavior were obtained for the Tyr145Ser and Lys149Ala mutants (Fig. 5). Tyr145Ser, in which the H-bond to propionate 2 is eliminated, required ~3,000 eq of H2O2 to turn over ≥95% of the coproheme substrate (Table S2). Neither harderoheme III nor heme b appeared as intermediates or products. Instead, the coproheme degraded without detectable products. The Tyr145Ser mutant readily bound harderoheme so its catalytic competence towards this stable intermediate was investigated. Harderoheme-Tyr145Ser mutant did not form observable heme b upon reaction with H2O2 (Fig. S12). The Lys149Ala mutant, in which the salt bridge to propionate 4 is removed, required ~300 eq of H2O2 to turn over ≥95% of the substrate. However, the major product was harderoheme III, with relatively little turnover of this species to heme b. This suggests that Lys149 is essential specifically for the conversion of harderoheme to heme b, while Tyr145 appears to play a role in both decarboxylation reactions.
Figure 5. Mutations at Lys149 and Tyr145 prevent decarboxylation of one or both propionic acid substrates, respectively.

Unreacted coproheme (red), harderoheme III intermediate (green) and the heme b product (blue) of the coproheme decarboxylase reaction with increasing eq of H2O2 were quantified by HPLC for (top) WT enzyme and the mutants (center) Lys149Ala and (bottom) Tyr145Ser. HPLC traces illustrating the starting material, observable intermediates, and final products are shown as insets. Retention times for unreacted coproheme, the harderoheme III intermediate, and heme b product were 4.8, 7.1 and 8.9 min, respectively.
Substitutions of other potentially significant residues surrounding propionate 2 (Arg218Ala) or 4 (Tyr113Ser, Trp157Phe, Trp198Phe), or in the distal pocket (Gln185Ala, Arg131Ala) had far smaller effects, suggesting that none of these on its own is essential for catalysis. Instead, the Gln185Ala and Arg131Ala mutants brought about small increases in the amount of H2O2 required to effect substrate conversion (60 and 24 eq, respectively). The Gln185Ala mutant additionally saw exhibited a marked decrease in the amount of harderoheme III accrued, suggesting this mutation had a proportionally greater impact on the first decarboxylation than the second. Arg218Ala, Trp157Phe, and Trp198Phe all behaved very similarly to WT, while the heme b product of Tyr113Ser was slightly less stable toward H2O2, possibly due to weaker heme b binding (Fig. S13).
The excess H2O2 required for coproheme turnover can be largely accounted for by catalase activity. The Sa decarboxylase containing heme b was previously shown to have catalase activity:
| (6) |
| (7) |
This reaction (~50 turnovers) occurs concomitantly with heme b degradation.28 It is possible that some of the excess H2O2 required for coproheme conversion to heme b might be expended in the catalase reaction. Following addition of 12 eq of H2O2 (120 μM) to the decarboxylase-coproheme complex (10 μM), one eq of O2 (10 μM) (measured by O2 polarography) (Fig. 6A) was generated over the same time frame in which coproheme converted to heme b. In light of the mechanism above, 2 eq of H2O2 (likely not a physiologically relevant amount) can be accounted for via the catalase reaction, in addition to the 2 required for the decarboxylation reactions. The remaining 8 of the initial 12 eq, though well within the limits of detection, were not detected by HRP-based colorimetric assays as the end of the reaction. Control reactions in which the ligand-free/apo-protein (5 μM) was mixed with 12 eq H2O2 (60 μM) under the same conditions showed that ~8 eq of H2O2 (40 μM) were consumed in a heme-independent reaction with the apo-protein without concomitant production of O2, while 4 eq (22 μM) remained unreacted in solution (Fig. S14).
Figure 6. The catalase activity of coproheme decarboxylase is strongly enhanced in the Lys149Ala and Tyr145Ser mutants.

(A) The progress of the coproheme decarboxylase reaction over time [coproheme (red), harderoheme III (green), heme b (blue)] measured previously22 is shown superimposed on the time course for O2 production (orange) (10 μM enzyme-coproheme, 120 μM H2O2, 50 mM KPi, pH 7.4, 25 °C). The generation of ~10 μM O2 via the catalase reaction suggests that ~20 μM H2O2 was consumed during the same time window in which coproheme converted to heme b. (B) Initial rates of O2 production were measured as a function of [H2O2] and fit to eqn (5) for the WT (black), Tyr145Ser (cyan), and Lys149Ala (purple) decarboxylases. The resulting steady state parameters describing catalase activity are tabulated in the inset.
The large excess of H2O2 required to turn over the coproheme bound to the Tyr149Ser and Lys145Ala mutants could likewise be primarily accounted for via the catalase reaction (Fig. S15). Mutation of either residue increases the values of both kcat and kcat/KM(H2O2) relative to WT (Fig. 6B). This suggests that either mutation stabilizes the heme species, thereby enhancing their efficiency as catalases.
DISCUSSION
Coproheme decarboxylase catalyzes H2O2-dependent C-C bond cleavage reactions to liberate two molecules CO2 from a pair of propionic acids, converting each to a vinyl (Scheme 1). An analogous reaction is catalyzed by the bacterial cytochrome P450, OleT.48 Ferric OleT and H2O2 react to generate compound I, which in turn abstracts a hydrogen atom directly from the exogenous carboxylic acid substrate. Unique among P450s, this reaction is not followed by rebound between the substrate radical and Fe(IV)-OH.19 Instead, the reaction is more peroxidase-like, with the unpaired electron from the nearby substrate reducing the iron to Fe(III), resulting in CO2 and the alkene. A similar mechanism could be proposed for the coproheme decarboxylase, with the ferryl oxygen most likely interacting for steric reasons with the Cα-H of either propionate 2 or 4 (Scheme 1). However, the 1.8 A structure of the enzyme in complex with its substrate analog (Fig. 1–2) suggests two complications with any straightforward application of either the peroxidase or OleT model to the coproheme decarboxylase.
The first is that the apparatus for activating H2O2 found in most heme peroxidases, including CCP and APX (Fig. S1 and Results),12 is completely absent in the coproheme decarboxylase. The coproheme is instead ligated by an uncharged proximal His (Fig 2–4) and its open coordination position is flanked by a distal Gln. Both the Gln and the Arg to which it is connected via H-bonds through-water interact with a coproheme-bound CO. Although mutagenesis showed that neither Gln185 nor Arg131 was essential for catalysis, their interactions with bound CO suggests they could play activating roles toward H2O2. Some role for the protein is supported by prior studies of the reaction of the coproheme decarboxylase with peracetic acid: an oxygen atom donor that directly converts ferric hemes to Fe(IV)=O(por■+) or Fe(IV)=O(Trp■+) without the proton movement or heterolytic bond cleavage associated with H2O2.22 The ferric coproheme decarboxylase reacts rapidly with peracetic acid to form the expected harderoheme intermediate and heme b product. The second order rate constant for the same reaction with H2O2 is 10-fold slower and, in contrast with the peracid reaction, dependent on both pH and solvent isotope (D2O).22 These observations strongly suggest that the reaction proceeds through a ferryl intermediate and that its formation is facilitated by protein-mediated proton transfers (Scheme 2A).
Scheme 2. Proposed mechanisms consistent with the available data†.

†A. Formation of Fe(IV)=O(coproporphyrin■+) (coproheme compound I) (E = enzyme; Fe(III) in red and Fe(IV) in purple). B. Oxidative decarboxylation of propionate 2 requires Tyr145. The neutral Tyr145-O■ radical is depicted abstracting hydrogen (H■) from the propionate Cβ, though this is hypothetical. Transfer of an electron and proton to the ferryl of Compound II then leads to the ferric-OH complex of harderoheme III, from which water can ultimately depart. C. Following another round of H2O2-activation analogous to (A), a second Fe(IV)=O(harderoporphyrin■+) complex forms. Three possible redox-active amino acids are connected to propionate 4 via hydrogen bonding through water molecules, though only Lys149 appeared to play a role decarboxylating propionate 4. The identity of the mediator for proton coupled electron transfer from the propionate (designated E+■ or E■) is unclear. Net uptake of H■ by the enzyme is designated via E(H).
The second issue concerns the presumptive sites of substrate oxidation – the Cβ-H bonds of propionic acids 2 and 4 – which the structure shows are spatially removed from the site of H2O2 activation at the metal. This separation immediately suggests that, in contrast with OleT, the reaction cannot proceed by direct interaction of a ferryl or other iron/oxygen intermediate with either propionate. A similar steric problem was noted previously in studies of mammalian peroxidases and some cytochrome P450s in which the methyl side chains of heme b are oxidatively modified via a compound I intermediate. In each case, the steric relationships between the ferryl of compound I and the heme substituents imply the use of an amino acid side chain or multiple side chains as redox mediators.49–51 Inspection of the environment surrounding propionate 2 (Fig. 2C) immediately identified Tyr145 as a possible secondary site of oxidation following the initial generation of a coproheme compound I. The side chain of Tyr145 sits near to and forms an apparent H-bond with the carboxylate moiety of propionate 2, in a position analogous to the ascorbate binding site in APX (Fig. S1).8 This configuration suggested that Tyr145 might be well-positioned to be oxidized by the coproheme compound I (Scheme 2B), and that it may in fact act as the reactive species toward propionate 2. Consistent with that hypothesis, substitution of Tyr145 with Ser led to complete loss of coproheme decarboxylation. A concomitant large increase in catalase activity (Fig. 6) suggested that, in the absence of Tyr145, the enzyme readily forms Fe(IV)=O(coproporphyrin■+) with the electron hole localized on the porphyrin.
Neutral tyrosyl radicals (TyrO■) are implicated in a variety of reactions, including those catalyzed by class I ribonucleotide reductase,52 photosystem II,53 galactose oxidase,54 and prostaglandin-H synthase.55 In each case, a metal center is involved in generating the tyrosyl radical, which then acts as the primary recipient of an electron or hydrogen atom from a substrate. In the case of prostaglandin-H synthase and other cyclooxygenases, the metal center is heme b, which oxidizes a nearby tyrosine side chain via a compound I intermediate. The bond dissociation free energy associated with TyrO■/TyrOH in these systems as well as phenolic model compounds can be quite high17, 56 and, in the case of the tyrosine Z in photosystem II, thermodynamically sufficient for the oxidation of water. Net transfer of H■ from the propionate Cβ-H to Tyr145-O■ would result in a resonance-stabilized coproheme propionyl-Cβ radical, suggesting a proportionally lower requisite driving force for carrying out the reaction. Though mutagenesis showed it was not essential for coproheme turnover by the decarboxylase, the side chain of Arg218 is well-positioned to shuttle propionate-derived protons from the active site. A similarly situated arginine residue obligately serves this function in the oxidation of ascorbate by APX (Fig. S1).10 Transfer of a second electron from the Cβ■ through the porphyrin and to the coproheme Fe(IV) would result in the regeneration of Fe(III) and production of the vinyl and CO2 (Scheme 2B).
Decarboxylation at propionate 2 is followed by a second, slightly faster22 H2O2-dependent reaction at propionate 4. Inspection of the environment surrounding this group identified three readily oxidizable aromatic residues in its vicinity (Tyr114, Trp198, Trp157) as well as a lysine (Lys149) which, like Arg218, could potentially act as an initial recipient or conduit for protons (Fig. 2E). Individual mutations at each of these residues showed that only Lys149 appeared to be essential for coproheme turnover. Substitution of this residue with alanine resulted in the accumulation of the three-propionate intermediate, harderoheme III (Scheme 1). This result suggested that harderoheme maintains the same position as the coproheme substrate, as predicted by solution state rR, and loss of the harderoheme-Lys149 interaction specifically inhibits the decarboxylation of propionate 4.22 We therefore conclude that H2O2 activation by the harderoheme-decarboxylase proceeds in a similar environment as the coproheme complex, though the Fe(III)/(II) reduction potential22 and the degree of distal pocket interaction with the bound substrate (Fig. 4) have diminished. The pronounced increase in catalase activity in the Lys149Ala mutant likewise suggests that it reacts with H2O2 to form Fe(IV)=O(harderoporphyrin■+).
If this intermediate forms during catalytic turnover, then Tyr114, Trp198, and Trp157 all appear reasonably positioned to form Tyr-O■ or Trp■+ radicals by filling the electron hole on (harderoporphyrin■+). The side chain could then mediate a reaction with propionate 4, where the Cβ-H electron could migrate to the same (e.g., Tyr114-O■) or separate destinations. In the latter case, Lys149 could serve an essential role as the proton acceptor in a proton coupled electron transfer. However, mutation of any of these aromatic residues independently had virtually no effect on the efficiency of either propionate decarboxylation. This suggests one of two interpretations. First, it is possible that radical density is spread over two or more of these residues, and that the others can compensate in the event that one is lost. Alternatively, Tyr145-O■ could act as the enzyme’s sole redox mediator toward both propionates 2 and 4. Though the latter is farther from Tyr145, net proton coupled electron transfers can occur across long distances. The reactive Tyr-O■ in ribonucleotide reductases sets an impressive precedent, reacting with a cysteine-SH over 35 A away.52 Supporting this hypothesis in part, we noted that reaction of the Tyr145Ser-coproheme complex with H2O2 yielded neither harderoheme nor heme b, suggesting that both decarboxylations are impaired by this substitution. Moreover, when a complex of the same mutant was prepared with exogenously supplied harderoheme III, its reaction with H2O2 also yielded no heme b. Tyr145 is clearly essential for both propionate oxidations, though whether it acts as a radical mediator in either case is still unknown. How Tyr145 would form the Tyr-O■ radical in the absence of the conduit provided by propionate 2, once it has converted to vinyl, is an open question, though we note that there is a clear kinetic preference for oxidizing propionate 2 first. This oxidation could be facilitated by the existence of the propionate-Tyr145 interaction.
The chemistry, spectroscopy, and structure reported here constitute an internally consistent account of function and mechanism in this class of decarboxylases. Tyr145 and Lys149 are H-bonded to the reactive propionates 2 and 4, respectively, and play essential roles in the reactivity and regioselectivity of propionates 2 and 4 toward oxidation. These nonbonded interactions differ markedly from those recently reported for the Listeria monocytogenes enzyme (LmHemQ), in which coproheme III is oriented such that propionates 2 and 7 are oriented toward the surface of the enzyme while propionates 4 and 6 are directed into the pocket. In the LmHemQ structure, propionate 2 is H-bonded to Gln187, propionate 4 is H-bonded to Tyr147, and the unreactive propionate 6 is H-bonded to Lys151 (comparable to Gln185, Tyr145 and Lys149, respectively, in the Sa enzyme). Though there are some differences in crystallization parameters (Gs decarboxylase crystals were grown at pH 7.5, for example, whereas the Lm decarboxylase crystals were obtained at pH 9.0) the reasons for this different coproheme orientation are unclear. Though it seems unlikely that homologous decarboxylases from different organisms would utilize different coproheme orientations, this possibility cannot be eliminated at this time. More in-depth comparison of the two coproheme-decarboxylase structures is not possible at the time of writing because the LmHemQ structure has yet to be released.57
OleT and the coproheme decarboxylase described here are atypical for using H2O2 as the cosubstrate in an oxidative decarboxylation. Coproheme decarboxylase is further distinguished by its evident use of an extended relay of propionate and side chain mediators to convey electrons and protons away from its pair of substrates and toward H2O2, ultimately releasing CO2. This use of the metalloporphyrin as a primary site of oxidation, in conjunction with a series of stably positioned mediators, also suggests an alternative approach for synthetic or semi-synthetic catalysis of the oxidative decarboxylation of fatty acids to generate alkenes.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by funding from the National Institutes of Health, General Medical Sciences R01GM090260 (J.L.D.) and R15GM114787 (G.S.L.-R.). Structural work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-FG02-04ER15563 to J.W.P.
We thank the National Institutes of Health for supporting this work. Additionally, we thank the Department of Energy for support of the structural work, carried out at the Stanford Synchrotron Radiation Laboratory at the Stanford Linear Accelerator Center. This work will be presented at the Gordon Research Conference on Metals in Biology and at the ASBMB national meeting, 2017.
ABBREVIATIONS
- ABTS
2,2′-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt
- ACN
acetonitrile
- APX
ascorbate peroxidase
- CCP
cytochrome c peroxidase
- Cld
Chlorite dismutase
- Da
Dechloromonas aromatica
- ESI
electrospray ionization
- Gs
Geobacillus stearothermophilus
- HPLC
High Performance Liquid Chromatography
- HRP
horseradish peroxidase
- LnP
lignin peroxidase
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- Nd
Nitrospira defluvii
- PAA
peracetic acid
- QTOF
quadrupole time-of-flight
- rR
resonance Raman
- Sa
Staphyloccocus aureus
- TFA
trifluoroacetic acid
- LmHemQ
Listeria monocytogenes decarboxylase
- rR
resonance Raman
Footnotes
Author Contributions
The manuscript was written through contributions of all authors.
All authors have given approval to the final version of the manuscript.
A series of 15 data figures and 4 tables is included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Traylor TG, Traylor PS. Reactions of dioxygen and its reduced forms with heme proteins and model porphyrin complexes. In: Valentine JS, Foote CS, Greenberg A, Liebman JF, editors. Active Oxygen in Biochemistry. Springer Netherlands; 1995. pp. 84–187. [Google Scholar]
- 2.Dunford HB. Heme Peroxidases. Wiley-VCH; New York: 1999. [Google Scholar]
- 3.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism and Biochemistry. 3. Kluwer Academic/Plenum; New York: 2005. [Google Scholar]
- 4.Rittle J, Green MT. Science. 2010;330:933–7. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 5.Hammes-Schiffer S. J Am Chem Soc. 2015;137:8860–8871. doi: 10.1021/jacs.5b04087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Philos Trans R Soc Lond B Biol Sci. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macdonald IK, Badyal SK, Ghamsari L, Moody PC, Raven EL. Biochemistry. 2006;45:7808–7817. doi: 10.1021/bi0606849. [DOI] [PubMed] [Google Scholar]
- 8.Sharp KH, Mewies M, Moody PC, Raven EL. Nat Struct Biol. 2003;10:303–307. doi: 10.1038/nsb913. [DOI] [PubMed] [Google Scholar]
- 9.Warren JJ, Mayer JM. J Am Chem Soc. 2011;133:8544–8551. doi: 10.1021/ja201663p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efimov I, Badyal SK, Metcalfe CL, Macdonald I, Gumiero A, Raven EL, Moody PC. J Am Chem Soc. 2011;133:15376–15383. doi: 10.1021/ja2007017. [DOI] [PubMed] [Google Scholar]
- 11.Gumiero A, Metcalfe CL, Pearson AR, Raven EL, Moody PC. J Biol Chem. 2011;286:1260–1268. doi: 10.1074/jbc.M110.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Poulos TL. Chem Rev. 2014;114:3919–3962. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cherifi G, Baxter EL, Doukov T, Cohen AE, McPhillips SE, Song J, Meharenna YT, Soltis SM, Poulos TL. Proc Natl Acad Sci USA. 2016;112:1226–1231. doi: 10.1073/pnas.1521664113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelletier H, Kraut J. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 14.Choinowski T, Blodig W, Winterhalter KH, Piontek K. J Mol Biol. 1999;286:809–827. doi: 10.1006/jmbi.1998.2507. [DOI] [PubMed] [Google Scholar]
- 15.Colin J, Wiseman B, Switala J, Loewen P, Ivancich A. J Am Chem Soc. 2009;131:8557–8563. doi: 10.1021/ja901402v. [DOI] [PubMed] [Google Scholar]
- 16.Sivaraja M, Goodin DB, Smith M, Hoffman BM. Science. 1989;245:738–740. doi: 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- 17.Tommos C, Skalicky JJ, Pilloud DL, Wand AJ, Dutton PL. Biochemistry. 1999;38:9495–9507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 18.Guallar V. J Phys Chem B. 2008;112:13460–13464. doi: 10.1021/jp806435d. [DOI] [PubMed] [Google Scholar]
- 19.Grant JL, Mitchell ME, Makris TM. Proc Natl Acad Sci USA. 2016;113:10049–10054. doi: 10.1073/pnas.1606294113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dailey H, Gerdes S, Dailey T, Burch J, Phillips J. Proc Natl Acad Sci USA. 2015;112:2210–2215. doi: 10.1073/pnas.1416285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Celis AI, Streit BR, Moraski GC, Kant R, Lash TD, Lukat-Rodgers GS, Rodgers KR, DuBois JL. Biochemistry. 2015;54:4022–4032. doi: 10.1021/acs.biochem.5b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Streit BR, Celis AI, Shisler K, Rodgers KR, Lukat-Rodgers GD, DuBois JL. Biochemistry. doi: 10.1021/acs.biochem.6b00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilski M, Borek D, Chen Y, Collart F, Joachimiak A, Otwinowski Z. [accessed Dec 7, 2016];RCSB Protein Data Bank. http://www.rcsb.org/pdb/home/home.do. PDB ID 1T0T, released Aug 4, 2004.
- 24.Gonzalez A, Tsai Y. [accessed May 1, 2016]; http://smb.slac.stanford.edu/facilities/software/xds/#autoxds_script.
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayfield JA, Hammer ND, Kurker RC, Chen TK, Ojha S, Skaar EP, DuBois JL. J Biol Chem. 2013;288:23488–504. doi: 10.1074/jbc.M112.442335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Celis A, DuBois J. Arch Biochem Biophys. 2015;574:3–17. doi: 10.1016/j.abb.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goblirsch B, Kurker RC, Streit BR, Wilmot CM, DuBois JL. J Mol Biol. 2011;408:379–398. doi: 10.1016/j.jmb.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofbauer S, Dalla Sega M, Scheiblbrandner S, Jandova Z, Schaffner I, Mlynek G, Djinovic-Carugo K, Battistuzzi G, Furtmuller PG, Oostenbrink C, Obinger C. Biochemistry. 2016;55:5398–5412. doi: 10.1021/acs.biochem.6b00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofbauer S, Howes BD, Flego N, Pirker KF, Schaffner I, Mlynek G, Djinović-Carugo K, Furtmuller PG, Smulevich G, Obinger C. Biosci Rep. 2016;36:e00312. doi: 10.1042/BSR20150330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choudhury K, Sundaramoorthy M, Hickman A, Yonetani T, Woehl E, Dunn MF, Poulos TL. J Biol Chem. 1994;269:20239–20249. [PubMed] [Google Scholar]
- 34.Poulos TL, Fenna RE. Metal Ions In Biological Systems. Vol. 30. Wiley VCH; New York: 1994. Peroxidases - Structure, Function, And Engineering. [Google Scholar]
- 35.Blanc B, Mayfield JA, McDonald CA, Lukat-Rodgers GS, Rodgers KR, DuBois JL. Biochemistry. 2012;51:1895–1910. doi: 10.1021/bi2017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh R, Grigg JC, Armstrong Z, Murphy MEP, Eltis LD. J Biol Chem. 2012;287:10623–10630. doi: 10.1074/jbc.M111.332171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erman JE, Vitello LB, Miller MA, Kraut J. J Am Chem Soc. 1992;114:6592–6593. [Google Scholar]
- 38.Layer G, Pierik AJ, Trost M, Rigby SE, Leech HK, Grage K, Breckau D, Astner I, Jaensch L, Heathcote P, Warren MJ, Heinz DW, Jahn D. J Biol Chem. 2006;281:15727–34. doi: 10.1074/jbc.M512628200. [DOI] [PubMed] [Google Scholar]
- 39.Draganova EB, Adrian SA, Lukat-Rodgers GS, Keutcha CS, Schmitt MP, Rodgers KR, Dixon DW. J Biol Inorg Chem. 2016;21:875–86. doi: 10.1007/s00775-016-1386-3. [DOI] [PubMed] [Google Scholar]
- 40.Spiro TG, Soldatova AV, Balakrishnan G. Coord Chem Rev. 2013;257:511–527. doi: 10.1016/j.ccr.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lukat-Rodgers GS, Rodgers KR, Caillet-Saguy C, Izadi-Pruneyre N, Lecroisey A. Biochemistry. 2008;47:2087–2098. doi: 10.1021/bi7019518. [DOI] [PubMed] [Google Scholar]
- 42.Spiro TG, Wasbotten IH. J Biol Inorg Chem. 2005;99:34–44. doi: 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 43.Blanc B, Mayfield JA, McDonald CA, Lukat-Rodgers GS, Rodgers KR, DuBois JL. Biochemistry. 2012;51:1895–1910. doi: 10.1021/bi2017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hofbauer S, Howes BD, Flego N, Pirker KF, Schaffner I, Mlynek G, Djinovic-Carugo K, Furtmueller PG, Smulevich G, Obinger C. Biosci Rep. 2016;36:e00312. doi: 10.1042/BSR20150330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goblirsch BR, Streit BR, DuBois JL, Wilmot CM. J Biol Inorg Chem. 2010;15:879–888. doi: 10.1007/s00775-010-0651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Streit BR, Blanc B, Lukat-Rodgers GS, Rodgers KR, DuBois JL. J Am Chem Soc. 2010;132:5711–5724. doi: 10.1021/ja9082182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hofbauer S, Gruber C, Pirker K, Schaffner I, Furtmuller P, Obinger C, Hagmuller A, Gysel K, Mlynek G, Kostan J, Djinovic-Carugo K, Bellei M, Battistuzzi G, Daims H. J Biol Inorg Chem. 2014;19:S253–S253. [Google Scholar]
- 48.Grant JL, Hsieh CH, Makris TM. J Am Chem Soc. 2015;137:4940–4943. doi: 10.1021/jacs.5b01965. [DOI] [PubMed] [Google Scholar]
- 49.Zheng YM, Baer BR, Kneller MB, Henne KR, Kunze KL, Rettie AE. Biochem. 2003;42:4601–4906. doi: 10.1021/bi020667t. [DOI] [PubMed] [Google Scholar]
- 50.Colas C, Ortiz de Montellano PR. Chem Rev. 2003;103:2305–2332. doi: 10.1021/cr0204303. [DOI] [PubMed] [Google Scholar]
- 51.Brown KR, Brown BM, Hoagland E, Mayne CL, Hegg EL. Biochemistry. 2004;43:8616–8624. doi: 10.1021/bi049056m. [DOI] [PubMed] [Google Scholar]
- 52.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 53.Okamura MY, Paddock ML, Graige MS, Feher G. Biochim Biophys Acta. 2000;1458:148–163. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- 54.Whittaker JW. Chem Rev. 2003;103:2347–2363. doi: 10.1021/cr020425z. [DOI] [PubMed] [Google Scholar]
- 55.Tsai AL, Kulmacz RJ, Palmer G. J Biol Chem. 1995;270:10503–10508. doi: 10.1074/jbc.270.18.10503. [DOI] [PubMed] [Google Scholar]
- 56.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hofbauer S, Mlynek G, Millazo L, Puhringer D, Maresch D, Schaffner I, Furtmuller PG, Smulevich G, Djinović-Carugo K, Obinger C. Hydrogen Peroxide Mediated Conversion of Coproheme to Heme b by HemQ. Lessons from the First Crystal Structure and Kinetic Studies. [accessed Oct 30, 2016];FEBS J. doi: 10.1111/febs.13930. [Online early access] Published Online: Nov 14, 2016. http://onlinelibrary.wiley.com/wol1/doi/10.1111/febs.13930/abstract. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.