

Abstract
Background
Controlled mechanical ventilation (CMV) is associated with diaphragm dysfunction. Dysfunction results from muscle atrophy and injury of diaphragm muscle fibers. Enhanced proteolysis and reduced protein synthesis play an important role in the development of atrophy. The current study is to evaluate the effects of the calpains inhibitor calpeptin on the development of diaphragm atrophy and activation of key enzymes of the ubiquitin-proteasome pathway in rats under CMV.
Methods
Three groups of rats were studied: control animals (CON, n = 8), rats subjected to 24 h of MV (CMV, n = 8), and rats subjected to 24 h of MV after administration of the calpain inhibitor calpeptin (CMVC, n = 8). The diaphragm was analyzed for calpain activity, myosin heavy chain (MHC) content, and cross-sectional area (CSA) of diaphragmatic muscle fibers as a marker for muscle atrophy. In addition, key enzymes of the ubiquitin-proteasome pathway (MAFbx and MuRF1) were also studied.
Results
CMV resulted in loss of both MHCfast and MHCslow. Furthermore, the CSA of diaphragmatic muscle fibers was significantly decreased after 24 h of CMV. However, calpain inhibitor calpeptin prevented loss of MHC and CSA after CMV. In addition, calpeptin prevented the increase in protein expression of calpain1 and calpain2 and reduced calpain activity as indicated by reduced generation of the calpain cleavage product αII-spectrin in the diaphragm. CMV-induced upregulation of both MAFbx and MuRF1 protein levels was attenuated by treatment with calpeptin.
Conclusions
The calpain inhibitor calpeptin prevents MV-induced muscle atrophy. In addition, calpeptin attenuated the expression of key proteolytic enzymes known to be involved in ventilator-induced diaphragm atrophy, including MAFbx and MuRF1.
Keywords: Calpain, Calpeptin, Mechanical ventilation, Diaphragm atrophy
Background
Mechanical ventilation is a life-saving intervention for patients with acute respiratory failure. However, it is recognized that mechanical ventilation may be associated with side effects, in particular lung injury [1] and respiratory muscle dysfunction often described as ventilator-induced diaphragm dysfunction (VIDD) [2, 3]. Respiratory muscle weakness in ICU patients is associated with prolonged weaning from mechanical ventilation and mortality [4]. Today, no specific treatment for ICU acquired respiratory muscle weakness is available. Improved understanding of the pathophysiology of ICU acquired respiratory muscle weakness may help to develop specific treatment strategies.
Studies in animal models and critically ill patients have demonstrated that VIDD results from both the loss of muscle proteins and dysfunction of the remaining contractile proteins [3, 5]. Loss of muscle proteins may result from increased protein degradation or inhibition of protein synthesis. The proteolytic ubiquitin-proteasome pathway (UPP) is the main proteolytic pathway in skeletal muscles [6] and involves several key enzymes including MAFbx and MuRF1. Activation of different components of the UPP in the diaphragm has been demonstrated in animals subjected to MV [7, 8] but also in ICU patients [3, 9]. The UPP cannot degrade intact myofibrils because neither the large myofibril nor thick and thin filaments could enter the central catalytic chamber of the proteasome. Activation of other enzymes, such as calpains cleaves major cytoskeletal proteins such as titin and nebulin, which leads to the release of myofilaments suitable for degradation by the UPP [10–12]. Today, only one study assessed the role of calpains in VIDD [12]. In that study, inhibition of calpain prevented MV-induced diaphragm atrophy.
The aim of the current study was to evaluate the effect of calpeptin, a specific calpain inhibitor, on ventilator-induced diaphragm atrophy and activation of proteolytic pathways.
Methods
Animals and experimental design
The study was approved by the University of Tongji Animal Care and Use Committee. Rats were offered by the Experimental Animal Center of Tongji Medical College. Twenty-four male Sprague-Dawley rats (body weight 320–350 g) were randomly assigned to one of three groups (n = 8/group): (1) acutely anesthetized control (CON), (2) 24 h of controlled mechanical ventilation (CMV), and (3) CMV rats treated with the calpain inhibitor calpeptin (CMVC). CMV rats received corresponding volumes of vehicle (0.1% DMSO in saline for calpeptin). Calpeptin (Sigma, MO, USA) was administered subcutaneously (4 mg/kg) 2 h before and 8, 15, and 23 h after initiation of mechanical ventilation. The timing and dosage of calpeptin administration 2 h before initiation of mechanical ventilation were in line with the study of Fareed MU [13]. Animals in the CON group were anesthetized with pentobarbital sodium (60 mg/kg body weight; intraperitoneal), and the diaphragm was removed immediately. Segments of the ventral costal region of the diaphragm were quickly removed, frozen in liquid nitrogen, and stored at −80 °C for subsequent biochemical analysis.
In ventilated rats, all surgical procedures were performed aseptic as previously described [14]. Animals selected for CMV were anesthetized with an intraperitoneal injection of pentobarbital sodium (60 mg/kg), tracheotomized and ventilated with a dedicated small-animal ventilator (TOPO; Kent Scientific, Connecticut, USA) for 24 h with the following settings: tidal volume 1 ml/100 g body weight, respiratory rate 80 breaths/min, and positive end-expiratory pressure 1 cm H2O. No visible diaphragm contractions were observed under mechanical ventilation. A time frame of 24 h was chosen based on previous data demonstrating significant diaphragm atrophy, proteolysis, and oxidative stress after this interval [15]. A catheter was inserted into the femoral vein for continuous infusion of isotonic saline (2 ml kg−1 h−1) and pentobarbital sodium (~10mg kg−1 h−1). Additionally, animals received enteral nutrition with a nutrient composition of 15% proteins, 35% lipids, 50% carbohydrates, and vitamins and minerals through an oral-gastric tube with total daily volume of 69 ml. Both heart rate and blood pressure were continuously monitored noninvasively with tail cuff. The care throughout the experimental period included lubricating the eyes, expressing the bladder, removing airway mucus and passively moving the limbs, and using glycopyrrolate (0.04 mg kg−1 2h−1 intramuscular) to reduce bronchial secretions. Rectal temperature was monitored and maintained at 36–38 °C with a heating blanket. Arterial blood (100 μl per sample) from abdominal aorta was withdrawn at end of CMV and was analyzed for pH and the partial pressures of O2 and CO2 by an electronic blood-gas analyzer (radiometer, Copenhagen, Danmark). A research scientist provided round-the-clock coverage and animal care for the duration of the study. On completion of MV, segments of the ventral costal region were quickly removed and frozen in liquid nitrogen and stored at −80 °C for subsequent biochemical analysis, as described earlier [16].
Biochemical measurements
Proteolytic activity of both calpain system and key enzymes of the UPP were analyzed by measuring protein levels of calpain1, calpain2, and the muscle-specific E3 ligases MAFbx and MuRF1 [7, 17]. In addition, calpain activities were evaluated by measuring the content of cleaved 145 kDa calpain fragment, which is specific to calpain1/2 cleavage products of αII-spectrin. Calpain activity was direct related to the expression level of 145-kDa cleavage product [18]. All of these proteins were measured by Western blotting.
Western blotting
Section of the costal diaphragm was homogenized and assayed to quantitatively determine the levels of the proteins above. Tissue was homogenized 1:10 (5 mM Tris-HCl, pH = 7.5, 5 mM EDTA) and then the homogenate was centrifuged at 1500g for 10 min (4 °C). After determining total protein content, 50 μg of protein in samples were then individually separated by polyacrylamide gel electrophoresis. SDS-PAGE was performed on 6% (for MHCslow, MHC2A, and 145-kDa cleavage product) or 10% (for calpain1, calpain2, MuRF1, MAFbx, and glyceraldehyde-3-phosphate dehydrogenase (GADPH) polyacrylamide gels. After electrophoresis, the separated proteins were transferred electrophoretically using semi-dry transfer methodology to polyvinylidene fluoride membrane (Millipore). Membranes were stained with appropriate Marker (26616, Thermo Scientific) and visually inspected for equal protein loading and transfer. The membranes were then washed and blocked in 5% BSA for 1 h and subsequently incubated with primary antibodies directed against MHCslow, MHC2A, calpain1, calpain2, α-II spectrin 145-kDa cleavage product, MAFbx, MuRF1, and GAPDH. Primary antibodies were diluted 1:2000 (MHCslow, MHC2A, calpain1, calpain2) or 1:1,000 (145-kDa cleavage product, MAFbx, MuRF1) or 1:5000 (GAPDH) in blocking buffer and applied to the membranes overnight at 4 °C. This step was followed by incubation with a horseradish peroxidase-antibody conjugate (Abcam) directed against the primary antibody for 1 h. The membranes were then treated with chemiluminescent reagents (luminol and enhancer) and exposed to light-sensitive film. Images of these films were captured, and protein bands were quantified by using computerized image analysis (Gel Doc 2000, BioRad, Hercules, CA).
Primary antibodies used included Mouse monoclonal anti-rat MHCslow antibody (ab11083, Abcam, UK); Mouse monoclonal anti-rat MHC2A antibody (F18, DSHB, University of Iowa, USA); Rabbit polyclonal anti-calpain1 large Subunit antibody (ab28258, Abcam, UK); Rabbit polyclonal anti-calpain2 large Subunit antibody (ab39165, Abcam, UK); Mouse monoclonal anti-αII spectrin antibody (sc-46696, Santa Cruz, USA); Rabbit polyclonal anti-MAFbx antibody (ab74023, Abcam, UK); and Rabbit polyclonal Anti-MuRF1 antibody (ab172479, Abcam, UK) and mouse polyclonal anti-GAPDH antibody (Good Science, Shanghai, China). Secondary antibodies consisted of Horseradish peroxidase conjugated goat anti-mouse IgG antibody and Horseradish peroxidase conjugated goat anti-rabbit IgG antibody.
Histology experiments
Serial cryosections were cut from the frozen biopsies (8 μm thick). Microscope slides were rehydrated in phosphate buffer (PBS) and subsequently blocked with phosphate buffer containing 1% bovine serum albumin (PBS-1%BSA). Cryosections were incubated with antibody for MyHCslow (1:100, ab11083, Abcam) and for MyHCfast (1:100, ab51263,Abcam) followed by appropriate fluorescent-labeled secondary antibodies (Invitrogen). Fibers were visualized by an antibody reactive to laminin (1:100, ab11575, Abcam). Following each incubation, cryosections were washed four times for 5 min with PBS. The cross-sectional area (CSA) of diaphragm muscle fiber was determined from a sample of 25–30 fibers of each type per animal (eight animals per group). Sections were analyzed with a Leica DM6000B microscope (Leica Application Suite). CSA was calculated by ImageJ software.
Statistical analysis
Continuous data are reported as mean and standard error and were compared using unpaired Student’s t test or Wilcoxon rank-sum test after testing for normal distribution (Shapiro-Wilk). When comparing multiple independent means, a one-way analysis of variance (ANOVA) was first performed to confirm a difference across all groups prior to comparison of individual means. Results were considered significant if p values were less than 0.05. Statistical analysis was performed with the SPSS statistical package (v.19).
Results
Systemic and biologic response to mechanical ventilation
Blood pressure was maintained within physiologic ranges during the course of mechanical ventilation and did not differ significantly between groups (p > 0.05). Blood gas analysis after 24 h of MV is shown in Table 1. Initial body weights in CMV and CMVC were not significantly different from CON group (p > 0.05; Table 1). The total pentobarbital dose was similar in the two ventilated groups (98.37 ± 13.05 mg vs. 100.62 ± 12.79 mg in CMV and CMVC, respectively).
Table 1.
Initial body weight, arterial blood pressure, and blood gas data in CON, CMV, and CMVC
| N | Initial body weight, g | pH | PaCO2, mmHg | PaO2, mmHg | Heart rate, bpm | Blood pressure, mmHg | |
|---|---|---|---|---|---|---|---|
| CON | 8 | 363 ± 21 | NA | NA | NA | 343 ± 27 | 123 ± 12 |
| CMV | 8 | 353 ± 22 | 7.41 ± 0.03 | 41.5 ± 2.90 | 95.4 ± 8.18 | 346 ± 26 | 123 ± 12 |
| CMVC | 8 | 368 ± 27 | 7.40 ± 0.02* | 39.4 ± 3.16* | 93.4 ± 8.13* | 350 ± 22 | 121 ± 11 |
| F | – | 0.78 | – | – | – | 0.97 | 0.28 |
| p | – | >0.05 | – | – | – | >0.05 | >0.05 |
Values are mean ± SD
CON control animals, CMV controlled mechanical ventilation, CMVC CMV treated with calpeptin, F the value of ANOVA in the three groups, p values are for ANOVA
*p > 0.05 for CMVC vs. CMV
Diaphragm muscle myosin heavy chain content and cross-sectional area
CMV for 24 h resulted in a significant reduction in MHCslow and MHC2A. In calpeptin-treated animals, both MHCslow and MHC2A density were significantly higher compared with CMV (Fig. 1). As shown in Fig. 2, the CSA of diaphragmatic muscle fibers both MHCslow and MHCfast was decreased significantly by 34 and 37% in 24 h of CMV compared with CON group, respectively. However, administration of the calpain inhibitor calpeptin significantly prevented loss of CSA of MHCslow and MHCfast compared with CMV, indicating that calpeptin reduced ventilator-induced diaphragm atrophy.
Fig. 1.
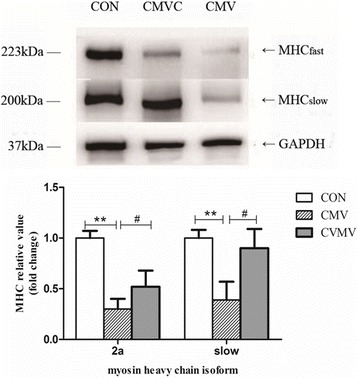
Western blot analyses of the two isoforms of the myosin heavy chain. GAPDH used as a reliable internal control. The results are expressed as a ratio of control group. CON control animals, CMV controlled mechanical ventilation, CMVC CMV treated with calpeptin. CMV rats received corresponding volumes of vehicle. **p < 0.01, #p < 0.05
Fig. 2.
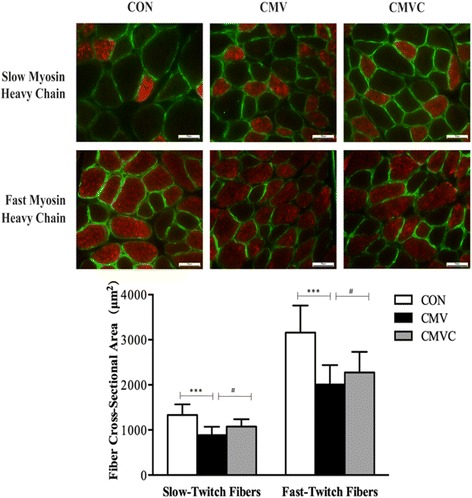
Diaphragm muscle cross-sectional area (CSA) in diaphragm skeletal muscle myofibers. In each of the sections, fibers reacting with the antibody appear red, whereas fibers not reacting with the antibody appear black. All fibers are outlined by an antibody reactive to laminin (ab11575, Abcam) which appear green. Scale bars represent 50 μm. CON control animals, CMV controlled mechanical ventilation, CMVC CMV treated with calpeptin. Each bar represents 200–240 fibers. CMV rats received corresponding volumes of vehicle. ***p < 0.001, #p < 0.05
Calpain activation
Calpain activation was assessed in diaphragm homogenates by the protein expression of calpain1 and calpain2 and calpain cleavage fragment. The 145-kDa protein fragment is specific to the calpain-dependent cleavage products of αII-spectrin. As shown in Fig. 3, 24 h of CMV resulted in a significant increase in calpain1 and calpain2 protein levels in the diaphragm compared to CON. In line with this, the protein level of αII-spectrin breakdown product in the diaphragm was significantly increased in CMV group compared with the CON (Fig. 4).
Fig. 3.
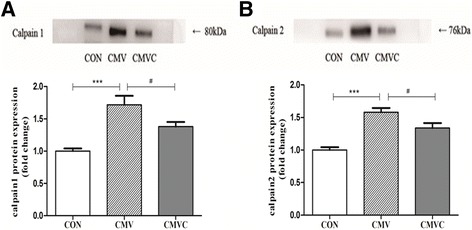
Protein levels of both calpain1 and calpain2 in the diaphragm. The images above the histograms in panels a and b are representative Western blots of data from the three experimental groups. The results are expressed as a ratio of control group. CON control animals, CMV controlled mechanical ventilation, CMVC CMV treated with calpeptin. CMV rats received corresponding volumes of vehicle. ***p < 0.001, #p < 0.05
Fig. 4.
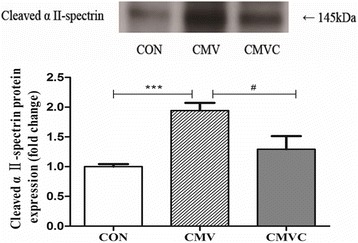
Levels of the 145 kDa α-II spectrin breakdown product in diaphragm. 145 kDa is a α-II -spectrin breakdown product that is specific to calpain1/2 cleavage of intact αII-spectrin. The images above the histograms are representative Western blots of data from the three experimental groups. The results are expressed as a ratio of control group. CON control animals, CMV controlled mechanical ventilation, CMVC CMV treated with calpeptin. CMV rats received corresponding volumes of vehicle. ***p < 0.001, #p < 0.05
Treatment with the calpain inhibitor calpeptin significantly reduced protein expression of calpain1 and calpain2 in CMV diaphragm (Fig. 3). In line with this, calpeptin significantly reduced αII-spectrin in ventilated rats (Fig. 4).
E3-ligase content
Compared with CON, CMV rats exhibited higher levels of both MAFbx and MuRF1 protein expression (Fig. 5) in the diaphragm. Both MAFbx and MuRF1 protein levels were lower in the diaphragm after treatment with calpeptin (Fig. 5).
Fig. 5.
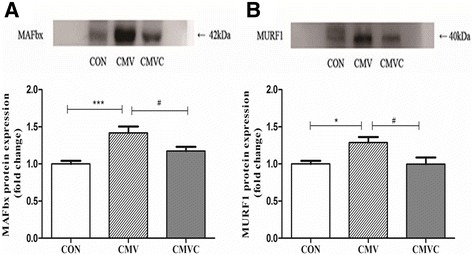
Protein levels of both MAFbx and MURF1 in the diaphragm. The images above the histograms in panels a and b are representative Western blots of data from the three experimental groups. The results are expressed as a ratio of control group. CON control animals, CMV controlled mechanical ventilation, CMVC CMV treated with calpeptin. CMV rats received corresponding volumes of vehicle. *p < 0.05, ***p < 0.001, #p < 0.05
Discussion
This study investigated the effects of calpain inhibition on ventilator-induced diaphragm atrophy and activation of proteolytic pathways. The main findings of our study can be summarized as follows: (1) calpain inhibitor calpeptin attenuates CMV-induced loss of myosin and CSA in the diaphragm muscle; (2) calpeptin treatment prevents the upregulation of calpain and the increase of calpain cleavage products; (3) calpeptin treatment reduces CMV-induced protein expression of the E3-ligases MAFbx and MuRF1. Collectively, our data indicate that calpain play an important role in CMV-induced diaphragm atrophy. Moreover, this study shows that inhibition of calpain activation prevents CMV-induced muscle atrophy.
Calpeptin is a dipeptide aldehyde and commonly used calpain inhibitor. Because calpeptin is membrane permeable, it is capable to penetrate into the cytosol of cells. There it binds to the critical cysteine residue in the active site of calpains [19]. Of note, Tsujinaka et al. showed that calpeptin was the most potent among synthesized inhibitors in terms of preventing the Ca2+-ionophore induced degradation of actin-binding protein and platelet talin in intact platelets. Furthermore, after 30-min incubation with intact platelets, calpeptin completely abolished calpain activity in platelets [20].
Previous studies in rats have demonstrated that CMV is associated with activation of the UPP, loss of contractile protein, and diaphragm dysfunction [5, 7]. For example, 12 h of CMV resulted in a decrease in force generation, promoted diaphragm fiber atrophy, and increased transcription of the muscle-specific E3-ligases MuRF1 and MaFbx in the diaphragm [5]. This is in line with our data, showing that 24 h of CMV induces loss of myosin and CSA in both slow and fast diaphragm fiber and an increase in protein expression of MuRF1 and MaFbx in the rat diaphragm. Recent studies in humans established that activation of the UPP is associated with loss of muscle protein in the diaphragm of patients receiving CMV [3, 8]. Levine and colleagues [8] reported that CMV for 18 to 69 h resulted in large decreases (i.e., 60%) of slow MyHC, fast MyHC, and a-actin in the diaphragm of brain dead organ donors. Also, critically ill patients receiving MV display atrophy and activation of UPP in both slow- and fast-diaphragm muscle fibers [3].
Although the UPP is activated during CMV, experimental inhibition of this proteolytic system has only been partially effective in preventing diaphragm muscle dysfunction [5, 21]. An explanation for this limited success is that other proteolytic systems act upstream of the UPP. Calpains are known to cleave myofilaments from the sarcomeric lattice [22] and thereby enable the UPP to degrade muscle protein into small peptides [23]. Immunolocalization studies showed that the calpains are concentrated in the Z-disk and I-band areas of the myofibril [22]. The Z-disk acts as anchor and mechanically links thin filaments from one sarcomere to the next along the myofibril [24]. One calpain substrate is the giant sarcomeric protein titin, which contains high-affinity calpain binding sites where the protein can be cleaved [25, 26]. Ex vivo experiments have shown that treatment of myofibrils from normal muscle with μ-calpain or m-calpain closely mimic structural disintegration as observed in rapidly atrophying [11, 22].
Several previous studies have evaluated the effect of calpain inhibition on preventing skeletal muscle atrophy [13, 27, 28]. For instance, inhibition of the calpain activity preserves sarcomere structure and attenuated the development of muscle weakness in hindlimbs of mice after 14 days of suspension [27]. During a 10-day unloading period of hindlimbs in mice, transgenic expression of calpastatin was also effective in reducing muscle atrophy by 30% [28].
Today, only one study assessed the role of calpain in the development of diaphragm dysfunction during CMV [12]. The authors showed that 12 h of MV of rats resulted in calpain activation in the diaphragm which was accompanied by diaphragm fiber atrophy and reduced force-generating capacity. This is in line with our data of 24 h of CMV. Treatment with the calpain inhibitor SJA-6017 in that study prevented the development of diaphragm fiber atrophy and weakness. Notably, diaphragm fiber force-generating capacity was expressed as specific force, i.e., tension normalized for cross-sectional area. So, the prevention of diaphragm weakness by calpain inhibition is only partially explained by attenuation of fiber atrophy, as calpain inhibition also increased specific force-generating capacity, independent of fiber size. The data of our study show that calpain inhibition prevented the loss of the major contractile protein myosin heavy chain. So, calpain inhibition not only prevents the loss of fiber size, but as our data show, also the loss of fiber contractile protein content. If the loss of contractile protein content exceeds the loss of fiber size, this would explain that calpain inhibition is able to prevent reduction of specific force-generating capacity in CMV animals. Interestingly, Nelson and colleagues also found that treatment with a calpain inhibitor attenuated activation of caspase-3 and vice versa [12]. This indicates that a regulatory cross talk exists between calpains and caspases for the modulation of CMV-induced diaphragmatic muscle atrophy. Our data suggest that a complimentary cross talk exists between the calpain system and the UPP. Because we showed that treatment with the calpain inhibitor calpeptin not only prevents calpain activation during CMV but also reduces the protein expression of the muscle-specific E3 ligases MuRF1 and MAFbx. To complete the circle of cross talks between calpain, caspase-3, and the UPP, a recent study showed that treatment of CMV rats with the proteasome inhibitor bortezomib partially protected the diaphragm against the development of weakness, most probably by indirect inhibition of caspase-3 [21]. The exact mechanism of these cross talks should be further investigated.
It is clear that prolonged MV results in diaphragmatic atrophy and contractile dysfunction in both animals and humans [2, 29]. However, in clinical settings, diaphragm dysfunction can also be caused or exacerbated by other factors, such as sepsis, malnutrition, and medicine. Sepsis is a major cause of mortality and long-term morbidity in ICU patients. Diaphragm dysfunction is strongly associated with sepsis [30]. Limited information exists concerning the combined impact of sepsis and MV on diaphragm function in humans or animals. In septic rats, Ebihara et al. [31] reported that 4 h CMV prevents diaphragmatic sarcolemmal injury and improves force-generating capacity of the diaphragm. However, Maes et al. [32] used an experimental rat model to simulate ICU settings in which patients with sepsis are MV in response to the development of multiple organ failure. In this protocol, CMV was applied 12 h after the initiation of sepsis. They found that 12 h of CMV in septic animals lead to worsening of diaphragm contractile dysfunction compared with CMV or sepsis alone but they found no exacerbation of muscle fiber atrophy. Future studies are needed to be investigated the interaction between the effects of mechanical ventilation and sepsis.
Clinical implications
Numerous data exists, even in humans [2, 3], that CMV could have deleterious effect on diaphragm structure and function. The most potential targets of pharmacological actors to prevent this seem to be upstream of the UPP. The current data, together with previous data in the literature, provides evidence that calpain inhibition might be an effective therapeutic target in patients with MV-induced muscle weakness. However, to our knowledge, no clinical study has reported the use of calpain inhibitors yet. Calpastatin is an endogenous calpain inhibitor and might therefore be the more safe option to start studies with. As in the current experimental study, we aimed to proof a role for calpains in the development of CMV-induced diaphragm atrophy; we administered the first dose of calpain inhibitor 2 h before the start of CMV. This strategy will not be feasible to implement in current daily clinical practice because diaphragm dysfunction is mostly recognized after a prolonged time of CMV. Nevertheless, recent studies have demonstrated that diaphragm dysfunction may also develop during elective surgery [33, 34]. In such patients prophylactic treatment may be of clinical relevance.
Conclusions
The current study showed that 24 h of CMV induced the activation of the calpain and UPP in the diaphragm. Specifically, treatment with a calpain inhibitor attenuated MV-induced muscle atrophy and calpain activation. Furthermore, calpain inhibition lead to reduced expression of muscle-specific E3-ligases suggesting a cross talk with the UPP. These finding provide more evidence that calpains are potential therapeutic target for treatment of CMV-induced diaphragm dysfunction.
Acknowledgements
This work was supported by a research grant from the National Natural Science Foundation of China (No. 81170074). We would like to thank Hong Guo and Xuelian Fu for excellent technical assistance.
Authors’ contributions
XPZ, LH, and SLM designed the study. XPZ, JRH, and LS performed the experiments. XPZ and SLM interpreted the data and performed analysis. XPZ, HWH, VanHee, SLM, and LH drafted the manuscript. FFW and LS performed the analysis. All authors participated in revising the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval
The study was approved by the University of Tongji Animal Care and Use Committee.
Abbreviations
- CMV
Controlled mechanical ventilation
- CMVC
CMV treated with calpeptin
- CON
Control animals
- CSA
Cross-sectional area
- GADPH
Glyceraldehyde-3- phosphate dehydrogenase
- MHC
Myosin heavy chain
- UPP
Ubiquitin-proteasome pathway
- VIDD
Ventilator-induced diaphragm dysfunction
Contributor Information
Xiaoping Zhu, Email: z_xping@hotmail.com.
Hieronymus W. H. van Hees, Email: Jeroen.vanHees@radboudumc.nl
Leo Heunks, Email: L.Heunks@vumc.nl.
Feifei Wang, Email: wangfefe2014@163.com.
Lei Shao, Email: surishao@163.com.
Jiaru Huang, Email: hjr0509@163.com.
Lei Shi, Email: shilei_peter@qq.com.
Shaolin Ma, Phone: xx 86 21 61569936, Email: m_slin@sina.com.
References
- 1.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2014;370:980. doi: 10.1056/NEJMc1400293. [DOI] [PubMed] [Google Scholar]
- 2.Levine S, Nguyen T, Taylor N, et al. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008;358:1327–1335. doi: 10.1056/NEJMoa070447. [DOI] [PubMed] [Google Scholar]
- 3.Hooijman PE, Beishuizen A, Witt CC, et al. Diaphragm muscle fiber weakness and ubiquitin-proteasome activation in critically ill patients. Am J Respir Crit Care Med. 2015;191:1126–1138. doi: 10.1164/rccm.201412-2214OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Jonghe B, Sylvie BJ, Durand MC, et al. Respiratory weakness is associated with limb weakness and delayed weaning in critical illness. Crit Care Med. 2007;35:2007–2015. doi: 10.1097/01.ccm.0000281450.01881.d8. [DOI] [PubMed] [Google Scholar]
- 5.Smuder AJ, Bradley N, Matthew HB, et al. Inhibition of the ubiquitin-proteasome pathway does not protect against ventilator-induced accelerated proteolysis or atrophy in the diaphragm. Anesthesiology. 2014;121:115–126. doi: 10.1097/ALN.0000000000000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell AP. Molecular regulation of skeletal muscle mass. Clin Exp Pharmacol Physiol. 2010;37:378–384. doi: 10.1111/j.1440-1681.2009.05265.x. [DOI] [PubMed] [Google Scholar]
- 7.DeRuisseau KC, Kavazis AN, Deering MA, et al. Mechanical ventilation induces alterations of the ubiquitin-proteasome pathway in the diaphragm. J Appl Physiol. 2005;98:1314–1321. doi: 10.1152/japplphysiol.00993.2004. [DOI] [PubMed] [Google Scholar]
- 8.Levine S, Biswas C, Dierov J, et al. Increased proteolysis, myosin depletion, and atrophic AKT-FOXO signaling in human diaphragm disuse. Am J Respir Crit Care Med. 2011;183:483–490. doi: 10.1164/rccm.200910-1487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaber S, Petrof BJ, Jung B, et al. Rapidly progressive diaphragmatic weakness and injury during mechanical ventilation in humans. Am J Respir Crit Care Med. 2011;183:364–371. doi: 10.1164/rccm.201004-0670OC. [DOI] [PubMed] [Google Scholar]
- 10.Shenkman BS, Belova SP, Lomonosova YN, et al. Calpain-dependent regulation of the skeletal muscle atrophy following unloading. Arch Biochem Biophys. 2015;584:36–41. doi: 10.1016/j.abb.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 11.Goll DE, Thompson VF, Li H, et al. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 12.Nelson WB, Smuder AJ, Hudson MB, et al. Cross-talk between the calpain and caspase-3 proteolytic systems in the diaphragm during prolonged mechanical ventilation. Crit Care Med. 2012;40:1857–1863. doi: 10.1097/CCM.0b013e318246bb5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fareed MU, Evenson AR, Wei W, et al. Treatment of rats with calpain inhibitors prevents sepsis-induced muscle proteolysis independent of atrogin-1/MAFbx and MuRF1 expression. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1589–1597. doi: 10.1152/ajpregu.00668.2005. [DOI] [PubMed] [Google Scholar]
- 14.Shanely RA, Zeregeroglu MA, Lennon SL, et al. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med. 2002;166:1369–1374. doi: 10.1164/rccm.200202-088OC. [DOI] [PubMed] [Google Scholar]
- 15.Powers SK, Shanely RA, Coombes JS, et al. Mechanical ventilation results progress contractile dysfunction in the diaphragm. J Appl Physiol. 2002;92:1851–1858. doi: 10.1152/japplphysiol.00881.2001. [DOI] [PubMed] [Google Scholar]
- 16.Zhu XP, Heunks LM, Versteeg EM, et al. Hypoxia-induced dysfunction of rat diaphragm: role of peroxynitrite. Am J Physiol (Lung Cell Physiol) 2005;288:L16–26. doi: 10.1152/ajplung.00412.2003. [DOI] [PubMed] [Google Scholar]
- 17.Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 18.Powers SK, Hudson MB, Nelson WB, et al. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med. 2011;39:1749–1759. doi: 10.1097/CCM.0b013e3182190b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welvaart WN, Paul MA, Stienen GJM, et al. Selective diaphragm muscle weakness after contractile inactivity during thoracic surgery. Ann Surg. 2011;254:1044–1049. doi: 10.1097/SLA.0b013e318232e75b. [DOI] [PubMed] [Google Scholar]
- 20.Tsujinaka T, Kajiwara Y, Kambayashi J, et al. Synthesis of a new cell penetrating calpain inhibitor (calpeptin) Biochem Biophys Res Commun. 1988;153:1201–1208. doi: 10.1016/S0006-291X(88)81355-X. [DOI] [PubMed] [Google Scholar]
- 21.Agten A, Maes K, Thomas D, et al. Bortezomib partially protects the rat diaphragm from ventilator-induced diaphragm dysfunction. Crit Care Med. 2012;40:2449–2455. doi: 10.1097/CCM.0b013e3182553a88. [DOI] [PubMed] [Google Scholar]
- 22.Bartoli M, Richard I. Calpains in muscle wasting. Int J Biochem Cell Biol. 2005;37:2115–2133. doi: 10.1016/j.biocel.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 23.Furuno K, Goodman MN, Goldberg AL. Role of different proteolytic systems in the degradation of muscle proteins during denervation atrophy. J Biol Chem. 1990;265:8550–8557. [PubMed] [Google Scholar]
- 24.Smith IJ, Dodd SL. Calpain activation causes a proteasome-dependent increase in protein degradation and inhibits the Akt signaling pathway in rat diaphragm muscle. Exp Physiol. 2007;92:561–573. doi: 10.1113/expphysiol.2006.035790. [DOI] [PubMed] [Google Scholar]
- 25.Solomon V, Lecker SH, Goldberg AL. The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J Biol Chem. 1998;273:25216–25222. doi: 10.1074/jbc.273.39.25216. [DOI] [PubMed] [Google Scholar]
- 26.Udaka J, Ohmori S, Terui T, et al. Disuse-induced preferential loss of the giant protein titin depresses muscle performance via abnormal sarcomeric organization. J Gen Physiol. 2008;131:33–41. doi: 10.1085/jgp.200709888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salazar JJ, Michele DE, Brooks SV. Inhibition of calpain prevents muscle weakness and disruption of sarcomere structure during hindlimb suspension. J Appl Physiol. 2010;108:120–127. doi: 10.1152/japplphysiol.01080.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tidball JG, Spencer MJ. Expression of a calpastatin transgene slows muscle wasting and obviates changes in myosin isoform expression during murine muscle disuse. J Physiol. 2002;545:819–828. doi: 10.1113/jphysiol.2002.024935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betters JL, Criswell DS, Shanely RA, et al. Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med. 2004;170:1179–1184. doi: 10.1164/rccm.200407-939OC. [DOI] [PubMed] [Google Scholar]
- 30.Demoule A, Jung B, Jaber S, et al. Diaphragm dysfunction on admission to the intensive care unit. Prevalence, risk factors, and prognostic impact-a prospective study. Am J Respir Crit Care Med. 2013;188:213–219. doi: 10.1164/rccm.201209-1668OC. [DOI] [PubMed] [Google Scholar]
- 31.Ebihara S, Hussain SNA, Danialou G, et al. Mechanical ventilation protects against diaphragm injury in sepsis. Am J Respir Crit Care Med. 2002;165:221–228. doi: 10.1164/ajrccm.165.2.2108041. [DOI] [PubMed] [Google Scholar]
- 32.Maes K, Stamiris A, Thomas D, et al. Effects of controlled mechanical ventilation on sepsis-induced diaphragm dysfunction in rats. Crit Care Med. 2014;42:e772–782. doi: 10.1097/CCM.0000000000000685. [DOI] [PubMed] [Google Scholar]
- 33.Ahn B, Beaver T, Ferreira LF, et al. Phrenic nerve stimulation increases human diaphragm fiber force after cardiothoracic surgery. Am J Respir Crit Care Med. 2014;190:837–839. doi: 10.1164/rccm.201405-0993LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welvaart WN, Paul MA, Stienen GJ, et al. Selective diaphragm muscle weakness after contractile inactivity during thoracic surgery. Ann Surg. 2011;254:1044–1049. doi: 10.1097/SLA.0b013e318232e75b. [DOI] [PubMed] [Google Scholar]