

Abstract
Idiopathic pulmonary fibrosis (IPF) has an unpredictable course and prognostic factors are incompletely understood. We aimed to identify prognostic factors, including multidimensional indices from a significant IPF cohort at the Bristol Interstitial Lung Disease Centre in the UK.
Patients diagnosed with IPF between 2007 and 2014 were identified. Longitudinal pulmonary physiology and exercise testing results were collated, with all-cause mortality used as the primary outcome. Factors influencing overall, 12- and 24-month survival were identified using Cox proportional hazards modelling and receiver operating characteristic curve analysis.
We found in this real-world cohort of 167 patients, diffusing capacity for carbon monoxide (DLCO) and initiation of long-term oxygen were independent markers of poor prognosis. Exercise testing results predicted 12-month mortality as well as DLCO, but did not perform as well for overall survival. The Composite Physiological Index was the best performing multidimensional index, but did not outperform DLCO. Our data confirmed that patients who experienced a fall in forced vital capacity (FVC) >10% had significantly worse survival after that point (p=0.024).
Our data from longitudinal follow-up in IPF show that DLCO is the best individual prognostic marker, outperforming FVC. Exercise testing is important in predicting early poor outcome. Regular and complete review should be conducted to ensure appropriate care is delivered in a timely fashion.
Short abstract
DLCO is a powerful prognostic marker in IPF http://ow.ly/EaEr307VTRN
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common and most aggressive of the idiopathic interstitial pneumonias. It is a distinct clinical entity, associated with unexplained chronic exertional dyspnoea, cough, bibasal crackles on auscultation of the chest and finger clubbing, with radiological and histopathological appearances consistent with usual interstitial pneumonia (UIP) [1].
The Bristol Interstitial Lung Disease (ILD) Service at North Bristol NHS Trust manages patients with IPF from around the south west of England. We performed a retrospective analysis of all patients diagnosed with IPF between 2007 and 2014. Our hypotheses were that exercise testing, and exertional desaturation in particular, significantly improves prognostication and that significant changes in lung function are detected with follow-up intervals of <4 months. In addition, we sought to compare published multidimensional indices for outcome prediction in this cohort and to compare the clinical course of patients following a decline of >10% in forced vital capacity (FVC) with those not suffering such a decline.
Our understanding of the natural history of IPF is limited by its highly variable clinical course and a lack of large-scale epidemiological studies [1]. Median survival in IPF ranges from 2.5 to 3.5 years [1, 2]; however, the clinical course of individual patients is highly variable.
Symptoms are recognised to precede diagnosis by 1–2 years [3]. More recently, incidental detection of subclinical IPF has become more frequent, resulting from thoracic imaging performed for other reasons. This has led to a wide range of reported mean and median survival estimates between 2 and 4 years from time of diagnosis [4–6]. There is a lack of consensus, and considerable debate surrounding appropriate outcome measures to assess treatment response in clinical trials [7–9] and to aid prognostication on routine clinical review.
The majority of studies examining prognostic indicators in IPF have been retrospective, or from clinical trial cohorts, which may be limited by their exclusion of those with more advanced disease [10–14]. Their findings have been heterogeneous, with conflicting data as to the most valuable variables in predicting outcome. Additionally, those studies examining the value of pulmonary function measures, including FVC, have not examined exercise testing variables in parallel [15, 16].
Several groups have developed multidimensional indices to help outcome prediction in IPF [10–12, 17]. No single scoring system has yet been widely accepted and these indices have not been compared in an independent cohort. The most accepted criterion for clinical trials remains a change in FVC.
Clinical guidelines have highlighted the lack of data to inform the ideal interval between clinical review for these patients [18]. In the UK there has been controversy surrounding the application of a “stopping rule” for use of novel antifibrotic agents following decline in FVC of >10%, and restriction of these new drugs based on FVC criteria [19, 20].
Methods
Patient and data identification
Patients were identified from review of the Bristol ILD Service multidisciplinary (MDT) team database and clinical records from September 1, 2007 to December 31, 2014. Inclusion criteria for this study were an MDT consensus diagnosis of IPF according to American Thoracic Society (ATS)/European Respiratory Society (ERS) criteria [1] made between these dates and ≥12 months of follow-up. Diagnoses made before publication of the 2011 criteria were reviewed at an MDT meeting for confirmation. Patients with a “working diagnosis” of IPF were diagnosed as such based on clinicoradiological parameters in the context of MDT discussion. Where a confident diagnosis could not be made, patients were referred for surgical lung biopsy following discussion of the risks and benefits of such an approach and considering the clinical condition of the patient, in accordance with national guidelines [18]. Ethical approval for this work was given by the East of England research ethics committee (reference 15/EE/0023).
Demographic data were collected in addition to all-cause mortality and use of pirfenidone. Computed tomography of the chest was classified as “definite” or “possible” UIP pattern. Where echocardiography was performed around the time of the first clinical assessment, incidence of pulmonary hypertension was noted. Initiation of long-term oxygen therapy (LTOT) was documented at the time of the first visit. This was initiated in accordance with national guidelines only for those patients with arterial oxygen tension of <7.3 kPa or <8 kPa in the presence of evidence of pulmonary hypertension [21]. Comorbidities, specifically those including past or current history of cancer or cardiac disease, were noted.
Pulmonary function and exercise testing were all performed in the same physiology department according to international criteria [22, 23]. Results from all spirometry, diffusing capacity of the lung for carbon monoxide (DLCO) and 6-min walk testing (6MWT) were collated over the follow-up period. Relative changes in FVC and DLCO were calculated, as suggested by Richeldi et al. [24]. Change in FVC and DLCO on follow-up were noted, in addition to subsequent trends in spirometry results. The minimal clinically important difference (MCID) for IPF of 5% was used [13]. Desaturation was defined as a fall in pulse oximetry on exertion of ≥4% to a level of <88%. Disease progression was defined as death, respiratory hospitalisation or a fall in FVC of >10%.
Multidimensional indices
The indices compared were the Composite Physiological Index (CPI) [11], the Gender, Age, Physiology (GAP) score [10] and the distance–saturation product (DSP) [25]. The score described by du Bois et al. [12], which incorporates longitudinal change in physiology values, was also assessed. These indices were selected based on the diversity of their underlying methodologies and the variables incorporated. Scores were calculated according to the methodology described by the authors.
Statistical analysis
All data were tested for normality and are reported as mean (95% CI) or median (interquartile range).
An a priori statistical plan was approved, using a multivariable, proportional hazard Cox model with backward selection to determine which individual variables were independently predictive of survival from both baseline variables and after 12 months of follow-up. Separate analyses were performed for the cohort with follow-up values available, including for comparison of the du Bois multidimensional index. The full model contained all variables with a univariable p-value <0.2. Variables with a p-value >0.1 were removed stepwise with model comparison by Bayesian information criteria (BIC). Data were assessed for multicollinearity and no variables were of concern. The proportional hazards assumption was checked by testing for a non-zero slope in a generalised linear regression of the scaled Schoenfeld residuals on functions of time.
Individual variables and multidimensional indices were compared using Cox univariable analysis (BIC). Early poor outcome was assessed by comparison of the area under the receiver operating characteristic (ROC) curve (c-statistic) analysis for 12- and 24-month survival (for continuous variables). Multiple survival analysis methodologies were used to ensure robust comparison of scoring systems. Statistical analyses were conducted using Stata software (Stata/IC v14.1; StataCorp, College Station, TX, USA).
Results
167 patients were identified in the study period with ≥12 months of follow-up and included in the analysis. Demographic details are shown in table 1. Average follow-up was 22.6 months (range 12–87 months). 78% of patients had definite UIP on high-resolution computed tomography (HRCT), meaning 22% of patients had a “working diagnosis” of IPF, according to ATS/ERS criteria. Eight of these patients were diagnosed with IPF following surgical lung biopsy. The remaining patients were not deemed sufficiently robust to undergo this procedure, or declined biopsy following discussion. Pulmonary hypertension was observed in 18.6% of patients and this was associated with a significantly lower DLCO (between-group difference 10.98%, p=0.01). Overall mortality was 40.1%, with 13.8% 12-month mortality and a median survival of 35.4 (16.5–65.5) months. Repeat pulmonary physiology testing at 12 months was available for 126 patients.
TABLE 1.
Baseline demographic and physiological variables
| Male | 137 (82) |
| Current or ex-smoker | 116 (69.5) |
| Definite UIP on CT | 131 (78.4) |
| Pulmonary hypertension | 31 (18.6) |
| Exertional desaturation | 98 (68.1) |
| LTOT initiation | 31 (18.6) |
| Pirfenidone use at any time | 45 (26.9) |
| Overall mortality | 67 (40.1) |
| 6-month mortality | 15 (9.0) |
| 12-month mortality | 23 (13.8) |
| 24-month mortality# | 49 (48.5) |
| Age years | 73.5 (71.9–75.0) |
| BMI kg·m−2 | 26.7 (25.9–27.6) |
| FVC % pred | 83.0 (79.6–86.5) |
| FEV1 % pred | 84.5 (80.7–88.3) |
| DLCO % pred | 52.8 (49.2–56.4) |
| FEV1/FVC ratio % | 81.5 (80.2–82.9) |
| 6-min walking distance m | 283.8 (264.9–302.8) |
| 6-min walking distance % pred | 60.6 (56.7–64.5) |
| Borg on exertion | 2.7 (2.4–2.9) |
| Lowest SaO2 on exertion | 87.5 (86.3–88.6) |
Data are presented as n (%) or mean (95% CI). UIP: usual interstitial pneumonia; CT: computed tomography; LTOT: long-term oxygen therapy; BMI: body mass index; FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s; DLCO: diffusing capacity of the lung for carbon monoxide; SaO2: arterial oxygen saturation. #: n=101.
Survival analysis from individual variables
Univariable Cox proportional hazards modelling identified pulmonary hypertension, forced expiratory volume in 1s (FEV1)/FVC ratio, FVC, DLCO, inability to complete DLCO test, 6-min walking distance, exertional desaturation, baseline initiation of LTOT, interval change in DLCO and interval change in walking distance as having a significant effect on survival (p<0.05) (online supplementary table S1). Median survival after initiation of LTOT was 17.4 months (11.3–21.8), compared to 48.5 months where oxygen was not required (p<0.001 by log rank test) (figure 1). Pirfenidone initiation was associated with a worse survival by univariable proportional hazards modelling (hazard ratio 1.99, p=0.02), with a median survival of 20.4 months, compared to 44.7 months where this was not given. There was no difference in the rate of FVC decline >10% with pirfenidone use in this cohort (p=0.43).
FIGURE 1.
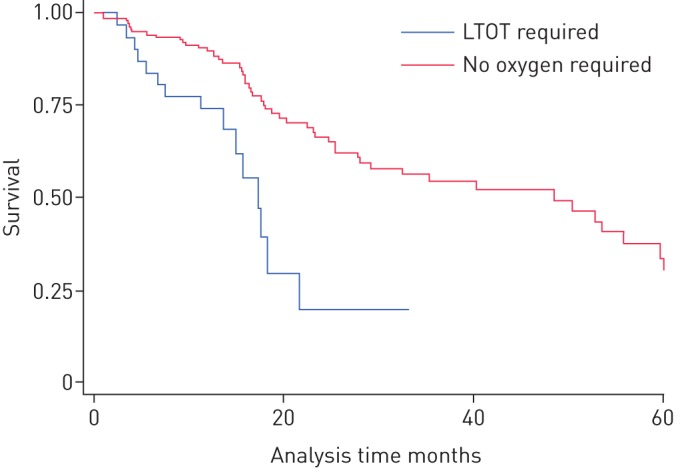
Kaplan–Meier survival curve examining long-term oxygen therapy (LTOT) initiation at baseline. p<0.001 by log-rank test.
On multivariable analysis using only baseline variables with p<0.2, male sex and DLCO were significant in the full model. The best performing model as assessed by BIC contained FVC, sex, DLCO, baseline initiation on LTOT and an inability to complete DLCO assessment (table 2).
TABLE 2.
Best-performing models on multivariable Cox proportional hazards analysis
| Hazard ratio (95% CI) | Significance | Model significance | |
| Baseline variables# | |||
| Male | 2.278 (1.126–4.608) | 0.022 | <0.001 |
| LTOT initiation | 2.050 (1.156–3.636) | 0.014 | |
| FVC | 1.020 (0.999–1.041) | 0.058 | |
| DLCO | 0.985 (0.970–1.000) | 0.044 | |
| Inability to perform DLCO assessment | 2.915 (1.380–6.158) | 0.005 | |
| 12-month follow-up¶ | |||
| DLCO | 0.953 (0.931–0.974) | <0.001 | <0.001 |
| Change in DLCO | 0.984 (0.972–0.997) | 0.014 | |
| Male | 1.933 (0.886–4.219) | 0.098 |
Data are presented as mean (95% CI), unless otherwise stated. LTOT: long-term oxygen therapy; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide. #: n=167; ¶: n=126.
When 12-month follow-up data were included in a multivariable model containing all variables with p<0.2, only baseline DLCO remained significant (table 2). The best performing model contained sex and baseline/change in DLCO.
Survival analysis from multidimensional indices
Baseline values and survival analysis results for CPI, GAP, DSP and du Bois scores are shown in table 3. Comparison of Cox models by BIC demonstrated that univariable models containing DLCO or CPI were the best performing. Using only FVC was worse than any of the multidimensional indices.
TABLE 3.
Multidimensional indices: baseline values and univariable Cox regression hazard ratios
| Mean (95% CI) | Hazard ratio (95% CI) | p-value | |
| CPI | 41.42 (38.64–44.21) | 1.064 (1.040–1.089) | <0.001 |
| DSP | 249 (232–266) | 0.996 (0.993–0.998) | 0.001 |
| GAP I | 21 (12.7) | Reference | |
| GAP II | 86 (51.8) | 1.501 (0.847–2.661) | 0.164 |
| GAP III | 59 (35.5) | 5.466 (2.604–11.474) | <0.001 |
| du Bois score# | 18.3 (16.4–20.2) | 1.039 (1.011–1.067) | 0.006 |
Data are presented as n (%), unless otherwise stated. CPI: Composite Physiological Index; DSP: distance–saturation product; GAP: Gender, Age, Physiology score. #: n=126.
The du Bois prognostic score includes 12-month follow-up variables. When comparing this within the cohort with follow-up data available, the best-performing univariable Cox models remained the DLCO and CPI.
Analysis of the area under ROC curves for both 12- and 24-month mortality, to investigate prediction of early poor outcome, showed that FVC performed worse than DLCO or CPI (table 4). It was no better at predicting early poor outcome than age alone. Exercise capacity, as measured by 6MWT, had equivalent performance to DLCO for 12-month mortality, but this was not seen at 24 months.
TABLE 4.
Comparison of multidimensional indices with individual variables by received operating characteristic (ROC) analysis at 12 and 24 months
| 12-month ROC c-statistic | p-value for comparison with FVC | 24-month ROC c-statistic | p-value for comparison with FVC | |
| Age | 0.694 | 0.213 | 0.639 | 0.942 |
| FVC | 0.564 | Ref. | 0.645 | Ref. |
| DLCO | 0.705 | 0.040 | 0.770 | 0.041 |
| Walking distance | 0.702 | 0.135 | 0.591 | 0.491 |
| Lowest SaO2 | 0.669 | 0.212 | 0.638 | 0.922 |
| CPI | 0.657 | 0.037 | 0.761 | 0.013 |
| GAP | 0.636 | 0.115 | 0.722 | 0.091 |
| DSP | 0.729 | 0.073 | 0.623 | 0.785 |
FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide; SaO2: arterial oxygen saturation; CPI: Composite Physiological Index; GAP: Gender, Age, Physiology score; DSP: distance–saturation product; Ref.: reference.
Progression-free survival
Overall median progression-free survival was 21.8 (11.1–53.5) months. Univariable Cox proportional hazards analysis showed that age, detection of pulmonary hypertension, LTOT initiation, exertional desaturation, walking distance and DLCO or inability to complete DLCO were significant variables (online supplementary table S2). Following multivariable analysis, age, LTOT initiation and DLCO remained significant.
Of the multidimensional indices, CPI, GAP and DSP were significantly predictive of disease progression on Cox proportional hazards analysis, with no substantial differences in model performance as compared by BIC. These indices did not perform better than individual variables of oxygen use and DLCO; however, they were superior to FVC.
Clinical course after FVC decline >10%
89 patients had ≥24 months of follow-up, providing sufficient data for analysis of their clinical course after FVC decline of >10% in a 12-month period. Median follow-up following the 12-month result for this cohort was 18.6 (11.1–32.5) months. Median survival by log rank comparison was significantly worse for those who experienced FVC decline >10% (p=0.024) (figure 2). However, the “new” baseline for this group was significantly lower following decline for both FVC (68.4% versus 81.6%, p=0.001) and DLCO (42.3% versus 53.5%, p=0.003). After a fall in FVC of >10%, patients went on deteriorate at a greater rate (4.5% per year versus 0.4% per year, p<0.001) (figure 3).
FIGURE 2.
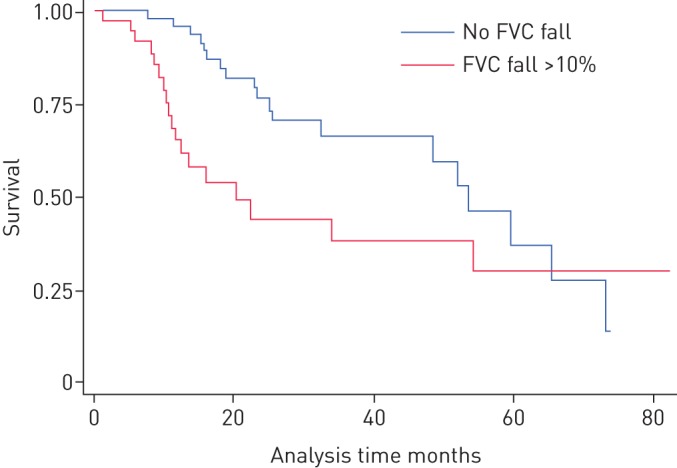
Kaplan–Meier survival curve comparing subjects with a fall in forced vital capacity (FVC) >10% with those without. p=0.024 by log-rank test.
FIGURE 3.
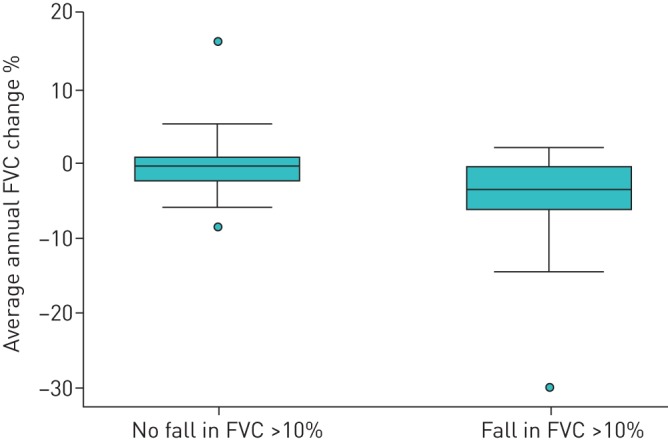
Comparison of average forced vital capacity (FVC) change following a fall in FVC >10%. Data are presented as median (interquartile range). p<0.001 by Mann–Whitney test.
Follow-up interval
860 individual clinic visits were recorded from our cohort in the study period. Median interval of follow-up between visits was 4.1 (3.1–6.1) months, with a median change in FVC between visits of −1.1% (−5.5–2.7%). 27.6% of recorded values had fallen by more than the MCID (>5%) from the previous visit (22.6% in those with follow-up <4 months, 32.3% with follow-up >4 months; p<0.001). Change in FVC was significantly, but weakly, correlated with interval of follow-up by (Spearman correlation co-efficient −0.135, p<0.001) (figure 4). Where a decline of >5% in FVC was observed, this was significantly related to the interval of follow-up (p<0.001 by Mann–Whitney U-test).
FIGURE 4.
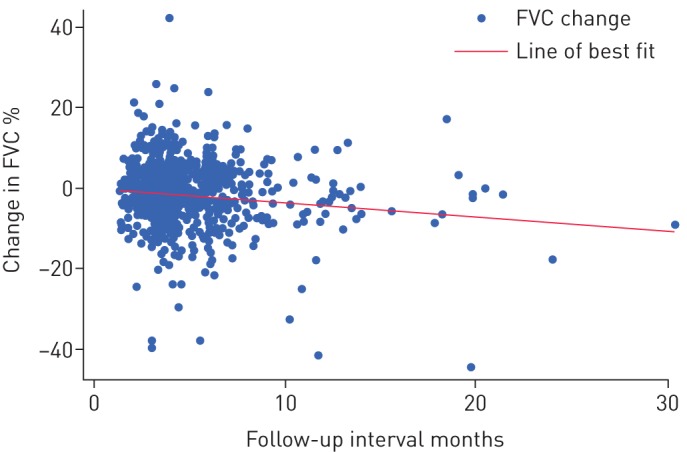
Change in forced vital capacity (FVC) by interval to last follow-up.
Discussion
These data show that best prediction of poor outcome in IPF comes from DLCO. FVC, despite its widespread use to guide prescribing and in clinical trials, does not perform well as a marker for prognosis, early poor outcome or progression-free survival. LTOT initiation is also a particular marker of poor prognosis, with a median survival of <18 months after this event. Multidimensional indices are predictive; however, only the CPI performs as well and as consistently as DLCO. Exercise testing variables, including exertional desaturation, are good markers for early poor outcome and perform as consistently as the multidimensional indices tested.
In addition, we have demonstrated that following a fall in FVC of >10%, further deterioration does occur and this is a marker of worse survival. This may be accounted for by the association of FVC fall with lower DLCO values. In order to detect these changes in pulmonary function in a timely fashion, regular follow-up is required; over 20% of occasions at which clinically significant decline in FVC had occurred were at <4 months’ interval since the previous visit. While we cannot generalise from this limited cohort study, it would seem in this cohort that regular review at an interval more frequent than 6 months is needed to promptly detect clinically significant changes. Approaches such as home spirometry [26] may enable monitoring of patients without the need for clinic visits in the future, but this requires further investigation.
These data are derived from a well-characterised, real-world cohort of IPF patients, with significant follow-up. We were able to conduct analyses both at baseline and after 12 months of follow-up in a significant number of patients. We have assessed the comparative strengths of indices for which no previous head-to-head comparison has been performed in an independent population. The use of a variety of methodologies gives strength to our conclusions, with consistency of results between the different approaches to survival analysis.
Our work also addresses an important question highlighted by guidelines, reporting the pattern of changes in FVC seen for different follow-up intervals. We have been able to observe the nature of FVC changes following a decline of 10%, which should inform policy decisions related to eligibility criteria for novel antifibrotic agents.
Our data are limited by their retrospective nature and resultant heterogeneity and attrition to follow-up. However, this is unavoidable in this area when attempting to gather such information about the real-world experience of IPF. In addition, there are issues related to the period covered by this study, during which antifibrotic treatment with pirfenidone became available. The group given this drug appeared to have a worse survival; however, we have interpreted this to reflect the eligibility criteria for pirfenidone in England, limiting its use to those with more severe disease (FVC 50–80% predicted) [19]. There did not appear to be any significant difference in the rate of FVC decline between the treated group and the remaining patients in this small cohort. It must be emphasised that this cohort was a real-world group and therefore not analogous to the groups used in randomised controlled studies of antifibrotic medications.
There has been controversy surrounding the use of prognostic indicators in clinical trials in IPF [8, 9]. Some have criticised the use of FVC, advocating the use of all-cause mortality or hospitalisation. There is heterogeneity in the findings of published studies exploring which variables are robust prognostic indicators, despite consistent methodologies.
A complete overview of all such studies is beyond our scope here and readers are directed to review articles [27, 28]. These studies have been conducted in cohorts from North America and South East Asia, in contrast to the demographics of the work presented here. This difference in population make-up may explain some of the contrasts in our findings. Where studies have examined pulmonary function measures as prognostic variables, they have not included exercise testing results [15, 16]. Where such results have been included, previous work has indicated the prognostic importance of both distance walked and desaturation during the 6MWT [14, 25, 29–31]. There is a consistent signal for DLCO, walking distance and change in FVC to be useful metrics; however, this is not uniform.
Our data support the routine use of exercise testing in clinical assessment of IPF and also lend weight to the use of DLCO. There are concerns surrounding the heterogeneity of test performance, limiting the use of these measures in clinical trials; however, this should not prevent its application as a prognostic indicator for IPF.
In IPF, studies have shown only moderate correlation between exercise capacity, dyspnoea on exertion and pulmonary function measurements [32, 33]. This highlights the multifactorial contribution to the symptoms of exertional dyspnoea and exercise capacity. Similarly, it has been observed that pulmonary function measurements do not robustly correlate with the degree of parenchymal involvement on HRCT [34–36]. A well-validated staging system in IPF should improve prognostication in disease, improve future study design and also facilitate decision making around medications and lung transplantation.
Several groups have developed multidimensional indices for IPF [10–12, 17, 25]; however, no single scoring system has yet been widely accepted. In other diseases, multidimensional indices have been superior to individual predictors; the BODE (body mass index (BMI), airflow obstruction, dyspnoea and exercise capacity) index in chronic obstructive pulmonary disease includes BMI, FEV1/FVC ratio, 6-min walking distance and Medical Research Council dyspnoea score [37], while the CURB-65 score (confusion, urea >7 mmol·L-1, respiratory rate ≥30 breaths·min-1, blood pressure <90 mmHg (systolic) or ≤60 mmHg (diastolic), age ≥65 years) is widely used in community acquired pneumonia [38]. Both of these indices are superior to individual components in isolation.
The ideal multidimensional index for IPF would be simple, making use of routinely collected clinical data that is easy to apply in day-to-day clinical practice. The indices compared in this work all fulfil these criteria. Our data suggest that, while performing well, these indices in IPF do not yet have superior performance to isolated elements of the clinical and physiological assessment.
This study confirms the validity of the DSP, GAP, CPI and du Bois multidimensional indices in prognostication for IPF; however, our data suggest that these do not consistently outperform exercise testing and DLCO. In addition, we have highlighted LTOT initiation as a significant prognostic indicator. Our data support clinical review of IPF patients at regular intervals and confirms that FVC decline >10%, while often an isolated event, is a harbinger of poor outcome in IPF. These data should inform the development of guidelines and policy decisions related to novel antifibrotic agents.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary data 00096-2016_Supplementary_data (19.3KB, docx)
Acknowledgements
We would like to thank Paul White (University of the West of England, Bristol, UK) for his statistical advice and Giles Dixon (University Hospitals Bristol NHS Foundation Trust, Bristol) for his help in data entry.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Conflict of interest: None declared.
References
- 1.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 431–440. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson AG, Colby TV, du Bois RM, et al. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162: 2213–2217. [DOI] [PubMed] [Google Scholar]
- 4.Martinez FJ, Safrin S, Weycker D, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 2005; 142: 963–967. [DOI] [PubMed] [Google Scholar]
- 5.Selman M, Carrillo G, Estrada A, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One 2007; 2: e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King TE Jr, Tooze JA, Schwarz MI, et al. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med 2001; 164: 1171–1181. [DOI] [PubMed] [Google Scholar]
- 7.Saketkoo LA, Mittoo S, Huscher D, et al. Connective tissue disease related interstitial lung diseases and idiopathic pulmonary fibrosis: provisional core sets of domains and instruments for use in clinical trials. Thorax 2014; 69: 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raghu G, Collard HR, Anstrom KJ, et al. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med 2012; 185: 1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.du Bois RM, Nathan SD, Richeldi L, et al. Idiopathic pulmonary fibrosis: lung function is a clinically meaningful endpoint for phase III trials. Am J Respir Crit Care Med 2012; 186: 712–715. [DOI] [PubMed] [Google Scholar]
- 10.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156: 684–691. [DOI] [PubMed] [Google Scholar]
- 11.Wells AU, Desai SR, Rubens MB, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 2003; 167: 962–969. [DOI] [PubMed] [Google Scholar]
- 12.du Bois RM, Weycker D, Albera C, et al. Ascertainment of individual risk of mortality for patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 184: 459–466. [DOI] [PubMed] [Google Scholar]
- 13.du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011; 184: 1382–1389. [DOI] [PubMed] [Google Scholar]
- 14.du Bois RM, Weycker D, Albera C, et al. Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal clinically important difference. Am J Respir Crit Care Med 2011; 183: 1231–1237. [DOI] [PubMed] [Google Scholar]
- 15.Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171: 639–644. [DOI] [PubMed] [Google Scholar]
- 16.Zappala CJ, Latsi PI, Nicholson AG, et al. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J 2010; 35: 830–836. [DOI] [PubMed] [Google Scholar]
- 17.Mura M, Porretta MA, Bargagli E, et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J 2012; 40: 101–109. [DOI] [PubMed] [Google Scholar]
- 18.National Institute for Health and Care Excellence (NICE). Idiopathic Pulmonary Fibrosis: the Diagnosis and Management of Suspected Idiopathic Pulmonary Fibrosis. CG163. www.nice.org.uk/guidance/cg163 Date last accessed: June 6, 2015. Date last updated: June 2013. [PubMed]
- 19.National Institute for Health and Care Excellence (NICE). Pirfenidone for Treating Idiopathic Pulmonary Fibrosis. TA282. www.nice.org.uk/guidance/ta282 Date last accessed: December 1, 2016. Date last updated: April 24, 2013.
- 20.National Institute for Health and Care Excellence (NICE). Nintedanib for Treating Idiopathic Pulmonary Fibrosis. TA379. www.nice.org.uk/guidance/ta379 Date last accessed: December 1, 2016. Date last updated: January 27, 2016.
- 21.Hardinge M, Annandale J, Bourne S, et al. British Thoracic Society guidelines for home oxygen use in adults. Thorax 2015; 70: i1–i43. [DOI] [PubMed] [Google Scholar]
- 22.Holland AE, Spruit MA, Troosters T, et al. An official European Respiratory Society/American Thoracic Society technical standard: field walking tests in chronic respiratory disease. Eur Respir J 2014; 44: 1428–1446. [DOI] [PubMed] [Google Scholar]
- 23.Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J 2005; 26: 319–338. [DOI] [PubMed] [Google Scholar]
- 24.Richeldi L, Ryerson CJ, Lee JS, et al. Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax 2012; 67: 407–411. [DOI] [PubMed] [Google Scholar]
- 25.Lettieri CJ, Nathan SD, Browning RF, et al. The distance-saturation product predicts mortality in idiopathic pulmonary fibrosis. Respir Med 2006; 100: 1734–1741. [DOI] [PubMed] [Google Scholar]
- 26.Russell AM, Adamali H, Molyneaux PL, et al. Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016; 194: 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rozanski C, Mura M. Multi-dimensional indices to stage idiopathic pulmonary fibrosis: a systematic review. Sarcoidosis Vasc Diffuse Lung Dis 2014; 31: 8–18. [PubMed] [Google Scholar]
- 28.Kolb M, Collard HR. Staging of idiopathic pulmonary fibrosis: past, present and future. Eur Respir Rev 2014; 23: 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lama VN, Flaherty KR, Toews GB, et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003; 168: 1084–1090. [DOI] [PubMed] [Google Scholar]
- 30.Caminati A, Bianchi A, Cassandro R, et al. Walking distance on 6-MWT is a prognostic factor in idiopathic pulmonary fibrosis. Respir Med 2009; 103: 117–123. [DOI] [PubMed] [Google Scholar]
- 31.du Bois RM, Albera C, Bradford WZ, et al. 6-minute walk distance is an independent predictor of mortality in patients with idiopathic pulmonary fibrosis. Eur Respir J 2014; 43: 1421–1429. [DOI] [PubMed] [Google Scholar]
- 32.Papiris SA, Daniil ZD, Malagari K, et al. The Medical Research Council dyspnea scale in the estimation of disease severity in idiopathic pulmonary fibrosis. Respir Med 2005; 99: 755–761. [DOI] [PubMed] [Google Scholar]
- 33.Mura M, Ferretti A, Ferro O, et al. Functional predictors of exertional dyspnea, 6-min walking distance and HRCT fibrosis score in idiopathic pulmonary fibrosis. Respiration 2006; 73: 495–502. [DOI] [PubMed] [Google Scholar]
- 34.Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157: 1063–1072. [DOI] [PubMed] [Google Scholar]
- 35.Xaubet A, Agusti C, Luburich P, et al. Pulmonary function tests and CT scan in the management of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998; 158: 431–436. [DOI] [PubMed] [Google Scholar]
- 36.Wells AU, King AD, Rubens MB, et al. Lone cryptogenic fibrosing alveolitis: a functional-morphologic correlation based on extent of disease on thin-section computed tomography. Am J Respir Crit Care Med 1997; 155: 1367–1375. [DOI] [PubMed] [Google Scholar]
- 37.Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 1005–1012. [DOI] [PubMed] [Google Scholar]
- 38.Lim WS, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58: 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary data 00096-2016_Supplementary_data (19.3KB, docx)