

Abstract
Summary: Excitotoxicity, defined as excessive exposure to the neurotransmitter glutamate or overstimulation of its membrane receptors, has been implicated as one of the key factors contributing to neuronal injury and death in a wide range of both acute and chronic neurologic disorders. Excitotoxic cell death is due, at least in part, to excessive activation of N-methyl-d-aspartate (NMDA)-type glutamate receptors and hence excessive Ca2+ influx through the receptor’s associated ion channel. Physiological NMDA receptor activity, however, is also essential for normal neuronal function; potential neuroprotective agents that block virtually all NMDA receptor activity will very likely have unacceptable clinical side effects. For this reason many NMDA receptor antagonists have disappointingly failed advanced clinical trials for a number of diseases including stroke and neurodegenerative disorders such as Huntington’s disease. In contrast, studies in my laboratory were the first to show that memantine, an adamantane derivative, preferentially blocks excessive NMDA receptor activity without disrupting normal activity. Memantine does this through its action as an open-channel blocker; it enters the receptor-associated ion channel preferentially when it is excessively open, and, most importantly, its off-rate is relatively fast so that it does not substantially accumulate in the channel to interfere with normal synaptic transmission. Past clinical use for other indications has demonstrated that memantine is well tolerated, and it has recently been approved in both Europe and the USA for the treatment of dementia of the Alzheimer’s type. Clinical studies of the safety and efficacy of memantine for other neurological disorders, including glaucoma and other forms of dementia, are currently underway. A series of second-generation memantine derivatives are currently in development and may prove to have even greater neuroprotective properties than does memantine. These second-generation drugs take advantage of the fact that the NMDA receptor has other modulatory sites, in addition to its ion channel, that could potentially be used for safe but effective clinical intervention.
Keywords: Excitotoxicity, NMDA antagonist, open channel blocker, memantine
INTRODUCTION
Acute and chronic neurologic diseases are among the leading causes of death, disability, and economic expense in the world. For example, cerebral ischemia (stroke) and Alzheimer’s disease rank third and fourth as causes for mortality in the United States. In fact, it has been estimated by the National Institutes of Health that as the population continues to age, treatment of stroke and dementia will consume our entire gross national product by the latter decades of this century. Excitotoxic cell death is thought to contribute to neuronal cell injury and death in these and other neurodegenerative disorders. Excitotoxicity is due, at least in part, to excessive activation of N-methyl-d-aspartate (NMDA)-type glutamate receptors and hence excessive Ca2+ influx through the receptor’s associated ion channel. On the other hand, glutamate-mediated synaptic transmission is critical for the normal functioning of the nervous system. Glutamate is the major excitatory neurotransmitter in the brain. There are three classes of glutamate-gated ion (or ionotropic) channels, known as AMPA, kainate, and NMDA receptors. Among these, the ion channels coupled to classical NMDA receptors are generally the most permeable to Ca2+. Excessive activation of the NMDA receptor in particular leads to production of damaging free radicals and other enzymatic processes contributing to cell death.1,2 With the disruption of energy metabolism during acute and chronic neurodegenerative disorders, glutamate is not cleared properly and may even be inappropriately released. Moreover, energetically compromised neurons become depolarized (more positively charged) because in the absence of energy they cannot maintain ionic homeostasis; this depolarization relieves the normal Mg2+ block of NMDA receptor-coupled channels because the relatively positive charge in the cell repels positively-charged Mg2+ from the channel pore. Hence, during periods of ischemia and in many neurodegenerative diseases, excessive stimulation of glutamate receptors is thought to occur. These neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, HIV-associated dementia, multiple sclerosis, amyotrophic lateral sclerosis, and glaucoma, are caused by different mechanisms but may share a final common pathway to neuronal injury due to the overstimulation of glutamate receptors, especially of the NMDA subtype.1 Hence, NMDA receptor antagonists could potentially be of therapeutic benefit in a number of neurologic disorders manifesting excessive stimulation of NMDA receptors, including stroke, dementia, and neuropathic pain syndromes. NMDA receptors are made up of different subunits: NR1 (whose presence is mandatory), NR2A-D, and, in some cases, NR3A or B subunits. The receptor is probably composed of a tetramer of these subunits. The subunit composition determines the pharmacology and other parameters of the receptor-ion channel complex. Alternative splicing of some subunits, such as NR1, further contributes to the pharmacological properties of the receptor. The subunits are differentially expressed both regionally in the brain and temporally during development. For this reason some authorities have suggested developing antagonists selective for particular subunits, such as NR2B, which is present predominantly in the forebrain.3
THE QUEST FOR NMDA RECEPTOR ANTAGONISTS
Excitotoxicity is a particularly attractive target for neuroprotective efforts because it is implicated in the pathophysiology of a wide variety of acute and chronic neurodegenerative disorders.1 The challenge facing those trying to devise strategies for combating excitotoxicity is that the same processes that, in excess, lead to excitotoxic cell death are, at lower levels, absolutely critical for normal neuronal function. Until recently, all of the drugs that showed the most promise as inhibitors of excitotoxicity also blocked normal neuronal function and consequently had severe and unacceptable side effects, so clinical trials for stroke and traumatic brain injury failed.4–6
Recently, however, the well-tolerated but underappreciated drug memantine has been rediscovered to be not only capable of blocking excitotoxic cell death7 but most importantly, of also doing it in a clinically tolerated, non-toxic manner.8–11 Memantine was recently approved by the European Union and in the USA for the treatment of Alzheimer’s disease and may also show efficacy for vascular dementia.12,13 The drug is also under investigation as a potential treatment for other neurodegenerative disorders, including HIV-associated dementia, neuropathic pain, and glaucoma.
The purpose of the present review is to provide a brief and perhaps somewhat surprising primer on excitotoxicity as a promising target of neuroprotective strategies and to present a scientific and clinical overview of the excitotoxicity blocker memantine. Some preliminary information on second-generation memantine derivatives, termed NitroMemantines, is also provided.
EXCITOTOXICITY
Definition and clinical relevance
The ability of the nervous system to rapidly convey sensory information and complex motor commands from one part of the body to another, and to form thoughts and memories, is largely dependent on a single powerful excitatory neurotransmitter, glutamate. There are other excitatory neurotransmitters in the brain, but glutamate is the most common and widely distributed. Most neurons (and also glia) contain high concentrations of glutamate (∼10 mm)1; after sequestration inside synaptic vesicles, glutamate is released for very brief amounts of time (milliseconds) to communicate with other neurons via synaptic endings. Because glutamate is so powerful, however, its presence in excessive amounts or for excessive periods of time can literally excite cells to death. This phenomenon was first documented when Lucas and Newhouse14 observed that subcutaneously injected glutamate selectively damaged the inner layer of the retina (representing primarily the retinal ganglion cells). John Olney later coined the term “excitotoxicity” to describe this phenomenon.15,16
A large variety of insults can lead to the excessive release of glutamate within the nervous system and, thus, excitotoxic cell death. When the nervous system suffers a severe mechanical insult, as in head or spinal cord injury, large amounts of glutamate are released from injured cells. These high levels of glutamate wash over thousands of nearby cells that had survived the original trauma, causing them to depolarize, swell, lyse, and die by necrosis. The lysed cells release more glutamate, leading to a cascade of autodestructive events and progressive cell death that can continue for hours or even days after the original injury. A similar phenomenon occurs in stroke; the ischemic event deprives many neurons of the energy they need to maintain ionic homeostasis, causing them to depolarize, lyse, die, and propagate the same type of autodestructive events that are seen in traumatic injury.1,17 This acute form of cell death occurs by a necrotic-like mechanism, although a slower component leading to an apoptotic-like death can also be present, as well as a continuum of events somewhere between the two (see below).
A slower, subtler form of excitotoxicity is implicated in a variety of slowly progressing neurodegenerative disorders as well as in the penumbra of stroke damage. In disorders such as Huntington’s disease, Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, HIV-associated dementia, amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease), and glaucoma, it is hypothesized that chronic exposure to moderately elevated glutamate concentrations or glutamate receptor hyperactivity for longer periods of time than occur during normal neurotransmission trigger cellular processes in neurons that eventually lead to apoptotic-like cell death, a form of cell death related to the programmed cell death that occurs during normal development.2,18–24
Importantly, elevations in extracellular glutamate are not necessary to invoke an excitotoxic mechanism. Excitotoxicity can come into play even with normal levels of glutamate if NMDA receptor activity is increased, e.g., when neurons are injured and thus become depolarized (more positively charged); this condition relieves the normal block of the ion channel by Mg2+ and thus abnormally increases NMDA receptor activity.25
Increased activity of the enzyme nitric oxide synthase (NOS) is associated with excitotoxic cell death. The neuronal isoform of the enzyme is physically tethered to the NMDA receptor and activated by Ca2+ influx via the receptor-associated ion channel, and increased levels of nitric oxide (NO) have been detected in animal models of stroke and neurodegenerative diseases.
Pathophysiology of excitotoxicity: role of the NMDA receptor
Apoptotic-like excitotoxicity is caused in part by excessive stimulation of the NMDA subtype of glutamate receptor (FIG. 1). When activated, the NMDA receptor opens a channel that allows Ca2+ (and other cations) to move into the cell. In some areas of the brain, this activity is important for long-term potentiation (LTP), thought to be a cellular/electrophysiological correlate of learning and memory formation. Under normal conditions of synaptic transmission, the NMDA receptor channel is blocked by Mg2+ sitting in the channel and only activated for brief periods of time. Under pathological conditions, however, overactivation of the receptor causes an excessive amount of Ca2+ influx into the nerve cell, which then triggers a variety of processes that can lead to necrosis or apoptosis. The latter processes include Ca2+ overload of mitochondria, resulting in oxygen free radical formation and activation of caspases, Ca2+-dependent activation of neuronal NOS, leading to increased NO production and the formation of toxic peroxynitrite (ONOO−), and stimulation of mitogen-activated protein kinase p38 (MAPK p38), which activates transcription factors that can go into the nucleus and influence neuronal injury and apoptosis.18,26–32
FIG. 1.

Schematic illustration of some apoptotic pathways triggered by excessive NMDA receptor activity. The cascade of steps leading to neuronal cell death include: 1) NMDA receptor (NMDA-Rc) hyperactivation, 2) activation of the p38 MAPK-MEF2C (transcription factor) pathway (MEF2 is subsequently cleaved by caspases to form an endogenous dominant-interfering form that contributes to neuronal cell death),32 3) toxic effects of free radicals such as NO and reactive oxygen species (ROS), and 4) activation of apoptosis-inducing enzymes including caspases. cyt c, cytochrome c. (Adapted from the Lipton website at http://www.burnham.org).
As mentioned above, conventional NMDA receptors consist of two subunits (NR1 and NR2A-D), and more rarely NR3A or B subunits. There are binding sites for glutamate, the endogenous agonist, and glycine, which is required as a co-agonist for receptor activation33 (FIG. 2). NMDA is generally not thought to be an endogenous substance in the body; it is an experimental tool that is highly selective for this subtype of glutamate receptor and therefore became the source of its name. When glutamate and glycine bind and the cell is depolarized to remove Mg2+ block, the NMDA receptor channel opens with consequent influx of Ca2+ and Na+ into the cell, the amount of which can be altered by higher levels of agonists and by substances binding to one of the modulatory sites on the receptor. The two modulatory sites that are most relevant to this review are the magnesium (Mg2+) site within the ion channel and an S-nitrosylation site located toward the N terminus (and hence extracellular region) of the receptor. [Note that S-nitrosylation reactions represent transfer of NO to a thiol or sulfhydryl group (-SH) of a critical cysteine residue. This reaction modulates protein function, in this case decreasing channel activity associated with stimulation of the NMDA receptor. There are actually five cysteine residues on the NMDA receptor that can be potentially nitrosylated, although only the principal site is considered here.] Each of these sites can be considered as targets for therapeutic intervention to block excitotoxicity, as explained below. Moreover, other modulatory sites also exist on the NMDA receptor and may in the future prove to be of therapeutic value. These include binding sites for Zn2+, polyamines, the drug ifenprodil (the endogenous ligand remains unknown), and a pH (e.g., proton)-sensitive site.34 Additionally, three pairs of cysteine residues can modulate channel function by virtue of their redox sensitivity.35
FIG. 2.

NMDA receptor model illustrating important binding and modulatory sites. Glu or NMDA, glutamate or NMDA binding site; Gly, glycine binding site; Zn2+, zinc binding site; NR1, NMDA receptor subunit 1; NR2, NMDA receptor subunit 2A; SNO, cysteine sulfhydryl group (−SH) reacting with NO species; X, Mg2+, MK-801, and memantine binding sites within the ion channel pore region.
To be clinically acceptable, an anti-excitotoxic therapy must block excessive activation of the NMDA receptor while leaving normal function relatively intact to avoid side effects. Drugs that simply compete with glutamate or glycine at the agonist binding sites block normal function and therefore do not meet this requirement, and have thus failed in clinical trials to date because of side effects (drowsiness, hallucinations, and even coma).1,8,12,36–40 Competitive antagonists compete one-for-one with the agonist (glutamate or glycine) and therefore will block healthy areas of the brain (where lower, more physiological levels of these agonists exist) before they can affect pathological areas. Thus, in fact, such drugs would preferentially block normal activity and would most likely be displaced from the receptor by the high concentrations of glutamate for prolonged periods that can exist under excitotoxic conditions.
As a useful analogy, the NMDA receptor can be compared to a television set. The agonist sites are similar to the “on/off” switch of the television. Drugs that block here cut off all normal NMDA receptor function. What we need to find is the equivalent of the volume control (or in biophysical terms, the gain) of the NMDA receptor. Then, when excessive Ca2+ fluxes through the NMDA receptor-associated ion channel, we would simply turn down the “volume” of the Ca2+ flux more toward normal. A blocker that acts at the Mg2+ site within the channel could act in such a manner; however, in the case of Mg2+ itself, the block is too ephemeral, a so-called “flickery block,” and the cell continues to depolarize until the Mg2+ block is totally relieved. Hence, in most cases Mg2+ does not effectively block excessive Ca2+ influx to the degree needed to prevent neurotoxicity. If, on the other hand, a channel blocker binds too tightly or works too well at low levels of receptor activation, it will block normal as well as excessive activation and be clinically unacceptable. Following the television set analogy, turning the volume all the way down is as bad as turning off the “on/off” switch in terms of blocking normal functioning of the television. This is the case with MK-801; it is a very good excitotoxicity blocker, but because “dwell time” in the ion channel is so long (reflecting its slow “off-rate”) due to high affinity for the Mg2+ site, MK-801 accumulates in the channels and therefore blocks critical normal functions. A human taking a neuroprotective dose of MK-801 not only becomes drowsy, but lapses into a coma. Drugs with slightly shorter but still excessive dwell times (off-rates) make patients hallucinate (e.g., phencyclidine, also known as Angel Dust), or so drowsy that they act as anesthetics (e.g., ketamine).
A clinically tolerated NMDA receptor antagonist should not make a patient drowsy, hallucinate, or comatose, and in fact should spare normal neurotransmission while blocking the ravages of excessive NMDA receptor activation. In fact, one type of drug that could do this and block preferentially higher (pathological) levels of glutamate over normal (physiological) levels is an “uncompetitive” antagonist. An uncompetitive antagonist is distinct from a noncompetitive antagonist (which simply acts allosterically at a noncompetitive site, i.e., a site other than the agonist-binding site). An uncompetitive antagonist is defined as an inhibitor whose action is contingent upon prior activation of the receptor by the agonist. Hence, the same amount of antagonist blocks higher concentrations of agonist better than lower concentrations of agonist. This uncompetitive mechanism of action, coupled with a longer dwell time than Mg2+ in the channel (and consequently a slower “off-rate” from the channel) but a substantially shorter dwell time (faster off-rate) than MK-801, would yield a drug that blocks NMDA receptor-operated channels only when they are excessively open while relatively sparing normal neurotransmission. Evidence suggests that memantine is such a drug.
Note that our principle discovery, which led to the testing of a clinically successful NMDA receptor antagonist, was the kinetics of the drug in the NMDA receptor-associated ion channel8–11; we found that the dwell time in (or off-rate from) the channel is the major determinant of clinical tolerability of an open-channel blocker because excessive dwell time (associated with a slow off-rate) causes the drug to accumulate in the channels, interfere with normal neurotransmission and produce unacceptable adverse effects (as is the case with MK-801). In contrast, too short a dwell time (too fast an off-rate) yields a relatively ineffectual blockade, especially with membrane depolarization, which relieves the block of positively charged molecules (as seen with Mg2+). The apparent affinity of a positively charged channel blocker at a particular membrane voltage is related to its off-rate divided by its on-rate (derived from Scheme 1, in the Appendix). The on-rate is a property of the open probability of the channel and the diffusion rate and concentration of the drug. On the other hand, the off-rate is an intrinsic property of the drug-receptor complex, unrelated to drug concentration (see Chen and Lipton for a detailed quantitative discussion of these points).9 The relatively fast off-rate is a major contributor to the drug’s low affinity for the channel pore.
The memantine class of drugs represents a relatively low-affinity, open-channel blocker, i.e., these drugs only enter the channel when it is opened by agonist. In the case of memantine, at concentrations administered to patients the drug appears to enter the channel preferentially when it is (pathologically) activated for long periods of time, i.e., under conditions of excessive glutamate exposure. As we showed previously,9 memantine has favorable kinetics in the channel to provide neuroprotection while displaying minimal adverse effects (occasional restlessness or, in rare cases, slight dizziness at higher dosages).1,8
MEMANTINE
Background and pharmacology
Memantine was first synthesized by Eli Lilly and Company and patented in 1968, as documented in the Merck Index, as a derivative of amantadine, an anti-influenza agent. It has a three-ring (adamantane) structure with a bridgehead amine (−NH2) that under physiological conditions carries a positive charge (−NH3+) that binds at or near the Mg2+ site in the NMDA receptor-associated channel (FIG. 3). 8–11,41 Unlike amantadine, memantine has two methyl (−CH3) side-groups that prolong its dwell time in the channel (hence slowing the off-rate and increasing the affinity for the channel somewhat compared to amantadine). The reported efficacy of amantadine and memantine in Parkinson’s disease, which was discovered by serendipity in a patient taking amantadine for influenza, led scientists to believe that these compounds were dopaminergic or possibly anticholinergic drugs. It was not until the 1980s and early 1990s that memantine was found to be neither dopaminergic nor anticholinergic at its clinically used dosage, but rather, an NMDA receptor antagonist.42 However, work at the small German company Merz suggested that the drug was quite potent at the NMDA receptor,42 which in fact it is not. Rather, the drug is of such poor potency (affinity in the micromolar range rather than nanomolar or higher) that big Pharma initially thought that it was a poor drug as a neuroprotective candidate. However, one should not confuse affinity with selectivity; as long as a drug acts selectively and specifically on the target of interest and the effective concentration can be achieved, a high affinity per se is not the key issue. In fact, work in my laboratory [then located at Harvard Medical School; patents on which S. A. Lipton is the named inventor are assigned to Harvard-affiliated institutions, including Children’s Hospital of Boston] first showed why memantine could be clinically tolerated as an NMDA receptor antagonist; namely, it was an uncompetitive open-channel blocker with a dwell time/off-rate from the channel that limited pathological activity of the NMDA receptor while sparing normal synaptic activity.9–11 These findings led to a number of publications and U.S. and worldwide patents on the use of memantine for NMDA receptor-mediated disorders and spurred several successful clinical trials with the drug, as discussed below.
FIG. 3.

Chemical structure of memantine. Several important features are 1) the three-ring structure, 2) the bridgehead amine (−NH2 group), which is charged at the physiological pH of the body (−NH3+) and represents the region of memantine that binds at or near the Mg2+ binding site in the NMDA receptor-associated ion channel, and 3) the methyl group (−CH3) side-chains (unlike amantadine), which serve to stabilize the interaction of memantine in the channel region of the NMDA receptor.
To illustrate the blockade of NMDA-induced ionic currents by memantine, a sample experiment is shown in FIG. 4 in which the membrane voltage of a neuron was held at the resting potential. The concentration of memantine used in this experiment is similar to the level that can be achieved in human brain when the drug is used clinically. At such concentrations, memantine greatly reduces pathologically high levels of NMDA-induced current to near zero within approximately 1 second. Once the memantine application stops, the NMDA response returns to previous levels over a period of about 5 seconds. This indicates that memantine is an effective but temporary NMDA receptor blocker.
FIG. 4.

Blockade of NMDA current by memantine. At a holding potential of approximately −50 mV, whole-cell recording of NMDA-evoked current from a solitary neuron revealed that the on-time (time until peak blockade) of micromolar memantine was approximately 1 second, while the off-time (recovery time) from the effect was ∼5.5 seconds. The application of memantine produced an effective blockade only during NMDA receptor activation, consistent with the notion that its mechanism of action is open-channel block.10
Perhaps the most astonishing property of memantine is illustrated in FIG. 5. 8,10 In this experiment, the concentration of memantine was held constant (at a clinically achievable level of 1 μm) while the concentration of NMDA was increased over a wide range. It was found that the degree to which this fixed concentration of memantine blocked NMDA receptor activity actually increased as the NMDA concentration was increased to pathological levels. In fact, memantine was relatively ineffective at blocking the low levels of receptor activity associated with normal neurological function but became exceptionally effective at higher concentrations. This is classical “uncompetitive” antagonist behavior.
FIG. 5.

Paradoxically, a fixed dose of memantine (e.g., 1 μm) blocks the effect of increasing concentrations of NMDA to a greater degree than lower concentrations of NMDA. This finding is characteristic of an uncompetitive antagonist.10
Further studies indicate that memantine exerts its effect on NMDA receptor activity by binding at or near the Mg2+ site within the ion channel.8–11,41 This information and the pharmacological/kinetic data presented above suggest that memantine preferentially blocks NMDA receptor activity if the ion channel is excessively open. During normal synaptic activity, the channels are open on average for only several milliseconds, and memantine is unable to act or accumulate in the channels; hence, synaptic activity continues essentially unabated. In technical terms, the component of the excitatory postsynaptic current due to activation of NMDA receptors is inhibited by only 10% or less.11 During prolonged activation of the receptor, however, as occurs under excitotoxic conditions, memantine becomes a very effective blocker. In essence, memantine only acts under pathological conditions without much affecting normal function, thus relatively sparing synaptic transmission, preserving long-term potentiation and maintaining physiological function on behavioral tests such as the Morris water maze.11 The kinetics of memantine action in the NMDA receptor-associated ion channel explains the favorable clinical safety profile that has been seen to date.
Neuroprotective efficacy
The neuroprotective properties of memantine have been studied in a large number of in vitro and in vivo animal models by several laboratories (for a recent review, see Parsons et al.).43 Among neurons protected in this manner both in culture and in vivo are cerebrocortical neurons, cerebellar neurons, and retinal neurons.8–11,21,44–47 Additionally, in a rat model of stroke, memantine, given as long as 2 hours after the ischemic event, reduces the amount of brain damage by approximately 50%.10,11
A series of human clinical trials have been launched to investigate the efficacy of memantine for the treatment of Alzheimer’s disease, vascular dementia, HIV-associated dementia, diabetic neuropathic pain, as well as glaucoma. Some of these studies have only been recently completed and remain unpublished at this time except in abstract form. One very recent, high-profile publication reported the results of a U.S. phase III (final) clinical study showing that memantine (20 mg/day) is efficacious for moderate-to-severe Alzheimer’s disease.12 Another study reported in abstract form that, in combination with Aricept, memantine treatment actually improves memory and function in moderate-to-severe Alzheimer’s patients. The results of clinical studies for Alzheimer’s disease were sufficiently positive to prompt the European Union to approve memantine for the treatment of this form of dementia last year, and the FDA recently approved the drug in the U.S. One full-length European publication of a multi-center, randomized controlled trial reported that memantine was beneficial to severely demented patients, probably representing Alzheimer’s disease and vascular dementia.48 Still another recent publication of a randomized, placebo-controlled clinical trial described significant benefit from memantine therapy (20 mg/day) in mild-to-moderate vascular dementia.13 These clinical trials are summarized in Table 1. Most trials have reported minimal adverse effects of memantine. In those trials reporting adverse effects, the only memantine-induced side effects encountered were rare dizziness and occasional restlessness/agitation at higher doses (40 mg/day), but these effects were mild and dose-related.
TABLE 1.
Clinical Trials with Memantine
| • German/Merz phase 3 trial for Vascular Dementia and Alzheimer’s Disease | + |
| • Karolinska/Italian phase 3 trial for Vascular Dementia | + |
| • Two USA Multicenter phase 3 trials for Alzheimer’s Disease | + |
| • United Kingdom phase 3 trial for Vascular Dementia | + |
| • French phase 3 trial for Vascular Dementia | + |
| • USA phase 2 trials for Neuropathic pain and HIV-Associated Dementia | +/− |
Importantly, as outlined above, the anti-intuitive aspects of memantine action that we discovered show that more glutamate receptor activity is blocked better than less activity at a fixed dose of memantine. Hence, memantine is expected to work better for severe conditions, e.g., excessive glutamate receptor activity to the point of causing cell death, than more mild conditions manifested by slightly elevated synaptic transmission. Bearing this out, recent studies suggest that memantine has a greater effect in patients with moderate-to-severe dementia than those with mild dementia. Another case in point is neuropathic pain, which is currently thought to be mediated at least in part by excessive NMDA receptor activity. Given the mode of action described for memantine, more severe pain, e.g., the nocturnal pain of diabetic neuropathy, is expected to benefit from memantine to a greater extent than milder forms of neuropathic pain. In fact, phase II clinical trials have suggested that this is indeed the case, whereas more mild pain conditions were not statistically benefited by memantine in phase II/III clinical trials. Along these lines, one could predict that a higher concentration of memantine is needed to combat pain than to prevent neuronal cell death because greater NMDA receptor activity is associated with cell death. Again, clinical trials have suggested that this is indeed the case because 40 mg/day of memantine were needed in successful pain studies but only 20 mg/day in dementia studies. However, further clinical trials will be necessary to prove the efficacy of memantine for severe neuropathic pain. Prior work predicted that only severe cases of neuropathic pain are positively affected by memantine at any reasonable dose, although at higher doses, normal or slightly increased NMDA receptor activity is also affected because an increase in drug concentration will accelerate the “on-rate” of memantine entering into the channel, as explained above. This fact also suggests that many cases of neuropathic pain are too mild for the drug to work.
As promising as the results with memantine are, we are continuing to pursue ways to use additional modulatory sites (the “volume controls”) on the NMDA receptor to safely block excitotoxicity even more effectively and safely than memantine alone. New approaches in this regard are explored below.
NITROMEMANTINES
NitroMemantines are second-generation memantine derivatives that were designed to have enhanced neuroprotective efficacy without sacrificing safety. As mentioned earlier, a nitrosylation site is located on the N terminus or extracellular domain of the NMDA receptor, and S-nitrosylation of this site (NO reaction with the sulfhydryl group of the cysteine residue) down-regulates receptor activity (FIG. 2). The drug nitroglycerin, which generates NO-related species, can act at this site to limit excessive NMDA receptor activity. In fact, in rodent models, nitroglycerin can limit ischemic damage,49 and there is some evidence that patients taking nitroglycerin for other medical reasons may be resistant to glaucomatous visual field loss.50
From crystal structure models and electrophysiological experiments, we found that NO binding to the NMDA receptor at the major S-nitrosylation site apparently induces a conformational change in the receptor protein that makes glutamate and Zn2+ bind more tightly to the receptor. The enhanced binding of glutamate and Zn2+ in turn causes the receptor to desensitize and, consequently, causes the ion channel to close.35 Electrophysiological studies have demonstrated this effect of NO on the NMDA channel.28,51,52
Unfortunately, nitroglycerin is not very attractive as a neuroprotective agent. The same cardiovascular vasodilator effect that makes it useful in the treatment of angina could cause dangerously large drops in blood pressure in stroke, traumatic injury, or glaucoma patients. Consequently, we carefully characterized S-nitrosylation sites on the NMDA receptor to determine if we could design a nitroglycerin-like drug that would more specifically target the NMDA receptor.
In brief, five different cysteine residues on the NMDA receptor were found that could interact with NO. One of these, located at cysteine residue 399 on the NR2A subunit of the NMDA receptor, mediates approximately 90% of the effect of NO under our experimental conditions. Using this kind of information, we created modified memantine molecules called NitroMemantines that will interact with both the memantine site within the NMDA receptor-associated ion channel and the predominant nitrosylation site. Two sites of modulation are analogous to having two volume controls on a television set for fine-tuning the audio signal.
Preliminary studies have shown NitroMemantines to be highly neuroprotective in both in vitro and in vivo animal models. In fact, it appears to be substantially more effective than memantine. Moreover, because the memantine portion makes these drugs specific for the NMDA receptor, NitroMemantines appear to lack the blood-pressure-lowering effect typical of nitroglycerin by targeting the NO group to the nitrosylation site on the NMDA receptor.
More research still needs to be performed on NitroMemantine drugs, but the fact that they chemically combine two clinically tolerated drugs (memantine and nitroglycerin) enhances their promise as second-generation memantine-derivatives that are both clinically safe and neuroprotective.
SUMMARY
Necrosis- and apoptosis-mediated excitotoxic cell death is implicated in the pathophysiology of many neurologic diseases, including stroke, CNS trauma, dementia, glaucoma, polyglutamine (triple-repeat) disorders, and other neurodegenerative conditions. This type of excitotoxicity is caused, at least in part, by excessive activation of NMDA-type glutamate receptors. Intense insults, such as that occurring in the ischemic core after a stroke, leads to massive stimulation of NMDA receptors because of increased glutamate and energy failure, leading to membrane depolarization, relief of Mg2+ block of NMDA channels, and disruption of ionic homeostasis. The fulminant buildup of ions results in neuronal cell swelling and lysis (necrosis). In contrast, more moderate NMDA receptor hyperactivity, such as that occurring in the ischemic penumbra of a stroke and in many slow-onset neurodegenerative diseases, results in moderately excessive influx of calcium ions into nerve cells which, in turn, triggers free radical formation and multiple pathways leading to the initiation of apoptotic-like damage.18,53 Blockade of NMDA receptor activity prevents to a large degree both necrosis- and apoptosis-related excitotoxicity. NMDA receptor activity is also required for normal neural function, however, so only those NMDA blockers that selectively reduce excessive receptor activation without affecting normal function will be clinically acceptable; memantine is such a drug. It has been shown in laboratory tests to only block excessive NMDA receptor activation but not normal, low-level activation. Importantly, the author, in collaboration with Vincent Chen (a graduate student at the time, and now a faculty member at the Burnham Institute and U.C. San Diego) reported that memantine had a relatively short dwell time in (and hence fast off-rate from) the NMDA-associated ion channel, in part explaining the drug’s relatively low apparent affinity as an antagonist.8–10 We realized that the relatively short dwell time/rapid off-rate from the channel was the predominant factor in determining the clinical tolerability as well as the neuroprotective profile of the drug.8 Most importantly, this mode of action meant that memantine blocked high (pathological) levels of glutamate at the NMDA receptor while relatively sparing the effects of low (physiological) levels of glutamate seen during normal neurotransmission because the drug does not accumulate in the channel during normal synaptic activity. The discovery that memantine, a low-affinity but still highly selective agent with a mechanism of uncompetitive antagonism, is neuroprotective yet clinically tolerated triggered a paradigm shift in the history of drug development by the pharmaceutical industry.8–11 Before that discovery, low-affinity drugs were thought to be inferior and not clinically useful. In particular, the relatively rapid off-rate from the NMDA receptor-associated ion channel of the memantine class of drugs largely accounts for its clinical tolerability as well as its low affinity. Clinical studies have borne out our hypothesis that low-affinity/fast off-rate memantine is a safe NMDA receptor antagonist in humans and beneficial in the treatment of neurological disorders mediated, at least in part, by excitotoxicity.
The NitroMemantines are second generation NMDA receptor antagonists that may work even better than memantine by using the memantine binding site for the targeted delivery of NO to a second modulatory site on the NMDA receptor. Work is progressing rapidly in this area of investigation.
Clinical studies of the efficacy of memantine in the treatment of stroke, Alzheimer’s disease, vascular dementia, HIV-associated dementia, glaucoma, and severe neuropathic pain, are currently underway, and there is every reason to expect the results to be positive although this is, of course, not yet proven except in the case of Alzheimer’s disease and possibly vascular dementia in which phase III (final) clinical trials have proven successful (Table 1).12,13,54 The efficacy of memantine in these neurodegenerative diseases and its ability to protect neurons in animal models of both acute and chronic neurologic disorders suggest that memantine and drugs acting in a similar manner could become very important new weapons in the fight against neuronal damage.
Acknowledgments
I would like to thank my colleagues, in particular Vincent Chen, Yun-Beom Choi, and especially Jonathan Stamler, for contributing to this work.
APPENDIX
SCHEME 1
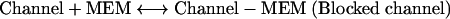 |
(Eq. 1) |
This simple bimolecular scheme predicts that the macroscopic blocking and unblocking actions of memantine (MEM) proceed with exponential relaxation. The macroscopic pseudo-first-order rate constant of blocking (kon) depends linearly on memantine concentration (as well as a constant, A), and the macroscopic unblocking rate (koff) is independent of memantine concentration ([MEM]):
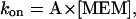 |
(Eq. 2) |
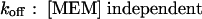 |
(Eq. 3) |
These predictions were borne out experimentally.9 Both the macroscopic blocking and unblocking processes could be well-fitted by a single exponential function. The macroscopic on-rate constant is the reciprocal of the measured time constant for onset (τon) and is the sum of the pseudo-first-order blocking rate constant (kon) and unblocking constant (koff). The unblocking rate constant (koff) is the reciprocal of the measured macroscopic unblocking time constant (τoff). These transformations lead to Eqs. 4 and 5:
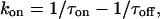 |
(Eq. 4) |
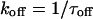 |
(Eq. 5) |
The kon calculated from Eq. 4 increased linearly with memantine concentration with a slope factor of 0.4 ± 0.03 106 M-1s-1 (mean ± SD), while the koff from Eq. 5 remained relatively constant with a y-axis intercept of 0.44 ± 0.1 s−1.9 A rapid method to validate this result was obtained by estimating the equilibrium apparent dissociation constant (Ki) for memantine action from the following equation:
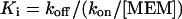 |
(Eq. 6) |
Here we found empirically that memantine was a relatively low-affinity (apparent affinity, ∼1 μm) open-channel blocker of the NMDA receptor-coupled ion channel, and a major component of the affinity was determined by koff at clinically relevant concentrations in the low micromolar range.
REFERENCES
- 1.Lipton SA, Rosenberg RA. Mechanisms of disease: excitatory amino acids as a final common pathway in neurologic disorders. N Engl J Med 330: 613–622, 1994. [DOI] [PubMed] [Google Scholar]
- 2.Lipton SA, Nicotera P. Calcium, free radicals and excitotoxins in neuronal apoptosis. Cell Calcium 23: 165–171, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nature Neurosci 5[Suppl]: 1039–1042, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Kemp JA, Kew JN, Gill R. Handbook of experimental pharmacology, Vol 141 (Jonas P, Monyer H, eds), pp 495–527. Berlin: Springer, 1999.
- 5.Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Kaste M et al. Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomized controlled trial. GAIN International Investigators. Lancet 355: 1949–1954, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Sacco RL, DeRosa JT, Haley EC Jr, Levin B, Ordronneau P, Phillips SJ et al. Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial. JAMA 28: 1719–1728, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Seif el Nasr M, Peruche B, Rossberg C, Mennel HD, Krieglstein J. Neuroprotective effect of memantine demonstrated in vivo and in vitro. Eur J Pharmacol 185: 19–24, 1990. [DOI] [PubMed] [Google Scholar]
- 8.Lipton SA. Prospects for clinically-tolerated NMDA antagonists: open-channel blockers and alternative redox states of nitric oxide. Trends Neurosci 16: 527–532, 1993. [DOI] [PubMed] [Google Scholar]
- 9.Chen H-SV, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol (Lond) 499: 27–46, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE et al. Open-channel block of NMDA responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci 12: 4427–4436, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen HS, Wang YF, Rayudu PV, Edgecomb P, Neill JC, Segal MM et al. Neuroprotective concentration of the NMDA open-channel blocker memantine are effective without cytoplasmic vacuolization following post-ischemic administration and do not block maze learning or LTP. Neuroscience 86: 1121–1132, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348: 1333–1341, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Orgogozo JM, Rigaud AS, Stoffler A, Mobius HJ, Forette F. Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300). Stroke 33: 1834–1839, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Lucas DR, Newhouse JP. The toxic effect of sodium l-glutamate on the inner layers of the retina. Am Med Assoc Arch Ophthalmol 58: 193–201, 1957. [DOI] [PubMed] [Google Scholar]
- 15.Olney JW. Glutamate-induced retinal degeneration in neonatal mice: electron microscopy of the acutely evolving lesion. J Neuropathol Exp Neurol 28: 455–474, 1969. [DOI] [PubMed] [Google Scholar]
- 16.Olney JW, Ho OL. Brain damage in infant mice following oral intake of glutamate, aspartate or cysteine. Nature 227: 609–611, 1970. [DOI] [PubMed] [Google Scholar]
- 17.Lipton SA. Molecular mechanisms of trauma-induced neuronal degeneration. Curr Opin Neurol Neurosurg 6: 588–596, 1993. [PubMed] [Google Scholar]
- 18.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced respectively by mild and intense insults with NMDA or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 92: 7162–7166, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreyer EB, Zhang D, Lipton SA. Transcriptional or translational inhibition blocks low dose NMDA-mediated cell death. Neuroreport 6: 942–944, 1995. [DOI] [PubMed] [Google Scholar]
- 20.Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci 36: 774–786, 1995. [PubMed] [Google Scholar]
- 21.Vorwerk CK, Lipton SA, Zurakowski D, Hyman BT, Sabel BA, Dreyer EB. Chronic low dose glutamate is toxic to retinal ganglion cells: toxicity blocked by memantine. Invest Ophthalmol Vis Sci 37: 1618–1624, 1996. [PubMed] [Google Scholar]
- 22.Dreyer EB, Grosskreutz CL. Excitatory mechanisms in retinal ganglion cell death in primary open angle glaucoma (POAG). Clin Neurosci 4: 270–273, 1997. [PubMed] [Google Scholar]
- 23.Dreyer EB, Lipton SA. New perspectives on glaucoma. JAMA 281: 306–308, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Naskar R, Vorwerk CK, Dreyer EB. Saving the nerve from glaucoma: memantine to caspases. Semin Ophthalmol 4: 152–158, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Zeevalk GD, Nicklas WJ. Evidence that the loss of the voltage-dependent Mg2+ block of the N-methyl-d-aspartate receptor underlies receptor activation during inhibition of neuronal metabolism. J Neurochem 59: 1211:1220, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA 88: 6368–6371, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci 13: 2651–2661, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lipton SA, Choi Y-B, Pan Z-H, Lei SZ, Chen H-SV, Sucher NJ, Singel DJ, Loscalzo J, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364: 626–632, 1993. [DOI] [PubMed] [Google Scholar]
- 29.Tenneti L, D’Emilia DM, Troy CM, Lipton SA. Role of caspases in N-methyl-d-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem 71: 946–959, 1998. [DOI] [PubMed] [Google Scholar]
- 30.Yun HY. Gonzalez-Zulueta M, Dawson VL et al. Nitric oxide mediates N-methyl-d-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci USA 95: 5773–5778, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci USA 97: 6161–6166, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okamoto S-I, Li Z, Ju C, Schölzke MN, Matthews E, Cui J, Salvesen GS, Bossy-Wetzel E, Lipton SA. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA 99: 3974–3979, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson J, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325: 529–531, 1987. [DOI] [PubMed] [Google Scholar]
- 34.McBain CJ, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev 74: 723–760, 1994. [DOI] [PubMed] [Google Scholar]
- 35.Lipton SA, Choi Y-B, Takahashi T, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci 25: 474–490, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Koroshetz WJ, Moskowitz MA. Emerging treatments for stroke in humans. Trends Pharmacol Sci 17: 227–233, 1996. [DOI] [PubMed] [Google Scholar]
- 37.Hickenbottom SL, Grotta J. Neuroprotective therapy. Semin Neurol 18: 485–492, 1998. [DOI] [PubMed] [Google Scholar]
- 38.Lutsep HL, Clark WM. Neuroprotection in acute ischaemic stroke: current status and future potential. Drug Res Dev 1: 3–8, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Rogawski MA. Low-affinity channel blocking (uncompetitive) NMDA receptor antagonists as therapeutic agents—toward an understanding of their favorable tolerability. Amino Acids 19: 133–149, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Palmer GC. Neuroprotection by NMDA receptor antagonists in a variety of neuropathologies. Curr Drug Targ 2: 241–271, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Chen H-SV, Rastogi, Lipton SA. Q/R/N site mutations in the M2 region of NMDAR1/NMDAR2A receptors reveal a non-specific site for memantine action. Soc Neurosci Abstr 24: 342, 1998. [Google Scholar]
- 42.Bormann J. Memantine is a potent blocker of N-methyl-d-aspartate (NMDA) receptor channels. Eur J Pharmacol 166: 591–592, 1989. [DOI] [PubMed] [Google Scholar]
- 43.Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist—a review of preclinical data. Neuropharmacology 38: 735–767, 1999. [DOI] [PubMed] [Google Scholar]
- 44.Lipton SA. Memantine prevents HIV coat protein-induced neuronal injury in vitro. Neurology 42: 1403–1405, 1992. [DOI] [PubMed] [Google Scholar]
- 45.Pellegrini JW, Lipton SA. Delayed administration of memantine prevents NMDA receptor-mediated neurotoxicity. Ann Neurol 33: 403–407, 1993. [DOI] [PubMed] [Google Scholar]
- 46.Sucher NJ, Lipton SA, Dreyer EB. Molecular basis of glutamate toxicity in retinal ganglion cells. Vision Res 37: 3483–3493, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Osborne NN. Memantine reduces alterations to the mammalian retina, in situ, induced by ischemia. Vis Neurosci 16: 45–52, 1999. [DOI] [PubMed] [Google Scholar]
- 48.Winblad B, Poritis N. Memantine in severe dementia: results of the 9M-best study (benefit and efficacy in severely demented patients during treatment with memantine). Int J Geriat Psych 14: 135–146, 1999. [DOI] [PubMed] [Google Scholar]
- 49.Lipton SA, Wang YF. NO-related species can protect from focal cerebral ischemia/reperfusion. In: Pharmacology of cerebral ischemia (Krieglstein J, Oberpichler-Schwenk H, eds), pp 183–191. Stuttgart: Wissenschaftliche Verlagsgesellschaft mbH, 1996.
- 50.Zurakowski D, Vorwerk CK, Gorla M, Kanellopoulos AJ, Chaturvedi N, Grosskreutz CL et al. Nitrate therapy may retard glaucomatous optic neuropathy, perhaps through modulation of glutamate receptors. Vision Res 38: 1489–1494, 1998. [DOI] [PubMed] [Google Scholar]
- 51.Lipton SA, Rayudu, PV, Choi Y-B, Sucher NJ, Chen HSV. Redox modulation of the NMDA receptor by NO-related species. In: Progress in brain research, Vol 118 (Mize RR, Dawson V, Dawson TM, Friedlander M, eds), pp 73–82. Amsterdam: Elsevier, 1998. [DOI] [PubMed]
- 52.Choi Y-B, Tenneti L, Le DA, Ortiz J, Bai G, Chen H-SV, Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nature Neurosci 3: 15–21, 2000. [DOI] [PubMed] [Google Scholar]
- 53.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial integrity. Neuron 15: 961–973, 1995. [DOI] [PubMed] [Google Scholar]
- 54.Le DA, Lipton SA. Potential and current use of N-methyl-d-aspartate (NMDA) receptor antagonists in diseases of aging. Drugs Aging 18: 717–724, 2001. [DOI] [PubMed] [Google Scholar]