

Graphical abstract

Keywords: Androgen receptor, CDC6, Chk1, ATR, TOPBP1, DNA damage, prostate cancer, enzalutamide, AZD7762
Summary
Cell division cycle 6 (CDC6), an androgen receptor (AR) target gene, is implicated in regulating DNA replication and checkpoint mechanisms. CDC6 is increased during prostate cancer (PCa) progression and positively correlates with AR in PCa tissues. AR or CDC6 knockdown together with AZD7762, a Chk1/2 inhibitor, results in decreased TopBP1-ATR-Chk1 signaling and markedly increased ataxia-telangiectasia mutated (ATM) phosphorylation, a biomarker of DNA damage, and synergistically increases treatment efficacy. Combination treatment of AR with enzalutamide (ENZ) and Chk1/2 inhibition with AZD7762 demonstrates synergy with regard to inhibition of AR/CDC6-ATR-Chk1 signaling, ATM phosphorylation induction, and apoptosis in VCaP (mutant p53) and LNCaP-C4-2b (wild-type p53) cells. CDC6 overexpression significantly reduced ENZ and AZD7762-induced apoptosis. Additive or synergistic therapeutic activities are demonstrated in AR-positive animal xenograft models. These findings have important clinical implications since they introduce a therapeutic strategy for AR-positive metastatic castration-resistant PCa, regardless of p53 status, through targeting AR/CDC6-ATR-Chk1 signaling.
Introduction
Metastatic prostate cancer remains an incurable disease with variable prognosis (Wu et al., 2014). After an initial period of response to systemic hormone therapy, the disease inexorably progresses to a state known as metastatic castration-resistant prostate cancer (mCRPC) (Karanika et al., 2014). The therapeutic armamentarium for mCRPC is limited to chemotherapy and novel inhibitors of AR signaling, such as abiraterone acetate and enzalutamide (ENZ), which provide only moderate survival benefits (Ryan et al., 2013, Scher et al., 2012).
DNA damage response (DDR) refers to coordinated cellular mechanisms that prevent DNA damage accumulation and maintain genomic integrity (Karanika et al., 2014) and plays a central role in PCa initiation, development and progression (Tapia-Laliena et al., 2014). AR signaling in PCa cells has been associated with numerous aspects of DDR pathways including regulation of ATM-Chk2 signaling for initiation of DDR (Ide et al., 2012), poly(ADP-ribose) polymerase function (Schiewer et al., 2012) and non-homologous end joining recombination (Polkinghorn et al., 2013). AR is reported to regulate TopBP1-ATR-Chk1 signaling (Li et al., 2014) whereas ENZ decreases CHEK1 expression in PCa cells (Li et al., submitted).
One of the main types of DNA damage istan and Lim, 2000 DNA strand breakage, which activates a cascade of intracellular events that promote cell-cycle arrest and DDR ensuring genomic integrity which can be particularly critical for cell survival in patients with aggressive malignancies accumulating a myriad of genetic errors (Karanika et al., 2014). DNA strand breaks activate ATR via upstream mediators such as TopBP1 leading to Chk1-mediated checkpoint activation and cell-cycle arrest. Cells are thereby able to repair DNA damage alleviating replication stress and genomic instability. Chk1 pathway inhibition results in DNA damage accumulation and thus increased ATM auto-phosphorylation, which mediates apoptosis of cells with incompletely replicated DNA (d'Adda di Fagagna, 2008, Sarmento et al., 2015). These findings demonstrate that ATR-Chk1 signaling is crucial for the prevention of DNA damage-induced cell death associated with increased ATM phosphorylation/activation.
CDC6 is an essential regulator of DNA replication in eukaryotic cells and its best-characterized function is pre-replicative complex assembly at origins of replication during G1 phase (Borlado and Mendez, 2008). Furthermore, CDC6 overexpression during G2 phase blocks mitotic entry by activating Chk1 (Clay-Farrace et al., 2003, Yoshida et al., 2010) which inhibits G2/M progression. CDC6 is required for Chk1 activation upon replication inhibition (Oehlmann et al., 2004) and human CDC6 interacts with ATR promoting activation of the replication checkpoint (Yoshida et al., 2010). Consequently, CDC6 decreases genomic instability which is vital for cancer cell survival. CDC6 can also manifest oncogenic activities via regulation of DNA replication and repression of tumor suppressors (Gonzalez et al., 2006). Targeting ATR-Chk1 signaling increases the sensitivity to treatment with DNA-damaging agents (Bartucci et al., 2012) making this approach particularly attractive for development of cancer therapies. Notably, Chk1 knockdown increases the sensitivity of PCa stem cells to radiotherapy through increased DNA damage (Fokas et al., 2012). AZD7762, a Chk1/2 inhibitor, synergizes with DNA-damaging agents and radiation to induce apoptosis via DNA double strand breaks mediated by ATM activation in many cell types (Mitchell et al., 2010, Sausville et al., 2014). Furthermore, Brooks et al found that Chk1 inhibition can selectively induce apoptosis in melanoma cells in proportion to the level of endogenous DNA damage related to replicative stress without further induction of DNA damage by chemotherapy (Brooks et al., 2013).
Taking the results of these studies into account, a potential approach to treatment of mCRPC is inhibition of more than one level of a specific DDR signaling cascade with the goal of completely abolishing a specific signaling pathway. This approach would take advantage of the fact that cancer cells eventually accumulate more DNA damage than normal cells do, eluding adverse effects of chemotherapy. Targeting the ATR-Chk1 pathway at multiple levels to inhibit the repair of DNA damage induced by replication stress in cancer cells may represent an effective strategy for more complete DDR pathway inhibition. Because CDC6 is an androgen receptor (AR) target gene (Jin and Fondell, 2009, Bai et al., 2005), and is also involved with ATR-Chk1 signaling this is a particularly intriguing strategy for AR-positive PCa. In addition, this approach may be effective under conditions of wild-type p53 which is involved in multiple DDR pathways and can mitigate the response to some DNA-damaging agents (Ma et al., 2012).
The aim of the present study was to determine the role of CDC6 in regulation of ATR-Chk1 signaling and to test combined inhibition of AR and Chk1 signaling as a therapeutic approach for AR-positive mCRPC. Through this study we also aimed to introduce a therapeutic approach that effectively targets a DDR pathway that promotes sufficient DNA damage accumulation in PCa cells to induce cell death, regardless of p53 status.
Results
CDC6 is induced during PCa progression and is positively correlated with AR expression
We first evaluated the CDC6 expression and phosphorylation, and analyzed its correlation with AR expression in normal human prostates, primary and metastatic prostate tumor specimens. In our immunohistochemistry (IHC) analysis, we found that AR expression (P=0.0057), and CDC6 expression (P<0.001) and phosphorylation (P=0.0083) increased significantly during PCa development and progression (Figure 1A). It was reported that AR regulates CDC6 expression (Jin and Fondell, 2009, Bai et al., 2005) and protein stability (Bai et al., 2005) indicating functional associations between these two molecules. To determine the mechanism of AR regulation of CDC6 in our experimental models, we performed qRT-PCR and protein stability analysis for Cdc6 using ARsi- and negative control (NC)si-transfected VCaP and LNCaP C4-2b (C4-2b) cells. Our results demonstrated that AR regulation of CDC6 occurs at the mRNA level, and not through regulation of protein stability (Figure 1B-D).
Figure 1. CDC6 is induced during PCa progression and is positively correlated with AR expression.
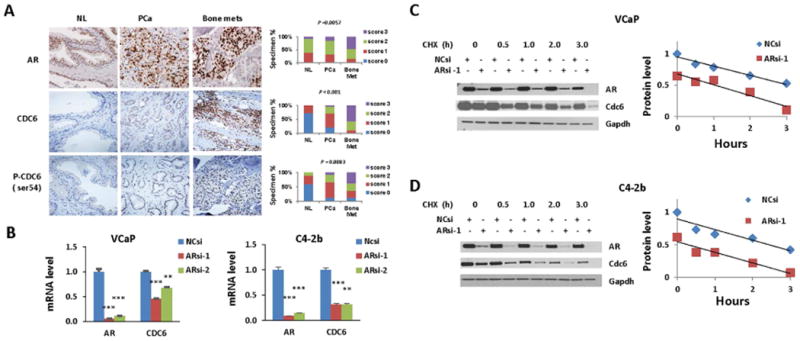
(A) Immunohistochemical analysis of AR, CDC6 and P-CDC6 (S54) in normal prostate, primary prostate tumor, and bone metastases. NL, normal prostate; PCa, prostate cancer; Bone met, bone metastasis. Treatment information of patient with metastasis is available in Table S1. (B) qRT-PCR analysis of CDC6 mRNA levels. VCaP and C4-2b cells were transfected with 20 nM ARsi or NCsi for 48 h. ** p<0.01, *** p<0.0001. (C-D) Protein stability analysis of Cdc6. VCaP (C) and C4-2b (D) cells were transfected with 20 nM Arsi-1 or NCsi for 48 h prior to the treatment with 100 microg/ml cycloheximide (CHX) for indicated time. Left panels, Western blotting analysis; right panels, densitometry analysis for Cdc6 protein stability in ARsi (red) and NCsi (blue) transfected cells.
AR or CDC6 knockdown increases the sensitivity of PCa cells to the Chk1/2 inhibitor AZD7762 through inhibition of TopBP1-ATR-Chk1 signaling
Targeting Chk1/2 with AZD7762 increases the efficacy of DNA-damaging modalities such as chemotherapy and radiation therapy in patients with multiple malignancies (Mitchell et al., 2010, Sausville et al., 2014). Thus, we tested our hypothesis that the combination treatment with AR or CDC6 downregulation and AZD7762 results in synergistic activities in VCaP and C4-2b. The combination of ARsi and AZD7762 reduced protein levels of CDC6, TopBP1, ATR, and Chk1; and reduced phosphorylation of ATR and Cdc25C in VCaP and C4-2b (Figure 2A, 2B). These signaling effects were accompanied by synergistically increased ATM S1981 phosphorylation, a marker of DNA damage. Phosphorylation of Chk1 S317, the site phosphorylated by ATR or ATM (Canman, 2001, Kastan and Lim, 2000, Gatei et al., 2003), was markedly elevated by AZD7762. Interestingly, Chk1 S317 phosphorylation was positively correlated with AZD7762-mediated, DNA damage-induced phosphorylation of ATM S1981, yet phosphorylation of Cdc25C S216, a Chk1 target, was substantially reduced (Figure 2A, 2B). To further evaluate the contribution of ATM and ATR to the phosphorylation of Chk1 at S317 in these specific cell contexts, we treated VCaP and C4-2b cells with ATM inhibitor KU-60019 or ATR inhibitor VE-821, in the absence and presence of AZD7762. The results showed that both ATM inhibitor and ATR inhibitor can reduce basal and AZD7762-induced phosphorylation of Chk1 S317 (Figure 2C-D). Since ATR and P-ATR were unchanged or reduced, it is unlikely that ATR activities are related to the substantially increased Chk1 S317 phosphorylation in response to AZD7762, instead the increased Chk1 phosphorylation at S317 is most likely due to DNA damage-induced ATM phosphorylation of Chk1 S317.
Fig. 2. Effect of AR knockdown and Chk1/2 inhibitor AZD7762 (AZD) on TopBP1-ATR-Chk1 signaling and PCa cell survival.
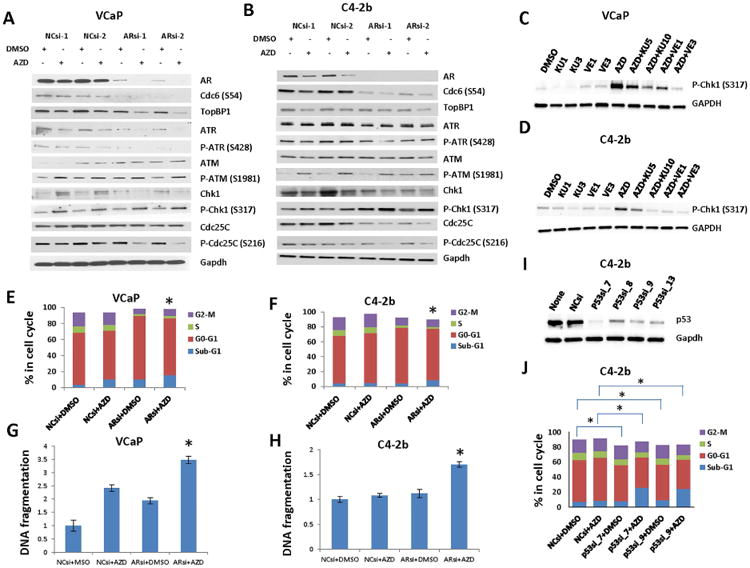
(A-B and E-H) VCaP and C4-2b cells were transfected with 20 mM ARsi or NCsi 24 h prior to the treatment with 200 nM AZD for 48 h. (A-B) Western blotting analysis of Cdc6 and TopBP1-ATR-Chk1 signaling molecules in ARsi, AZD and ARsi+AZD treated VCaP (A) and C4-2b (B) cells. (C-D) Western blotting analysis of P-Chk1 (S317) in VCaP and C4-2b cells that were treated with 200 nM AZD, 5 and 10 μM KU-60019 (KU), an ATM inhibitor, 1 and 3 μM VE-821 (VE), an ATR inhibitor, or combination AZD and KU or VE for 48 h. (E-F) Cell cycle analyses of ARsi, AZD and ARsi+AZD treated VCaP (E) and C4-2b (F) cells. (G-H) DNA fragmentation analyses of ARsi, AZD and ARsi+AZD treated VCaP (G) and C4-2b (H) cells. (I) p53 wild-type C4-2b cells were transfected with 20 nM p53si or NCsi for 48 h prior to Western blotting analysis. (J) C4-2b cells were transfected with 20 nM p53si or NCsi for 48 h prior to the treatment with 200 nM AZD for 48 h, followed by cell cycle analysis. *Statistically significant (p<0.05) in sub-G1 cell distribution (E and F) or in DNA fragmentation (G and H) compared ARsi and AZD combination to ARsi or AZD alone, or in G0-G1 and S cell distribution compared p53si+DMSO to NCsi+DMSO (J), or in sub-G1, G0-G1, S, and G2-M cell distribution compared p53si+AZD to NCsi+AZD (J). Detail statistical information on panel J is available in Table S2.
Flow cytometric analysis demonstrated that the combination treatment with AR knockdown and AZD7762 resulted in a greater apoptotic effect than did AR knockdown (P<0.001 in both cell lines) or AZD7762 alone (P=0.01-VCaP, P<0.001-C4-2b) (Figure 2E, 2F). DNA fragmentation analysis demonstrated that ARsi and AZD7762 combination increased the rate of apoptosis over AR knockdown (P<0.001-VCaP, P<0.001-C4-2b) and AZD7762 (P=0.013-VCaP, P<0.001-C4-2b) alone (Figure 2G, H). The differing responses of VCaP and C4-2b cells to AZD7762 treatment are notable. In addition to higher treatment efficacy from the combination of ARsi and AZD7762, inhibition of AR with ARsi caused G1 arrest and reduction of cells in S phase in both cell lines, however, and inhibition of Chk1 by AZD7762 led to significantly increased sub-G1 cells and DNA fragmentation in VCaP, but had very little effect on C4-2b (Figure 2E-H), which may be largely due to different p53 status in the two cell lines (VCaP, p53 mutant; C4-2b p53 wild-type). It was reported that p53-mediated G1 arrest in response to DNA damage can spare cells from AZD7762 action that dominantly occurs during G2 (Castedo et al., 2004, Zhou and Bartek, 2004, Benada and Macurek, 2015). To address this possibility, we knocked down p53 using siRNA in p53 wild-type C4-2b cells and analyzed the effect on cell cycle distribution. As expected, knockdown of p53 significantly reduced G0-G1 and S cell fractions and the combination of p53 knockdown and AZD7762 resulted in significantly reduced G0-G1, S, and G2-M cells and significantly increased sub-G1 cells (Figure 2I-J and Table S3).
Interestingly, we found that CDC6 knockdown and combination treatment with AZD7762 also markedly reduced TopBP1 protein levels, ATR S428 and Cdc25C S216 phosphorylation in both cell lines (Figure 3A and 3B). We also found that the combination treatment resulted in markedly greater ATM auto-phosphorylation than did treatment with both agents alone, suggesting a synergistic increase in DNA damage (Figure 3A and B). Additionally, the combination treatment increased Chk1 S317 phosphorylation in both cell lines (Figure 3A and 3B).
Figure 3. CDC6 knockdown increases the sensitivity of PCa cells to treatment with Chk1/2 inhibitor AZD through inhibition of TopBP1-ATR-Chk1 signaling.

VCaP and C4-2b cells were transfected with 20 μM CDC6si or NCsi 24 h prior to the treatment with 200 nM AZD for 48 h. (A and B) Western analysis of TopBP1-ATR- Chk1 signaling proteins in VCaP (A) and C4-2b (B) cells. (C and D) Flow cytometry analysis of CDC6si7 and AZD treated VCaP (C) and C4-2b (D) cells. Top panels, representative cell cycle profiles, bottom panels, quantitative analysis of cell cycle distribution. Red arrows point to sub-G1 and blue arrows point to S phase. (E and F) DNA fragmentation analysis of CDC6si7 and AZD treated VCaP (E) and C4-2b (F) cells. *Statistically significant in sub-G1 cell distribution (B and E) or in DNA fragmentation (C and F) when compare ARsi and AZD7762 combination to ARsi or AZD7762 alone.
To determine the biological effects of combination treatment with CDC6si and AZD7762, we examined apoptotic activities using flow cytometry and a DNA fragmentation assay. Combination treatment increased the percentage of sub-G1 (apoptotic) cells more than did CDC6 knockdown (P=0.01-VCaP, P<0.001-C4-2b), or AZD7762 (P=0.0048-VCaP, P<0.001-C4-2b) alone (Figure 3C, D). The combination treatment also resulted in greater DNA fragmentation/apoptosis than did CDC6 knockdown (P<0.001-VCaP, P=0.004-C4-2b) and AZD7762 (P=0.03-VCaP, P=0.03-C4-2b) alone (Figure 3E, F).
TopBp1 is an essential activator for the ATR-Chk1 signaling pathway (Cimprich and Cortez, 2008, Wardlaw et al., 2014). Combination of AR knockdown or CDC6 knockdown with AZD7762 led to markedly reduced TopBP1 protein levels (Figure 2A, B and Figure 3A, B). These observations prompt us to test whether knockdown of TOPBP1 could also synergize with AZD7762 in induction of PCa apoptosis and cell death. Western blotting analysis showed that TOPBP1knockdown reduced Chk1 S317 phosphorylation, Cdc25C expression, and Cdc25C S216 phosphorylation, and that TOPBP1 knockdown together with AZD7762 further reduced Cdc25C S216 phosphorylation (Figure 4 A, B). Flow cytometric analysis demonstrated that the combination treatment with TOPBP1 knockdown and AZD7762 also resulted in a greater apoptotic effect than did TOPBP1 knockdown (TOPBP1si_sc P=0.0023 and TOPBP1si_3 P=0.0066 in VCaP; TOPBP1si_sc P=0.0142 and TOPBP1si_3 P=0.0037 in C4-2b) and AZD7762 alone (TOPBP1si_sc P=0.0014 and TOPBP1si_3 P=0.0063 in VCaP; TOPBP1si_sc P=0.0006 and TOPBP1si_3 P=0.0002 in C4-2b) (Figure 4C, D). DNA fragmentation analysis demonstrated that this combination increased the rate of apoptosis over TOPBP1 knockdown (TOPBP1si_sc P<0.0001 and TOPBP1si_3 P=0.0047 in VCaP; TOPBP1si_sc P=0.0028 and TOPBP1si_3 P=0.0368 in C4-2b) and AZD7762 (TOPBP1si_sc P<0.0001 and TOPBP1si_3 P=0.0027 in VCaP; TOPBP1si_sc P=0.0003 and TOPBP1si_3 P=0.0011 in C4-2b) alone (Figure 4E, F).
Figure 4. TOPBP1 knockdown increases the sensitivity of PCa cells to treatment with Chk1/2 inhibitor AZD.

VCaP and C4-2b cells were transfected with 20 μM TOPBP1si or NCsi 24 h prior to the treatment with 200 nM AZD for 48 h. (A and B) Western analysis of TopBP1, Chk1, P-Chk1 (S317), Cdc25C and P-Cdc25C after TOPBP1 knockdown and treatment of AZD7762 in VCaP (A) and C4-2b (B) cells. (C and D) Flow cytometry analysis for sub-G1 cell distribution in TOPBP1si and AZD treated VCaP (C) and C4-2b (D) cells. (E and F) DNA fragmentation analysis of TOPBP1si and AZD treated VCaP (E) and C4-2b (F) cells. *Statistically significant in sub-G1 cell distribution (C and D) or in DNA fragmentation (E and F) when compare TOPBP1si and AZD7762 combination to TOPBP1si or AZD alone.
Treatment with ENZ and AZD7762 inhibits CDC6-TopBP1-ATR-Chk1 signaling and promotes DNA damage and apoptosis in PCa cells
ENZ is a potent AR signaling inhibitor approved for treatment of mCRPC (Scher et al., 2012), and by combining it with AZD7762, we can translate our findings into a viable therapeutic approach for prostate cancer. We initially treated VCaP and C4-2b with ENZ and/or AZD7762. We found that the combination treatment markedly reduced Cdc6 phosphorylation and protein levels, TopBP1 protein levels, ATR and Chk1 phosphorylation (Figure 5A). Importantly, the combination treatment synergistically increased ATM phosphorylation, and reduced Cdc25C phosphorylation, markers for DNA damage and abrogation of G2/M checkpoint, respectively (Figure 5A).
Figure 5. Combination treatment with ENZ and AZD inhibits CDC6-TopBP1-ATR-Chk1 signaling promoting DNA damage and apoptosis in PCa cells.
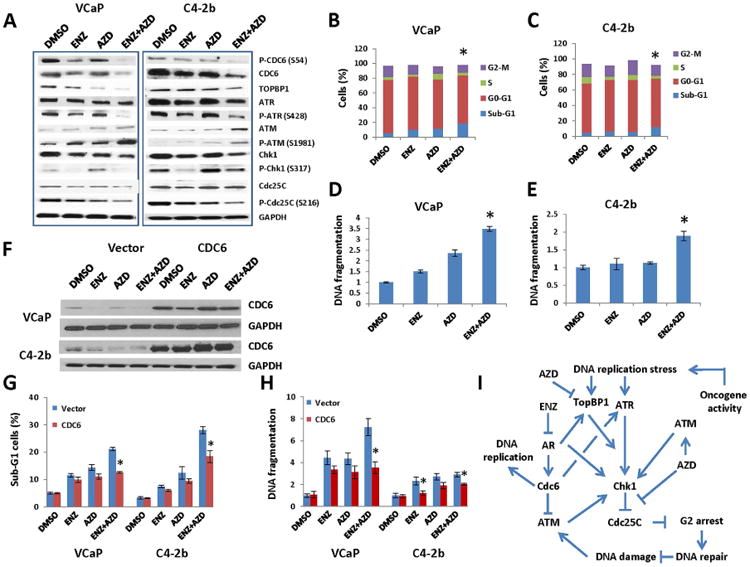
(A-E) VCaP and C4-2b cells were transfected with DMSO, 1 μM ENZ, 200 nM AZD or ENZ+AZD for 48 h. (A) ENZ and AZD combination treatment reduced phosphorylation and protein levels of CDC6, TopBP1 protein levels, and phosphorylations of ATR, Chk1 and Cdc25C, leading to increased ATM phosphorylation in both VCaP and C4-2b cells. (B and C) Flow cytometric analysis. ENZ and AZD combination treatment increased the percentage of apoptotic (sub-G1) cells in VCaP (B) more than treatment with ENZ (P=0.0053) or AZD7762 (P=0.012) alone did, and also increased the percentage of apoptotic (sub-G1) cells in C4-2b (C) more than treatment with ENZ (P=0.009) or AZD7762 (P=0.008) alone did. (D and E) DNA fragmentation assays. The combination treatment increased apoptosis in VCaP cells (D) more than treatment with ENZ alone (P<0.001) or AZD7762 (P<0.001) alone did, and also increased apoptosis in C4-b cells (E) more than treatment with ENZ (P=0.018) or AZD7762 (P=0.0046) alone did. (F-H) Overexpression of CDC6 reduces ENZ- and/or AZD-induced apoptotic cell death. VCaP and C4-2b cells were transfected with 1 micro g of CDC6 plasmid DNA or control empty vector DNA for 24 h prior to the treatment with DMSO, 1 mM ENZ, 200 nM AZD or ENZ+AZD for 48 h. (F) CDC6 protein levels markedly increased after enforced CDC6 expression in VCaP and C4-2b cells. (G) Flow cytometric analysis for apoptotic cell death (Sub-G1 cells). (H) DNA fragmentation assay. Data in (H) are presented as fold of DMSO control. *statistically significant when comparing combination treatment to single agent (B-E) and comparing CDC6 overexpression to empty vector (G-H). (I) Proposed signaling schema.
Next, we examined the apoptotic effect of combination treatment with ENZ and AZD7762 using flow cytometry and a DNA fragmentation assay. The results of flow cytometric analysis demonstrated that the combination treatment significantly increased the percentage of apoptotic (sub-G1) cells over that resulting from ENZ (P=0.0053-VCaP, P=0.009-C4-2b) and AZD7762 (P=0.012-VCaP, P=0.008-C4-2b) alone (Figure 5B, C). The results of DNA fragmentation analysis demonstrated that the combination treatment increased the apoptotic effect over that induced by ENZ (P<0.001-VCaP, P=0.018-C4-2b) and AZD7762 (P<0.001-VCaP, P=0.0046-C4-2b) alone (Figure 5D, E).
To strengthen our finding regarding the role of CDC6 in the combination therapy using ENZ and AZD7762, we tested whether overexpression of CDC6 can overcome ENZ and AZD7762 induced PCa cell death. Our data demonstrated that overexpression of CDC6 (Figure 5F) significantly reduced sub-G1 cells (Figure 5G) and apoptotic DNA fragmentation (Figure 5H) in both VCaP and C4-2b.
Overall, our data demonstrated that combination treatment with ENZ and AZD7762 treatment downregulated CDC6 and TopBP1-ATR-Chk1 signaling, released Cdc25C from inactivating phosphorylation by Chk1, abolished G2/M checkpoint, resulting in increased DNA damage and apoptosis (Figure 5I).
Combination treatment with enzalutamide and AZD7762 inhibits the growth of prostate tumor xenografts
To test our hypothesis that combination treatment with ENZ and AZD7762 is a potential therapeutic approach for mCRPC, we used three different animal models: VCaP, C4-2b xenografts, and patient-derived xenograft (PDX) MDA-133-4, which harbors a missense p53 mutation (Lee et al., 2011). We administered treatment to mice with orthotopic VCaP xenografts and monitored their tumor progression. Treatment with ENZ alone and the combination of ENZ and AZD7762 reduced tumor growth compared to control mice (P=0.05 and P=0.02, respectively) but the differences between combination and single-agent treatment were not statistically significant according to assessment using IVIS (Figure 6A and Figure S2A). However, ENZ is an inducer of CYP450, it increases luciferin metabolism and, through this activity, reduces bioluminescence. In comparison, AZD7762 is an ATP-competitive Chk1/2 inhibitor and it binds to their respective ATP-binding sites, increases the availability of ATP which is free to react with D-luciferin to produce light and, through this activity, increase bioluminescence leading to false increased signal. When we evaluated tumor wet weights, we found that single-agent treatment reduced tumor growth significantly (P=0.009-ENZ, P<0.001-AZD7762), whereas the combination treatment significantly reduced weights more than that resulting from treatment with ENZ (P<0.001) or AZD7762 (P=0.008) alone. Remarkably, the combination treatment was synergistic with regard to tumor wet weight (P=0.0097) (Figure 6B) according to two-way ANOVA (Slinker, 1998).
Figure 6. Combination treatment with ENZ and AZD7762 inhibited the growth of prostate tumor xenografts.
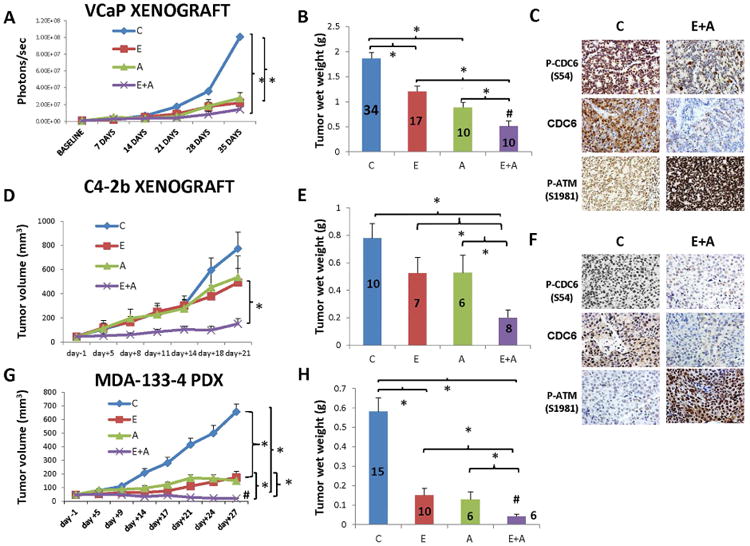
VCaP orthotopic xenografts, C4-2b subcutaneous xenografts, and MDA-133-4 PDX model were treated with vehicle control (C), enzalutamide (E), AZD7762 (A), or enzalutamide and AZD7762 (A+E) for 35, 21, and 28 days respectively. A-C. VCaP xenografts. ENZ alone and combined with AZD7762 reduced tumor growth more than the control treatment (P=0.05 and P=0.02, respectively) via IVIS measurements (A). Treatment with ENZ and AZD7762 as single agents had significant effects (P=0.009 and P<0.001, respectively) on tumor wet weights, and mice given the combination had tumors with significantly lower wet weight than mice given ENZ (P<0.001) or AZD7762 (P=0.008) alone did (B). The combination treatment had synergistic effects on wet weights as determined using two-way ANOVA (P=0.0097, indicated by # in the figure). C. IHC analysis demonstrated that the combination of ENZ and AZD7762 significantly decreased CDC6 phosphorylation (P=0.01208), Cdc6 protein levels (P=0.012) and significantly increased ATM phosphorylation (P=0.01208) compared to the control treatment did in VCaP xenografts. Full IHC analysis results including comparison of combination treatment with single agent treatment and quantitative analysis results can be found in Fig. S2. D-F. C4-2b xenografts. Neither of the single agents had a significant effect on tumor volume, whereas the combination treatment resulted in significantly lower tumor volumes at 8 days than the control treatment (P=0.004) and enzalutamide (P=0.047) and at 11 days than AZD7762 (P=0.04). These differences continued to be statistically significant over 21 days (D). Neither of the single agents had a significant effect on tumor wet weights, but the combination treatment produced significantly lower tumor wet weights than the control treatment (P<0.001), ENZ (P=0.02), or AZD7762 (P=0.022) did (E). F. IHC analysis demonstrated that the combination of ENZ and AZD7762 significantly decreased CDC6 phosphorylation (P=0.01208), CDC6 protein levels (P=0.01208) and significantly increased ATM phosphorylation (P=0.01208) compared to the control treatment. Full IHC analysis results including comparison of combination treatment with single agent treatment and quantitative analysis results can be found in Fig. S2. G, H. The subcutaneous MDA-133-4 PDX model. The combination treatment had a greater effect on tumor volume than ENZ alone at 21 (P=0.016), 24 (P=0.004), and 27 (P=0.015) days or AZD7762 alone at 9 (P=0.04), 14 (P=0.014), 17 (P=0.005), 21 (P=0.009), 24 (P=0.006), and 27 (P<0.001) days (G). The combination treatment also had a greater effect on tumor wet weights than did ENZ (P=0.036) and AZD7762 (P=0.034) alone did (H). Synergism was evaluated using ANOVA (P=0.0004 for tumor volume and P=0.0107 for tumor wet weight, indicated by # in the figures). *Statistically significant.
To further validate our in vivo data and establish associations with our WB data, we analyzed CDC6 and ATM expression and phosphorylation in mice. Our in vitro studies demonstrated that combination treatment with ENZ and AZD7762 significantly reduced CDC6 expression and phosphorylation and increased ATM phosphorylation. IHC analysis of VCaP xenografts demonstrated that ENZ reduced CDC6 phosphorylation significantly compared to control (P=0.036) whereas AZD7762 did not produce significant effects (P=0.06) (Figure S3A). However, the combination treatment significantly reduced CDC6 phosphorylation compared to control (P=0.012), ENZ-treated (P=0.0366) and AZD776-treated (P=0.036) mice (Figure 6C and Figure S3A). Similarly, CDC6 expression was reduced to a greater extent in ENZ-treated mice than in control mice (P=0.036) whereas the difference in AZD7762-treated and control mice was not statistically significant (P=0.4) (Figure S3A). The combination treatment reduced CDC6 expression significantly compared to the control (P=0.012), ENZ (P=0.0214) and AZD7762 (P=0.0214) did. Treatment with ENZ or AZD7762 alone did not have a significant effect on ATM phosphorylation whereas the combination treatment significantly increased this phosphorylation over that in control (P=0.021), ENZ-treated (P=0.036) and AZD7762-treated (P=0.036) mice (Figure 6C and Figure S3A).
In subcutaneous C4-2b xenografts, we found that ENZ and AZD7762 as single agents did not significantly affect tumor volume or wet weight compared to the control treatment (Figure 6D, E). In contrast, the combination treatment produced significantly lower tumor volumes at 8 days than in control (P=0.004) and enzalutamide-treated (P=0.047) and at 11 days than in AZD7762-treated mice (P=0.04). The differences continued to be statistically significant throughout the 21-day treatment period (Figure 6D). Moreover, the combination treatment produced significantly lower tumor wet weights than those in the control (P<0.001), ENZ-treated (P=0.02) and AZD7762-treated (P=0.022) mice (Figure 6E and Figure S2B).
IHC analysis of C4-2b xenograft tumors, show that single-agent treatment did not significantly reduce CDC6 phosphorylation, whereas the combination treatment significantly reduced it to a greater extent than that in control (P=0.012), ENZ-treated (P=0.021) and AZD7762-treated (P=0.021) mice (Figure 6F and Figure S3B). CDC6 protein expression was reduced by single–agent treatment but differences did not reach statistical significance compared to control (P=0.4) mice. In comparison, the combination treatment significantly reduced CDC6 expression compared to the control (P=0.012), ENZ-treated (P=0.036) and AZD7762-treated (P=0.012) mice (Figure 6F and Figure S3B). Moreover, we found that neither of the single-agent treatments had a greater effect on ATM phosphorylation than the control treatment did. However, the combination treatment significantly increased ATM phosphorylation over that with control (P=0.012), ENZ (P=0.036) and AZD7762 (P=0.036) treatment (Figure 6F), indicating that this combination treatment synergistically increases the incidence of DNA damage in PCa cells.
We also used the MDA-133-4 PDX, which was shown to harbor a frameshift mutation of p53 that results in a truncated protein, as an additional model ((Lee et al., 2011); data not shown). We found that the combination treatment had a greater effect than ENZ did at 21 (P=0.016), 24 (P=0.004) and 28 (P=0.015) days, than AZD7762 did at 9 (P=0.04), 14 (P=0.014), 17 (P=0.005), 21 (P=0.009), 24 (P=0.006) and 27 (P<0.001) days (Figure 6G). Similar to the mice with VCaP xenografts, single-agent treatment had a greater effect on tumor wet weight than did the control treatment, but the combination treatment further reduced wet weights more so than ENZ (P=0.036) and AZD7762 (P=0.034) alone did (Figure 6H). These data suggested that ENZ and AZD7762 can synergistically inhibit tumor growth in a mCRPC model such as MDA-133-4 PDX, which we confirmed via Two-way ANOVA (P=0.0004-tumor volume and P=0.0107-wet weight). No significant differences in body weight were found between drug-treated and vehicle control-treated mice in all three models.
Discussion
For this study, we hypothesized that targeting of ATR-Chk1 signaling in PCa cells is an effective approach. To maximize the therapy effect, we evaluated the impact of targeting CDC6 on ATR-Chk1 signaling. Gonzalez at al. claimed that CDC6 exerts oncogenic activity though repression of the INK4/ARF locus (Gonzalez et al., 2006). Additionally, Sideridou et al. showed that not only p14, p15 and p16, but also E-cadherin is downregulated at transcriptional level by increased CDC6 (Sideridou et al., 2011). Interestingly, p14ARF can repress AR transactivation in prostate cancer cells (Lu et al., 2013). CDC6 can protect genomic integrity via activation of DDR (Clay-Farrace et al., 2003, Yoshida et al., 2010, Oehlmann et al., 2004), yet deregulated overexpression of CDC6 can lead to rereplication, a form of replication stress that can result in genomic instability (Liontos et al., 2007). Within this context the role of CDC6 in PCa is particularly intriguing, given that CDC6 is a direct AR target gene (Jin and Fondell, 2009, Bai et al., 2005) and that AR upregulates the expression of genes involved in DNA repair and DDR (Polkinghorn et al., 2013, Li et al., 2014). Indeed, we found that CDC6 was upregulated during PCa progression and CDC6 downregulation synergized with the dual Chk1/2 inhibitor, AZD7762, to inhibit TopBP1-ATR-Chk1 signaling in VCaP and C4-2b cells and to increase its cytotoxic effects. It was notable that in marked contrast to treatment with single agents, the combination of CDC6 knockdown and AZD7762 markedly affected the expression of TopBP1, ATR (Figure 3A, B) and downstream biological effects (Figure 3C-F). To monitor increased DNA damage, we utilized the ATM S1981 phosphorylation as a DNA damage marker, which is increased under Chk1 inhibition-mediated accumulation of DNA damage (Sarmento et al., 2015). Experimentally, we demonstrated that ATM S1981 phosphorylation was increased following CDC6 knockdown and further increased by combination treatment of CDC6si together with AZD7762 (Figure 3A-B). Importantly, CDC6 knockdown and AZD7762 combination treatment significantly increased the apoptotic response to DNA damage in C4-2b cells. Although it was not a major focus of this study, we infer that in p53 wild-type C4-2b cells, the combination of CDC6 knockdown and AZD7762 treatment will increase ATM-dependent p53 phosphorylation/activation, leading to increased Bax and p21 protein expressions and subsequent p53-dependent G1 arrest, which may spare C4-2b cells from AZD-mediated DNA damage-induced cell death. We used p53 knockdown to analyze the role of p53 in C4-2b cells following AZD7762 treatment, and demonstrated that reduction of p53 levels significantly reduced G0-G1 and S cell fractions, and significantly increased sub-G1 cells in AZD7762 treated cells (Figure 2I-J and Table S3).
Previous publications have shown that TopBp1 plays an important role in activation of ATR-Chk1 pathway (Cimprich and Cortez, 2008, Wardlaw et al., 2014, Li et al., 2014). Our results show AR or CDC6 knockdown combined with AZD7762 treatment coordinately downregulated TopBP1 in both VCaP and C4-2b models, and that similar to AR or CDC6 knockdown, knockdown of TOPBP1 synergizes with AZD7762 in induction of apoptotic cell death in VCaP and C4-2b PCa cells. These data together with the results of ENZ and AZD7762 combination experiments demonstrated that synergistic therapeutic effect can be reached by targeting AR and Chk1 simultaneously through inhibition/downregulation of TopBP1-ATR-Chk1 signaling.
To validate our in vitro results, we selected VCaP and C4-2b xenograft and human MDA-133-4 PDX AR-positive models for our studies on the basis of their phenotypic characteristics which allow us to evaluate specific drug activities within the context of androgen-dependence and variable p53 status. We found that ENZ and AZD7762 synergistically inhibit tumor growth in VCaP xenografts compared to single agents. Regarding C4-2b xenografts, we found that AZD7762 was not effective as it was in the other models owing to its p53wild-type status (Ma et al., 2012). Reduced response to ENZ and to ENZ and AZD7762 combination treatment was anticipated since this model is AR-positive but androgen-independent (Nguyen et al., 2014). However, even though this model is androgen-independent, the combination of AR and Chk1/2 inhibition approach exhibited marked activity. This suggests that PCa that is AR-positive, yet fails to respond to ENZ treatment alone (Nguyen et al., 2014) will respond to this combination therapeutic approach. Similar to VCaP, in MDA-PCa-133-4 model, combination treatment of ENZ and AZD7762 synergistically inhibited tumor growth in this model despite the marked activities of single-agent treatment. IHC analysis demonstrated that this combination reduced CDC6 phosphorylation and expression and increased the levels of ATM phosphorylation, a DNA damage biomarker, in VCaP and C4-2b xenografts.
In summary, we demonstrated that CDC6 is upregulated during progression of PCa and is positively associated with AR expression. Our results indicated that targeting of AR/CDC6, via gene knockdown or ENZ, together with inhibition of Chk1/2 signaling by AZD7762 resulted in maximal suppression of TopBP1-ATR-Chk1 signaling, and induction of DNA damage and apoptosis in vitro. Furthermore, this therapeutic strategy exhibited additive or synergistic therapeutic activities in xenograft and PDX models in vivo. Importantly, one of the models we used, C4-2b, is androgen-positive but androgen-independent and wild-type for p53. Its marked response to the combination treatment indicates that the combined inhibition of androgen-signaling and Chk1/2 can be effective in the absence of intact AR signaling and the presence of wild-type p53. Additional studies are required to confirm the efficacy of this approach and evaluate alternative methods of targeting DDR using AR signaling inhibitors combined with DDR-targeted agents in patients with mCRPC.
Experimental Procedures
Cell Lines and Reagents
The human PCa cell line VCaP was validated as described previously (Li et al., 2014), and LNCaP C4-2b (C4-2b) was recently validated in MD Anderson's Characterized Cell Line Core Facility using the same method. Cycloheximide (239763) was purchased from Calbiochem and AZD7762, enzalutamide (MDV3100), KU-60019 and VE-821 were purchased from SelleckChem.
RNA interference
CDC6si6 (SI04218389), CDC6si7 (SI04254782), TOPBP1si3 (SI00749553), TP53si_7 (SI026233764), TP53si_8 (SI02623754), TP53si_9 (SI02655170), TP53si_13 (SI4384079) and NCsi (1022076) were purchased from Qiagen, ARsi1 (s1538) and ARsi2 (s1540) were purchased from Life Technologies, and TOPBP1si_sc (sc-41068) was purchased from Santa Cruz. Gene knockdown experiments were performed using the Lipofectamine RNAiMax transfection reagent (Life Technologies). PCa cells were seeded at desired densities (VCaP: 1.0×106/well and C4-2b: 5×105/well in 6-well plates, one fifth or one thirtieth of these densities in 24-or 96-well plates, respectively). Cells were transfected with 20 nM siRNA in the following day. 24 hours later, VCaP and C4-2b cells were treated with dimethyl sulfoxide (DMSO) or AZD772 for 24 hours for Western blot (WB) analysis or 48 hours for DNA fragmentation assay or flow cytometric analysis.
WB analysis
For WB analysis of the effects of CDC6si, ARsi, TOPBP1si, AZD7762 and combinations of them, cells were transfected with siRNA for 24 hours and then, treated with DMSO or AZD7762 for 24 hours. Afterward, cells were treated with a serum-free medium overnight and then with full serum for 4 hours (synchronization) before protein extract preparation. For WB analysis of ENZ and AZD7762 effects, cells were treated with DMSO or ENZ for 24 hours. DMSO or AZD7762 was then added for 24 hours. Synchronization was achieved as above. Antibodies against CDC6 (3387), ATR (2790), P-ATR Ser428 (2853), Chk1 (2360), P-Chk1Ser317 (12302), Cdc25C (4688), P-Cdc25C Ser216 (9528), ATM (2873), P-ATM Ser1981 (13050) were purchased from Cell Signaling Technology. Antibodies against P-CDC6 Ser54 (ab75809), P-Chk1ser296 (ab79758), and TopBp1 (ab2402) were purchased from Abcam. Antibodies against GAPDH (365062) and AR (816) were purchased from Santa Cruz Biotechnology. When indicated, densitometric analysis was performed and quantification of integrated density was assessed using the NIS-Elements-AR software program (version 3.0; Nikon) followed by GAPDH normalization.
DNA fragmentation assay
A DNA fragmentation assay was performed for apoptotic evaluation of siRNA and/dug effects. Cells were transfected with CDC6si, ARsi, TOPBPlsi or NCsi and treated with DMSO or ENZ for 24 hours and then, treated with DMSO, AZD7762, or ENZ+AZD7762 for 48 hours. DNA fragmentation analysis was performed as described previously (Karantanos et al., 2014).
Flow cytometry analysis
Cells were treated as described above and were prepared flow cytometric analysis as described previously (Li et al, 2014).
Protein stability assay
VCaP and C4-2b cells were treated with NCsi or ARsi for 48 hours and then, incubated with 100 μg/ml cycloheximide for the indicated time periods. Cell extracts were obtained via lysis in a modified RIPA buffer.
Animal studies
Orthotopic VCaP xenografts
VCaP xenografts were generated in mice as described previously (Li et al, 2014). The experimental groups received DMSO, ENZ (1Omg/kg daily), AZD7762 (25 mg/kg, twice daily every third day) or ENZ and AZD7762 combination for 35 days. Tumor size was monitored weekly according to luminescence signal using IVIS 200 Imaging System (PerkinElmer). The mice were sacrificed and their tumors were collected.
Subcutaneous C4-2b xenografts
Aliquots of 6×106 C4-2b cells in 100 μI of 10% FCS and T medium+50% matrigel were injected subcutaneously into the right flanks of athymic nude male mice (Taconic) to induce subcutaneous tumors. Tumors were allowed to grow for 24 days before treatment. The experimental groups received treatment for 21 days in doses as described above. Tumor size was monitored by measuring diameters of 3 dimensions (length/2×width/2×height/2×π×4/3). The mice were sacrificed and their tumors were collected.
MDA-PCa-133-4 patient-derived xenografts
Subcutaneous patient-derived xenografts (PDXs) of MDA-PCa-133-4 were generated by implanting 0.125 cm3 tumor fragment into the left flanks of previously castrated severe combined immunodeficiency mice (SCID) mice (Charles River Laboratories). RNASeq analysis showed that this tumor harbors a missense p53 mutation (P72R). Tumors were allowed to grow until they reached a volume of 50 mm3. The experimental groups received treatment for 28 days in doses as described above. Tumor size was monitored as described for the C4-2b model. The mice were sacrificed and tumors were collected.
Patient-derived xenografts
The MD Anderson Cancer Center PCa PDXs were developed with the suitable support as described previously (Li et al, 2014)
Immunohistochemical analysis
Eleven human normal prostate specimens, twenty eight human primary PCa (not previously treated) obtained after radical prostatectomy and nine metastatic PCa specimens (submitted to various previous treatments, Table SI) were obtained after patients provided informed consent and used to analyze P-CDC6 and CDC6 expression. Antibodies against AR (816), P-CDC6 S54 (12920) and CDC6 (8341) obtained from Santa Cruz Biotechnology were used for IHC. CDC6 and AR immunostainings were also performed and scored on tissue microarray slides composed of 34 tumor xenografts generated from different histologic types of PCa (Table S2). For IHC analysis of tumor xenografts, specimens were prepared as described previously (Likun et al., 2014). Antibodies against P-CDC6 S54 and CDC6 (Santa Cruz Biotechnology), P-ATM S1981 (32420) and ATM (1292; Abcam) were used. Immunostaining scoring was performed as described previously (Li et al., 2014).
Statistical analysis
The results are presented as the mean ± standard error from at least three independent experiments. Comparisons of groups were appropriately analyzed using the Student-t test, the Mann-Whitney U test, Spearman's Rho or the Kruskal-Wallis rank test. P values less than 0.05 were considered statistically significant and all tests were two-tailed. Synergism was determined using two-way analysis of variance (ANOVA) test (Slinker, 1998, Li et al., 2014).
Supplementary Material
Acknowledgments
We thank Xinhai Wan and Jun Yang for guiding in animal experiments and Donald Norwood and Linda Bohannon for editing the manuscript.
Funding: This work was supported in part by National Cancer Institute grant R0150588 (to T.C.T.); National Cancer Institute grant P50140388, the Prostate Cancer Specialized Program of Research Excellence at The University of Texas MD Anderson Cancer Center; the NIH through MD Anderson's Cancer Center Support Grant, CA16672; and Tony's Prostate Cancer Research Foundation.
Footnotes
Author Contributions: T.C.T., S.K., T.K. and L.L. conceived and designed the study and wrote the paper. In vitro studies, including siRNA and drug treatments, Western blotting analysis, flow cytometric, DNA fragmentation experiments, protein stability assays, statistic and synergy analysis: L.L., S.K., T.K., J.W., X.Z., W. Z., S.L.; IHC and tissue microarray analysis: G.Y., S.K., T.K.; Xenograft model studies: S.P., S.K., T.K., J.W., J.S. and G.G. P.G.C. contributed to manuscript preparation. S.N.M, A.M.A., P.T. and N.N. contributed to pathological analysis of human samples and the establishment of PDX lines. B.B and G.C.M. performed bioinformatics analyses of RNASeq data.
Conflict of Interest: The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai VU, Cifuentes E, Menon M, Barrack ER, Reddy GP. Androgen receptor regulate Cdc6 in synchronized LNCaP cells progressing from G1 to S phase. J Cell Physiol. 2005;204:381–7. doi: 10.1002/jcp.20422. [DOI] [PubMed] [Google Scholar]
- Bartucci M, Svensson S, Romania P, Dattilo R, Patrizii M, Signore M, Navarra S, Lotti F, Biffoni M, Pilozzi E, Duranti E, Martinelli S, Rinaldo C, Zeuner A, Maugeri-sacca M, Eramo A, De Maria R. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012;19:768–78. doi: 10.1038/cdd.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benada J, Macurek L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules. 2015;5:1912–37. doi: 10.3390/biom5031912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlado LR, Mendez J. CDC6: from DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis. 2008;29:237–43. doi: 10.1093/carcin/bgm268. [DOI] [PubMed] [Google Scholar]
- Brooks K, Oakes V, Edwards B, Ranall M, Leo P, Pavey S, Pinder A, Beamish H, Mukhopadhyay P, Lambie D, Gabrielli B. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene. 2013;32:788–96. doi: 10.1038/onc.2012.72. [DOI] [PubMed] [Google Scholar]
- Canman CE. Replication checkpoint: preventing mitotic catastrophe. Curr Biol. 2001;11:R121–4. doi: 10.1016/s0960-9822(01)00057-4. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–37. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay-farrace L, Pelizon C, Santamaria D, Pines J, Laskey RA. Human replication protein Cdc6 prevents mitosis through a checkpoint mechanism that implicates Chk1. EMBO J. 2003;22:704–12. doi: 10.1093/emboj/cdg046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'adda Di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, Vallis KA, Hammond EM, Olcina MM, Gillies Mckenna W, Muschel RJ, Brunner TB. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J, Khanna KK. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003;278:14806–11. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Klatt P, Delgado S, Conde E, Lopez-rios F, Sanchez-cespedes M, Mendez J, Antequera F, Serrano M. Oncogenic activity of Cdc6 through repression of the INK4/ARF locus. Nature. 2006;440:702–6. doi: 10.1038/nature04585. [DOI] [PubMed] [Google Scholar]
- Ide H, Lu Y, Yu J, China T, Kumamoto T, Koseki T, Yamaguchi R, Muto S, Horie S. Testosterone promotes DNA damage response under oxidative stress in prostate cancer cell lines. Prostate. 2012;72:1407–11. doi: 10.1002/pros.22492. [DOI] [PubMed] [Google Scholar]
- Jin F, Fondell JD. A novel androgen receptor-binding element modulates Cdc6 transcription in prostate cancer cells during cell-cycle progression. Nucleic Acids Res. 2009;37:4826–38. doi: 10.1093/nar/gkp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanika S, Karantanos T, Li L, Corn PG, Thompson TC. DNA damage response and prostate cancer: defects, regulation and therapeutic implications. Oncogene. 2014;0 doi: 10.1038/onc.2014.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- Lee YC, Cheng CJ, Bilen MA, Lu JF, Satcher RL, Yu-Lee LY, Gallick GE, Maity SN, Lin SH. BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Research. 2011;71:5194–203. doi: 10.1158/0008-5472.CAN-10-4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chang W, Yang G, Ren C, Park S, Karantanos T, Karanika S, Wang J, Yin J, Shah PK, Takahiro H, Dobashi M, Zhang W, Efstathiou E, Maity SN, Aparicio AM, Li Ning, Tapia EM, Troncoso P, Broom B, Xiao L, Lee HS, Lee JS, Corn PG, Navone N, Thompson TC. Targeting poly(ADP-ribose) polymerase and the c-Myb-regulated DNA damage response pathway in castration-resistant prostate cancer. Sci Signal. 2014;7:ra47. doi: 10.1126/scisignal.2005070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liontos M, Koutsami M, Sideridou M, Evangelou K, Kletsas D, Levy B, Kotsinas A, Nahum O, Zoumpourlis V, Kouloukoussa M, Lygerou Z, Taraviras S, Kittas C, Bartkova J, Papavassiliou AG, Bartek J, Halazonetis TD, Gorgoulis VG. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007;67:10899–909. doi: 10.1158/0008-5472.CAN-07-2837. [DOI] [PubMed] [Google Scholar]
- Lu W, Xie Y, Ma Y, Matusik RJ, Chen Z. ARF represses androgen receptor transactivation in prostate cancer. Mol Endocrinol. 2013;27:635–48. doi: 10.1210/me.2012-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, Lin L, Hoog J, Goiffon RJ, Prat A, Aft RL, Ellis MJ, Piwnica-worms H. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest. 2012;122:1541–52. doi: 10.1172/JCI58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JB, Choudhuri R, Fabre K, Sowers AL, Citrin D, Zabludoff SD, Cook JA. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin Cancer Res. 2010;16:2076–84. doi: 10.1158/1078-0432.CCR-09-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HG, Yang JC, kung HJ, Shi XB, Tilki D, Lara PN, JR, Devere White RW, Gao AC, Evans CP. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014;33:4521–30. doi: 10.1038/onc.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlmann M, Score AJ, Blow JJ. The role of Cdc6 in ensuring complete genome licensing and S phase checkpoint activation. J Cell Biol. 2004;165:181–90. doi: 10.1083/jcb.200311044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, Carver BS, Chen Y, Watson PA, Shah NP, Fujisawa S, Goglia AG, Gopalan A, Hieronymus H, Wongvipat J, Scardino PT, Zelefsky MJ, Jasin M, ChaudhurI J, Powell SN, Sawyers CL. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CJ, Molina A, Griffin T. Abiraterone in metastatic prostate cancer. N Engl J Med. 2013;368:1458–9. doi: 10.1056/NEJMc1301594. [DOI] [PubMed] [Google Scholar]
- Sarmento LM, Povoa V, Nascimento R, Real G, Antunes I, Martins LR, Moita C, Alves PM, Abecasis M, Moita LF, Parkhouse RM, Meijerink JP, Barata JT. CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene. 2015;34:2978–90. doi: 10.1038/onc.2014.248. [DOI] [PubMed] [Google Scholar]
- Sausville E, Lorusso P, Carducci M, Carter J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, Zabludoff S, Agbo F, Oakes P, Senderowicz A. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;73:539–49. doi: 10.1007/s00280-014-2380-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, De Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, De Bono JS, Investigators A. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM, Mccue P, Gomella LG, Raj GV, Dicker AP, BrodY JR, Pascal JM, Centenera MM, Butler LM, Tilley WD, Feng FY, knudsen KE. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- sideridou M, Zakopoulou R, Evangelou K, Liontos M, kotsinas A, Rampakakis E, Gagos S, Kahata K, Grabusic K, Gkouskou K, Trougakos IP, Kolettas E, Georgakilas AG, Volarevic S, Eliopoulos AG, Zannis-hadjopoulos M, Moustakas A, Gorgoulis VG. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J Cell Biol. 2011;195:1123–40. doi: 10.1083/jcb.201108121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30:723–31. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- Tapia-laliena MA, Korzeniewski N, Hohenfellner M, Duensing S. High-risk prostate cancer: a disease of genomic instability. Urol Oncol. 2014;32:1101–7. doi: 10.1016/j.urolonc.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Wardlaw CP, Carr AM, Oliver AW. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst) 2014;22:165–74. doi: 10.1016/j.dnarep.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Wu JN, Fish KM, Evans CP, Devere white RW, Dall'era MA. No improvement noted in overall or cause-specific survival for men presenting with metastatic prostate cancer over a 20-year period. Cancer. 2014;120:818–23. doi: 10.1002/cncr.28485. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sugimoto N, Iwahori S, Yugawa T, Narisawa-saito M, Kiyono T, Fujita M. CDC6 interaction with ATR regulates activation of a replication checkpoint in higher eukaryotic cells. J Cell Sci. 2010;123:225–35. doi: 10.1242/jcs.058693. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer. 2004;4:216–25. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.