

Abstract
Background
Colorectal adenocarcinoma is the second leading cause of cancer-related death in the world. The stage of the disease is related to the survival of the patient, and in early phases surgery is the main modality of treatment. The main aim of modern medicinal chemistry is to synthesize small molecules via drug designing, especially by targeting tumor cells.
Material/Methods
A new series of 19 compounds containing benzothiazole and thiazole were designed. Molecular docking studies were performed on the designed series of molecules. Compounds showing good binding affinity towards the EGFR receptor were selected for synthetic studies. Characterization of the synthesized compounds was done by FTIR, 1HNMR, Mass and C, H, N, analysis.
Results
The anticancer evaluation of the synthesized compounds was done at NIC, USA at a single dose against colon cancer cell lines HCT 116, HCT15, and HC 29. The active compounds were further evaluated for the 5-dose testing. Compounds were designed by using docking analysis. To ascertain the interaction of EGFR tyrosine kinase binding, energy calculation was used.
Conclusions
The results of the present study indicate that the designed compounds show good activity against colon cancer cell lines, which may be further studied to design new potential molecules.
MeSH Keywords: Antineoplastic Agents, Benzothiazoles, Colonic Neoplasms, Thiazoles
Background
Colorectal adenocarcinoma is the second leading cause of cancer-related death in the world. The stage of the disease is related to the survival of the patient [1], and in early phases surgery is the main modality of treatment. The main aim of modern medical chemistry is to synthesize small molecules via drug designing, especially by targeting the tumor cell. Clinical trials reported that the substitution pattern to the different heterocycles also influences carcinogenesis. Some of the characteristic properties of the malignant phenotype of colorectal cancer are mediated by overexpression and/or activation of receptor tyrosine kinases, pointing to these molecules as attractive anticancer treatment targets. One of these approaches is the inhibition of epidermal growth factor (EGF) receptor (EGFR) either by EGFR tyrosine kinase inhibition or antibody-induced receptor blockade. Inhibition of EGFR signaling significantly inhibits tumor growth in numerous preclinical models, including colon cancer models [2–5].
Targeted drug design for anticancer studies proves protein kinases as a feasible target for drug development [6,7]. It is next most important target after G-protein linked receptor. The role of certain tyrosin kinases, particularly epidermal GFR, plays an important role in regulating cell proliferation. Overexpression of EGFR has been reported in 25–82% of colorectal cancers. Some new studies report protein overexpression in 35–49% of total cases [8–12]. The wide range of EFFR expression in colorectal cancer is reported in many studies, and EGFR expression as a prognostic indicator may be related to the methodology used to detect EGFR [13–15]. Therefore, the design of inhibitors that target EFFR-TK is a better approach to new drug development (Figure 1). Lapatinib, Imatinib, and Erlotinib are widely used as EGFR-TK inhibitors for the treatment of cancer [16,17].
Figure 1.

ATP binding site showing hydrophobic region, sugar pocket, adenine region, and hydrophobic region.
Benzothiazole is a versatile nucleus and widely reported to have various biological activities, such as antitumor [18], antimicrobial [19], antitubercular [20], antioxidant [21], and antidepressant [22] activities. Moreover, thiazole and its derivatives are reported to have various pharmacological activities, such as antitubercular [23], antibacterial [24], antidepressant [25], and antitumor [26] activities. Benzothiazole derivatives bearing thiazole moiety were reported to have antiproliferative activity against colon cancer cell lines HCT-116, HT29, HCT-15, and caco-2 [27].
Rational and design
It is well known that quinazoline is a versatile moiety for the inhibition of an extensive range of tyrosine kinases, and EGFR is the most studied receptor of tyrosine kinases [28]. So, in the present work we tried to design a new series of compounds using molecular modeling studies which contain a benzothiazole core as the EGFR-TK inhibitor. The designed molecules are the structural analogs of 4-anilino-quinazoline of ‘tinibs’ (Erlotinib, Lapatinib, and Gefinitib) (Figure 2) with varied chemical properties. Isosteric replacement of the quinazoline ring with benzothiazole may mimic the ATP competitive binding regions of EGFR-TK [26–28]. We were guided by information obtained in our literature search and by the fact that benzothiazole acts in the catalytic domain (EGFR-TK) for ATP binding, showing interaction with the adenine ring with N1 and N6 through the interaction of hydrophilic and hydrophobic regions and channels (Figure 2) [26].
Figure 2.
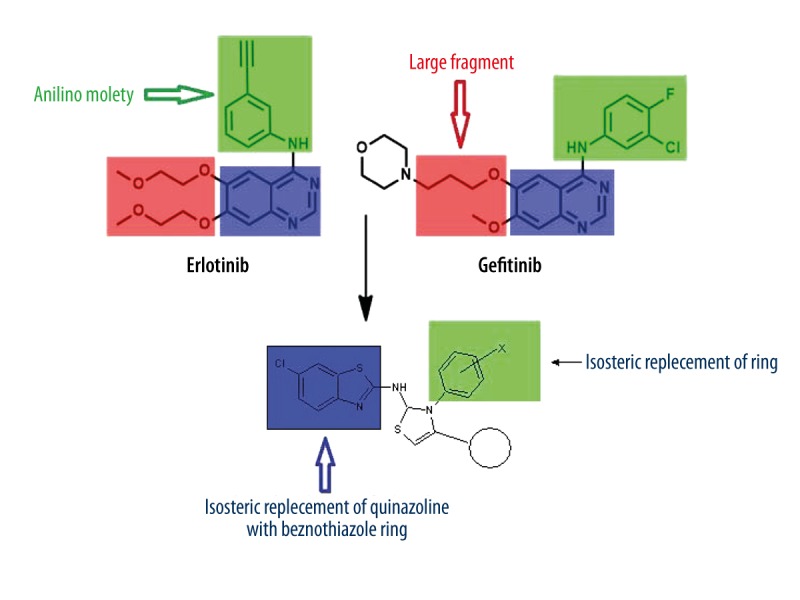
Reported anticancer compounds and designed benzothiazole derivatives showing bioisosteric replacement.
It is well known that overexpression of the RTK signaling pathway is mostly associated with occurrence of cancer. Therefore, deactivation of the tyrosine kinase signaling pathway may be a better target for the treatment of cancer. Based on these facts, molecular modeling studies were used to ascertain the interaction of designed moieties with active site of EGFR-TK. For this study, lapatinib was used as a reference compound. We use similarity-based virtual screening because it is a simple and widely used method for ligand-based virtual screening of chemical databases, where bio-isosteric compounds are used [26].
Material and Methods
Some new benzothiazole thiazole derivatives were designed. The structures were docked using Accelyres Drug Discovery Studio 3.5 [29]. The structure of the enzyme EGFR-TK was obtained from the Protein Data Bank [30] (PDB code: 1XKK: Figure 3) and was used for docking. The enzyme exists in tetrameric form in the crystal associated with water molecules, which were not considered in docking as they are not found in the ligand zone of the crystal structure. The crystal structure was cleaned by deleting the ligand and cofactors, followed by adding hydrogen atoms to the protein. The ligand structure was imported to the new window with proper names and the program was run to prepare the ligand. Finally, the input ligands were selected (Molecules: All) on the protein molecular window and docked for 1XKK. The setup for side-chain flexibility by selection of the all-visible option and the setting for the other selected chain during the docking and other parameters were kept in default. The binding energy and electrostatic interaction energy were calculated (docking analysis Table 1).
Figure 3.
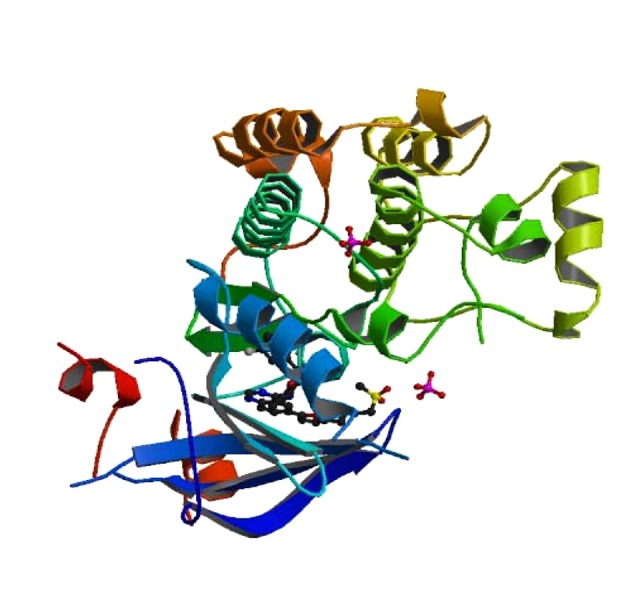
The crystal structure of EGFR kinase domain (pdb code 1XKK) in complex with lapatinib.
Table 1.
Docking analysis of compounds.
| Sr. No. | Compound | Amino acid | Bond | Binding energy | Inhibitory constant (uM) | Inter molecular energy | Electrostatic energy |
|---|---|---|---|---|---|---|---|
| 1 | LEPATINIB | LYS806, ILE938, Gly–512 | HO, HH, NH | −10.26 | 125.89 | −10.54 | −3.5 |
| 2 | BT1 | LYS806; SER912; ILE938 | HH; HH; HN | −6.64 | 13.55 | −7.83 | −0.32 |
| 3 | BT2 | LYS806; LYS806 | HN; HN | −5.94 | 44.42 | −7.13 | −0.22 |
| 4 | BT3 | LYS806 | HH | −5.31 | 128.27 | −6.5 | 0.09 |
| 5 | BT4 | LYS806; Leu–305 | HH; OH | −6.12 | 39.98 | −7.19 | −0.24 |
| 6 | BT5 | LYS806 | – | −5.35 | 119.49 | −6.54 | −0.08 |
| 7 | BT6 | LYS806 | – | −5.72 | 63.69 | −6.92 | −0.05 |
| 8 | BT7 | LYS806 | HN | −4.88 | 265.76 | −6.07 | −0.09 |
| 9 | BT8 | LYS806, SER912; Glu–514 | HH, NH, HH | −6.23 | 27.02 | −7.43 | −0.02 |
| 10 | BT9 | ARG803 | HO | −5.87 | 49.59 | −7.07 | −0.18 |
| 11 | BT10 | LYS806; LYS806 | HO; HN | −5.62 | 76.57 | −7.11 | −0.25 |
| 12 | BT11 | LYS806 | – | −5.65 | 72.3 | −7.14 | −0.15 |
| 13 | BT12 | LYS806 | HO | −5.66 | 70.71 | −6.86 | −0.1 |
| 14 | BT13 | – | – | −5.38 | 113.31 | −6.58 | 0.03 |
| 15 | BT14 | – | – | −5.47 | 98.3 | −6.66 | 0.01 |
| 16 | BT15 | LYS806 | HN | −5.43 | 104.74 | −6.62 | −0.09 |
| 17 | BT16 | LYS806, ASP807 | HO, HN | −6.36 | 21.54 | −7.86 | −1.36 |
| 18 | BT17 | LYS806, ASN808 | HO, NH | −6.41 | 20.16 | −7.9 | −1.25 |
| 19 | BT18 | ASP807; ASN808 | HN; HN | −6.36 | 21.86 | −7.85 | 0.11 |
| 20 | BT19 | LYS806 | HO | −5.94 | 44.1 | −7.73 | −0.86 |
The bolded compounds show good binding and were used for synthetic studies.
Ligand preparation
The ligand structures were drawn in 2D using Marvin Sketch (ChemAxon), then the 2D structures were converted into 3D by using the same software. Optimization of structure and energy minimization were done to confirm the stable ligands.
Protein preparation
The protein was prepared by using the standard protocol. In the first step, missing residue was added, followed by removal of water molecules, and atom names were standardized. All the related energies (e.g., TE, PE, and VDW energy) were noted before and after minimization.
Synthetic studies
Based on the result of dockings, the compounds showing good binding affinity with the receptor were selected for the synthetic studies.
Scheme 1: Synthesis of 1- (6-chloro-1,3-benzothiazol-2-yl)-3-substituted thiourea
Scheme 1.

Synthesis of Compounds 2.a–2.c
In the mixture of (1) (0.02 mol) in EtOH (50 mL), substituted isothiocyanate (0.02 mol) was put in and the resultant solution was heated under reflux for 10 h. The resultant reaction mixture was then distilled under vacuum for removal of solvent, if any. The solid product was obtained and washed with Et2O and recrystallized using rectified spirit to obtain a pale yellow product (2.a–2.c).
2.a: 1-(6-chloro-1,3-benzothiazol-2-yl)-3-(3,5-difluorophenyl) thiourea: mp 210–212° (CH3OH); Rf 0.56 (ACN: Me 1:1); FTIR (KBr) cm−1: 3215, 3229 (amine), 1310 (C=S stretch) cm−1; 1H NMR (CDCl3): δ, 6.66–7.72 (m, 8H, Ar-H), 7.56 (s, 1H, Amide), 9.98 (s, 1H, NH), 10.79 (s, 1H, NH-Ar); FABMS (m/z, 100%): 355 ([M+1], 100%). C (47.21%) H (2.22%) N (11.81%), calculated for C14H8ClF2N3S2 C (47.27%) H (2.27%) N (11.81%).
2.b: 1-(6-chloro-1,3-benzothiazol-2-yl)-3-(3,5-dichlorophenyl) thiourea: mp 201–204° (CH3OH); Rf 0.59 (ACN: Me 1:1); FTIR (KBr) cm−1: 3210, 3227 (NH), 735 (C-Cl stretching)1312 (C=S stretching) cm−1; 1H NMR (CDCl3): δ 6.55–7.23 (m, 7H, Ar-H), 7.50 (s, 1H, CO-NH), 0.40 (s, 1H, NH), 10.92 (s, 1H, NH-Ar); FABMS (m/z, 100%): 388 ([M+2], 100%). C(43.26%) H(2.02%) N(10.81%), calculated for C14H8Cl32N3S2 C(43.26%) H(2.07%) N(10.81%).
2.c: 1-(6-chloro-1,3-benzothiazol-2-yl)-3-(4-nitrophenyl)thiourea: mp 218–220° (CH3OH); Rf 0.7 (CAN: Me 1:1); FTIR (KBr) cm−1: 3210, 3229 (NH), 735 (C-Cl stretching)1320 (C=S stretching) cm−1; 1510 (NO2-Ar Stretching) 1HNMR (CDCl3): δ 6.23–7.49 (m, 7H, Ar-H), 7.74 (s, 1H, CO-NH), 0.39 (s, 1H, NH), 10.92 (s, 1H, NH-Ar); FABMS (m/z, 100%): 364 ([M+2], 100%). C(46.09%) H (2.42%) N (15.36%), calculated for C14H9ClN4O2S2 C(46.09%) H(2.47%) N(15.36%).
Scheme 2: Synthesis of 6-chloro-N-[3,4-disubstituted-1,3-thiazol-2(3H)-ylidene]-1,3-benzothiazol-2-amine
Scheme 2.

Synthesis of compounds BT1-BT19.
A mixture of the (2.a–2.c) (0.01 mole) with different substituted phenacyl bromide (0.01 mole) and sodium acetate (1.3 g, 0.1 mole) in EtOH (30 ml) was heated under reflux for 7 h. The reaction mixture was stabilized for 1 h and then water was added to develop cloudiness. The resultant mixture was kept overnight for complete reaction. The solid product obtained was filtered, washed with water, and recrystallized from spirit to obtain the final product.
As discuss earlier, the compounds showing good binding affinity towards the receptor were used for the synthetic studies.
BT1: 3-{2-[(6-chloro-1,3-benzothiazol-2-yl)amino]-3-(2,4-difluorophenyl)-2,3-dihydro-1,3-thiazol-4-yl}phenol: Rf: 0.60 (ACN: Me 1:1), MP 370–373°C FTIR, 3133.44 (Ar C-H), 1640 (C=N),1550 (C-C),1280 (CN), 3540(N-H), 1HNMR (CDCl3), 4.1 (s, 1H,NH), 6.4–7.9 (m,7H, Ar-H), 6.9–7.9 (m,3H,2-benzothiazole),5.35 (s,Ar-OH), FABMS (m/z 100%) 473; anal. Found C(55.25%) H(2.97%), N(8.85%) for C22H14ClF2N3OS2, calculated C(55.75%) H(2.98%) N(8.87%).
BT4: N-[4-(3-bromophenyl)-3-(2,4-difluorophenyl)-2,3-dihydro-1,3-thiazol-2-yl]-6-chloro-1,3-benzothiazol-2-amine: Rf: 0.56 (ACN: Me 1:1), MP 375–380°C IR, 3136.44 (Ar C-H), 1670.88(C=N),1558.38, C-Br 790, (C-C), 1320.18 (CN),3534.31(N-H),1HNMR (CDCl3), 4.08 (s, 1H, NH), 6.50–7.30 (m,7H, Ar-H), 6.9–7.9 (m,3H,2-benzothiazole), FABMS (m/z 100%) 536; anal. Found C(49.11%) H(2.43%), N(7.79%) for calculated C22H13BrClF2N3S2 C(49.22%) H(2.44%) N(7.83%)
BT8: 6-chloro-N-[4-(3-chlorophenyl)-3-(2,4-dichlorophenyl)-2,3-dihydro-1,3-thiazol-2-yl]-1,3-benzothiazol-2-amine: Rf: 0.54 (ACN: Methanol 1:1), MP 315–320°C IR, 3144.44 (Ar C-H), 1641.12(C=N), 1558.38, C-Cl 693, (C-C),1340.18 (CN),3520.31(N-H), 1HNMR (CDCl3), 4.11 (m, 2H, NH), 6.56–7.48 (m,7H, Aryl-H), 7.5–8.2(m,3H,2-benzothiazole), FABMS (m/z 100%) 525; anal. Found C(50.30%) H(2.49%), N(8.00%) for calculated C22H13Cl4N3S2 C(50.30%) H(2.49%) N(8.00%).
BT16: 6-chloro-N-[3-(4-nitrophenyl)-4-phenyl- 2,3-dihydro- 1,3-thiazol- 2-yl]-1,3-benzothiazol-2-amine: Rf: 0.74 (ACN: Methanol 1:1), MP 279–281°C IR, 3140.44 (Ar C-H), 1621.12 (C=N),1558.38, C-Cl 693, (C-C),1340.18 (CN),3550.31(N-H), 1HNMR (CDCl3), 4.21 (m, 2H, NH), 6.46–7.71 (m,8H, Aryl- H), 7.5–8.2(m,3H,2-benzothiazole), FABMS (m/z 100%) 466; anal. Found C(56.59%) H(3.25%), N(12.00%) for calculated C22H15ClN4O2S2 C(56.59%) H(3.24%) N(12.00%).
BT17: 6-chloro-N-[4-(3-chlorophenyl)-3-(4-nitrophenyl)-2,3-dihydro-1,3-thiazol-2-yl]-1,3-benzothiazol-2-amine: Rf: 0.66 (ACN: Methanol 1:1), MP 300–310°C IR, 3122.44 (Ar C-H), 1621.12(C=N), 1566.38, C-Cl 693, (C-C),1340.18 (CN),3540.31(N-H), 1HNMR (CDCl3), 4.10 (m, 2H, NH), 6.46–6.99 (m,8H, Ar-H), 7.3–7.9 (m,3H,2-benzothiazole), FABMS (m/z 100%) 501; anal. Found C(52.70%) H(2.81%), N(11.17%) For Calculated C22H14Cl2N4O2S2 C(52.70%) H(2.81%) N(11.17%).
BT18: 3-{2-[(6-chloro-1,3-benzothiazol-2-yl)amino]-3-(4-nitrophenyl)-2,3-dihydro-1,3-thiazol-4-yl}phenol: Rf: 0.6 (ACN: Methanol 1:1), MP 340–342°C IR, 3120.44 (Ar C-H), 1611.88 (C=N),1512.38 (C-C),1333.18 (CN),3533.31(N-H),1500–1600 (Ar-NO2), 1HNMR (CDCl3), 4.11 (s, 1H,NH), 6.4–7.11 (m,7H, Ar-H), 6.9–8.33(m,3H,2-benzothiazole),FABMS (m/z 100%) 482.90; anal. Found C(54.70%) H(3.13%), N(11.60%) For C22H15ClN4O3S2 Calculated C(54.71%) H(3.13%) N(11.60%).
Anticancer activity
The synthesized compounds were evaluated for their anticancer activity at the National Institute of Cancer (NCI).
Toxicity assays
The toxicity assay was performed by the standard protocol of the manufacturer. Toxi-light assay method was used to measure the toxicity of the compounds.
Results
Analysis of docking
The designed compounds were docked at the assigned site of the protein. The results of docking and the binding energy of every compound with the receptor are listed in Table 1. The ligands with good binding scores showed good interactions with the receptor. The ATP binding pocket is hydrophobic by nature, and the hydrophobic interaction play a vital role in affinity towards EGFR-TK binding. At least 2 hydrogen bonds of ligand and receptor are necessary for small molecules to act as EGFR inhibitors (Figures 4–9).
Figure 4.
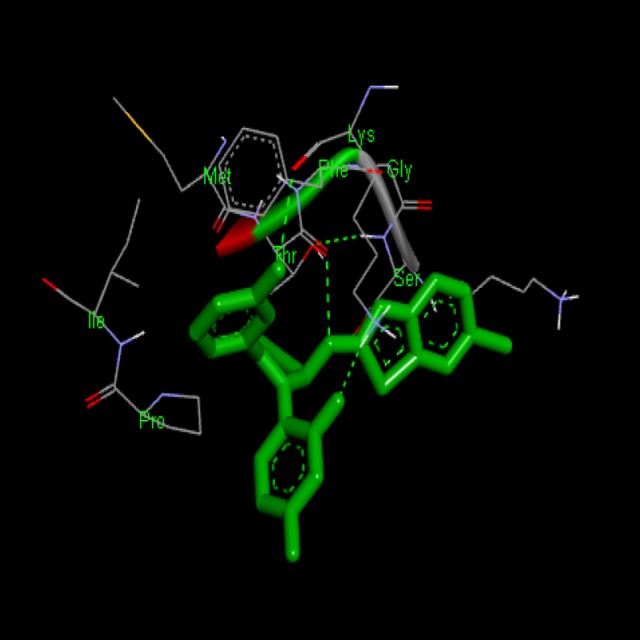
Docking interaction of Compound BT-1 with 1XKK. Dashed line represents H bonds.
Figure 5.
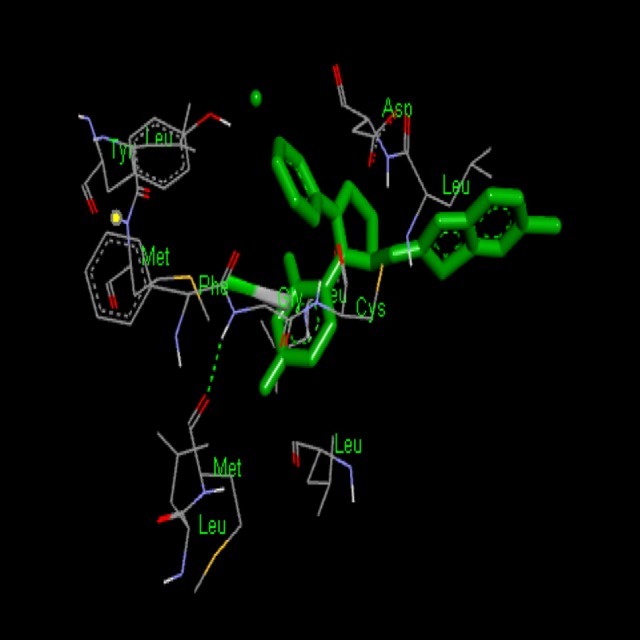
Docking interaction of compound BT-4 with 1XKK. Dashed line represents H bonds.
Figure 6.
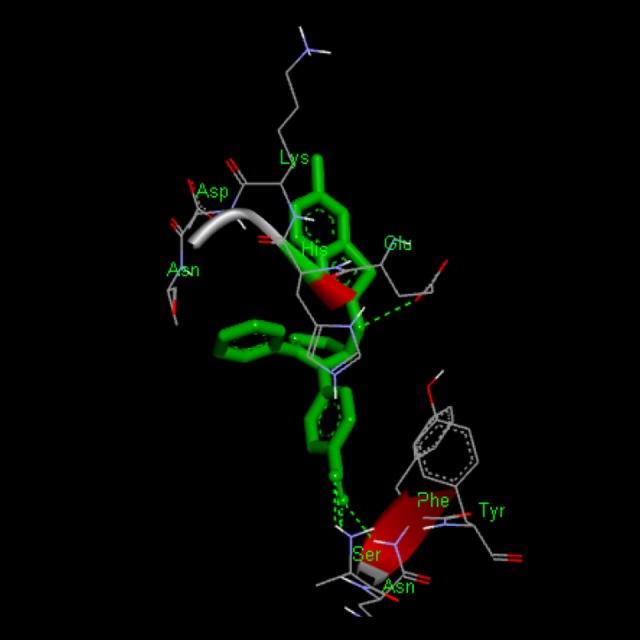
Docking interaction of compound BT-16 with 1XKK. Dashed line represents H bonds.
Figure 7.
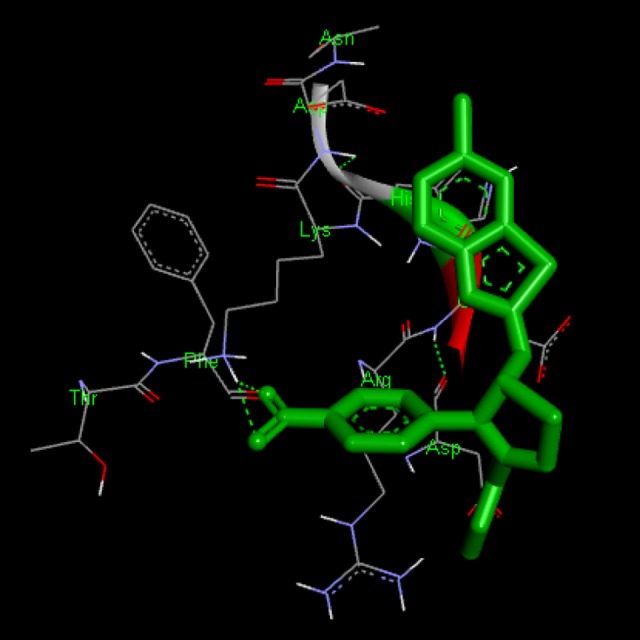
Docking interaction of compound BT-17 with 1XKK. Dashed line represents H bonds.
Figure 8.
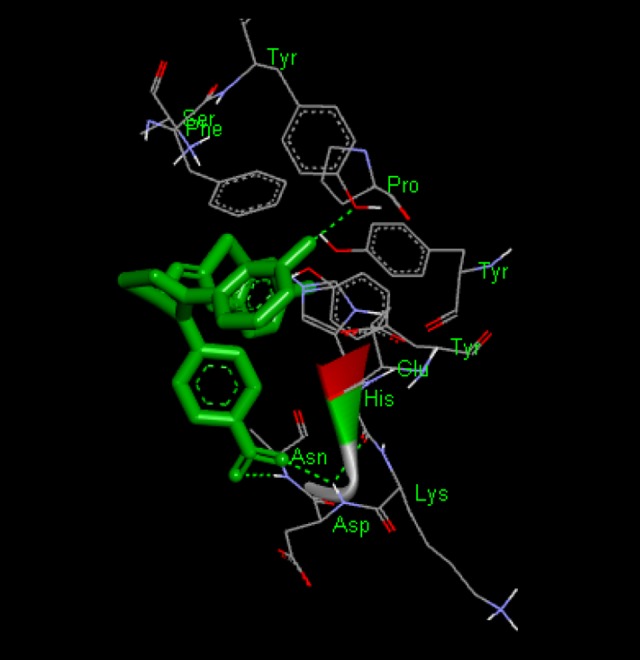
Docking interaction of compound BT-18 with 1XKK. Dashed line represents H bonds.
Figure 9.
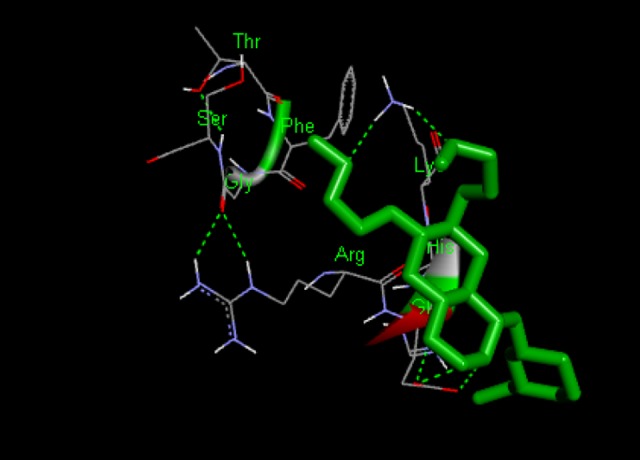
Docking interaction of Lepatinib with 1XKK. D dashed line represents H bonds.
Synthetic studies
The moieties were synthesized according to the above-mentioned procedure. The synthesized compounds were characterized by using FTIR, 1HMNR, Mass, and C, H, N, Analysis. Because the electron density is the crucial factor in the reactivity, the substitutions were performed at N2 as it is more electron-rich than N1 [31–33].
Antiproliferative activity
The anticancer evaluation of the synthesized compounds was performed at the Developmental Therapeutic Program (www.dtp.nci.nih.gov) of the US National Cancer Institute (NCI), using the standard protocol for anticancer evaluation. The compounds were evaluated for their action against non-small cell lung cancer and colon cancer (HCT116, HCT-15, and HT29) cell lines at NCI using standard protocol and the procedure is contained in their website. The compounds were firstly assessed for 1-dose testing. If they were found to be active at 1-dose testing, they were further evaluated for 5-dose testing. Inhibition of growth 50% (GI50) values were calculated for each cell line (Table 2).
Table 2.
Results for anticancer evaluation at concentration 10−5 M.
| Compound No. | Subpanel tumor cell lines colon cancer | Active (selected for 5-dose 60 cell lines assay) | ||
|---|---|---|---|---|
| Assay of cell lines in 1-dose 10−5 M | ||||
| HCT-116 | HCT-15 | HT29 | ||
| BT1 | 12.36 | 10.3 | 2.9 | Active |
| BT4 | 11.41 | 9.43 | 3.0 | Active |
| BT8 | 2.46 | 11.32 | 4.5 | Active |
| BT16 | 11.46 | 0.43 | 0.23 | Inactive |
| BT17 | 15.67 | 4.23 | 6.7 | Active |
| BT18 | 7.12 | 11.10 | 14.47 | Active |
| Lapatinib | 22.31 | 27.23 | 36.12 | – |
Discussion
The compounds (BT1, BT4, BT8, BT17, and BT18) were found to have substantial activity in 1-dose testing and were further evaluated for 5-dose testing using the SRB (sulforhodamine B) protein assay. The compound B16 showed moderate activity, so it was not selected for further testing. The dose-dependent parameters for 5-dose testing were calculated for 5-dose antiproliferative activity, and dose-response parameters (GI50,(TGI) and LC50) were evaluated for synthesized compounds against all cell lines. Furthermore, (MG_MID) was deliberated for every parameter, which gives an average activity parameter over all cell lines for tested compounds. The results of the present study indicate that the synthesized compounds exhibit good antiproliferative activity against colon cancer cell lines (HCT116, HCT15, and HT29) compared with the reference. The synthesized compounds showed notable selectivity against the tested cancer cell lines. The tested compounds (BT1, BT4, BT8, BT17, and BT18) showed good antiproliferative activity against the tested cancer cell lines with selectivity pattern. They showed better activity against selected colon cancer cell lines with log GI50 ranging from −7.20 to −4.80. (Table 2). The compounds showed a good selectivity pattern, so their activity against cancer cell lines was confirmed (Tables 3, 4).
Table 3.
Dose-dependent assay for anticancer evaluation.
| Compound No. | Na | LogGI50 | Log TGI | Log LC50 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N1b | Range | MG_MID | N2b | Range | MG_MID | N3b | Range | MG_MID | ||
| BT1 | 3 | 3 | −4.64 to −6.49 | −5.07 | 56 | −4.0 to −4.83 | −4.4 | 19 | −4.0 to −4.36 | −4.06 |
| BT4 | 3 | 3 | −4.64 to −6.52 | −5.33 | 37 | −4.0 to 5.08 | −4.36 | 09 | −4.0 to −4.28 | −4.02 |
| BT8 | 3 | 3 | −4.0 to −4.84 | −4.45 | 08 | −4.0 to −4.24 | −4.02 | 00 | −4.0 to −4.02 | −4.0 |
| BT17 | 3 | 3 | −4.42 to −5.02 | −4.71 | 25 | −4.0 to −4.32 | −4.07 | 03 | −4.0 to −4.08 | −4.0 |
| BT18 | 3 | 3 | −3.92 to −6.93 | −4.90 | 49 | −3.92 to −6.01 | −4.07 | 49 | −3.92 to −4.75 | −4.29 |
N – no. of human cancer cell lines tested at the IInd stage assay;
N1, N2, N3 – no. of sensitive cell lines, against which the compound possessed considerable growth inhibition as per the standard parameter (logGI50, logTGI, and log LC50 ≥4.00) (GI50 (50% of net cell growth inhibition at molar concentration of the compound.) TGI (Total inhibition at molar concentration of the compound) and LC50 (50% of net cell death at molar concentration of the compound).
Table 4.
The sensitivity pattern of synthesized compounds.
| Compd. No. | Cancer type | Most sensitive cell line | Log GI50 | LOG TGI | Log LC50 |
|---|---|---|---|---|---|
| BT1 | Colon cancer | HCT-116 | −5.31 | −4.64 | >−4.00 |
| HC-29 | −5.19 | −4.49 | >−4.00 | ||
| BT-4 | Colon cancer | HCT116 | −5.59 | −4.70 | >−4.00 |
| HCT15 | −5.71 | −5.28 | −4.27 | ||
| HC29 | −6.52 | −4.42 | >−4.00 | ||
| BT-8 | Colon cancer | HCT116 | −6.01 | >−4.00 | >−4.00 |
| HCT15 | −5.59 | −4.70 | >−4.00 | ||
| HC29 | −5.71 | −5.28 | −4.27 | ||
| BT-16 | Colon cancer | HCT116 | −5.59 | −4.70 | >−4.00 |
| HCT15 | −5.71 | −5.28 | −4.27 | ||
| HC29 | −5.58 | −5.07 | −4.06 | ||
| BT-17 | Colon cancer | HCT116 | −5.59 | −4.70 | >−4.00 |
| HCT15 | −5.71 | −5.28 | −4.27 | ||
| HC29 | −5.58 | −5.07 | −4.06 |
For SAR studies, common substituent pattern was followed. The different substituent in compounds BT1 to BT18 reveals that substitution on positions 3 and 4 of the thiazole ring plays an important role in the biological activity. Furthermore, the presence of electron withdrawing groups BT1, BT4, BT8, BT17, and BT18 showed better antiproliferative activity. It was previously reported that electron-withdrawing/donating groups mostly affect the biological activity because it significantly affects the lipophilicity of the compound, which is a key factor in crossing the barriers of the cell membrane [32–33].
On the basis of these results, the SAR study revealed that:
For better anticancer activity, position 3 of thiazole may be substituted by the electron-withdrawing group for good activity, which might impart its lipophilicity, strengthening their hydrophobic interactions with the receptor.
Hydroxyl group introduction may increase the activity, as it may be involved in hydrogen-binding with the receptor.
Position 2 of thiazole does not affect the activity.
Conclusions
The synthesized compounds exhibited good anti-tumor activity against different colon cancer cell lines. The compound BT17 shows potent activity against all the colon cell lines with the GIC50 values in the molecular range. It can be concluded from the present study that hydrophobic group substitution is the primary structural requirement for fitting into the active pocket of the receptor, with 2 hydrogen bond acceptors and 1 hydrogen bond donor. The results of the present study can be used for the discovery of some new antiproliferative agents. Moreover, these compounds may be further tested for their activity against other cancer types.
Footnotes
Source of support: Departmental sources
References
- 1.Boyle P, Langman JS. ABC of colorect al cancer: Epidemiology. BMJ. 2000;321:805–8. doi: 10.1136/bmj.321.7264.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Midgley R, Kerr D. Colorectal cancer. Lancet. 1999;353:391–99. doi: 10.1016/S0140-6736(98)07127-X. [DOI] [PubMed] [Google Scholar]
- 3.Rougier P, Mitry E. Epidemiology, treatment and chemoprevention in colorectal cancer. Ann Oncol. 2003;14(Suppl 2):ii3–5. doi: 10.1093/annonc/mdg722. [DOI] [PubMed] [Google Scholar]
- 4.Prewett MC, Hooper AT, Bassi R, et al. Enhanced antitumor activity of anti-epi dermal growth factor receptor monoclonal antibody IMC-C225 in combination with irinotecan (CPT-11) against human colorectal tumor xenografts. Clin Cancer Res. 2002;8:994–1003. [PubMed] [Google Scholar]
- 5.Ballard P, Bradbury RH, Harris CS, et al. Inhibitors of epidermal growth factor receptor tyrosine kinase: Optimization of potency and in vivo pharmacokinetics. Bioorg Med Chem Lett. 2006;16:4908–12. doi: 10.1016/j.bmcl.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 6.Cheng Y, Cui W, Chen Q, et al. The molecular mechanism studies of chirality effect of PHA-739358 on Aurora kinase A by molecular dynamics simulation and free energy calculations. J Comput Aided Mol Des. 2011;25:171–80. doi: 10.1007/s10822-010-9408-7. [DOI] [PubMed] [Google Scholar]
- 7.Cherry M, Williams DH. Recent kinase inhibitor Xray structures: Mechanisms of inhibition and selectivity insights. Curr Med Chem. 2004;11:663–73. doi: 10.2174/0929867043455792. [DOI] [PubMed] [Google Scholar]
- 8.Cohen P. Protein kinases: The major drug targets of the twenty-first century? Nat Rev Drug Discov. 2008;1:309–15. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 9.Cressier D, Prouillac C, Hernandez P, et al. Synthesis, antioxidant properties and radioprotective effects of new benzothiazoles and thiadiazoles. Bioorg Med Chem. 2009;17:5275–84. doi: 10.1016/j.bmc.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 10.El-Sherbeny MA. Synthesis of certain pyrimido[2,1-b]benzothiazole and Benzothiazolo [2,3-b]quinazoline derivatives for in vitro antitumor and antiviral activities. Arzneim Forsch. 2000;50:848–53. doi: 10.1055/s-0031-1300300. [DOI] [PubMed] [Google Scholar]
- 11.Elzahabi HAS. Synthesis, characterization of some benzazoles bearing pyridine moiety: Search for novel anticancer agents. Eur J Med Chem. 2011;46:4025–36. doi: 10.1016/j.ejmech.2011.05.075. [DOI] [PubMed] [Google Scholar]
- 12.Fabbro D, Ruetz S, Buchdunger E, et al. Protein kinases as targets for anticancer agents: From inhibitors to useful drugs. Pharmacol Ther. 93:79–98. doi: 10.1016/s0163-7258(02)00179-1. 200. [DOI] [PubMed] [Google Scholar]
- 13.Gabr MT, El-Gohary NS, El-Bendary ER, El-Kerdawy MM. Synthesis and in vitro antitumor activity of new series of benzothiazole and pyrimido[2,1-b]benzothiazole derivatives. Eur J Med Chem. 2014;85:576–92. doi: 10.1016/j.ejmech.2014.07.097. [DOI] [PubMed] [Google Scholar]
- 14.Grever MR, Schepartz SA, Chabner BA. The National Cancer Institute: Cancer drug discovery and development program. Semin Oncol. 1992;19:622–38. [PubMed] [Google Scholar]
- 15.Prasad PR, Bhuvaneswari K, Kumar KP, et al. Synthesis and biological activity evaluation of some fused pyrimido-benzothiazole derivatives. J Chem Pharm Res. 2002;4:1606–11. [Google Scholar]
- 16.Hu WP, Chen YK, Liao CC, et al. Synthesis and Biological evaluation of 2-(4-aminophenyl)benzothiazole derivatives as photosensitizing agents. Bioorg Med Chem. 2010;46:6197–207. doi: 10.1016/j.bmc.2010.04.082. [DOI] [PubMed] [Google Scholar]
- 17.Grünwald V, Hidalgo M. Developing inhibitors of the epidermal growth factor receptor for cancer treatment. J Natl Cancer Inst. 2003;95:851–67. doi: 10.1093/jnci/95.12.851. [DOI] [PubMed] [Google Scholar]
- 18.Kinnings SL, Jackson RM. ReverseScreen 3D: A structure-based ligand matching method to identify protein targets. J Chem Inf Model. 2011;51:624–34. doi: 10.1021/ci1003174. [DOI] [PubMed] [Google Scholar]
- 19.Kristi GB, Thomas S, Herald S. Defective down regulation of receptor tyrosine kinases in cancer. Eur Mol Biol Org. 2010;23:2707–12. [Google Scholar]
- 20.Chikhale R, Menghani S, Babu R, et al. Development of selective DprE1 inhibitors: Design, synthesis, crystal structure and antitubercular activity of benzothiazolylpyrimidine-5-carboxamides. Eur Jr Med Chem. 2015;96:30–46. doi: 10.1016/j.ejmech.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 21.Gabr MT, Gohary NH, Bendary ER. New series of benzothiazole and pyrimido[2,1-b]benzothiazole derivatives: Synthesis, antitumor activity, EGFR tyrosine kinase inhibitory activity and molecular modeling studies. Med Chem Res. 2015;24:860–76. [Google Scholar]
- 22.Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev. 2007;17:60–65. doi: 10.1016/j.gde.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 23.Mason JS, Good AC, Martin EJ. 3-D pharmacophores in drug discovery. Curr Pharm Design. 2001;7:567–97. doi: 10.2174/1381612013397843. [DOI] [PubMed] [Google Scholar]
- 24.Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst. 1991;83:757–66. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 25.Saeed S, Rashid N, Jones PG, et al. Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole moiety as potential antimicrobial and anticancer agents. Eur J Med Chem. 2010;45:1323–31. doi: 10.1016/j.ejmech.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 26.Sahu PK, Sahu PK, Gupta SK, et al. Synthesis and evaluation of antimicrobial activity of 4H-pyrimido[2,1-b]benzothiazole,pyrazole and benzylidene derivatives of curcumin. Eur J Med Chem. 2012;54:366–78. doi: 10.1016/j.ejmech.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 27.Sharma FA, Sharma R, Tyagi T. Receptor tyrosine kinase inhibitors as potent weapons in war against cancers. Curr Pharm Design. 2009;15:758–76. doi: 10.2174/138161209787582219. [DOI] [PubMed] [Google Scholar]
- 28.Shendarkar GR, Labhsetwar LB, Butle SR, et al. Synthesis and antimicrobial evaluation of some fused Iminopyrimidobenzothiazole derivatives. Int J Res Pharm Biomed Sci. 2011;2:1350–56. [Google Scholar]
- 29.http://accelrys.com/products/collaborative-science/biovia-discovery-studio/
- 30.www.pdb.com/www.rcsb.org/pdb/explore.do?structureId=1XKK
- 31.Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “Gatekeeper Door”: Exploiting the active kinase conformation. J Med Chem. 2010;53:2681–94. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- 32.Bonde CG, Gaikwad NJ. Synthesis and preliminary evaluation of some pyrazine containing thiazolines and thiazolidinones as antimicrobial agents. Bioorg Med Chem. 2006;12(9):2151–61. doi: 10.1016/j.bmc.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 33.Ma H, Deacon S, Horiuchi K. The challenge of selecting protein kinase assays for lead discovery optimization. Expert Opin Drug Discov. 2008;3:607–21. doi: 10.1517/17460441.3.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]