

SUMMARY
Tissue-specific gene expression is often thought to arise from spatially restricted transcriptional cascades. However, it is unclear how expression is established at the top of these cascades in the absence of pre-existing specificity. We generated a transcriptional network to explore how transcription factor expression is established in the Arabidopsis thaliana root ground tissue. Regulators of the SHORTROOT-SCARECROW transcriptional cascade were validated in planta. At the top of this cascade, we identified both activatorsand repressors ofSHORTROOT. The aggregate spatial expression of these regulators is not sufficient to predict transcriptional specificity. Instead, modeling, transcriptional reporters, and synthetic promoters support a mechanism whereby expression at the top of the SHORTROOT-SCARECROW cascade is established through opposing activities of activators and repressors.
In Brief
Sparks et al. investigate how tissue-specific expression is established at the top of transcriptional cascades. The authors generated a root-enriched eY1H network to dissect regulation at the top of the SHORTROOT-SCARECROW transcriptional cascade. Their results suggest that SHORTROOT expression is established through the opposing activities of multiple activators and repressors.
INTRODUCTION
A hallmark of multicellular organisms is tissue-specific gene expression. This expression is often described as arising from spatially restricted transcriptional cascades (e.g., Jäckle and Sauer, 1993). How these cascades are initiated is rarely addressed.
In plants, specificity of gene expression is mediated by transcriptional regulators binding to DNA flanking a gene’s coding region. Of the multitude of regulators, the differential expression and binding of transcription factors (TFs) is considered to be most central to achieve transcriptional specificity (Levine, 2010). TFs bind a specific, often degenerate, DNA sequence (motif) that can be found millions of times in the genome. However, not all of these motif instances are bound and it remains unclear how motif instances are distinguished by TFs (White et al., 2013). Furthermore, for motifs that are bound, not all of these associations are functional (MacNeil et al., 2015). One model suggests that TF-DNA interactions are not necessarily “optimized solutions,” but are instead the result of the “path of least resistance” (Sorrells and Johnson, 2015). In other words, if an interaction has no deleterious effect, it may occur without having a regulatory consequence. This suggests that neither the expression of a TF within a tissue nor the presence of a TF binding motif within the regulatory DNA is sufficient to predict in vivo TF-DNA regulation.
Enhanced yeast-one-hybrid (eY1H) assays are an experimental alternative to predictive models, and have been utilized to identify TF-DNA interactions in multiple systems (Gaudinier et al., 2011; Reece-Hoyes et al., 2011a, 2011b). A significant overlap in eY1H interactions and TF-centric chromatin immunoprecipitation (ChIP) peaks has been shown (Fuxman Bass et al., 2015; Reece-Hoyes et al., 2013). In plants, results from eY1H assays have been used to generate large-scale interaction networks that uncover biologically relevant gene-regulatory interactions (Li et al., 2014; Taylor-Teeples et al., 2015; De Lucas et al., 2016). A key finding from network approaches has been that the range of tissues in which TFs are expressed tends to be broader than that of the target genes they regulate (Brady et al., 2011). This suggests that cell-type-specific TF cascades may not be the only mechanism to control spatiotemporal gene expression.
Arabidopsis roots have been used extensively to gain a systems-level understanding of growth, differentiation, and morphogenesis due to their developmental simplicity and the vast genetic and genomic resources available (Benfey and Scheres, 2012). Two TFs, SHORTROOT (SHR) and SCARECROW (SCR), are required to determine the cell fates of the cortex and endodermis, which together form the ground tissue. The expression of both TFs is spatially restricted, with SHR transcribed in the immature vasculature (Helariutta et al., 2000) and SCR transcribed in the endodermis (Di Laurenzio et al., 1996). SHR protein moves from the vasculature into the cortex-endodermal initial (CEI) and CEI daughter. There, it interacts with SCR to upregulate SCR expression in a positive feedback loop and promote asymmetric cell division (Heidstra et al., 2004; Helariutta et al., 2000; Nakajima et al., 2001). Although strong SCR expression is established through the spatially restricted expression of SHR, low levels of SCR are present in the ground tissue of shr mutants (Helariutta et al., 2000), suggesting that additional TFs are required to regulate SCR expression. In contrast, forward genetic screens have failed to identify upstream regulators of SHR expression (unpublished data). This suggests that multiple TFs may be involved in establishing tissue-specific expression at the top of the SHR-SCR transcriptional cascade.
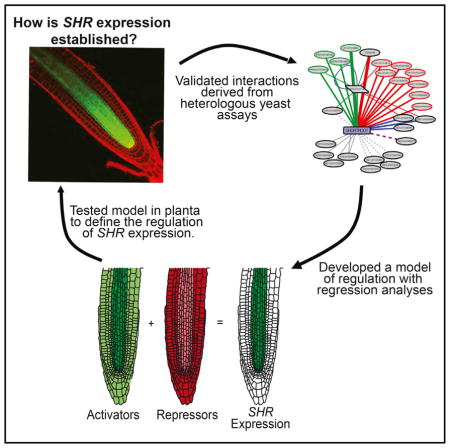
To dissect the upstream regulation of ground-tissue-specific TFs, we generated a transcriptional network with eY1H assays. For network analysis, we developed a clustering approach based on node similarity, which suggested common regulators of SHR and SCR. The SHR and SCR subnetworks were validated in planta, and TFs that regulate the expression of both SHR and SCR were confirmed. At the top of the transcriptional cascade, the SHR expression domain can be predicted by the expression of activators and repressors within a logistic regression. Based on these results, we developed a model in which SHR expression is established through opposing expression domains of activators and repressors. We validated this model in planta with SHR reporter analyses and the generation of synthetic promoters containing repressor motifs from the SHR promoter. These results highlight the complex mechanisms regulating TF expression at the top of transcriptional cascades.
RESULTS
Root Transcription Factors Appear to Function Broadly across Cell Types
We mined cell-type-specific microarray data to determine the number of TFs expressed in one or more cell types of the root (Brady et al., 2007; Sozzani et al., 2010). The root consists of approximately 15 cell types, including the cortex, endodermis, and vascular tissues. Expression profiles have been generated for each of these cell types, as well as specific developmental stages, resulting in a total of 23 transcriptome profiles. We defined a TF by its inclusion in the Plant Transcription Factor Database (PlnTFDB; Guo et al., 2008) or the Database of Arabidopsis Transcription Factors (DATF; Guo et al., 2005). Of the estimated 2,266 Arabidopsis TFs, 1,300 are expressed at a mean normalized expression value of >1 in at least one root cell type by microarray analysis, with the majority expressed in more than one cell type (Figure 1A and Table S1). As the threshold for defining expression is increased, the total number of TFs expressed in root cells is reduced (n = 396 for >5 and n = 153 for >10) and the distribution of TFs skews toward more tissue-specific expression (Figure 1A and Table S1). This suggests that while the majority of root TFs are expressed in many cell types at low levels, the more highly expressed factors tend to have high expression in one or a few cell types. We then asked whether the expression distribution for TFs is different than what might be expected from a random selection of genes (Figure 1A and Table S1). Analyzing five random subsets of 1,300 root-expressed genes, we found significant differences in the mean (Mann-Whitney U, p ≤ 0.01) and distribution (χ2, p ≤ 0.01) of these data. Upon closer inspection, the TFs were skewed toward more specific expression (e.g., in the >1 threshold dataset, 232 of the 1,300 TFs were expressed in one or two profiles versus 185 in the random selection). This suggests that while root TFs show a wide range of tissue specificities, their expression is more tissue specific than would be expected by chance.
Figure 1. Transcription Factors Are Expressed Broadly across Root Cell Types.

(A) Microarray profiles were mined to determine the specificity of TF expression (top panels). The majority of TFs expressed at a mean normalized expression value >1 are found in more than one transcriptome profile (white, gray, or black). When considering a higher threshold for expression, mean value >5 (gray or black) and mean value >10 (black), we see a shift toward more specific expression patterns. Performing the same analysis on random subsets of root-expressed genes yields a significantly different distribution. While the distribution appears similar, we see a shift toward more specific expression in the TF-only analysis.
(B) The distribution of TSS peak shapes across TF promoters is the same as genome wide. Together, these data indicate that TFs are expressed and likely function in multiple tissues.
See also Table S1.
Another potential indication of tissue-specific transcription is the distribution of transcription start site (TSS) locations for a given gene. Genome-wide mapping of TSS locations has identified specific patterns associated with different types of gene function and/or specificity in both plants and animals (Carninci et al., 2006; Morton et al., 2014; Rach et al., 2009). In Arabidopsis, narrow distributions of TSS are correlated with tissue-specific gene expression, although the mechanism underlying this correlation is unclear (Morton et al., 2014). The prevailing theory is that tissue-specific gene promoters are bound by TFs that more precisely regulate the site of transcription initiation to mediate tissue specificity. Analysis of root TF promoters identified a similar distribution of peak shapes as is present throughout the genome (Figure 1B and Table S1), suggesting that root TFs show a range of specific and broad expression. Together, these observations suggest that while some TFs may be enriched in specific cell types, many root TFs are expressed in multiple cell types.
eY1H-Based Networks Can Be Used for Hypothesis Generation
To gain insight into the regulation of spatially restricted TFs during ground tissue development, we generated a gene-regulatory network (GRN) using eY1H assays. We identified 148 ground tissue-enriched TFs based on prior publications or because they exhibited a 1.2-fold expression enrichment over other cell types (Figure 2A and Table S2). The DNA upstream of the start codon (3 kb upstream or to the nearest gene) was successfully cloned for 111 of these TFs and used as eY1H bait. For prey, we considered any TF expressed in the cortex, endodermis, or immature vasculature at a mean normalized value of ≥1, which includes the aforementioned ground tissue-enriched TFs, for a total of 1,079 TFs. We then developed a TF prey collection for 51% of these TFs containing 511 previously available TFs and 44 TFs cloned de novo. This prey collection was screened in a pairwise fashion against the DNA of the 111 TF-encoding gene promoters (Figure 2A and Table S2). One hundred TFs were present in both bait and prey collections. The resulting GRN consists of 871 interactions (edges) between 267 TFs (nodes) (Figure 2B and Table S2, http://root-transcriptional-network.herokuapp.com/index.html). Of the nodes, 178 are TFs only (Figure 2B, green), 59 are TF-encoding gene promoters only (Figure 2B, blue), and 30 are present as both TFs and TF-encoding gene promoters (Figure 2B, red). The latter come from the 100 TFs present in both the bait and prey collections.
Figure 2. eY1H Assays Define a Network of Ground Tissue Transcription Factor Interactions.

A ground tissue-enriched gene-regulatory network was generated by eY1H assays.
(A) Experimental design of eY1H assays. The 555 prey open reading frames and 111 bait promoters were transformed into yeast mating strains, diploid selected, and transferred onto reporter plates.
(B) Hierarchical representation of the 871 interactions (edges) between 267 TFs (nodes) in the ground tissue transcriptional network. The network can be explored online at http://root-transcriptional-network.herokuapp.com/index.html.
We looked for enrichment of TF binding sites in the bait promoters, which has been previously suggested to correlate with the confidence that an eY1H interaction represents a functional regulatory event in vivo (Reece-Hoyes et al., 2013). Recently, binding motifs for 745 Arabidopsis TFs were identified or inferred (Weirauch et al., 2014). Of the 208 TFs that are part of our GRN, motifs for 110 have been identified. These TFs participate in 448 interactions with 77 TF-encoding gene promoters (Table S2).
The binding motifs were primarily identified from direct and indirect analysis of protein binding microarray (PBM) data (Berger et al., 2006) (Table S2). To ensure the robustness of our analysis, we used the Seed-and-Wobble algorithm (Berger and Bulyk, 2009) to analyze raw PBM data for the motifs published by Weirauch et al. (2014) (Table S2). Using the Finding Individual Motif Occurrences (FIMO) software, we determined the log-likelihood ratio that a given TF motif is found within these 77 promoters (Table S2). With a p value cutoff of 10−3, only ten interactions were not predicted when a motif was present (Figure S1). However, the overlap in eY1H interactions and TF binding predictions is not enriched over random. We performed the same analysis with a more stringent p value threshold of 10−4, and still observed no enrichment of eY1H interactions over random.
Gene Expression Patterns Are Not Sufficient to Predict the Similarity of eY1H Connections
We clustered genes based on node similarity to see whether shared network connections could provide insight into biological function (Figure S2 and Table S3, http://root-transcriptional-network.herokuapp.com/3A_TF_Clusters.html). In a GRN, it is not surprising to find two nodes that share a common neighbor if this neighbor is highly connected. Therefore, we used an inverse log-weighted similarity measurement, which models the assumption that two nodes are more similar if they share common neighbors that are of low degree (number of connections). This clustering considers two types of connections, incoming (in-degree) and outgoing (out-degree). In-degree and out-degree clustering were combined to define 38 independent clusters (Figure S2).
To determine whether these clusters correlate with cell-type-specific expression patterns or response to stimuli, ground tissue GRN TFs were independently clustered by k means using root cell-type-specific microarray data (Brady et al., 2007; Sozzani et al., 2010) and the AtGenExpress development and stress-response expression atlas (Schmid et al., 2005; Goda et al., 2008; Kilian et al., 2007). Pearson correlation coefficients were calculated to compare cluster assignments. The clusters based on node similarity were not correlated with the cell-type-specific expression clusters (r = 0.01) and only mildly correlated with clusters from the AtGenExpress data (r = 0.39). We used support vector machine learning to ask if either of these datasets is sufficient to predict the node similarity-based cluster assignment. Using 10-fold cross-validation, we were able to predict 21.3% and 21.7% of the node similarity-based cluster assignments from cell-type-specific and AtGenExpress data, respectively. These results indicate that gene expression patterns are not sufficient to predict the similarity of eY1H connections.
SCARECROW and SHORTROOT Expression Is Regulated by Multiple TFs
Although SHR and SCR are known to function together, their mRNA expression patterns are distinct, with SHR expressed in vascular tissue and SCR expressed in the ground tissue and its stem cells. Therefore, we were surprised to find them clustered together in the node similarity-based clustering analysis (Figure S2). In total, 16 TFs bound the SCR promoter and 27 TFs bound the SHR promoter (Figure 3, http://root-transcriptional-network.herokuapp.com/3B_SCR_SHR_All.html). Five of these bound both, which resulted in co-clustering and suggests that SCR and SHR share common upstream regulators.
Figure 3. Unique and Shared TFs Bind the SHORTROOT and SCARECROW Promoters.

Visualization of the SHORTROOT and SCARECROW subnetwork reveals five upstream interactions in common. This suggests that common TFs regulate the expression of SHORTROOT and SCARECROW. See also Table S3 and Figure S2.
To test this hypothesis, we validated SCR and SHR subnetworks in planta using available overexpression, single, and/or higher-order mutants from our candidate upstream TFs (Table S4). We tested alleles for 11 of the 16 SCR interactions and 23 of the 27 SHR interactions through analysis of root growth and cellular patterning. Since mutation of either SCR or SHR results in shorter roots with radial patterning defects, we predicted that upstream regulators might show similar defects. However, mutation of the 29 different TFs (including the five that bind both) resulted in only low penetrance defects of radial patterning. We examined root growth over 10 days and observed that several of the mutants had a reduction in root growth; however, the degree of reduction was small and did not phenocopy the shr or scr mutant (Table S4). In addition, none of the mutants had a single ground tissue layer, which would again phenocopy the loss-of-function shr or scr mutant. Instead, in the mutant lines we observed that between 0% and 33% of the plants have precocious division of the endodermis (Table S4). These low penetrance effects could indicate subtle modulations of gene expression, consistent with the dose-dependent effect of SHR on ground tissue patterning (Koizumi et al., 2012). This is consistent with the hypothesis that multiple TFs contribute to establish and maintain gene expression such that alteration in a single gene is not sufficient to exhibit severe morphological changes. An alternative hypothesis is that, despite binding, the 29 TFs do not regulate SHR or SCR expression under the conditions tested.
To determine the effects of these putative upstream TFs at the molecular level, we used qRT-PCR analysis of target gene expression in whole roots of the aforementioned mutant and overexpression lines. Of the regulators tested, we observed molecular phenotypes for 18 of 23 (78%) of the SHR putative upstream regulators (Figures 4A and 4D; Table S4) and 5 of 11 (45%) of the SCR putative upstream regulators (Figures 4B and 4D; Table S4; http://root-transcriptional-network.herokuapp.com/4D_SCR_SHR_Validated.html). These results indicate that multiple TFs are required to establish and maintain gene expression in these subnetworks.
Figure 4. Subnetwork Validation In Planta Reveals Complex and Combinatorial Control of SHORTROOT and SCARECROW Expression.

(A and B) Subnetworks were validated by obtaining mutants or overexpression lines of upstream TFs and assaying whole-root RNA for changes in (A) SHR or (B) SCR expression. Colored bars indicate assays achieving statistical significance (t test, p < 0.05) along with the inferred relationship (red, repressor; green, activator). Expression values were normalized by primer efficiency and ecotype control, represented by the horizontal bar at 1. Error bars represent the standard deviation between biological replicates. Horizontal lines under the graph indicate multiple alleles of the same gene. Capitalized gene names followed by OX indicate an overexpression allele, whereas lowercase gene names indicate a mutant allele.
(C) Additional validation was achieved by transient inducible overexpression of upstream TFs in root protoplasts. Protoplasts with TFs fused to the glucocorticoid receptor (35S:TF-GR) were subjected to a 4-hr induction with dexamethasone prior to analysis. Five TFs that bound SHR (triangles) or SCR (circles) were analyzed. Three activate (left panel) and two repress (right panel) the expression of their target.
(D) Network summary of validated interactions from in planta assays. Green lines indicate activating TFs and red lines indicate repressing TFs. Blue lines indicate non-linear relationships. Purple dotted lines indicate allele-dependent results. Solid gray lines represent untested relationships, and dotted gray lines indicate relationships that were tested but not validated. Thick edges demonstrate validation by both assays. Thin edges are validation for the regulators upstream of CRF8.
See also Table S4.
For the SCR subnetwork, we validated three activators (Figures 4B and 4D, green) and one repressor (Figures 4B and 4D, red). dewax, storekeeper01 (stk01), and homeobox34 (hb34) mutants have decreased SCR expression in whole roots (Figures 4B and 4D, green) indicating that they function as activators. The activity of a repressor, PHYTOCHROME INTERACTING FACTOR 3 (PIF3), was inferred by an increase in SCR expression in the quadruple PIF mutant (pif1;pif3;pif4;pif5 or pifq) (Figures 4B and 4D, red). The results from DEHYDRATION RESPONSIVE ELEMENT BINDING 2A (DREB2A) were inconsistent between alleles (Figures 4B and 4D).
For the SHR subnetwork, five activators were identified: CYTOKININ RESPONSE FACTOR 8 (CRF8), DREB2A, STK01, DF1, and ABSCISIC ACID RESPONSIVE ELEMENTS-BINDING FACTOR 2 (AREB2) (Figures 4A and 4D, green). Ten repressors of SHR were also identified: DEWAX, AT2G44730, BZIP17, ETHYLENE RESPONSE FACTOR 15 (ERF15), ERF6, DWARF AND DELAYED FLOWERING 1 (DDF1), HEAT SHOCK FACTOR C1 (HSFC1), ZIM-LIKE 1 (ZML1), GBF1, and GROWTH REGULATING FACTOR 1 (GRF1) (Figures 4A and 4D, red). Lastly, we identified two TFs with a non-linear relationship to SHR expression (Figures 4A and 4D): HB13 and ETHYLENE RESPONSE DNA BINDING FACTOR 3 (EDF3). The non-linear relationship is defined by SHR expression being induced in both a mutant and an overexpression allele (Figures 4A and 4D). These results suggest that HB13 and EDF3 regulate SHR expression but that the regulation is dose dependent, possibly due to cooperativity or stoichiometric constraints. The results for JUMONJI DOMAIN-CONTAINING PROTEIN 18 (JMJ18) were inconsistent between alleles (Figures 4A and 4D).
To confirm the robustness of our in planta validation strategy, we used a transient induction system in root protoplasts to assay the effect of TF overexpression on SCR or SHR expression (Bargmann et al., 2013). In this assay, a plasmid containing the 35S promoter driving a TF fused to the glucocorticoid receptor (35S:TF-GR) and 35S:RFP is transfected into root protoplasts. Protoplasts are incubated overnight before a 4-hr induction with dexamethasone, and fluorescence-activated cell sorting for RFP-positive transfected cells. The RNA from transfected protoplasts is analyzed by qRT-PCR for changes in target gene expression compared with a mock induction. As a positive control for the assay, we expressed SCR-GR and assayed changes in the MYB36 target (Liberman et al., 2015). In this control, MYB36 was induced to varying degrees in two of three replicates (Table S4), suggesting that this assay can be used as a Boolean measure of regulation, but cannot determine the strength or extent of regulation. To validate our in planta results we induced three SHR regulators, DF1 (activator), AT2G44730 (repressor), and bZIP17 (repressor), and assayed the effect on SHR expression. Both DF1 and AT2G44730 showed the same relationship in protoplasts as previously determined (Figures 4C and 4D). In contrast, bZIP17 acts as an activator of SHR expression at high levels, suggesting a non-linear relationship between this TF and its target (Figures 4C and 4D). We also used this approach to validate two previously untested regulators of SCR expression, B-BOX 21 (BBX21) and bZIP30. Upon overexpression, BBX21 functions as an activator and bZIP30 functions as a repressor of SCR (Figures 4C and 4D). This increased our validation rate to 7 of 13 (54%) for the SCR putative upstream regulators.
Together, these data suggest that multiple TFs contribute to establish and maintain gene expression. Of the five TFs that bind both SCR and SHR, three were validated in both subnetworks (Figure 4D). These results indicate complex, coordinated regulation of SCR and SHR expression.
SHORTROOT Expression Is Established through Opposing Expression of Activators and Repressors
The validated SHR subnetwork suggests that expression is established through the combination of multiple regulators. To understand how these regulators might function, we looked at the spatial expression patterns of the upstream regulators obtained from cell-type-specific microarray data (Brady et al., 2007; Sozzani et al., 2010). We found that in every cell type profiled, one activator (DREB2A) is expressed higher than any other regulator (Figure S3A). From our validation, a constitutively active, overexpression DREB2A allele altered the expression of SHR, but the two mutant alleles had no effect (Figure 4A). Therefore, it remains possible that the regulation of SHR by DREB2A occurs only under this extreme circumstance (i.e., constitutively active over-expression). If we remove the expression of DREB2A from the analysis, a repressor becomes the most highly expressed TF in all of the tissues except the immature xylem (S4, where SHR expression is the highest) and CYCD6 (capturing expression in the CEI; Figure S3B). These results suggest that combinatorial regulation among TFs is required to establish specificity.
To overcome our limited knowledge of combinatorial regulation, we used multiple predictor regression modeling. Specifically, we used a logistic regression to determine whether the spatial expression of the upstream regulators is sufficient to explain SHR expression. Modeling was based on cell-type-specific mRNA expression patterns of the TFs as predictors, with SHR expression as the outcome (Table S5). Consistent with our previous results, no single TF was able to generate the SHR expression pattern (Figure 5A and Table S5), suggesting that multiple TFs are required to establish SHR expression. Using the expression of all activators and repressors as predictors in the model was sufficient to predict the spatial pattern of SHR expression (Figure 5A and Table S5). We next asked whether the expression of any subset of these genes could generate the expression domain of SHR. Using activators only, we were able to generate the SHR expression pattern. This is unsurprising, and our interpretation is that these activators are most highly expressed in the SHR domain. Given that the cumulative expression of repressors was high across cell types, we hypothesized that repressors would be broadly expressed and thus unable to generate the SHR expression pattern. Instead, we were surprised to find that a set of only repressors was also able to generate the SHR expression pattern. While, theoretically, either set of TFs is able to generate the SHR expression pattern, our in planta validation indicates that both types of interactions are required. Since the logistic regression does not take into account our predetermined directionality (i.e., activation or repression), our interpretation of this result is that activators and repressors are likely expressed in opposing domains, either of which would model the SHR domain (Figure 5B). The opposing activities of the repressors and activators are likely to provide a measure of robustness to establish stable expression of SHR.
Figure 5. SHORTROOT Expression Is Established through Opposing Expression of Activators and Repressors.

We used logistic regression modeling to determine whether the spatial expression of upstream regulators can recapitulate SHR expression.
(A) The 23 microarray profiles were ordered based on the level of SHR expression (top). The two tissues considered to have SHR expressed in this model are S4 (immature xylem) and xylem_2501. No single TF was able to recover the pattern of SHR expression, suggesting that multiple TFs are required to establish SHR expression (middle). The expression of all regulators, only activators, or only repressors is, in each case, sufficient to recover the pattern of SHR expression (bottom).
(B) A model in which SHR expression is established and maintained through opposing expression of activators and repressors.
(C) This model predicts that activators function within the central vasculature and repressors function in all other tissues. Three SHR repressors (red) and two activators (green) were crossed to a transcriptional reporter (pSHR::GFP, blue) and fluorescence intensity was measured in the vasculature proximal to the QC. The three repressor mutants do not show a difference in reporter expression, whereas the activator mutants show a decrease (t test, p ≤ 0.05). Error bars represent the standard deviation between biological replicates. These results are consistent with a model in which repressors function in tissues other than the vasculature.
The model predicts that activators function collectively within the immature vasculature and repressors function outside this domain. Therefore, activator mutants should show a reduction of SHR expression within the immature vasculature and repressor mutants should not. To test this hypothesis, we crossed two activator mutants (crf8 and stk01) and three repressor mutants (at2g44730, bzip17, and erf6) into an SHR transcriptional reporter (pSHR::erGFP). We measured the fluorescence intensity of pSHR::erGFP from the sum projection of a z stack through vasculature cells adjacent to the quiescent center (QC) and found that, consistent with our model, pSHR::erGFP is significantly reduced in activator mutants and unchanged in the repressor mutants (Figure 5C).
Furthermore, our model suggests that repressors function outside of the central vasculature. Single or double mutants for repressors show minor and inconsistent expansion of the pSHR::erGFP reporter, likely due to redundancy and buffering by other repressors. As an alternative, we tested the functionality of repressor motifs found in the SHR promoter. Based on our FIMO binding analysis, two repressors, DDF1 and ZML1, are predicted to bind within the first 500 bp of the translational start of the SHR gene (Figure 6A and Table S2). To test the functionality of these sequences as repressors, we generated synthetic promoters with these motifs inserted into a ubiquitously expressed promoter. Specifically, we took the sequence of the highest-scoring motif for each repressor and inserted this sequence upstream of the TSS of the promoter of RCH1, which is ubiquitously expressed in root tips (Galinha et al., 2007; Figure 6B). We inserted three or six copies of the ZML1 motif or DDF1 motif, with a random 10-bp spacer between motifs. Plants from pRCH1-ZML1::GAL4-UAS::ER-GFP show stochastic reduction of GFP expression in external cell files, with no apparent differences between three or six copies of the motif (Figure 6C, arrowheads). Interestingly, although we observed repression in single cells, we did not observe consistent repression within one or more cell types. Plants from pRCH1-DDF1::GAL4-UAS::ER-GFP with three copies of the DDF1 motif show single-cell repression of GFP expression, but six copies drive ectopic expression in the QC (Figure 6C, arrows). One explanation could be that DDF1 is expressed more highly in the QC. However, in the cell-type-specific transcriptomic data, DDF1 has its highest expression in the phloem pole pericycle (S17, Table S5). Alternatively, the increased number of binding sites could promote combinatorial binding with other TFs, resulting in an expanded expression domain. These results suggest that DDF1 regulation of gene expression is tissue dependent and influenced by the number of binding sites, again highlighting the importance of combinatorial relationships in gene regulation.
Figure 6. Repressor Motifs within the SHORTROOT Promoter Function outside the Vasculature.

(A) Two repressors, ZML1 and DDF1, are predicted to bind within the first 500 bp of the SHR gene. The highest-scoring motifs (red arrowheads) for each were inserted into the RCH1 promoter.
(B) pRCH1::GAL4-UAS::ER-GFP is expressed broadly across the root tip, excluding the QC and root cap.
(C) Synthetic promoters were generated with the ZML1 and DDF1 motifs from the SHR promoter. T2 plants with three or six copies of the ZML1 motif show stochastic reduction of GFP expression in external cell files (white arrowheads), with no apparent differences between three or six copies of the motif (top panels). Three copies of the DDF1 motif also show reduced GFP expression (white arrowheads), but six copies drive ectopic expression in the QC (white arrows, middle panels). To assay the synergy of binding sites, we inserted two repeats of each motif in tandem once or twice in the RCH1 promoter (bottom panels). In these lines, we observe an overall reduction of GFP expression, and stochastic loss of GFP within a cell file (white arrowheads). These results suggest that the ZML1 and DDF1 motifs within the SHR promoter function to repress gene expression outside of the central vasculature.
To determine whether we could obtain synergy with the repressor motifs, we inserted two repeats of each in tandem once or twice in the RCH1 promoter. In these lines, pRCH1-DDF1-ZML1::GAL4-UAS::ER-GFP, we again observed an overall reduction of GFP expression and stochastic loss of GFP within single cells (Figure 6C). However, no synergy was observed between DDF1 and ZML1, as the addition of the two TF repressor motifs did not show a more severe reduction in GFP than the individual TF motifs. These results suggest that ZML1 and DDF1 motifs within the SHR promoter function to repress gene expression outside of the central vasculature. None of these combinations was sufficient to completely abrogate GFP expression, supporting the hypothesis that the combination of multiple repressors is required to establish stable repression. Together, these results support a model in which SHR expression is established through opposing activities of activators and repressors.
DISCUSSION
A central question in developmental biology is how tissue-specific transcriptional cascades are initiated and maintained. Using eY1H assays, we generated a large-scale transcriptional network for the root ground tissue. This network led to the development of hypotheses about gene regulation and gene function. Our results indicate that expression at the top of a transcriptional cascade can be achieved by combinatorial control through activators and repressors.
One of the great mysteries in gene regulation is how to predict binding specificity and regulation. While common experimental approaches, such as the TF-centric ChIP assays and DNA-centric eY1H assays, are sufficient to identify binding interactions, neither is able to accurately predict the functional relevance of that interaction. To increase the probability that an eY1H interaction is functional, one approach is to consider only those interactions that show an enrichment of TF binding sites in the relevant promoter. Our analysis did not find such enrichment. The reason is likely attributable to the limitations of each assay. False negatives are common in eY1H assays for a variety of reasons, including the requirement of a co-factor for TF binding or because the interaction does not exceed the auto-activation level of the reporter. On the other hand, the binding site analysis is based on the presence of a DNA motif, which is not sufficient to predict binding (White et al., 2013). It is likely a combination of these assay limitations that led to the lack of enrichment. Because we validated a subset of the interactions in planta, we were able to show that the p value of a predicted binding site is not a predictor of which interactions are likely to be validated. Indeed, we were unable to find a metric that would indicate which eY1H interactions are most likely to occur in planta under our conditions. Despite these limitations, the eY1H network proved to be a good resource for hypothesis generation as evidenced by the fact that we were able to validate many of the interactions in planta.
The SHR-SCR transcriptional cascade has been well studied in cortex/endodermal cell-fate determination, and the predominant mechanism to establish SCR expression is through activation by SHR with subsequent auto-regulatory feedback on the SCR promoter (Heidstra et al., 2004; Helariutta et al., 2000). In the absence of SHR, the endodermis has residual SCR expression, which is required for initiation of the feedback loop (Helariutta et al., 2000). We identified several additional regulators of SCR expression that likely coordinate to maintain the low levels of SCR required for feedback initiation. Interestingly, three of these regulators function to regulate the expression of both SHR and SCR.
We identified several non-linear relationships between a TF and its target. Non-linear relationships are proposed to be a common feature of dynamic biological systems. For example, Arrieta-Ortiz et al. (2015) examined the relationship between the expression of 50 Bacillus subtilis TFs and two to four of their target genes. Of the 136 TF-target relationships examined, only 28% showed a linear relationship (R2 > 0.7) (Arrieta-Ortiz et al., 2015). Non-linear relationships have been best documented in bacteria, and the extent of these relationships in higher-order organisms has yet to be established. Overexpression and mutant alleles were available for ten of our TF-target relationships, and we found non-linear relationships for three of these. Mechanistically, non-linear relationships can arise due to cooperativity or stoichiometry among TFs. For example, when overexpressed, a TF might multimerize in such a way as to prevent DNA binding, thereby resulting in a molecular phenotype similar to that in the mutant. Other explanations include post-transcriptional modification of the TF or a requirement for a transcriptional co-factor. Alternatively, feedforward loops can lead to non-linear relationships. Any of these mechanisms would result in a non-linear relationship between the TF and target, as seen here. The exact nature of this regulation at the individual gene level is the subject of future investigation.
At the top of the SHR-SCR transcriptional cascade, our results indicate that SHR expression is established by the combination of activators and repressors (Figure 5B). The expression pattern of these regulators indicates that most are expressed across a wide range of tissues. If we consider a low threshold for expression (mean normalized expression >1, Figure 1A), all regulators are expressed in at least two of the microarray profiles, with expression in an average of 16 populations. However, mean normalized expression in any one cell type exceeds 10 for only two of the five activators and two of the nine repressors (Figure 1A). The differential, combinatorial expression of these TFs across tissues is likely one of the driving forces behind differential SHR expression.
Regarding the mechanistic basis of transcriptional repression, two major categories have been proposed: physically blocking general TFs or activators from binding, and protein-protein interactions between an activator and a repressor preventing the activator from functioning (Levine and Manley, 1989). In the context of the SHR promoter, it is likely that both mechanisms are contributing to the regulation. Binding site predictions indicate an overlap in the predicted binding sites of activators and repressors, suggesting that competition for binding sites or tandem binding may contribute to SHR expression. Furthermore, ZML1 and DDF1 cannot stably disrupt gene expression in the context of the RCH1 promoter, suggesting that a single mechanism is not sufficient to maintain stable repression.
Our work highlights the importance of using network-based approaches to understand the complexities of gene regulation. Many important developmental regulators tend to have a key activator, e.g., the role of SHR in activating SCR expression. However, at the top of these activator cascades, regulation is likely to be more complex. The outcome of this complex regulation is buffered gene expression, with the use of multiple regulators reducing the possibility that mutation of a single gene will significantly alter SHR expression. This mechanism is consistent with the failure of forward genetics to identify SHR upstream regulators. As we delve deeper into the complexities of gene-regulatory control, it has become increasingly important to utilize network-based approaches that aim to model the whole system. This study highlights an application of system-wide approaches to understand regulation at the top of a transcriptional cascade.
EXPERIMENTAL PROCEDURES
The data and computational resources used in this article are available for download at http://github.com/eesparks/Sparks_Root_Network. Seed stocks generated in this article will be submitted to the ABRC. All eY1H interaction data will be submitted to Virtual Plant, AGRIS, and the BAR Arabidopsis Interactions Viewer.
Enhanced Yeast-One-Hybrid
eY1H assays were performed as previously described (Gaudinier et al., 2011; Taylor-Teeples et al., 2015). Screens were performed by sequentially mating one promoter clone against 555 TFs present in duplicate in 384-well format. His and LacZ assays were performed separately, and positive results were called on the consensus of at least two authors.
Node Clustering Analysis
Node similarity clustering was used to identify nodes with similar neighbors using igraph (http://igraph.org/) and R script.
In Silico Prediction of Binding Sites
Bait DNA sequences were scanned for the presence of TF binding sites using the FIMO package (Grant et al., 2011) and a p value cutoff of 10−3 or 10−4. Background nucleotide frequency was calculated from the upstream 3 kb sequence of all nuclear encoded genes. We only considered sequences of TF promoters that were involved in an eY1H interaction with a TF that had a binding motif. Enrichment analyses were performed by edge switching with the number of switches equal to 10% of the total number of interactions for 500 randomly generated datasets.
Plant Materials and Growth Conditions
Mutant and overexpression seed lines were obtained from the ABRC, Transplanta collection (Coego et al., 2014) or from other laboratories. Arabidopsis accession Columbia-0 (Col-0) or Landsberg erecta (LER) were used as controls where indicated (Table S4). For phenotyping and qRT-PCR experiments, seeds were surface sterilized, stratified, and imbibed for 48 hr at 4°C before plating on 1 × Murashige-Skoog (MS), 1% sucrose, 1% agar plates. Plates were sealed with parafilm and grown vertically under long-day conditions, and roots were analyzed at 7 days after induction.
qRT-PCR from Whole Roots
Two or three biological replicates and three technical replicates were used for each experiment. Standard curves were run for each primer pair, and values are represented by the efficiency corrected quantification (ECQ) model. PP2A was used as reference.
Transient Transfection of Root Protoplasts
Transient transfections were performed using the TARGET approach, as previously described (Bargmann et al., 2013). Values are represented by the ECQ model with each dexamethasone-induced sample normalized to the corresponding mock induction.
Fluorescent Quantification of pSHR::erGFP Reporter
z stack images were obtained through the vasculature on a LSM510 confocal microscope. Twenty slices were obtained at a 2.17-μm interval. Sum projections were generated in Fiji, and raw integrated density was obtained from a 236-μm area proximal to the QC.
Logistic Regression
Regressions were performed in MATLAB. The intercept (β0) and feature coefficients (β1, ..., βn) were calculated using the glmfit function. The probability of SHR expression was calculated as a function of these parameters using the logistic equation.
Generation of Synthetic Promoters
Repressor motifs within the SHR promoter were identified with FIMO. DNA synthesis was used to generate repeat copies of motifs separated by a random 10-bp spacer. Synthesized fragments were inserted one or two times into a PacI site located 211 bp upstream of the translational start of the RCH1 promoter. Sequences are listed in Supplemental Experimental Procedures. Images were obtained at 5 days after induction on an LSM510 Inverted confocal (Zeiss) with identical settings.
Supplementary Material
Highlights.
A gene-regulatory network reveals regulation of transcription factor expression
Spatially opposed activators and repressors regulate SHORTROOT expression
The top of a transcriptional cascade is regulated by many transcription factors
Acknowledgments
We thank K. Yamaguchi-Shinozaki, J. Mizoi, L. Ma, C. Breuer, and M. de Lucas for seeds, R. Heidstra for the pRCH1:GAL4-UAS:ER-GFP construct, T. Jeffers for assistance with TARGET, the Duke Light Microscopy Core Facility, and the Duke Cancer Center Flow Cytometry Shared Resource. This research was funded by NSF Arabidopsis 2010 (IOS-1021619), NIH (R01-GM043778), and Howard Hughes Medical Institute and Gordon and Betty Moore Foundation (GBMF3405) awards to P.N.B. Additional support was provided by NIH K99-R00 Pathway to Independence Award (GM097188) to M.M. and Virginia Tech College of Agriculture and Life Sciences start-up fund to S.L. G.T. is supported by an NSF Graduate Research Fellowship (DGE-1148897), M.A. is supported by start-up funds from Oregon State University, and J.J.P. was supported by a NIH Ruth L. Kirschstein NRSA F32 fellowship (GM086976).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures, and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2016.09.031.
AUTHOR CONTRIBUTIONS
Conceptualization, E.E.S. and P.N.B.; Methodology, E.E.S., C.D., A.G., S.L., A.J.H., R.G., M.M., S.M.B., and P.N.B.; Investigation, E.E.S., C.D., A.G., S.L., M.A., N.S., J.H.H., J.Z., G.T., J.J.P., J.F., and S.L.; Writing – Original Draft, E.E.S. and P.N.B.; Writing – Review & Editing, all authors.
References
- Arrieta-Ortiz ML, Hafemeister C, Bate AR, Chu T, Greenfield A, Shuster B, Barry SN, Gallitto M, Liu B, Kacmarczyk T, et al. An experimentally supported model of the Bacillus subtilis global transcriptional regulatory network. Mol Syst Biol. 2015;11:1744–4292. doi: 10.15252/msb.20156236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann BOR, Marshall-Colon A, Efroni I, Ruffel S, Birnbaum KD, Coruzzi GM, Krouk G. TARGET: a transient transformation system for genome-wide transcription factor target discovery. Mol Plant. 2013;6:978–980. doi: 10.1093/mp/sst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey PN, Scheres B. Arabidopsis as a model for systems biology. In: Walhout M, Vidal M, Dekker J, editors. Handbook of Systems Biology Concepts and Insights. Elsevier; 2012. pp. 391–406. [Google Scholar]
- Berger MF, Bulyk ML. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc. 2009;4:393–411. doi: 10.1038/nprot.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SM, Orlando DA, Lee JY, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U, Benfey PN. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science. 2007;318:801–806. doi: 10.1126/science.1146265. [DOI] [PubMed] [Google Scholar]
- Brady SM, Zhang L, Megraw M, Martinez NJ, Jiang E, Yi CS, Liu W, Zeng A, Taylor-Teeples M, Kim D, et al. A stele-enriched gene regulatory network in the Arabidopsis root. Mol Syst Biol. 2011;7:459. doi: 10.1038/msb.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CAM, Taylor MS, Engström PG, Frith MC, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Coego A, Brizuela E, Castillejo P, Ruíz S, Koncz C, del Pozo JC, Piñeiro M, Jarillo JA, Paz-Ares J, León J The TRANSPLANTA Consortium. The TRANSPLANTA collection of Arabidopsis lines: a resource for functional analysis of transcription factors based on their conditional overexpression. Plant J. 2014;77:944–953. doi: 10.1111/tpj.12443. [DOI] [PubMed] [Google Scholar]
- De Lucas M, Pu L, Turco GM, Gaudinier A, Marao AK, Harashima H, Kim D, Ron M, Sugimoto K, Roudier FM, Brady SM. Transcriptional Regulation of Arabidopsis Polycomb Repressive Complex 2 Coordinates Cell Type Proliferation and Differentiation. Plant Cell. 2016 doi: 10.1105/tpc.15.00744. http://dx.doi.org/10.1105/tpc.15.00744. [DOI] [PMC free article] [PubMed]
- Di Laurenzio L, Wysocka-Diller J, Malamy JE, Pysh L, Helariutta Y, Freshour G, Hahn MG, Feldmann KA, Benfey PN. The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the Arabidopsis root. Cell. 1996;86:423–433. doi: 10.1016/s0092-8674(00)80115-4. [DOI] [PubMed] [Google Scholar]
- Fuxman Bass JI, Sahni N, Shrestha S, Garcia-Gonzalez A, Mori A, Bhat N, Yi S, Hill DE, Vidal M, Walhout AJM. Human gene-centered transcription factor networks for enhancer and disease variants. Cell. 2015;161:661–673. doi: 10.1016/j.cell.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinha C, Hofhuis H, Luijten M, Willemsen V, Blilou I, Heidstra R, Scheres B. PLETHORA proteins as dose-dependent master regulators of Arabidopsis root development. Nature. 2007;449:1053–1057. doi: 10.1038/nature06206. [DOI] [PubMed] [Google Scholar]
- Gaudinier A, Zhang L, Reece-Hoyes JS, Taylor-Teeples M, Pu L, Liu Z, Breton G, Pruneda-Paz JL, Kim D, Kay SA, et al. Enhanced Y1H assays for Arabidopsis. Nat. Methods. 2011;8:1053–1055. doi: 10.1038/nmeth.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda H, Sasaki E, Akiyama K, Maruyama-Nakashita A, Nakabayashi K, Li W, Ogawa M, Yamauchi Y, Preston J, Aoki K, et al. The AtGenExpress hormone and chemical treatment data set: experimental design, data evaluation, model data analysis and data access. Plant J. 2008;55:526–542. doi: 10.1111/j.0960-7412.2008.03510.x. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, He K, Liu D, Bai S, Gu X, Wei L, Luo J. DATF: a database of Arabidopsis transcription factors. Bioinformatics. 2005;21:2568–2569. doi: 10.1093/bioinformatics/bti334. [DOI] [PubMed] [Google Scholar]
- Guo A-Y, Chen X, Gao G, Zhang H, Zhu Q-H, Liu X-C, Zhong Y-F, Gu X, He K, Luo J. PlantTFDB: a comprehensive plant transcription factor database. Nucleic Acids Res. 2008;36:D966–D969. doi: 10.1093/nar/gkm841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidstra R, Welch D, Scheres B. Mosaic analyses using marked activation and deletion clones dissect Arabidopsis SCARECROW action in asymmetric cell division. Genes Dev. 2004;18:1964–1969. doi: 10.1101/gad.305504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helariutta Y, Fukaki H, Wysocka-Diller J, Nakajima K, Jung J, Sena G, Hauser MT, Benfey PN. The SHORT-ROOT gene controls radial patterning of the Arabidopsis root through radial signaling. Cell. 2000;101:555–567. doi: 10.1016/s0092-8674(00)80865-x. [DOI] [PubMed] [Google Scholar]
- Jäckle H, Sauer F. Transcriptional cascades in Drosophila. Curr Opin Cell Biol. 1993;5:505–512. doi: 10.1016/0955-0674(93)90017-k. [DOI] [PubMed] [Google Scholar]
- Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D’Angelo C, Bornberg-Bauer E, Kudla J, Harter K. The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007;50:347–363. doi: 10.1111/j.1365-313X.2007.03052.x. [DOI] [PubMed] [Google Scholar]
- Koizumi K, Hayashi T, Wu S, Gallagher KL. The SHORT-ROOT protein acts as a mobile, dose-dependent signal in patterning the ground tissue. Proc Natl Acad Sci USA. 2012;109:13010–13015. doi: 10.1073/pnas.1205579109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M. Transcriptional enhancers in animal development and evolution. Curr Biol. 2010;20:R754–R763. doi: 10.1016/j.cub.2010.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Manley JL. Transcriptional repression of eukaryotic promoters. Cell. 1989;59:405–408. doi: 10.1016/0092-8674(89)90024-x. [DOI] [PubMed] [Google Scholar]
- Li B, Gaudinier A, Tang M, Taylor-Teeples M, Nham NT, Ghaffari C, Benson DS, Steinmann M, Gray JA, Brady SM, Kliebenstein DJ. Promoter-based integration in plant defense regulation. Plant Physiol. 2014;166:1803–1820. doi: 10.1104/pp.114.248716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman LM, Sparks EE, Moreno-Risueno MA, Petricka JJ, Benfey PN. MYB36 regulates the transition from proliferation to differentiation in the Arabidopsis root. Proc. Natl. Acad. Sci. USA. 2015;112:12099–12104. doi: 10.1073/pnas.1515576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil LT, Pons C, Arda E, Giese GE, Myers CL, Walhout AJM. Transcription factor activity mapping of a tissue specific in vivo gene regulatory network. Cell Syst. 2015;1:152–162. doi: 10.1016/j.cels.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton T, Petricka J, Corcoran DL, Li S, Winter CM, Carda A, Benfey PN, Ohler U, Megraw M. Paired-end analysis of transcription start sites in Arabidopsis reveals plant-specific promoter signatures. Plant Cell. 2014;26:2746–2760. doi: 10.1105/tpc.114.125617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Sena G, Nawy T, Benfey PN. Intercellular movement of the putative transcription factor SHR in root patterning. Nature. 2001;413:307–311. doi: 10.1038/35095061. [DOI] [PubMed] [Google Scholar]
- Rach EA, Yuan H-Y, Majoros WH, Tomancak P, Ohler U. Motif composition, conservation and condition-specificity of single and alternative transcription start sites in the Drosophila genome. Genome Biol. 2009;10:R73. doi: 10.1186/gb-2009-10-7-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Barutcu AR, McCord RP, Jeong JS, Jiang L, MacWilliams A, Yang X, Salehi-Ashtiani K, Hill DE, Blackshaw S, et al. Yeast one-hybrid assays for gene-centered human gene regulatory network mapping. Nat Methods. 2011a;8:1050–1052. doi: 10.1038/nmeth.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Diallo A, Lajoie B, Kent A, Shrestha S, Kadreppa S, Pesyna C, Dekker J, Myers CL, Walhout AJM. Enhanced yeast one-hybrid assays for high-throughput gene-centered regulatory network mapping. Nat Methods. 2011b;8:1059–1064. doi: 10.1038/nmeth.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Pons C, Diallo A, Mori A, Shrestha S, Kadreppa S, Nelson J, DiPrima S, Dricot A, Lajoie BR, et al. Extensive rewiring and complex evolutionary dynamics in a C. elegans multiparameter transcription factor network. Mol. Cell. 2013;51:116–127. doi: 10.1016/j.molcel.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann JU. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37:501–506. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- Sorrells TR, Johnson AD. Making sense of transcription networks. Cell. 2015;161:714–723. doi: 10.1016/j.cell.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzani R, Cui H, Moreno-Risueno MA, Busch W, Van Norman JM, Vernoux T, Brady SM, Dewitte W, Murray JAH, Benfey PN. Spatiotemporal regulation of cell-cycle genes by SHORTROOT links patterning and growth. Nature. 2010;466:128–132. doi: 10.1038/nature09143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Teeples M, Lin L, de Lucas M, Turco G, Toal TW, Gaudinier A, Young NF, Trabucco GM, Veling MT, Lamothe R, et al. An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature. 2015;517:571–575. doi: 10.1038/nature14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Myers CA, Corbo JC, Cohen BA. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc Natl Acad Sci USA. 2013;110:11952–11957. doi: 10.1073/pnas.1307449110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.