

Abstract
The nutritional curcumin (CUR) is beneficial in cell-mediated autoimmune diseases. The molecular mechanisms underlying this food-mediated silencing of inflammatory immune responses are poorly understood. By investigating antigen-specific immune responses we found that dietary CUR impairs the differentiation of Th1/Th17 cells in vivo during encephalomyelitis and instead promoted Th2 cells. In contrast, feeding CUR had no inhibitory effect on ovalbumin-induced airway inflammation. Mechanistically, we found that CUR induces an anti-inflammatory phenotype in dendritic cells (DC) with enhanced STAT3 phosphorylation and suppressed expression of Il12b and Il23a. On the molecular level CUR readily induced NRF2-sensitive heme oxygenase 1 (HO-1) mRNA and protein in LPS-activated DC. HO-1 enhanced STAT3 phosphorylation, which enriched to Il12b and Il23a loci and negatively regulated their transcription. These findings demonstrate the underlying mechanism through which a nutritional can interfere with the immune response. CUR silences IL-23/Th17-mediated pathology by enhancing HO-1/STAT3 interaction in DC.
Silencing inflammatory pathways by nutritionals may have sustainable impact on treatment strategies for chronic immune-mediated diseases. Despite the direct role of nutritionals as allergens or inducers of oral tolerance, increasing data suggests that dietary factors or their metabolites can affect the nature of T cell-mediated immune responses1. This has been intensively studied for dietary fibre metabolites like short-chain fatty acids in experimental models of colitis2 or allergic airway inflammation3. Another group of nutritional factors modifying the course of chronic inflammatory disorders consists of phytochemicals. Some phytochemicals seem to be beneficial in the treatment of cancer but also in certain settings of chronic inflammation. Among the well characterized phytochemicals are sulforaphane, resveratrol and curcumin. The isothiocyanate sulforaphane that naturally occurs in cruciferous vegetables has been reported to protect from cancer and from inflammatory autoimmune disease4,5. Other examples of nutritional compounds with anti-tumoral and anti-inflammatory activities are the natural phenol resveratrol6,7, and the polyphenol diferuloylmethane (curcumin; CUR). This component of turmeric isolated from the rhizome of Curcuma longa has been reported to have anti-tumor properties and to dampen inflammatory conditions8. As a nutritional product, CUR has been tested in several preclinical settings of T cell-dependent organ-specific inflammatory diseases including arthritis9, colitis10, diabetes11 or graft-versus-host disease12. Similarly, some studies reported a beneficial effect of CUR treatment in rodent models of multiple sclerosis13,14,15,16.
Different mechanisms have been proposed to explain the beneficial effects of CUR. While its anti-tumoral effects seem to be mediated by the regulation of tumor suppressor genes, cellular apoptosis and oxidative stress, its anti-inflammatory mechanisms are not fully understood. CUR has been suggested to affect the phenotype of immune cells like T cells and dendritic cells (DC) and to interfere with different signaling pathways like NF-κB, NRF2 and JAK/STAT10,13,17,18,19. The effects of CUR on DC and DC-associated signaling pathways are of special interest since DC are the most relevant cell type for initiating inflammatory autoimmune responses and for priming T cells. Yet, the exact mechanism by which CUR improves T cell-mediated autoimmune disease remains enigmatic.
Here we aimed to unravel the molecular mechanisms, by which CUR affects inflammatory T cell-mediated immune responses. Although CUR is part of the daily dietary intake, most investigators have used an artificial route of administration.
Therefore, we first show that the nutritional compound CUR in food pellets administrated orally protects from experimental encephalomyelitis in a murine model of multiple sclerosis. The protective effect of CUR was associated with a targeted suppression of the encephalomyelitis-inducing Th17 response in vivo. In contrast, CUR did not suppress airway inflammation induced by an in vivo Th2 response. To understand the mechanism responsible for the effects of CUR on Th17-mediated inflammation, we focused on the cellular and molecular events in T cells and antigen-presenting cells (APC). Non-toxic doses of CUR did not directly affect T cell cytokine expression, but instead strongly affected the phenotype of DC, as CUR preferentially enhanced phosphorylation of STAT3 in LPS-stimulated DC. Activated STAT3 binds to the promoter loci of Il23a and Il12b and negatively regulates their transcription. Interestingly, the activation of STAT3 by CUR in DC is mediated through heme oxygenase 1 (HO-1), a stress-response protein readily induced by CUR in vitro and in vivo.
Results
Feeding CUR protects from Th17 cell-mediated encephalomyelitis
The nutritional CUR has been reported to improve the outcome of experimental autoimmune encephalomyelitis (EAE) in rodents13,14,15,16 by using artificial and parenteral routes of administration. CUR was either dissolved in DMSO and injected intraperitoneally13,16 or encapsulated and delivered intranasally15. We asked if CUR also protects mice from EAE when given orally, the natural route of administration of CUR and other food additives like sulforaphane5 or fumarates20. Therefore mice were fed with 2% CUR-containing food pellets or received a control diet (CON) and were then immunized for the induction of EAE. The average daily consumption of CUR was 80.5 mg (Supplementary Fig. S1). Mice on CON diet immunized with PLP139-151 peptide developed a severe encephalomyelitis with clinical symptoms starting on day 12 after immunization and an average EAE score of 3.0 ± 0.31 SEM (Fig. 1a). In contrast, mice on oral CUR diet developed only mild symptoms of encephalomyelitis (average EAE score 1.3 ± 0.25 SEM). Accordingly, analysis of the central nervous system tissue of immunized mice on CUR diet showed less infiltration with mononuclear cells compared to mice on CON diet (Supplementary Fig. S2). Thus, the oral administration of this nutritional also protected from severe encephalomyelitis in vivo. To explain the clinical improvement of EAE by oral CUR administration, we first analyzed the influence of this dietary phytochemical on the activation of autoantigen-specific T cells. CUR diet had no significant impact on the proliferation of in vivo primed PLP139-151 peptide-specific CD4+ T cells (Fig. 1b). We therefore analyzed the cytokine phenotype of the CD4+ T cell responses in draining lymph nodes on day 7 after immunization with PLP139-151 peptide. Intracellular cytokine analysis of CD4+ T cells revealed a significant decrease of the pro-inflammatory cytokines IL-17, IFN-γ and IL-2 in mice receiving CUR diet (Fig. 1c,d), and an increase of the Th2 cytokine IL-4. T cell-derived IL-10 and TNF production remained unaffected. Thus, oral CUR administration impaired the development of Th17 and Th1 responses in vivo, while it promoted antigen-specific Th2 cells.
Figure 1. CUR protects mice from severe encephalomyelitis and inhibits IL-17 and IFN-γ production in vivo.

(a) Mice were fed with 2% CUR or control diet (CON), immunized for EAE and followed for disease symptoms. The results show mean EAE score ± SEM from 2 independent experiments with n = 16 mice (*P < 0.05 day 16 to 22; Wilcoxon test). (b) Mice were fed with CUR as in (a) and immunized for EAE. On day 7 after immunization, CD4+ T cells were purified from draining lymph nodes and co-cultured with DC and PLP peptide. T cell proliferation was assessed by [3H] thymidine incorporation. NS: not significant, Mann-Whitney test. (c,d) Mice were treated as in (a). On day seven after immunization draining lymph nodes were isolated to analyze intracellular cytokine production in PMA/ionomycin-stimulated CD4+ T cells. First, a FSC/SSC plot was made and all lymph node cells were gated. The CD4+ T cell population was gated by a SSC/CD4 (left panel) plot and then analyzed for CD4+ T cell-specific cytokine expression. Dot plots from single mice are depicted in (c), pooled data from control mice (CON; n = 5) and CUR-treated mice (CUR; n = 5) are shown in (d). Bars represent the mean ± SEM (*P < 0.05, Mann-Whitney test).
Oral CUR does not improve Th2-mediated airway inflammation
To study whether oral CUR impairs an on going T cell activation or instead, selectively affects T cell polarization in vivo, we next tested the effect of CUR in ovalbumin (OVA)-specific Th2-dominated allergic airway inflammation models. First, airway hyperresponsiveness (AHR) of mice on either CON diet or CUR diet was measured to assess the effect of oral CUR diet on allergic airway inflammation. We found no significant difference in AHR induced by OVA/ALUM immunization followed by OVA inhalation between the mice on CON diet and the mice on CUR diet (Fig. 2a). Consistently, after inhaled antigen challenge, the mice on oral CUR diet showed a similar increase in infiltrating cells including T cells, eosinophils and neutrophils in the bronchoalveolar lavage (BAL) fluid as mice on CON diet (Fig. 2b–f). Histological analysis revealed that CUR diet affected neither the numbers of infiltrated mononuclear cells into the peribronchiolar regions of the lungs (Fig. 2g, upper panel and Supplementary Fig. S3a), nor the production of mucus as shown by periodic acid-Schiff (PAS) staining (Fig. 2g, lower panel and Supplementary Fig. S3b). In accordance with these results, we found equivalent levels of the Th2 cytokines IL-4, IL-5 and IL-13 and enhanced IL-10 in the BAL fluid from the mice challenged with OVA inhalation after CUR diet as in CON-fed mice (Fig. 2h). Altogether, oral administration of CUR showed no suppression of Th2-dominated allergic airway inflammation in vivo.
Figure 2. Oral administration of CUR showed little effects on OVA-induced allergic airway inflammation.

(a) AHR was assessed as lung resistance. Mean values (5 to 6 mice per group) are shown with SEM (square symbols represent OVA challenge, circles represent PBS challenge) (b–f) The cell number of total mononuclear cells (b), eosinophils (c), neutrophils (d), lymphocytes (e), and macrophages (f) in the BAL fluid are shown. Mean values with SEM (5 to 6 mice per group) are shown. (g) Antigen-induced leukocyte infiltration into the lungs was evaluated by H&E staining (upper panel). Mucus production induced by OVA was examined by histological analysis (PAS staining) (lower panel). (h) The concentration of the indicated cytokines in the BAL fluid were measured by cytometric bead array. Mean values with SEM (5 to 6 mice per group) are shown. (a–f), no significant differences were found between CUR/OVA and CON/OVA; h, *P < 0.05, Mann-Whitney test).
CUR regulates APC-dependent T cell differentiation
The results obtained in the EAE and the allergic airway inflammation in vivo models indicate that CUR does not generally suppress antigen-reactive T cell activation but selectively impairs Th17 and Th1 differentiation (Figs 1 to 3). To explore the mechanisms causing these immune-modulating effects of CUR on Th cell polarization we first studied T cell differentiation in an APC-independent system. Therefore we activated freshly isolated CD4+ T cells with plate-bound anti-CD3 and anti-CD28 monoclonal antibodies in the presence or absence of CUR. After three days of TCR stimulation and further expansion with IL-2, T cells were analyzed on day seven for the production of IFN-γ, IL-17, IL-4, IL-10, IL-2 and TNF (Fig. 3a). We found no effect of CUR on T cell cytokine production, indicating that the effects of CUR on T cell polarization in vivo were not mediated by a direct action of this nutritional on T cells. Since CUR has been reported to impair IL-12 production by APC like DC13, CUR may affect T cell differentiation by acting primarily on DC. Non-activated DC were therefore loaded with OVA peptide and co-cultured with OVA-specific T cell receptor-transgenic (OT-II) T cells. On day seven of the co-culture, we found modest expression of IFN-γ, IL-17, IL-4, IL-10 and high production of IL-2 and TNF. The treatment of non-activated DC with CUR had no significant impact on the cytokine phenotype of T cells (Fig. 3b,c). Next, we activated OVA-loaded DC for 1 hour with LPS before initiating the co-culture with OT-II T cells. As expected, LPS-activated DC induced CD4 T cells that were capable to produce large amounts of the lineage-defining cytokines IFN-γ, IL-17 and of IL-4 in OVA-reactive T cells. In sharp contrast, when DC were treated with CUR for 2 hours and subsequently activated with LPS, the DC lost their capacity to strongly induce IFN-γ- and IL-17-producing OT-II T cells, while the induction of IL-4-producing OT-II T cells was significantly enhanced. In contrast to the effects on lineage-defining cytokines, CUR-treated DC had only a minor impact on IL-2, IL-10 or TNF production by OT-II T cells (Fig. 3b,c). The characteristics of the Th cell differentiation induced by CUR with LPS-activated DC in vitro reflected those findings on Th cells in vivo when mice were immunized in the presence of CUR-containing diet (Fig. 1c).
Figure 3. CUR alters the polarization of activated T cells in the presence of DC.
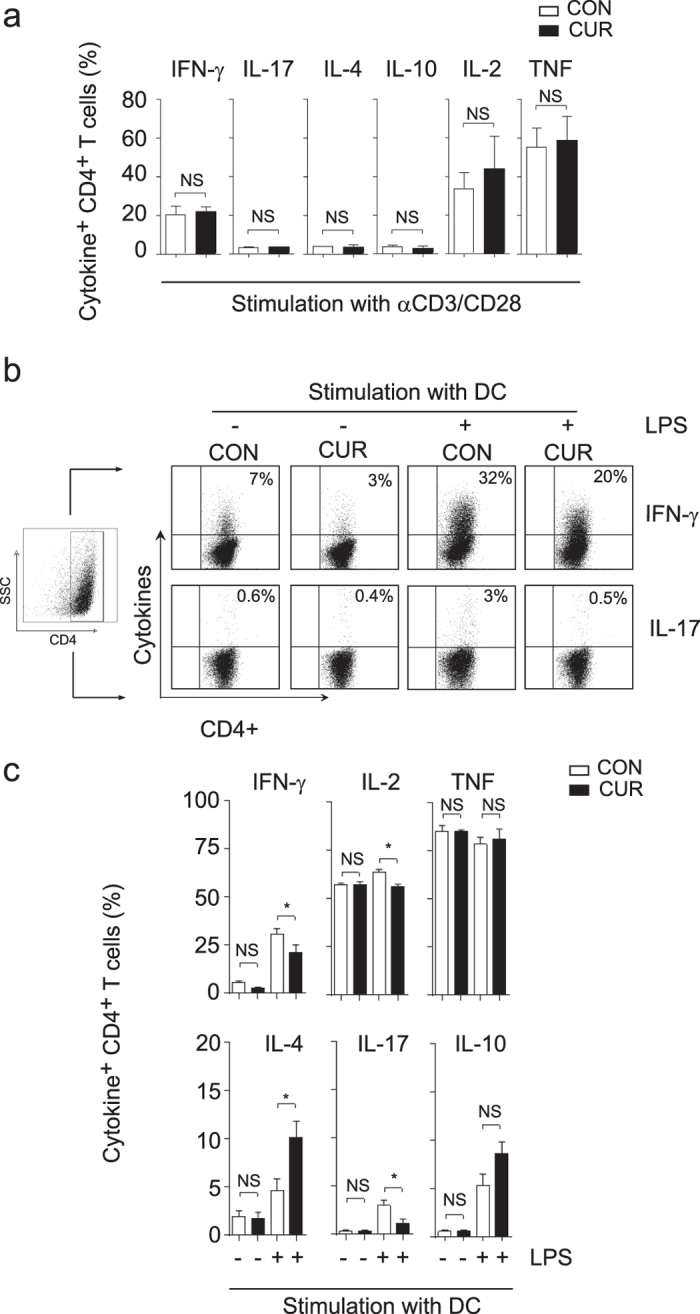
(a) Isolated CD4+ T cells were activated with plate bound anti-CD3 and anti-CD28 Ab and cultivated with DMSO (CON) or curcumin in DMSO (CUR). Cytokine expression was determined after stimulation with PMA/ionomycin by intracellular staining and flow cytometry. Data show mean ± SEM of 4 independent experiments, NS: not significant, Mann-Whitney test. (b and c) DC were treated with DMSO (CON) or curcumin in DMSO (CUR) for 2 hours, loaded with OVA peptide, left untreated or activated with LPS and used for stimulation of OT II CD4+ T cells. On day 7, cells were stimulated with PMA/ionomycin, stained for CD4 and intracellular cytokine production. First, a FSC/SSC plot was made and all leukocytes were gated. The CD4+ T cell population was gated by a SSC/CD4 plot (left panel) and then analyzed for CD4+ T cell-specific cytokine expression. Dot plots from single experiments are depicted in (b), pooled data are shown in (c). Bars represent mean ± SEM of three independent experiments (*P < 0.05; One Way ANOVA, Tukey’s post-hoc test).
CUR selectively inhibits IL-12 and IL-23 production by activated DC
Since our data and data from other groups argued that CUR affects T cell differentiation by acting on APC10,11, we explored the effects of CUR on DC differentiation. First, we re-evaluated possible toxic or apoptosis-inducing effects of CUR on DC. CUR concentrations from 1 up to 15 μM did not increase cell death in DC as determined by fixable viability dye staining as well as apoptosis staining with propidium iodide and annexin V (data not shown). Next, we compared the maturation status of DC treated with CUR to control DC and determined the expression of MHC class II and co-stimulatory molecules CD80, CD86 and CD83. As expected, LPS activation induced the up-regulation of MHC class II, CD80, CD86 and CD83 expression by DC. CUR treatment with concentrations ≤15 μM neither affected the maturation status of quiescent DC nor the maturation status of LPS-activated DC (Fig. 4a). From these experiments we concluded that the altered Th cell response by CUR observed in vitro and in vivo is not due to changes in DC maturation. But, CUR-mediated changes in DC cytokine expression could explain the impaired Th17 and Th1 response. Therefore we first studied the secreted protein levels of IL-12, IL-23, IL-10 and IL-6 after 18 hours of cultivation in medium alone or in presence of LPS. Activation of DC with LPS induced certain amount of IL-12, IL-23 and IL-6 protein, and slight production of IL-10. CUR treatment of non-activated DC did not stimulate cytokine secretion. In sharp contrast, CUR treatment of LPS-activated DC resulted in a dose-dependent suppression of IL-12 and IL-23 production, while the secretion of IL-6 and IL-10 by DC was not affected by CUR treatment (Fig. 4b). Inhibition of IL-12 production from DC by CUR had been reported earlier11,13. But, even more importantly, we identified that low-dose CUR is also a potent suppressor of IL-23 production by DC. We next aimed to understand the molecular mechanism by which CUR provides an anti-inflammatory phenotype in DC with low expression of IL-12 and IL-23.
Figure 4. Curcumin affects the DC phenotype without interfering with cell maturation or viability and inhibits IL-12 and suppresses IL-23 production in vitro.

(a) DC were treated with DMSO (CON) or increasing doses of curcumin in DMSO (CUR; 1 to 30 μM) for 2 hours before resting in medium alone or activating with LPS. After 18 hours cells were stained for the expression of CD80, CD83, CD86 and MHC II. Flow cytometry data are depicted as mean ± SEM of 3 independent experiments. (b) DC were treated and activated with LPS. Supernatant was collected after 18 hours and analysed for IL-12p70, IL-23, IL-10 and IL-6 secretion by ELISA. The results show mean ± SEM of three biological replicates and represent one out of three independent experiments with similar results.
Treatment of DC with CUR enhances STAT3 phosphorylation
Anti-inflammatory type II DC have been associated with alterations in different signaling pathways21. One of these concepts is based on the activation of STAT transcription factors. While the activation of STAT1 in DC is linked to a pro-inflammatory DC phenotype, the activation of STAT3 in DC is associated with an anti-inflammatory or even tolerogenic DC phenotype22. We followed the activation status of both STAT1 and STAT3 in our experimental setting since CUR treatment has been suggested to interfere with STAT activation in different cell types23. To analyze the balance of STAT1 and STAT3 activation in DC, we quantified the phosphorylated forms of both transcription factors in quiescent and LPS-activated DC. Non-activated DC showed low levels of phosphorylation of both STAT1 and STAT3, irrespective of CUR treatment. As reported, LPS activation induced STAT1 and STAT3 activation in 10–20% of DC20,24 (Fig. 5a,b). Surprisingly, treatment of DC with CUR and subsequent activation with LPS significantly enhanced phosphorylation of STAT3 without affecting the phosphorylation status of STAT1 (Fig. 5a,b). To investigate if the enhanced phosphorylation of STAT3 by CUR treatment results in functional alteration of the transcriptional activity of STAT3, we quantified the DNA protein binding by STAT3 electromobility shift assay (EMSA). We activated DC with LPS and isolated nuclear extracts, which were tested by EMSA for binding to a STAT3 consensus sequence. As a positive control we treated DC with the STAT3-activating cytokine IL-6 before LPS activation. The nuclear interaction between STAT3-oligos and proteins was weaker in untreated control DC activated with LPS when compared to nuclear extracts from LPS-activated DC that were either treated with CUR or IL-6 (Fig. 5c). These data suggest that CUR significantly increased STAT3 activity in DC after LPS activation. Moreover, Western blot analysis of DC lysates revealed that CUR enhanced LPS-induced STAT3 phosphorylation without affecting total STAT3 protein level (Fig. 5d).
Figure 5. CUR enhances STAT3 phosphorylation in activated DC.

(a,b) DC were treated with DMSO (CON) or curcumin (CUR) in DMSO (7.5 μM) for 2 hours before activation with LPS for 1 hour. STAT1 and STAT3 phosphorylation was determined by intracellular staining and flow cytometry. First, a FSC/SSC plot was made and all DC were gated. The CD11c+ DC population was gated by a SSC/CD11c plot (left panel) and then analyzed for CD11c+ DC-specific STAT phosphorylation. Histogram plots from single experiments are depicted in (a), pooled data are shown in (b). Bars represent mean ± SEM of three independent experiments (*P < 0.05, **P < 0.01). (c) EMSA analysis with STAT3 promotor oligonucleotides. DC were treated as in (a) or with recombinant IL-6 in medium or medium alone (CON) and then activated with LPS. Nuclei were extracted and mixed with flourochrome labelled STAT3 EMSA probes. Complexes were separated on a TAE-gel and the band intensity was quantified with an OdysseySA Infrared Imaging System. One representative EMSA (left panel) and pooled data from 3 independent experiments (right panel) are shown (mean ± SEM, *P < 0.05; Mann-Whitney test). The samples were run on the same gel, the lanes of interest were cut and re-ordered. (d) DC were treated with DMSO (CON) or CUR in DMSO (CUR) as in (a) before activation with LPS. Expression of pSTAT3 (upper panel) and total STAT3 protein (lower panel) was analyzed by Western blotting. One representative immunoblot and pooled data from 3 independent experiments are shown. The samples for STAT3/β-actin or pSTAT3/β-actin were run on the same gels and the blot images were cropped based on the molecular weight (79/86 kDa for STAT3/pSTAT3 and 43 kDa for β-actin). Bars represent mean ± SEM of three independent experiments (*P < 0.05, **P < 0.01; One Way ANOVA, Tukey’s post-hoc test). The complete EMSA (c) and Western blot (d) figures can be found as Supplementary Figs S7 to S10.
Activated STAT3 negatively regulates Il23a and Il12b expression in DC
We next asked if the enhanced STAT3 activation by CUR is responsible for the regulation of IL-12 and IL-23 in DC. Since activated STAT3 had been reported previously to negatively control Il12b promoter activity25, we first asked if CUR treatment directly affects Il23a promoter activity. To answer this question, we used RAW 246.7 macrophages transfected with an Il23a luciferase reporter plasmid. At steady state RAW 246.7 cells showed low basal reporter activity, while LPS activation induced Il23a reporter activity. Treatment of macrophages with CUR impaired Il23a reporter activity (Fig. 6a). Similar results were obtained, when treating macrophages with the STAT3-activating cytokine IL-6 before LPS activation (Fig. 6a).
Figure 6. CUR inhibits LPS induced Il23a promotor activity.

(a) Luciferase reporter activity was measured in RAW 264.7 macrophages after 2 hours of incubation or with DMSO (CON) or CUR in DMSO (CUR) and subsequent incubation in the absence or presence of LPS. In addition Luciferase reporter activity was measured in RAW 264.7 macrophages after 1 hour of incubation with recombinant IL-6 (50 ng/ml) or PBS (CON) and subsequent incubation in the absence or presence of LPS. The results show mean ± SEM of quadruplicates. Three independent experiments showed similar results (*P < 0.01; One Way ANOVA, Tukey’s post-hoc test). (b) IL-6 suppresses Il23a and Il12b expression in DC after LPS activation. DC were incubated with PBS (CON) or IL-6 (50 ng/ml) and activated with LPS for 1 hour. Expression of the indicated genes was determined by quantitative RT-PCR. The results show mean ± SEM of 4 independent experiments. Data were normalized to ß-actin and expression before LPS activation was set as 1.0 (*P < 0.05; One Way ANOVA, Tukey’s post-hoc test). (c) Treatment of DC with CUR inhibits IL-12 and suppresses IL-23 production in vitro. DC were incubated with DMSO (CON) or increasing doses of CUR (1 to 30 μM) in DMSO (CUR) for 2 hours and activated with LPS for 1 hour. Expression of the indicated genes was determined by quantitative RT-PCR. The results show mean ± SEM of 4 independent experiments. Data were normalized to ß-actin and expression before LPS activation was set as 1.0.
To further confirm our findings on the participation of STAT3 in the regulation of IL-23 expression, we next studied the mRNA expression of Il12a, Il12b and Il23a in DC that were treated with IL-6. Non-activated DC did not express Il12a, Il12b or Il23a mRNA. IL-6 induced some Il12a expression, without affecting Il12b or Il23a expression. LPS activation of DC strongly induced Il12a, Il12b and Il23a expression. Treatment of DC with STAT3-activating IL-6 prior to LPS activation significantly inhibited Il12b and Il23a induction, while Il12a expression was not affected (Fig. 6b). We then tested the regulation of IL-12 and IL-23 mRNA expression by CUR. Treatment of DC with increasing doses of CUR prior to LPS activation resulted in a dose-dependent suppression of Il12b and IL23a expression, but had no significant impact on Il12a expression (Fig. 6c). Thus, CUR impairs IL-12 and especially IL-23 protein production by activated DC (Fig. 4b) through inhibition of the transcriptional expression of Il12b and Il23a (Fig. 6c) and this inhibition is linked to STAT3 phosphorylation (Fig. 5a–c).
To further prove that STAT3 directly regulates Il12b and Il23a expression independently of DC cytokine production or CUR treatment, we infected DC with adenoviral vectors containing a control construct (Ad CON) or construct encoding constitutive active STAT3 (Ad CA STAT3) prior to LPS activation. Neither Ad CON nor Ad CA STAT3 constructs influenced IL-12 or IL-23 expression in non-activated DC (Fig. 7a). Cells treated with Ad CON and activated with LPS showed strong induction of Il12a, Il12b and Il23a. Importantly, Ad CA STAT3 significantly suppressed Il12b and Il23a expression of LPS-activated DC, while Il12a was not affected (Fig. 7a).
Figure 7. Overexpression of CA STAT3 selectively inhibits Il12b and Il23a but not Il12a expression.
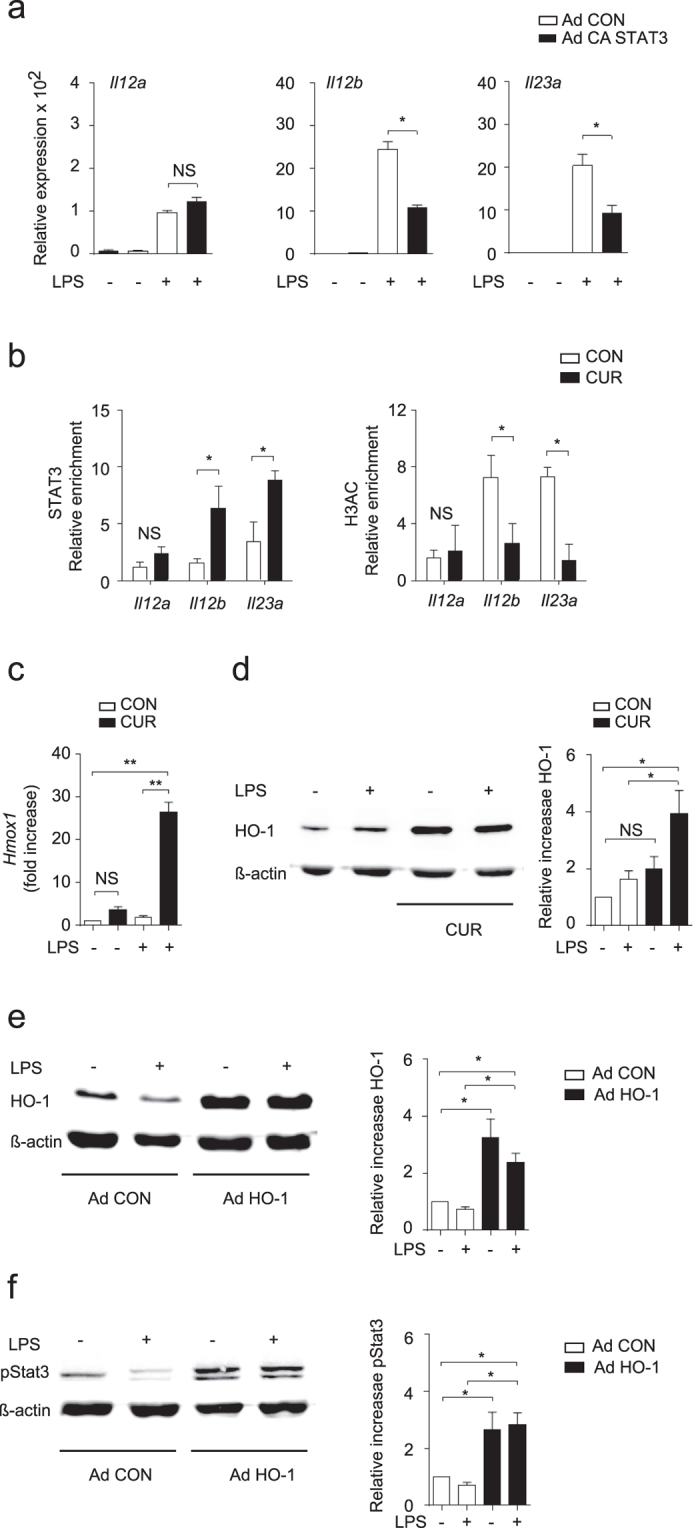
(a) DCs were infected with control adenovirus (Ad CON) or CA STAT3 (Ad CA STAT3) containing adenovirus and stimulated with LPS for 1 hour. Il12a, Il12b and Il23a mRNA expression was analyzed by quantitative RT-PCR. Data were normalized to ß-actin and expression before LPS activation was set as 1.0. The results show mean ± SEM of 3 independent experiments (*P < 0.01; One Way ANOVA, Tukey’s post-hoc test). (b) ChIP analysis of DC treated with DMSO (CON) or CUR and stimulated with LPS, followed by crosslinking and immunoprecipitation with antibody to STAT3 (left panel) or acetylated histone H3 (H3Ac, right panel). Bound DNA was amplified by quantitative PCR for primer sites in Il12a, Il12b and Il23a promoter regions. Bars represent mean ± SEM of 3 independent experiments (*P < 0.05; One Way ANOVA, Tukey’s post-hoc test). (c) HO-1 is induced by CUR in DC in vitro. DC were treated with DMSO (CON) or CUR in DMSO (CUR) before activation with LPS for 1. Expression of Hmox1 mRNA was determined by quantitative RT-PCR (c) and HO-1 protein was analyzed by Western blotting (d). The results in (c,d) show mean ± SEM of three independent experiments (*P < 0.05, **P < 0.01; One Way ANOVA, Tukey’s post-hoc test), the blot shown in (d) is representative for three independent experiments with similar results. Actin expression served as control. (e,f) Overexpression of HO-1 increased STAT3 phosphorylation in DC. DCs were infected with control adenovirus (Ad CON) or HO-1 containing Ad HO-1 and stimulated with LPS for 1 h. Overexpression of HO-1 protein was analysed by Western blotting (e). The expression of phosphorylated STAT3 (pSTAT3) in DC treated as in (e) was analysed by Western blotting (f). One representative immunoblot and pooled data from 4 independent experiments are shown (mean ± SEM *P < 0.05; One Way ANOVA, Tukey’s post-hoc test). The Western blot samples for HO-1/β-actin or pSTAT3/β-actin were run on the same gels and the blot images were cropped based on the molecular weight (32 kDa for HO-1, 79/86 kDa for pSTAT3 and 43 kDa for β-actin). The complete Western blot figures (d–f) can be found as Supplementary Figs S11 to S13.
To further assess the direct regulation of IL-12 and IL-23 by CUR-activated STAT3, we performed chromatin immunoprecipitation (ChIP) using DC lysates and antibodies directed against STAT3 or histone 3 acetylation (H3Ac). We found enhanced binding of STAT3 to the Il12b and Il23a promoter in LPS-treated DC treated with CUR compared to DC treated with DMSO (CON) (Fig. 7b). The binding of STAT3 to the Il12a locus was not significantly affected by CUR treatment. Likewise, we found significantly less H3Ac within the Il12b and Il23a promoter regions but not the Il12a promoter region (Fig. 7b). Thus, CUR negatively regulated Il12b and IL23a expression through enhanced STAT3 activity. By using a pharmacological STAT3 inhibitor we could abolish the negative regulation of CUR on Il12b and IL23a expression in RAW 246.7 macrophages activated with LPS (Supplementary Fig. S4).
CUR enhances STAT3 phosphorylation through induction of HO-1
We further decided to unravel the mechanism by which CUR enhances STAT3 phosphorylation in LPS-activated DC. Since IL-6 and IL-10 production were minimally affected by CUR (Fig. 4b), a cell-intrinsic effect could explain the enhanced STAT3 activity. The expression of Src homology phosphatase-1 (SHP-1), a protein tyrosine phosphatase with negative impact on STAT3 activity, was not affected by CUR in LPS-activated DC (Supplementary Fig. S5). A well-documented effect of CUR is the induction of heme oxygenase 1 (HO-1) in various cell types and HO-1 has been reported to interact with the activity of transcription factors including STAT proteins26. Therefore, we investigated the regulation of HO-1 in DC by CUR treatment. Quantitative analysis showed that CUR enhanced Hmox1 mRNA and HO-1 protein expression in DC, especially after activation with LPS (Fig. 7c,d). To evaluate possible effects of HO-1 on STAT3 activation we overexpressed HO-1 by an adenoviral construct in DC (Fig. 7e) and studied the levels of phosphorylation of STAT3 by Western blotting. Strikingly, Ad HO-1 but not Ad CON transfection resulted in enhanced STAT3 phosphorylation, independently of LPS activation (Fig. 7f). To translate these findings to the in vivo situation we studied the expression of HO-1, IL-12 and IL-23 in draining lymph nodes of mice immunized for EAE on day 3. Mice on oral CUR diet showed a significant induction of Hmox1 expression and suppressed levels of Il12b and Il23a, while Il12a expression was not affected by CUR (Fig. 8).
Figure 8. CUR induces Hmox1 and suppresses Il12b and Il23a expression in vivo.
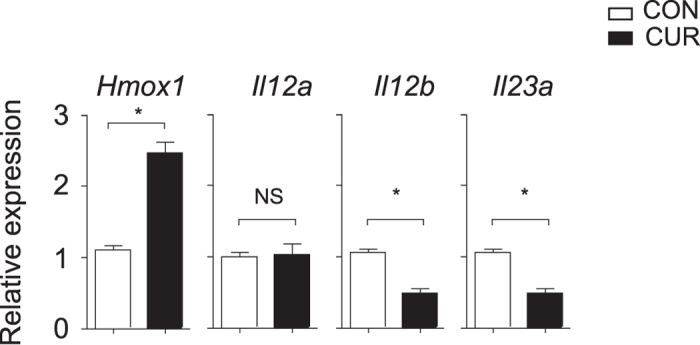
Mice were fed with CUR or a control diet (CON) and immunized for EAE. On day three after immunization draining lymph nodes were isolated and expression of Hmox1, Il12a, Il12b and Il23a was determined by quantitative RT-PCR. Bars represent mean ± SEM of control mice (n = 5) and CUR-fed mice (n = 5). Data were normalized to ß-actin (*P < 0.05, NS: not significant, Mann-Whitney test).
Taken together, we could show that the nutritional CUR negatively regulates Il12b and Il23a expression through HO-1 and STAT3 interaction (Supplementary Fig. S6). Our study shows how low concentrations of a classical nutritional factor can influence molecular events that link anti-oxidative and anti-inflammatory signalling pathways, and, that help to control T cell-mediated autoimmunity.
Discussion
Numerous studies have highlighted the beneficial effects of dietary CUR as anti-tumoral and anti-inflammatory phytochemical8,9,10,11,12,13,14,15,16. Mechanistically, CUR has been reported to interact with multiple signaling pathways18,19. Many of the molecular and cellular interactions observed, require high concentrations of CUR and some of these interactions are associated with toxic or apoptotic effects. For instance CUR at doses of 50 μM has been reported to inhibit NF-κB activation but in addition induces apoptotic cell death in myeloma cell lines studied. In contrast, CUR at low concentrations (≤10 μM) did not show such effects in this setting27. Similar results have been observed in other cell types treated with CUR in vitro demonstrating that only low concentrations of CUR (≤10 μM) allow study specific molecular events that are not linked to apoptotic pathways or loss of cell viability. This is of importance, since the in vivo absorption of dietary CUR is low and the tissue levels achieved are around 0.5 to 3 μM28,29. In clinical studies CUR is administrated orally using capsules, solid lipid nanoparticles or complex formulations30.
Here, we show that oral CUR diet prevents severe encephalomyelitis in mice, a model that strictly depends on IL-23 and Th17 responses31. The data on the clinical EAE parameters are in agreement with other reports demonstrating that injections of CUR improve EAE in rodents13,14,15,16. Though, we did not find any beneficial effects of dietary CUR in a Th2-dominated asthma model using OVA. Other groups have revealed an attenuation of airway inflammation in mice by CUR32,33,34. The discrepancy of the results between our study and others could be caused by the different route of CUR administration (intranasal or intraperitoneal administration versus oral feeding) and by the different protocols used for the induction of airway inflammation (repetitive OVA exposure and chronic airway inflammation versus acute airway inflammation)32,33. The airway inflammation model used by us is less sensitive to IL-23, a cytokine critically affected by oral CUR administration.
The main immune cell population directly affected by CUR seems to be the DC population. In our studies, CUR concentrations up to15 μM had no effect on DC surface receptors. Other reports demonstrate that CUR concentrations of ≥20 μM impair the expression of costimulatory molecules by DC17,35,36 and as a consequence inhibit their capacity to stimulate T cell proliferation17,35,36,37. Since in our experimental setting, CUR concentrations ≥20 μM started to affect DC viability, we used low CUR concentrations that neither affected DC viability nor DC maturation (Fig. 4). Importantly, no suppressive effects on T cell proliferation have been observed when using DC treated with CUR ≤ 10 μM in vitro10 nor by oral CUR diet in vivo (Figs 1b and 2b). This is in agreement with a report demonstrating that CUR supplemented in drinking water does not affect antigen-specific T cell proliferation in mice immunized for EAE38.
In this study, we found that low CUR concentrations equip DC with an anti-inflammatory profile. Treatment of DC with 7.5 μM CUR specifically suppressed the expression of LPS-induced IL-12p40 and IL-23p19 (Figs 4b and 6c) and by this attenuated inflammatory Th17 responses and instead promoted Th2 cell development (Fig. 1d). In the human setting dietary supplementation of CUR could be beneficial for patients with IL-23/Th17-dominated inflammatory autoimmune disease like multiple sclerosis. To unravel the molecular mechanisms we focused on signaling pathways that had been linked to CUR and to the inflammatory status of DC. One of these pathways is the NF-κB pathway, which is also implicated in the regulation of Il12b and Il23a20,39. In vitro, CUR concentrations ≥25 μM have been reported to affect NF-κB activity in activated DC and macrophages17,40,41. However, in vivo, CUR showed no direct effects on NF-κB activity, as demonstrated by using NF-κB-driven reporter gene transgenic mice under inflammatory conditions40,41. One other signaling pathway linked to the effects of CUR is the JAK/STAT pathway. In vitro, CUR has been reported to modulate cytokine-dependent STAT4 phosphorylation in human T cells and to inhibit STAT3 phosphorylation in multiple cell types when using doses of 20 to 50 μM13,23,42,43,44. As mentioned above, the concentrations used to study the effects of CUR on signaling pathways are critical, since concentrations of CUR ≥ 20 μM can impair cell viability and induce apoptosis43,44. To our surprise, we found enhanced STAT3 activation in DC treated with CUR < 10 μM, which resulted in a negative regulation of Il12b and Il23a in LPS-activated DC. Activated STAT3 has been reported previously to impair Il12b gene expression and IL-12 protein secretion by LPS-activated DC24,25,45,46,47. A similar mechanism seems to be valid for Il23a, but not for Il12a (Fig. 7a). Here we show that STAT3 binds to the promoter loci of Il12b and Il23a, suppresses the transcription of Il12b and Il23a accompanied with the negative regulation of H3Ac (Fig. 7b). DC themselves produce STAT3-activating cytokines like IL-6 or IL-10 upon LPS activation. We did not find significant changes in IL-6 or IL-10 secretion by CUR-treated DC in vitro, indicating that the mechanism of enhanced STAT3 activation by low-dose CUR is cell-intrinsic.
One of the molecular events readily induced by even minimal concentrations of CUR (2 μM) that can also be achieved in vivo is the induction of HO-1 as demonstrated in various cell types including macrophages48. We found a rapid induction of Hmox1 mRNA and HO-1 protein in DC by low-dose CUR (Fig. 7c,d). The expression of HO-1 is regulated by Nrf248,49 and CUR activates this signaling pathway. Whole genome expression profiling unraveled that oral CUR feeding regulates more than 800 NRF2-dependent genes in vivo with a >2-fold induction or suppression. Among these genes Hmox1 was one of the most highly induced (>75-fold induction in the small intestine) by oral CUR50. Likewise we found a significant increase of Hmox1 in draining lymph nodes of mice on CUR diet after immunization for EAE (Fig. 8).
As reported previously, HO-1 translocates into the nucleus where it interacts with the activity of classical transcription factors including NF-κB and STAT35,20,26, which are critically implicated in the regulation of Il12b and Il23a expression in DC and macrophages25,51. Here, we could identify that a nutritional affects IL-23-dominated autoimmune disease by influencing the HO-1/STAT3 axis. CUR induced HO-1 expression and STAT3 phosphorylation in DC resulting in an anti-inflammatory DC phenotype. By overexpression of HO-1 in DC, we could show an enhancement of STAT3 phosphorylation (Fig. 7f). This mechanism is also relevant in vivo. Recent studies demonstrated that feeding CUR enhances STAT3 activation as shown in rodent models of chemically-induced colitis52 and ischemia-reperfusion injury53,54. Nrf2/HO-1 is a key factor mediating the immune-regulatory properties of CUR and other Nrf2 activators5,20. The anti-inflammatory properties of HO-1/STAT3 interaction seem to be of general importance. As demonstrated recently, overexpression of HO-1 in Nrf2-deficient macrophages promotes STAT3 signaling and anti-inflammatory effects in a mouse model of liver ischemia/reperfusion injury55. Interestingly, the regulating effects of HO-1 on STAT3 activity seem to be cell-type specific. In keratinocytes, HO-1 activation by hemin or cobalt protoporphyrin inhibits STAT3 activity through the induction of SHP-156. This is different in DC, where HO-1 induction by CUR enhances STAT3 activity without affecting SHP-1 expression (Fig. 5 and Supplementary Fig. S5).
Taken together we could unravel the mechanism by which two proposed molecular targets of the nutritional CUR, namely HO-1 and STAT3 interact and determine the phenotype of DC controlling IL-23/Th17 responses (Supplementary Fig. S6).
Methods
Mice
SJL mice, 6–8 weeks of age, were purchased from Janvier, C57BL/6 mice, were purchased from Charles River or CLEA and maintained in the animal laboratories of the FORS Institute, University of Tübingen or in the animal facility of Chiba University. Animal experiments were approved by the Institutional Animal Care and Use Committee of the Regierungspräsidium Tübingen (HT01/08 and HT10/13) and the Chiba University Review Board for Animal Care. All animal procedures and methods were carried out in accordance with the approved guidelines and institutional regulations.
Experimental autoimmune encephalomyelitis
EAE was induced by subcutaneous immunization of female SJL mice with 37.5 μg PLP139-151 Peptide (Genaxxon bioscience) in Complete Freund’s Adjuvant (CFA) (Difco) and i.p. injection of 200 ng pertussis toxin (Calbiochem)20. SJL mice were fed with 2% curcumin (CUR)-containing food pellets or received a control diet (CON), starting 10 days prior to immunization. CUR (Sigma-Aldrich) was mixed into food pellets by a professional animal food manufacturer (Ssniff). CNS-infiltrating mononuclear cells were isolated on day 18 after immunization by using a commercial neural tissue dissociation kit (Miltenyi Biotec), followed by a Percoll® (Sigma-Aldrich) density gradient centrifugation, trypan blue staining and cell quantification using a Neubauer chamber. The clinical EAE score was followed and rated by the following scale: 0 - no disease; 1 - limp tail; 2 - hind limb weakness or partial paralysis; 3 - complete hind limb paralysis; 4 - fore limb and hind limb paralysis; 5 - moribund state20,57.
Airway inflammation model
C57BL/6 mice were fed with 2% CUR-containing food pellets or received a control diet from day 0. The mice were immunized with 250 μg OVA (Sigma-Aldrich) i.p. in 4 mg of aluminum hydroxide gel (alum) on days 8 and 15. The mice were nebulized with aerosolized 1% OVA in saline for 30 minutes using a supersonic nebulizer (model NE-U07; Omron) on days 22 and 24. Airway hyper responsiveness was assessed by measuring the changes in lung resistance in response to increasing doses of inhaled methacholine, as described previously58. For collection and anaylsis of bronchioalveolar lavage (BAL) fluid the lungs were lavaged with 1.0 ml of PBS one day after the last OVA inhalation (day 25). For the differential cell count, BAL fluid was cyto-centrifuged onto slides by a Cytospin 4 (Thermo Electron) and stained with Diff-Quick (Sysmex). For histology, the lung samples taken on day 25 and sections were stained with H&E and PAS reagents as previously described58. Slides were examined in a random blinded fashion. The numbers of infiltrated inflammatory cells at the peribronchial region were counted as visible on H&E stained lung sections. Infiltration of inflammatory cells in the lung was graded with a semiquantitative scoring system (5 = large widespread infiltrate around the majority of vessels and bronchioles to 1 = small number of inflammatory foci). Goblet cells were counted on Periodic Acid-Schiff (PAS)-stained lung sections. Percentage of PAS positive cells were calculated in each mouse (n = 3 in each group). The goblet cell hyperplasia was graded with a semiquantitative scoring system (0 = <5% goblet cells in airway epithelium; 1 = 5–25%; 2 = 25–50%; 3 = 50–75%; 4 = >75%) as described59. The average of the airway scores from each lung was expressed as mucus score in arbitrary units.
RNA isolation and gene expression
Total RNA was purified from cultured cells or from ex vivo isolated cells and reverse transcribed into cDNA using commercially available kits (Biozym). Relative gene expression levels were determined by quantitative realtime-PCR (qRT-PCR) using TaqMan probes (TIB MolBiol) for ßactin, Il6, Il10, Il12a, Il12b, Il23a and Hmox1 and the LightCycler480 system (Roche). The relative expression of the indicated genes was calculated relative to the expression of ß-actin. Control conditions were set as 1.0 as indicated.
ßactin for: ACCCACACTgTgCCCATCTA, rev: gCCACAggATTCCATACCCA
TM: 6FAM-CATCCTgCgTCTggACCTggC-BBQ
Il6 for: CggAggCTTAATTACACATgTTCTC, rev: ggTAgCTATggTACTCCAgAAgACCA
TM: 6FAM-ACgATgATgCACTTgCAgAAAACAATCTgA-BBQ
Il10 for: AgCTggACAACATACTgCTAAC, rev: CTCTTATTTTCACAggggAGAA
TM: 6FAM-CgCCTCAgCCgCATCCTGAgggTC-BBQ
Il12a for: CATggTgAAgACggCCAgA, rev: CCAggCAACTCTCgTTCTTgT
TM: 6FAM-AggTCTTCAATgTgCTggTTTggTCCC-BBQ
Il12b for: gCTCAgAgTCTCgCCTCCTT, rev: gAgCTggAgAAAgACgTTTATgTTg
TM: 6FAM-ACATggAgTCATAggCTCTggAAAgACCC-BBQ
Il23a for: CACCAgCgggACATATgAATC, rev:CAgAACTggCTgTTgTCCTTgA
TM: 6FAM-CACTggATACggggCACATTATTTTTAgTC-BBQ
Hmox1 for: TgCTCgAATgAACACTCTggAgA, rev: gTTTCCCTCggggTgTCT
TM: 6FAM-ACgAAgTgACgCCATCTgTgAgggA-BBQ
Ptpn6 for: ggTgTCCTCAgCTTTCTggAT, rev: AATgTCACAgTCTAgCCCCTTg
TM: 6FAM-CCCATCATTgTgCATTgCAgCg-BBQ
Cytokine analysis and flow cytometry
Commercially available ELISA Kits were used for the quantification of the cytokines IL-12p70, IL-23, IL-6 and IL-10 from cell culture supernatants. Immune cells were isolated from draining lymph nodes of immunized mice on day 3 or 7 as indicated and analzsed for cytokine production by qRT-PCR or flow cytometry. Intracellular cytokine staining was performed after stimulating cells with PMA and ionomycin (Sigma-Aldrich) in the presence of GolgiStop® (BD Biosciences) for 4 h20. Cells were fixed with 2% formaldehyde, permeabilized with saponin containing buffer and stained with fluorochrome-labeled antibodies directed against CD4 (Gk1.5, Biolegend) and IFN-γ (XMG1.2, eBioscience), IL-4 (11B11, eBioscience), IL-17A (TC11-18H10, BD Biosciences), IL-2 (JES6-5H4, eBioscience), IL-10 (JES5-16E3, eBioscience) or TNF (MP6-XT22, eBioscience). Fluorochrome-labeled anti-CD11c (HL3, BD Biosciences) antibodies were used for staining DC. Cells were analysed by flow cytometry (LSRII fow cytometer, BD Biosciences) and collected data were analysed by FLOWJO/FCS Express. Concentrations of indicated cytokines in BAL fluid were measured by Cytometric Bead Array (BD Biosciences, San Jose, CA, USA).
Cell culture
Bone marrow-derived DC were isolated and cultivated as described previously57,60. DC were harvested on day 7, stimulated with CUR (Sigma-Aldrich) in DMSO or DMSO alone as control and activated with 100 ng/ml LPS from E. coli O111:B4 (Sigma-Aldrich) for the indicated time points. CD4+ T cells were isolated from spleen and lymph nodes of naïve C57BL/6 mice by using CD4 microbeads and MACS® purification system (Miltenyi Biotec). Isolated T cells were activated for 3 days with 10 μg/ml plate-bound anti-CD3 and anti-CD28 antibodies (eBiosciences) with DMSO or 7.5 μM CUR in DMSO and expanded for 4 days with IL-2 (Chiron Therapeutics). For co-culture experiments, DC were first treated with 7.5 μM CUR in DMSO or DMSO for 1 h, left untreated or activated with 100 ng/ml LPS and loaded with 100 ng/ml ovalbumin (OVA323-339)-peptide (EMC microcollections) for another 1 h. Then, purified CD4+ T cells isolated from OT-II mice were cultured with these DC. On day 7 cytokine expression by T cells was determined after activation by intracellular staining and flow cytometry using a LSRII flow cytometer (BD Biosciences).
Il23p19 reporter assay
RAW 264.7 macrophages were transfected with a pGL3-Il23p19 reporter plasmid (bp −1180/+110 promoter fragment) using LipofectamineTM 2000 (Invitrogen). The p19 luciferase construct was a gift from Y. Chen (University of Pennsylvania, Philadelphia, PA)61. Twenty-four hours post transfection cells were pre-treated with 7.5 μM curcumin in DMSO or DMSO alone for 1 h and activated with 100 ng/ml LPS. After 6 h cells were lysed with passive lysis buffer (Promega) and luciferase activity was measured using a luminescence microplate reader (Berthold Technologies). Protein concentration of each sample was determined by a protein assay (Roth) and luciferase activity was calculated in relation to the protein content.
Western blotting
DC were incubated with 7.5 μM curcumin in DMSO or DMSO alone and activated with 100 ng/ml LPS for the indicated time points. Cells were washed in ice-cold PBS, lysed and heated in SDS sample buffer [125 mM Tris-HCl (pH 6.8), 2% w/v SDS, 10% glycerol, 100 mM DTT, and 0.01% w/v bromphenol blue] at 95 °C for 5 min. Equal amounts of samples were loaded onto a 12% PAA gel and transferred to a PVDF membrane or to a nitrocellulose membrane. After blocking with blocking buffer for 1 h the membrane was incubated with anti-actin (Millipore), anti-pStat3, anti-Stat3, anti-SHP-1 (Cell Signaling Technology) or anti-HO-1 (Enzo Life Sciences) antibodies overnight at 4 °C, followed by incubation with IRDye680RD or IRDye800CW-conjugated secondary antibody (Licor) for 1 h. Proteins were detected by OdysseySA Infrared Imaging System (Licor). The band size was quantified using the OdysseySA software version 1.1. Western blot images were cropped based on the molecular weight of indicated proteins.
Overexpression of HO-1 and STAT3
E1- and E3-deleted adenoviral vectors (Ad) were expressing EGFP, CA STAT3 or mouse HO-1-EGFP were gifts from T. Tüting (University of Bonn, Germany), H. Inoue (Kanazawa University, Ishikawa, Japan) and J.W. Kupiec-Weglinski (University of California, Los Angeles, USA). All recombinant adenoviruses were based on Ad5 and contained the transgene under the control of the CMV immediate–early promoter. Viruses were propagated in 293 cells, purified by cesium chloride density gradient centrifugation and subsequent dialysis according to standard protocols, and stored at 70 °C. Adenoviruses were added to DC cultures on day 8 at a multiplicity of infection of 300. Cells were incubated for 24 h at 37 °C and 7.5% CO2 before stimulation with LPS for indicated time points. Infection efficiency was confirmed by flow cytometry and Western blot analysis of HO-1. For inhibition of STAT3 activity in RAW 264.7 macrophages a pharmacological inhibitor (STAT3 inhibitor V, Calbiochem) was used.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously20. In brief, DC were treated with curcumin in DMSO (7.5 μM) or DMSO alone for 1 h and activated with 100 ng/ml LPS for 1.5 h. Cells were cross-linked, harvested, and lysed by sonication. Subsequently, DC lysates were immunoprecipitated with anti-Stat3 (Cell Signaling Technology) or anti-acetyl histone H3 (Millipore) antibodies. The precipitated and eluted DNA was analyzed by qRT-PCR. Primers were designed to amplify 150–250 bp fragments from selected genomic regions. The qRT-PCR was performed in duplicate on each sample and input DNA using LightCycler480 (Roche) and TaqMan probes (TIB MolBiol) according to the manufacturers’ instructions. To account for differences in DNA quantity, for every genomic sequence studied, a ΔCt value was calculated for each sample by subtracting the Ct value of the chromatin-immunoprecipitated sample from the Ct value obtained for the input and no antibody ChIP, respectively. Calculating 2ΔΔCt yielded the relative amount of PCR product (relative enrichment). Binding sites for Stat3 within the Il23a, Il12b and Il12a promoter regions were identified with Matinspector (Genomatix). Primers encompassing putative STAT3-binding sites in the indicated promoter regions were designed by TIB MolBiol.
Il23a for: AggCAggTggATTTCTgAgTTC, rev: ggTTgCTCTTCCAgAggTCCT
TM: 6FAM-CAgCCAAggCTATACAgAgAAACCCTgT-BBQ,
Il12b for: gggCCTgTAACACCTACTTATTTgA; rev: ggCTAACCTCTCCCgATTCC
TM: 6FAM-ACCgAgTTCATggTTCATgCCACA-BBQ,
Il12a for: TTTTAAgACAAgCTCTCATgTCATTg,
rev: TAgATgCTgAgCCAgTgATgAgTACA
TM: 6FAM-AgCTATgTTTATggCTgAggCACAAgTTCA-BBQ
T cell proliferation assay
For determining proliferation of in vivo activated T cells during EAE, CD4+ cells were isolated from draining lymph nodes of immunized mice and co-cultured with PLP-pulsed DC for 3 days. 3H-thymidine (0,25 μCi/well; PerkinElmer) was added for 16 h prior to harvest. Incorporation of radioactivity was determined by scintillation counting. Data are shown as means of triplicate cultures. The standard deviation was less than 10% of the mean cpm.
Electrophoretic Mobility Shift Assay
DC were treated with curcumin in DMSO (7.5 μM) or DMSO alone for 1 h and activated with 100 ng/ml LPS for 1 h. Nuclei were extracted using nuclear and cytoplasmic extraction reagents (Thermo Scientific). The binding mixture (20 μl) consisting of 5 μg protein of nuclear extract, 25 mM DTT, 2,5% Tween-10, 1%NP40, 100 mM MgCl2, 1 μg poly(di-dC) and 10 ng of a IRDye 700 STAT3 Consensus Oligonucleotide was incubated for 1 h at RT. The DNA-protein complexes were separated on native 15% TAE and detected by a OdysseySA Infrared Imaging System (Licor). The band intensity was quantified using the OdysseySA software version 1.1 (Licor).
Statistics
All data were analyzed and plotted using GraphPad Prism 6 software. Statistical analyses were performed by using the Mann-Whitney test, the Wilcoxon test or the Tukey’s post-hoc test after One-way ANOVA as indicated. Values of P < 0.05 were considered significant.
Additional Information
How to cite this article: Brück, J. et al. Nutritional control of IL-23/Th17-mediated autoimmune disease through HO-1/STAT3 activation. Sci. Rep. 7, 44482; doi: 10.1038/srep44482 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We thank Thomas Tüting, Hiroshi Inoue and Jerzy W. Kupiec-Weglinski for providing the indicated vectors. We thank Eva Müller-Hermelink for excellent technical support. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) Sonderforschungsbereich (SFB) transregio (TR-)156 TPA06/B07/B06 (to K.G., F.C.E., M.R., and A.S.Y.), the SFB 685 (to K.G. and A.S.Y.), DFG Ro 764/14-1 (to M.R.) and the Bundesministerium für Bildung und Forschung Grant 0315079 (to K.G.).
Footnotes
The authors declare no competing financial interests.
Author Contributions Conceptualization and designed experiments: J.B., K.H. and K.G. Performed EAE and in vitro experiments: J.B., J.H., I.G., U.S., J.G., K.W., T.S. Performed asthma experiments: T.K., J.K., N.M. and S.S. J.B. Data analysis and interpretation: J.B., A.S.Y., F.C.E., K.H. and K.G. Writing, review and editing: J.B., A.S.Y., F.C.E., K.H., T.N., M.R. and K.G. Supervision: K.G.
References
- De Rosa V. et al. Nutritional control of immunity: Balancing the metabolic requirements with an appropriate immune function. Semin Immunol 27, 300–309 (2015). [DOI] [PubMed] [Google Scholar]
- Furusawa Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013). [DOI] [PubMed] [Google Scholar]
- Trompette A. et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20, 159–166 (2014). [DOI] [PubMed] [Google Scholar]
- Tortorella S. M., Royce S. G., Licciardi P. V. & Karagiannis T. C. Dietary Sulforaphane in Cancer Chemoprevention: The Role of Epigenetic Regulation and HDAC Inhibition. Antioxid Redox Signal 22, 1382–1424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisel J. et al. Sulforaphane protects from T cell-mediated autoimmune disease by inhibition of IL-23 and IL-12 in dendritic cells. J Immunol 192, 3530–3539 (2014). [DOI] [PubMed] [Google Scholar]
- Jang M. et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275, 218–220 (1997). [DOI] [PubMed] [Google Scholar]
- Xuzhu G. et al. Resveratrol modulates murine collagen-induced arthritis by inhibiting Th17 and B-cell function. Ann Rheum Dis 71, 129–135 (2012). [DOI] [PubMed] [Google Scholar]
- Kawamori T. et al. Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res 59, 597–601 (1999). [PubMed] [Google Scholar]
- Capini C. et al. Antigen-specific suppression of inflammatory arthritis using liposomes. J Immunol 182, 3556–3565 (2009). [DOI] [PubMed] [Google Scholar]
- Cong Y. et al. Curcumin induces the tolerogenic dendritic cell that promotes differentiation of intestine-protective regulatory T cells. Eur J Immunol 39, 3134–3146 (2009). [DOI] [PubMed] [Google Scholar]
- Castro C. N. et al. Curcumin ameliorates autoimmune diabetes. Evidence in accelerated murine models of type 1 diabetes. Clin Exp Immunol 177, 149–160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M. J. et al. Curcumin attenuates acute graft-versus-host disease severity via in vivo regulations on Th1, Th17 and regulatory T cells. PLoS One 8, e67171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan C. & Bright J. J. Curcumin inhibits experimental allergic encephalomyelitis by blocking IL-12 signaling through Janus kinase-STAT pathway in T lymphocytes. J Immunol 168, 6506–6513 (2002). [DOI] [PubMed] [Google Scholar]
- Xie L. et al. Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL-17 production. Int Immunopharmacol 9, 575–581 (2009). [DOI] [PubMed] [Google Scholar]
- Zhuang X. et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther 19, 1769–1779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanakasabai S. et al. Differential regulation of CD4(+) T helper cell responses by curcumin in experimental autoimmune encephalomyelitis. J Nutr Biochem 23, 1498–1507 (2012). [DOI] [PubMed] [Google Scholar]
- Kim G. Y. et al. Curcumin inhibits immunostimulatory function of dendritic cells: MAPKs and translocation of NF-kappa B as potential targets. J Immunol 174, 8116–8124 (2005). [DOI] [PubMed] [Google Scholar]
- Shehzad A. & Lee Y. S. Molecular mechanisms of curcumin action: signal transduction. Biofactors 39, 27–36 (2013). [DOI] [PubMed] [Google Scholar]
- Panahi Y. et al. Molecular mechanisms of curcumins suppressing effects on tumorigenesis, angiogenesis and metastasis, focusing on NF-kappaB pathway. Cytokine Growth Factor Rev (2016). [DOI] [PubMed] [Google Scholar]
- Ghoreschi K. et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med 208, 2291–2303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B., Tang H. & Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol 11, 647–655 (2010). [DOI] [PubMed] [Google Scholar]
- Li H. S. & Watowich S. S. Diversification of dendritic cell subsets: Emerging roles for STAT proteins. JAKSTAT 2, e25112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y., Park E. J., Joe E. H. & Jou I. Curcumin suppresses Janus kinase-STAT inflammatory signaling through activation of Src homology 2 domain-containing tyrosine phosphatase 2 in brain microglia. J Immunol 171, 6072–6079 (2003). [DOI] [PubMed] [Google Scholar]
- Hutchins A. P., Takahashi Y. & Miranda-Saavedra D. Genomic analysis of LPS-stimulated myeloid cells identifies a common pro-inflammatory response but divergent IL-10 anti-inflammatory responses. Sci Rep 5, 9100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoentjen F., Sartor R. B., Ozaki M. & Jobin C. STAT3 regulates NF-kappaB recruitment to the IL-12p40 promoter in dendritic cells. Blood 105, 689–696 (2005). [DOI] [PubMed] [Google Scholar]
- Lin Q. et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem 282, 20621–20633 (2007). [DOI] [PubMed] [Google Scholar]
- Bharti A. C., Donato N., Singh S. & Aggarwal B. B. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 101, 1053–1062 (2003). [DOI] [PubMed] [Google Scholar]
- Begum A. N. et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther 326, 196–208 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. S., Sang S., Lambert J. D. & Lee M. J. Bioavailability issues in studying the health effects of plant polyphenolic compounds. Mol Nutr Food Res 52 Suppl 1, S139–151 (2008). [DOI] [PubMed] [Google Scholar]
- Kunnumakkara A. B. et al. Curcumin, the golden nutraceutical: multitargeting for multiple chronic diseases. Br J Pharmacol, in press (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K. et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467, 967–971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman M. et al. Anti-inflammatory effects of curcumin in a murine model of chronic asthma. Allergol Immunopathol (Madr) 40, 210–214 (2012). [DOI] [PubMed] [Google Scholar]
- Chauhan P. S., Subhashini, Dash D. & Singh R. Intranasal curcumin attenuates airway remodeling in murine model of chronic asthma. Int Immunopharmacol 21, 63–75 (2014). [DOI] [PubMed] [Google Scholar]
- Chong L. et al. Protective effect of curcumin on acute airway inflammation of allergic asthma in mice through Notch1-GATA3 signaling pathway. Inflammation 37, 1476–1485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley S. A., Montpetit A. J., Lockey R. F. & Mohapatra S. S. Curcumin prevents human dendritic cell response to immune stimulants. Biochem Biophys Res Commun 374, 431–436 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers N. M., Kireta S. & Coates P. T. Curcumin induces maturation-arrested dendritic cells that expand regulatory T cells in vitro and in vivo. Clin Exp Immunol 162, 460–473 (2010).21070208 [Google Scholar]
- Jeong Y. I. et al. Curcumin suppresses the induction of indoleamine 2,3-dioxygenase by blocking the Janus-activated kinase-protein kinase Cdelta-STAT1 signaling pathway in interferon-gamma-stimulated murine dendritic cells. J Biol Chem 284, 3700–3708 (2009). [DOI] [PubMed] [Google Scholar]
- Verbeek R., van Tol E. A. & van Noort J. M. Oral flavonoids delay recovery from experimental autoimmune encephalomyelitis in SJL mice. Biochem Pharmacol 70, 220–228 (2005). [DOI] [PubMed] [Google Scholar]
- Mise-Omata S. et al. A proximal kappaB site in the IL-23 p19 promoter is responsible for RelA- and c-Rel-dependent transcription. J Immunol 179, 6596–6603 (2007). [DOI] [PubMed] [Google Scholar]
- Larmonier C. B. et al. Limited effects of dietary curcumin on Th-1 driven colitis in IL-10 deficient mice suggest an IL-10-dependent mechanism of protection. Am J Physiol Gastrointest Liver Physiol 295, G1079–1091 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. A. et al. Oral administration of nano-emulsion curcumin in mice suppresses inflammatory-induced NFkappaB signaling and macrophage migration. PLoS One 9, e111559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey A. J., Adrian Robins R. & Constantinescu C. S. Curcumin modulation of IFN-beta and IL-12 signalling and cytokine induction in human T cells. J Cell Mol Med 11, 1129–1137 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill M. A. et al. Structurally modified curcumin analogs inhibit STAT3 phosphorylation and promote apoptosis of human renal cell carcinoma and melanoma cell lines. PLoS One 7, e40724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenberger J. et al. Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1,2/STAT3 signaling pathway. Clin Cancer Res 16, 5781–5795 (2010). [DOI] [PubMed] [Google Scholar]
- Kortylewski M. et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11, 1314–1321 (2005). [DOI] [PubMed] [Google Scholar]
- Melillo J. A. et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol 184, 2638–2645 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assi H. et al. Assessing the role of STAT3 in DC differentiation and autologous DC immunotherapy in mouse models of GBM. PLoS One 9, e96318 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S. O. et al. Dimethoxycurcumin, a Synthetic Curcumin Analogue, Induces Heme Oxygenase-1 Expression through Nrf2 Activation in RAW264.7 Macrophages. J Clin Biochem Nutr 44, 79–84 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. et al. Curcumin ameliorates asthmatic airway inflammation by activating nuclear factor-E2-related factor 2/haem oxygenase (HO)-1 signalling pathway. Clin Exp Pharmacol Physiol 42, 520–529 (2015). [DOI] [PubMed] [Google Scholar]
- Shen G. et al. Modulation of nuclear factor E2-related factor 2-mediated gene expression in mice liver and small intestine by cancer chemopreventive agent curcumin. Mol Cancer Ther 5, 39–51 (2006). [DOI] [PubMed] [Google Scholar]
- Kortylewski M. et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15, 114–123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Q. et al. Dietary fish oil and curcumin combine to modulate colonic cytokinetics and gene expression in dextran sodium sulphate-treated mice. Br J Nutr 106, 519–529 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W. et al. The effects of curcumin post-treatment against myocardial ischemia and reperfusion by activation of the JAK2/STAT3 signaling pathway. Basic Res Cardiol 107, 263 (2012). [DOI] [PubMed] [Google Scholar]
- Li L., Li H. & Li M. Curcumin protects against cerebral ischemia-reperfusion injury by activating JAK2/STAT3 signaling pathway in rats. Int J Clin Exp Med 8, 14985–14991 (2015). [PMC free article] [PubMed] [Google Scholar]
- Huang J. et al. Adoptive transfer of heme oxygenase-1 (HO-1)-modified macrophages rescues the nuclear factor erythroid 2-related factor (Nrf2) antiinflammatory phenotype in liver ischemia/reperfusion injury. Mol Med 20, 448–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. et al. Heme oxygenase-1 induction attenuates imiquimod-induced psoriasiform inflammation by negative regulation of Stat3 signaling. Sci Rep 6, 21132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brück J. et al. Cholesterol Modification of p40-Specific Small Interfering RNA Enables Therapeutic Targeting of Dendritic Cells. J Immunol 195, 2216–2223 (2015). [DOI] [PubMed] [Google Scholar]
- Hirahara K. et al. Repressor of GATA regulates TH2-driven allergic airway inflammation and airway hyperresponsiveness. J Allergy Clin Immunol 122, 512–520 e511 (2008). [DOI] [PubMed] [Google Scholar]
- Matthews S. P., McMillan S. J., Colbert J. D., Lawrence R. A. & Watts C. Cystatin F Ensures Eosinophil Survival by Regulating Granule Biogenesis. Immunity 44, 795–806 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz M. B. et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223, 77–92 (1999). [DOI] [PubMed] [Google Scholar]
- Carmody R. J., Ruan Q., Liou H. C. & Chen Y. H. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol 178, 186–191 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.