

Abstract
Growth of Salmonella enterica serovar Typhimurium strain 14028 with myo-inositol (MI) is characterized by a bistable phenotype that manifests with an extraordinarily long (34 h) and variable lag phase. When cells were pre-grown in minimal medium with MI, however, the lag phase shortened drastically to eight hours, and to six hours in the absence of the regulator IolR. To unravel the molecular mechanism behind this phenomenon, we investigated this repressor in more detail. Flow cytometry analysis of the iolR promoter at a single cell level demonstrated bistability of its transcriptional activation. Electrophoretic mobility shift assays were used to narrow the potential binding region of IolR and identified at least two binding sites in most iol gene promoters. Surface plasmon resonance spectroscopy quantified IolR binding and indicated its putative oligomerization and high binding affinity towards specific iol gene promoters. In competitive assays, the iolR deletion mutant, in which iol gene repression is abolished, showed a severe growth disadvantage of ~15% relative to the parental strain in rich medium. We hypothesize that the strong repression of iol gene transcription is required to maintain a balance between metabolic flexibility and fitness costs, which follow the inopportune induction of an unusual metabolic pathway.
Bacterial pathogens often encounter limited availability of carbon, nitrogen and energy sources during their life cycles due to competition with the host organism and the commensal microbiota for nutrients. Increasing evidence suggests that enteric pathogens have developed specific metabolic strategies to overcome those restrictions and thus increase their fitness in the environment and in vivo. Metabolic adaptations of salmonellae during infection include the degradation of ethanolamine cleaved from phospholipids, fucose derived from mucosal glycoconjugates, 1,2-propanediol, the fermentation product of fucose or rhamnose1, and other potential substrates not accessible to commensal bacteria or present in competition-free microenvironments.
The ability of Salmonella enterica serovar Typhimurium (S. Typhimurium) to grow with myo-inositol (MI) as its sole carbon and energy source provides another example of an adaptive metabolic pathway with a potential role in vivo. In S. Typhimurium 14028, the iol genes responsible for MI utilization are located on a 22.6-kb genomic island (GEI4417/4436)2. The catabolic pathway requires the IolG- and IolE-mediated convertion of MI to 3D-trihydroxycyclohexane-1,2-dione, which is further hydrolysed by IolD. Next, 5-deoxy-glucuronic acid is isomerized by IolB to 2-deoxy-5-keto-D-gluconic acid, which is in turn phosphorylated by the kinase IolC and degraded to dihydroxyacetone phosphate, acetyl coenzyme A and CO2. The activator ReiD has been shown to interact with the promoter of iolE and thus to positively regulate iol gene expression3.
A remarkable and unique property of S. Typhimurium 14028 cultivated in minimal medium (MM) with MI is the manifestation of a heterogeneous growth phenotype that is abolished in the absence of the iol gene repressor IolR or the presence of at least 0.55% CO24. This phenotypic heterogeneity correlates with the bistable expression of iolE and iolG, the products of which catalyze the first steps in MI degradation. At the single cell level, fluorescence microscopy and flow cytometry (FC) analyses revealed that increasing numbers of cells exhibit PiolE activation during growth with MI4. More recently, we demonstrated that a shift from MI medium to LB medium is characterized by a “memory” of salmonellae for the former medium, and that this hysteresis is successively lost after 8 hours in a medium without MI5.
A key player in MI utilization is the negative autoregulator IolR that represses in rich medium all but one promoter of the iol genes involved in MI degradation, including its own promoter2 and that of the iolT1 gene encoding the predominant MI transporter in S. Typhimurium 140286. An intermediate of MI degradation, 2-deoxy-5-keto-D-gluconic acid 6-phosphate (DKGP), has been shown to antagonize IolR binding, thus inducing the expression of iol genes7,8. Here, we investigated IolR in greater detail, as this repressor plays a pivotal role in the coordinated control of MI degradation and the observed phenotypic heterogeneity. Using quantitative growth data of the wildtype and the iolR deletion mutant, the key parameters of growth with MI were delineated. The transcriptional activity of the iolR promoter was analyzed at a single cell level via FC. Electrophoretic mobility shift assays (EMSAs) and surface plasmon resonance (SPR) spectroscopy qualitatively characterized the binding properties of IolR towards its target promoters. Finally, competitive growth assays suggested that induced cells suffer a growth disadvantage in the absence of MI, a constraint that might contribute to the tight repression of iol gene transcription in rich medium.
Results
IolR contributes to lag-phase length during growth with MI
The lag phase of S. Typhimurium in the presence of MI has not previously been evaluated in detail. Therefore, we measured cultures that were inoculated with cells pregrown either in LB medium or on solid MM/MI. An overnight culture of 14028 in LB medium was adjusted to an OD600 = 0.8 and then diluted 1:500 into MM/MI. When this dilution was further cultivated under standard conditions, a lag phase of approximately 34 h was observed (Fig. 1A). The length of this lag phase was characterized by a low level reproducibility, which was reflective of the stochastic and heterogeneous growth behaviours under this condition as indicated by high standard deviations4. In contrast, when strain 14028 was pregrown on MM/MI agar plates, adjusted to an OD600 = 0.8 and diluted as described, the resulting culture exhibited a much shorter lag phase of 8 h, which could be attributed to the fact that the cells had already adapted to MI utilization. As a control, a growth curve of strain 14028 pregrown in LB medium that was subsequently diluted into fresh LB medium showed a lag phase of <2 h (Fig. 1A). The standard deviations of data from the experiment using 14028 pregrown on MM/MI were similar to those observed in the control, indicating that strain 14028 grows homogeneously in MM after preadaptation to this medium.
Figure 1. Lag phases of S. Typhimurium during growth with MI.
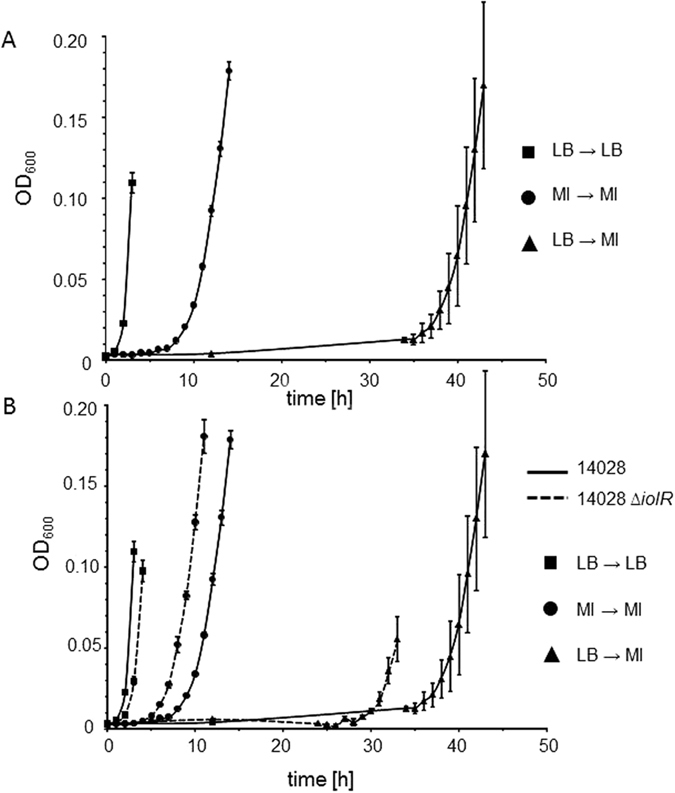
(A) Strain 14028 pre-grown in MM/MI and then diluted 1:500 into MM/MI exhibited a much shorter lag phase in comparison with that of a culture inoculated with a pre-culture in LB medium. The growth curve of a culture taken from LB medium and re-inoculated into the same medium is shown as a control. (B) Lag phases were shortened by deletion of the repressor gene iolR. Growth curves of 14028 from (A) are displayed in black, and those of 14028 ∆iolR are indicated by dashed lines. Growth curves were derived as described in (A). Standard deviations were calculated from three biologically independent measurements.
Next, mutant 14028 ∆iolR, which lacks the repressor IolR, was investigated under the experimental settings described above. When an LB culture was used to inoculate a MM/MI culture, we observed a reduction in the lag phase to 30 h, as well as a reduced population heterogeneity, as indicated by the low standard deviation, relative to the growth of the parental strain (Fig. 1B). When the mutant was pre-grown on MM/MI prior to the dilution into fresh MM/MI, a lag phase of 6 h was observed compared with 8 h observed for strain 14028 under the same conditions. In the LB control experiment, the lag phase of the ∆iolR mutant was one hour longer than that of strain 14028, possibly because the targeted induction of the MI degradation pathway in 14028 ΔiolR led to weak growth attenuation (see below).
Taken together, the lag phase of S. Typhimurium growth in MM/MI ranges between 6 h and 34 h under the conditions used herein.
Temporal analysis of P iolR transcription by FC
We have previously demonstrated at the single cell level that the promoter of iolE is bistable4. The promoter of iolR, PiolR, is transcriptionally active in the presence of both glucose and MI2. To investigate the transcription of iolR in more detail, we performed a temporal FC analysis of strain MvP101 with the transcriptional fusion PiolR::gfp during growth in MM with MI. Figure 2A shows that all cells exhibit fluorescence below the threshold level at time point zero (i. e. when LB-cultured cells were inoculated into MM/MI medium). During cultivation until the end of the lag phase, the number of cells exhibiting a higher level of fluorescence steadily increased, and two subpopulations with a low and a high expression level were detected. 14 hours after inoculation, a temporary increase of the fluorescence activity was visible. At this time point, the division rate of the cells was still low, possibly resulting in an accumulation of GFP within the cells. To test whether the translation of iolR is also subject to bistability, we measured the fluorescence of strain MvP101 harboring the translational fusion iolR::gfp in the presence of MI. The data shown in Fig. 2B generally resemble those obtained with a transcriptional reporter fusion protein, indicating that the transcriptional bistability is not abolished during IolR production by strain MvP101. Samples of the control strain MvP101 PrpsM::gfp with constitutive expression were collected and measured for up to 48 h of growth in MM/MI, during which time we observed averages of approximately 93% cells with and 7% cells without gfp expression (Fig. 2C). As the fusion PrpsM::gfp is expected to be active in viable cells only, the data demonstrate that the majority of cells without significant fluorescence activity are not dead cells.
Figure 2. Temporal FC analysis of PiolR activity in MM with MI.

(A) MvP101 PiolR::gfp or (B) MvP101 iolR::gfp grown in LB medium was adjusted to an OD600 of 0.8, diluted 1:500 into MM/MI and incubated at 37 °C without agitation. At the indicated time points during growth, samples were collected and GFP-expressing cells were quantified by FC. The abscissa of each histogram represents the green fluorescence intensity at 515–545 nm on a bi-exponential scale, and the ordinate represents the numbers of bacteria relative to the maximal cell counts. Each histogram shows one representative data set of three biologically independent measurements. The inset illustrates the growth phase of the population during sample acquisition. (C) FC measurement of MvP101 PrpsM::gfp as a control. The percentages of gfp-expressing cells (left) and non-fluorescent cells (right) are indicated. Samples were collected during a 48-h growth period after inoculation, and the average values and standard deviations of three independent growth experiments are shown; ***p < 0.001.
As an indicator for heterogeneous gene expression within a population, we calculated the variability in the fluorescence intensity, or the noise-level9, over all the growth phases in MM/MI of the two reporter strains described above (Table 1). The noise-levels of both MvP101 PiolR::gfp and MvP101 iolR::gfp were highest (1.67 ± 0.13) at the beginning of the lag phase and decreased to minimum levels (0.08 ± 0.0) during the stationary phase, when the cell populations showed more homogeneous gene expression. In contrast, the control strain MvP101 PrpsM::gfp exhibited an average noise-level of 0.01 ± 0.01 during all growth stages.
Table 1. Fluorescence variance (noise level) of MvP101 P iolR ::gfp and MvP101 iolR::gfp from three independent growth experiments in MM/MI.
| Hours | noise-level with s.d. | growth phase | |
|---|---|---|---|
| MvP101 PiolR::gfp | 0 | 1.41 ± 1.05 | lag phase |
| 4 | 0.45 ± 0.02 | ||
| 10 | 0.16 ± 0.01 | ||
| 14 | 0.06 ± 0.02 | log phase | |
| 18 | 0.19 ± 0.06 | ||
| 38 | 0.12 ± 0.00 | stationary phase | |
| 48 | 0.08 ± 0.00 | ||
| MvP101 iolR::gfp | 0 | 1.67 ± 0.13 | lag phase |
| 24 | 1.68 ± 0.58 | ||
| 26 | 1.49 ± 0.95 | ||
| 28 | 0.41 ± 0.29 | log phase | |
| 30 | 0.17 ± 0.10 | ||
| 32 | 0.12 ± 0.07 | ||
| 34 | 0.13 ± 0.08 |
s. d., standard deviation.
To summarize, these findings demonstrate a bistable transcriptional activity of PiolR similar to that of PiolE.
Interaction of IolR-His6 with iol gene promoters
The binding of IolR to most iol promotor regions was demonstrated previously2,3,6. To delineate a molecular model for the interaction of IolR and its target sites, we here performed EMSAs using different-length promoter fragments that represented possible IolR binding regions. The regulator was overproduced in E. coli BL21(DE3) and purified as described in the Methods. Putative promoter fragments of iolR, iolT1, iolT2, iolA, reiD, iolC1 and iolD1 were incubated without or with increasing amounts of purified IolR-His6 protein, and the resulting protein-DNA complexes were separated on a polyacrylamide (PAA) gel10. The EMSAs demonstrated the specificity of IolR-His6 to its target DNA and narrowed the binding region of IolR within the promoters PiolR, PiolT1, PiolT2, PiolA, PreiD, PiolC1 and PiolD1 to a length of approximately 100 bp (Fig. 3). The molecular excess of protein over DNA for quantitative binding of most promoters tested was in a range between 5 to 21 and thus very low in comparison with other repressors tested under similar conditions11,12, pointing to a strong affinity of IolR to its target sequences. The bandshift experiments with the promoters PiolT1, PiolD1, PiolR, PiolC1 and PreiD exhibited at least two retarded protein/DNA complexes, suggesting an oligomerization of IolR with increasing protein concentration. We then investigated two fragments of 74 and 81 bp from PreiD with an overlap of only 7 nucleotides by EMSA and observed a binding of IolR-His6 to both sequences, indicating the presence of two independent IolR target sites (Fig. 3). Moreover, the molar excesses of approximately 20 and 5, respectively, for a complete shift of these two PreiD fragments demonstrate different affinities of IolR to the two binding sites.
Figure 3. IolR binding to promoter regions within GEI4417/4436.

The genomic regions of the island are highlighted, and the different fragments used for EMSAs are shown. Black fragments were bound by IolR, grey fragments not. The molar excess of protein over DNA is indicated above the gels. The argS (100 ng) promoter fragment of served as a negative control and competitor (c) DNA, and the first lane in each EMSA was loaded without IolR. Asterisks indicate fragments selected for SPR spectroscopy.
Quantification of DNA-binding by IolR-His6
To analyse the interaction of IolR with its target sites identified above in more detail, we performed a kinetic analysis of IolR-His6 binding to PiolT1, PiolR and PreiD via SPR spectroscopy using fragments selected from the EMSAs. IolR was confirmed to specifically interact with PiolT1, PiolR and PreiD, whereas no interaction was detected with the control promoter PpcfA (Fig. 4A–D). Because the maximal binding capacity (Rmax) for each sensorgram is >1.000 responsive units (RU) and the Rmax for an 1:1 interaction is expected to be approximately 20 RU, these data suggest that IolR binds as a higher oligomer and/or that multiple binding sites for IolR exist within the iol promoter regions. The finding that none of the sensorgrams followed 1:1 binding kinetics, as indicated by the hyperbolic curve shape, underscores the assumption that the sensorgrams represent the sums of different binding events. To calculate reliable binding constants and kinetic parameters, we performed an IM analysis and determined the individual binding events for the SPR spectroscopy curves of IolR-PiolT1. Based on the sensorgrams, the IM analysis identified two clearly separated peak values (Fig. 4E) that represented a high-affinity interaction, and an interaction with an approximately 400-fold lower affinity. The first peak (blue) had a peak weight of 40.5%, an ON-rate of 6.95 × 105 M−1s−1 and an OFF-rate of 5.78 × 10−4s−1, resulting in an overall high affinity of 0.81 nM (Fig. 4E and F, upper panel). The second peak (green) had a peak weight of 57.6% and an ON-rate of 1.05 × 105 M−1s−1. The OFF-rate for this interaction was determined to be 3.2 × 10−2s−1, which was 50-fold higher than the first peak (depicted in blue), corresponding to a lower KD of 312 nM (Fig. 4E and F, lower panel). We therefore conclude that two IolR binding sites with different affinities exist in the PiolT1 promoter region, which are occupied by a IolR oligomer that influences target gene expression. Because each binding peak accounts for approximately 50%, it is hypothesized that both DNA-binding sites are bound by equal numbers of IolR molecules.
Figure 4. SPR spectroscopy of IolR binding to PiolT1, PiolR and PreiD.

The biotin-labeled DNA fragments PiolT1 (A), PiolR (B), and PreiD (C) and control fragment PpcfA (D) were captured on a streptavidin-coated sensor chip, and purified IolR was passed over the chip at a flow rate of 30 μl/min and temperature of 25 °C [concentrations of 0, 0.066, 0.165, 0.33, 0.66, 1.65, 3.3, 4.95, 6.6, 13.2, 19.8, 26.4, 33, 49.5, 66 (black and purple lines, internal reference), 165 and 330 nM] using a contact (association) time of 180 sec, followed by a 300-sec dissociation phase. The increase in RU correlates with an increasing IolR concentration. (E) IM analysis of the IolR-PiolT1 interaction. The green and blue spots represent both interactions of IolR with the PiolR DNA. The separate sensorgrams with the specific KD values calculated from ON/OFF-rates map are shown in (F), together with the quantification of the in silico binding kinetics, e. g. the calculated association (ka) and dissociation (kd) rates as well as the quantitative portion of the total peak.
According to the OneToTwo evaluation algorithm, the binding kinetics underlying the sensorgrams of IolR-PiolR and IolR-PreiD interactions were nearly identical to that of the IolR-PiolT1 complex. The ON-rates for the IolR-PiolR and IolR-PreiD interaction were determined to be ka1 = 1.3 × 105 M−1s−1 and ka2 = 2.4 × 105 M−1s−1, and ka1 = 1.5 × 105 M−1s−1 and ka2 = 2.1 × 105 M−1s−1, respectively. The corresponding OFF-rates were kd1 = 5.5 × 10−2 s−1 and kd2 = 1.4 × 10−3 s−1 and of kd1 = 4.9 × 10−2/s and kd2 = 1.4 × 10−3/s, respectively. For both promoters, these data yielded to two binding events that differed with respect to the overall affinity (KD 1 = 425 nM; KD2 = 6.0 nM, and KD1 = 321 nM; KD2 = 6.6 nM, respectively).
Taken together, IolR binds at two sites within the respective iol gene promoter region, with one site bound with very high affinity in the low nanomolar range, and one site with lower affinity. The binding kinetics of the higher affinity-site with a high ON-rate and a very slow OFF-rate are comparable to those of antigen-antibody interactions13,14, thus demonstrating a remarkable high affinity of IolR to the promoter regions of iolT1, iolR, and reiD.
Fitness of cells with constitutive iol gene expression
The high binding affinity of IolR prompted us to speculate that in media lacking MI, cells with an activated MI utilization pathway would exhibit attenuated growth compared to those with silenced iol genes. As demonstrated previously through a promoter probe and FC analysis2,4, deletion of iolR for results in constitutive iol gene transcription regardless of the growth conditions. Therefore, we investigated the relative growth fitness of strain 14028 iolR::KanR in comparison with the kanamycin-sensitive (KanS) strain 14028 in a competitive growth experiment. Both strains were grown separately in LB medium to an OD600 of 0.5 and subsequently mixed in a 1:10 ratio (14028:14028 iolR::KanR). To control the ratio of the two strains, aliquots were plated to determine the numbers of colony forming units (cfu)/ml. The mixture (“inoculum”) was appropriately diluted into LB medium. After incubation for 24 h without agitation, the numbers of viable KanR and KanS cells were determined by plating dilutions of the culture on solid LB medium with or without kanamycin. To determine the longer-term strain fitness, 0.1 ml of the 24-h culture was inoculated 1:500 into fresh LB medium and incubated for 1 day. Then, the cfu/ml of each strain was determined and an aliquot of this culture was used to inoculate fresh medium. This third culture was also incubated for 24 h and analyzed as described above. As a measure of the relative fitness of each strain, we calculated the cfu/ml of each strain after each of the three passages. The experiments started with a roughly 10-fold excess of strain 14028 iolR::KanR versus 14028 (89.6%, ±0.62%). Then, the percentage of the iolR-negative strain in the mixture decreased drastically to 29.2% (±7.46%) after the first passage, to 11.8% (±2.36%) after the second passage and to 4.2% (±0.51%) after the third passage (Fig. 5A), indicating a significant reduction in the fitness of strain 14028 iolR::KanR relative to strain 14028.
Figure 5. Competitive fitness assays of 14028 strains versus their iolR mutants.

Strains were grown separately in LB medium to an OD600 of 0.5 and mixed together in a ratio of approximately 1:10 for 14028 vs. iolR::KanR (A) and 14028 dacB::KanR vs. ∆iolR (B). This mixture was used to inoculate 50 ml LB medium in an appropriate dilution of 1:250 to 1:1000. After a 24-h incubation, the cfu/ml were determined by plating culture samples on LB medium with or without kanamycin, and an aliquot of each culture was diluted 1:500 into fresh LB medium. This step was repeated twice. For each passage, the percentages of strains within the cultivated mixture are shown. (C) Competitive growth experiment with complemented strain 14028 iolR::kanR/pBR-iolR against 14028/pBR322, and with strains 14028 and 14028 iolR::KanR Δ4418–4436; 14028 and 14028 iolR::KanR were used as a control. The inoculum was diluted 1:500. Cell numbers were calculated only after the third passage. Standard deviations were calculated from triplicate measurements of three independent experiments. Significance values were below 1% (p < 0.01*) or below 0.01% (p < 0,0001***).
To exclude the possibility that the decrease of the iolR mutant fitness was related to the energetic cost of the KanR cassette, we generated a mutant (14028 dacB::KanR) in which dacB was replaced with a KanR cassette. DacB encodes penicillin binding protein 5, which exhibits DD-carboxypeptidase activity, and does not affect S. Typhimurium growth15. Using the kanamycin-sensitive, non-polar deletion mutant 14028 ∆iolR as a competitor strain, we repeated the described experiment using a strain ratio as mentioned above. Again, we observed a strong growth retardation of the iolR mutant compared to strain 14028 dacB::KanR, in which iol gene transcription was silenced by IolR (Fig. 5B). The initial percentage of strain 14028 ∆iolR in the mixture was 91.5% (±1.04%) and decreased to 49.7% (±6.25%), 19.9% (±7.16%) and 4.6% (±0.73%) after the first, second and third passages, respectively. To quantify the fitness cost, we compared the division rates of 14028 (0.39 h−1) and 14028 iolR::KanR (0.33 h−1) during the third passage and calculated a 15.4% reduction of the growth rate.
We then tested 14028 iolR::KanR/pBR-iolR against 14028/pBR322 in a similar manner. The complementation of iolR strongly reduced the growth advantage of strain 14028 over the mutant 14028 iolR::KanR (initial ratio 2.4:1 versus 441:1 after three passages) to a ratio of 29:1 (Fig. 5C). This data refute the hypothesis that incidental mutations outside the iolR gene played a role to the growth phenotype observed above.
Recently, IolR was reported to activate the expression of genes encoding components of the reversible lysine acetylation system16. To therefore exclude the possibility that any function beside iol gene repression contributes to the growth attenuation of 14028 iolR::KanR, we performed a competitive growth experiment with strains 14028 and 14028 iolR::KanR Δ4418–4436. Here, the initial ratio of 1:3 between the strains shifted to a ratio of 2.3:1 after three passages (Fig. 5C) in contrast to the control experiment with strains 14028 and 14028 iolR::KanR (ratio 2.4:1 versus 441:1), demonstrating that the manifestation of fitness costs of a iolR mutant requires the presence of all genes of the genomic island. As an additional control, the growth of each strain in LB medium was monitored to exclude non-specific growth deficiencies, but no differences in growth were observed (Figure S1).
Taken together, both 14028 ∆iolR and 14028 iolR::KanR, which feature artificially arrested iol gene expression, exhibited a fitness disadvantage in rich medium in comparison with strains 14028 dacB::KanR or 14028, in which the iol genes are tightly repressed. We conclude that tight repression of the MI utilization pathway in S. Typhimurium is likely a consequence of the fitness costs of inappropriately relaxed iol genes, resulting in a constraint of the number of cells with activated iol genes within an isogenic population.
Discussion
Recent findings suggested that the MI utilisation pathway of S. Typhimurium is very tightly repressed in the absence of MI, and that this repression is mainly achieved by the repressor IolR2. This explanation is biologically reasonable as S. Typhimurium can utilize a number of carbon sources, among which MI is not the most preferable1. Strong iol gene repression is phenotypically manifested by the start of logarithmic growth of cells that were pre-grown in LB medium diluted 1:500 into MM with MI after only 35 h in culture. In contrast, a much shorter lag phase similar to that observed in rich medium was obtained when Salmonella cells had been pre-grown in MI-containing MM and were thus adapted to the MI-utilisation metabolic pathway. Under this experimental condition the bistable phenotype was abolished, as indicated by homogeneous growth behavior4 and low variability in the lag phase duration, which resembled that observed in rich medium. We therefore investigated the repressor IolR here in more detail.
The SPR spectroscopy performed in this study demonstrated high binding affinities of IolR to the promoters PiolT1 and PreiD, which control the genes encoding the main MI transporter and the activator of the MI degradation cascade. Additionally, this study revealed the oligomerization of IolR as indicated by the retarded protein/DNA complexes (Fig. 3). IolR oligomerization was also suggested by SPR spectroscopy. Calculating the max binding capacity of the chip surface at a simple one-to one interaction would result in an RU value of ~20. Since a maximal RU of 1200 RU was measured, we conclude that IolR binds as a higher oligomer even if two binding sites are present in a promoter fragment. Analysis of the SPR sensorgrams then identified one high ON/high OFF and one high ON/slow OFF binding sites of IolR within PiolT1, PiolR and PreiD with different affinities. The detection of these two binding sites with high-affinity (A-site) and low-affinity (B-site) suggests a complex, cooperative DNA-binding mechanism of IolR similar to the so-called AB-BA mechanism that was recently described for the bacterial response regulator YpdB17. In this example, subsequent and cooperative promoter binding by several YpdB copies and a rapid successive promotor clearance results in a pulse-like gene expression.
Using the data obtained in this study, we developed a model for iol gene transcription (Fig. 6). We propose that the heterogeneous phenotype of S. Typhimurium growth with MI is essentially related to the mode how IolR interacts with the promoters of those genes that are required for MI utilization. The iolR promoter exhibits a non-linear response to growth in MI with two subpopulations of low and high expression level as indicator for bistability18. The bistable growth phenotype might be caused by varying copy numbers of heterogeneously transcribed IolR in a sense that IolR dimers do not bind or bind to one or to both of the binding sites, depending on the copy numbers of the repressor. FC analysis demonstrated that during growth with MI, iolR expression increases. The IolR molecules, however, are not able to bind iol promoters, possibly due to the presence of the catabolic intermediate DKGP that may induce the release of IolR from its target promoters and fine-regulate the repression by IolR. Then, the high amount of inactived IolR permits rapid re-silencing of the iol genes upon DKGP dissociation if additional MI is not available or has been replaced by a more optimal carbon and energy source, thus allowing a quick response to changing environments.
Figure 6. Model of IolR interaction with its promoters.

Two different binding sites for IolR with high-affinity (A-site) and low-affinity (B-site) are present in the iol promoters exemplified here by PreiD. (A) Initial binding of IolR dimers to the A-site of the promoter hampering RNA polymerase (RNAP) activity. (B) In case of higher copy numbers of IolR, also the B-site with lower affinity will be occupied by the repressor. (C) Oligomerization of IolR dimers result in trapping of the RNAP and completely inhibits reiD transcription. (D) Binding of the putative inducer (ligand) DKGP abolishes IolR binding, allowing the RNAP to start transcription. In this model, the binding or release of the B-site by IolR depends on the IolR copy numbers and the concentration of the inducer, leading to a pulse-like and highly reversible gene expression; together with the bistable autoregulation of iolR, the equilibrium between status (B) and (C) is decisive for iol gene transcription.
The average number of S. Typhimurium cells harbouring PiolE in the ON status is remarkably low in the absence of MI4. Therefore, in light of the high binding affinity of IolR, we speculated that a strong selective force acts on the cells. To test this hypothesis, we used a deletion mutant of iolR in which the repression of most iol genes is abolished, a situation that at least partially mimics that of cells with an activated iolE promoter. In competitive growth experiments with cross-over labelled strains, strain 14028 clearly outcompeted 14028 ΔiolR (Fig. 3). The outcome of this experiment led us to conclude that cells that do not use MI but express iol genes have a significant fitness disadvantage and are outgrown by cells in which IolR tightly represses expression of these genes. We calculated a high growth rate reduction by ~15% of 14028 iolR::KanR. This is in line with the finding that a misfolded YFP protein of resulted in growth disadvantage of 3.2% in yeast19, because up to 16 enzymes are untimely produced in a iolR-negative strain.
In a cross-over labelled experiment involving kanamycin-resistant strain 14028 dacB::KanR and kanamycin-sensitive strain 14028 ∆iolR, we excluded the possibility that reduced fitness is attributable to the antibiotic resistance20. Interestingly, when macrophage infection assays were performed21, the survival rate of a iolR knockout mutant was reduced to only 32% in comparison with the parental strain 14028 (data unpublished). This phenotype may be attributed to the growth disadvantage of the repressor mutant revealed here.
While biological costs of antibiotic resistances or reporter proteins have been studied in detail22,23, much less reports are available on costly metabolic pathways in case of untimely regulation. Experiments with the lactose operon led the assumption that those costs are associated with transcription and/or translation, but not with the products themselves24. More recently, however, this statement was challenged by the hypothesis that the lac permease activity is the main physiological burden to E. coli25. A recent example of biological costs of gene induction in a pathogen was studied in a fraction of cells harbouring an active type three secretion system (T3SS), which is involved in the virulence of S. Typhimurium. The authors reported the retarded growth of cells exhibiting the T3SS-positive phenotype relative to those not expressing the T3SS26. A similar observation is the growth restriction of Yersinia strains under T3SS-inducing conditions after two generations27.
Regulatory mechanisms have probably been evolved to manage tradeoffs for the fitness costs imposed by gene expression and enzymatic activities28. We therefore hypothesize here that the strength of the transcriptional repression of the iol genes is an evolutionary result of the balance between the expression of a metabolic capability that would be useful in niche occupation, and the fitness cost imposed by such a pathway when an alternative to the respective substrate is present.
Methods
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 2. Strain MvP101 was chosen for safety reasons. Comparison with a shotgun sequence of MvP101 confirmed that its GEI4417/4436 sequence is identical to that of ATCC strain 14028. S. Typhimurium strains were grown at 37 °C in Luria-Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract and 5 g/L NaCl) or minimal medium (MM). MM comprised M9 medium supplemented with 2 mM MgSO4, 0.1 mM CaCl2 and 55.5 mM (1%, wt/vol) MI or 25.2 mM (0.5%, wt/vol) glucose. A solution of 1.5% agar (wt/vol) was supplemented to yield solid media. For all FC experiments, bacterial strains were grown overnight at 37 °C in LB medium or MM and subsequently diluted to 1:500 into fresh medium. Bacterial growth curves were obtained from cultures incubated at 37 °C without agitation in 250-ml bulb flasks containing 50 ml of medium. The optical density at 600 nm (OD600) was measured at the indicated time points.
Table 2. Strains and plasmids used in this study.
| Bacterial strains | Description and relevant features | Source or literature |
|---|---|---|
| 14028 | S. Typhimurium strain ATCC14028 | ATCC |
| 14028 iolR::KanR | Allelic-exchange mutant | This study |
| 14028 ∆iolR | In-frame iolR (STM4417) deletion mutant | This study |
| 14028 dacB::KanR | Allelic-exchange mutant | This study |
| 14028 iolR::KanR ∆4418–4436 | Deletion of iolT1 (STM4418) to iolH (STM4436) in 14028 iolR::KanR | This study |
| MvP101 | 14028 with sseD::aphT, KanR; allelic-exchange mutant | 37 |
| MvP101 ∆iolR | In-frame iolR (STM4417) deletion mutant of MvP101 | This study |
| E. coli BL21(DE3) | F−, ompT, hsdSB (rB−mB−), gal, lon, dcm, rne131, λ (DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5] | 38 |
| Plasmids | ||
| pKD3 | pir-dependent, FRT sites, CmR | 29 |
| pKD4 | pir-dependent, FRT sites, KanR | 29 |
| pKD46 | Lambda-Red helper plasmid; AmpR | 29 |
| pCP20 | FLP recombinase plasmid; AmpR | 29 |
| pUTs-gfp(CmR) | Replacement of lux with gfp from pPROBE-NT in a transposase-negatvive derivate of pUT mini-Tn5 luxCDABE Km2; suicide plasmid, mobRP4, ori R6K, gfp, CmR | 39 |
| pUTs-PiolR::gfp | pUTs-gfp(CmR) with the 500 bp region upstream of iolR (transcriptional fusion to gfp) | This study |
| pUTs-iolR::gfp | pUTs-gfp(CmR) with the last 500 bp of iolR (translational fusion to gfp) | This study |
| pUTs-PrpsM::gfp | pUTs-gfp(CmR) with the 500 bp region upstream of rpsM (STM3418) in front of gfp | This study |
| pBR-iolR | iolR with promoter region cloned into pBR322 | 2 |
| pET28b-iolR | Expression vector, T7lac promoter; KanR | 2 |
Standard procedures
DNA manipulations and chromosomal or plasmid DNA isolation were performed according to standard protocols10 and the relevant manufacturers’ instructions. Plasmid DNA was transformed via electroporation using a Bio-Rad Gene Pulser II as recommended by the manufacturer (Bio-Rad, Hercules, CA, USA) and as described previously21. Polymerase chain reactions (PCRs) were performed using Taq polymerase (Fermentas, St. Leon-Rot, Germany). Chromosomal DNA, plasmid DNA or cells from a single colony were used as a PCR template. S. Typhimurium 14028 gene numbers refer to the LT2 annotation (NC 003197). Statistical analyses were performed with ANOVA or t-test; three levels of significance were considered (p < 0.05, p < 0.01 and p < 0.001).
Cloning of gfp reporter strains
The rpsM and iolR promoters defined as the 500 bp upstream of the start codons, and the last 500 bp of the iolR coding sequence were amplified from the chromosomal DNA of strain 14028 by PCR using the oligonucleotides listed in Table S1. The fragments were cloned via KpnI (Fermentas) upstream of gfp into the multiple cloning site of the suicide vector pUTs-gfp(CmR) and amplified in E. coli S17.1. Plasmids containing the correct gfp-fusions were isolated, verified by PCR and transferred into S. Typhimurium 14028 via conjugation. Strains with chromosomal insertions were selected, and validated by DNA sequencing (GATC, Konstanz, Germany).
Generation of an iolR deletion mutant
The one-step method based on phage λ Red recombinase cassette2,29 was used for the allelic replacement of iolR by a kanamycin-resistance cassette (KanR). To exclude unspecific recombination events, 5 ml (OD600) of 14028 donor cells harbouring the KanR were mixed with 5 μl of P22 phage suspension and incubated for 6 h without agitation. The culture was stored for 2 h at 4 °C, after which the cells were sedimented via centrifugation at 7000 rpm and 4 °C for 10 min. The supernatant was filter sterilized through a 0.2-μm pore filter and stored at 4 °C. Recipient MvP101 cells were prepared from an overnight culture, mixed at a 1:20 ratio with P22 lysate and incubated for 1 h at 37 °C without agitation. After incubation, the mixture was plated on selective agar-plates and incubated at 37 °C. To obtain phage-free colonies, bacteria were cultivated on green indicator plates30. A nonpolar deletion mutant was then obtained upon transformation with pCP20. Gene deletions were verified by PCR analysis and DNA sequencing.
Flow cytometry (FC)
Single cell analysis was performed on a FACSAria II analyzer (Becton Dickinson, San Jose, CA, USA) at an excitation wavelength of 488 nm. Light emission from green fluorescent protein (GFP) was measured between 515 and 545 nm. For all assays, independent overnight cultures of S. Typhimurium strain MvP101 carrying a chromosomal fusion of gfp with PiolR, iolR or the promoter of the house-keeping gene rpsM (PrpsM) were diluted 1:500 into 50 ml of MM with MI or glucose and incubated in 250-ml bulb flasks. Samples were collected at the indicated time points and diluted in 1% phosphate-buffered saline (PBS) containing 2% formaldehyde to ensure GFP stability31. For all FC experiments, 10,000 events were recorded and the collected data were analyzed using Flowing Software (v 2.5.1; http://www.uskonaskel.fi/flowingsoftware/). To exclude doublets, we applied the Area Scaling function of the FACS Aria II that adjusts the pulse area with the pulse level of the forward scatter signal. Thus, cells of the same size exhibit a similar pulse area and level, and can be identified in a scatter diagram. To separate the single cells from the doublets, we used a gate on the diagonal of the diagram.
Overproduction and purification of IolR
IolR-His6 was overproduced from the plasmid pET28b-IolR in E. coli BL21 (DE3) and S. Typhimurium strain 140282. Overnight culture of the strains were diluted 1:100 into 400 ml of LB medium supplemented with 50 μg/ml kanamycin and incubated for 3 h at 37 °C with rotation at 180 rpm. At an OD600 of 0.6, IolR production was induced by adding 0.1 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG). After an overnight incubation at 37 °C and 180 rpm, the cells were harvested by centrifugation at 4 °C and 7500 rpm for 20 min. The pellets were each resuspended in 5 ml of native lysis buffer (50 mM NaH2PO4, 300 mM NaCl and 10 mM imidazole at pH 8.0) and lysed by three passages through a French press (SLM Aminca Instruments, Rochester, NY, USA) at 900 psi; residual cell debris was removed twice by centrifugation at 4 °C and 9000 rpm for 15 min. Following filtration, IolR was isolated using a HisTrap HP colunm with an ÄKTA purifier 10 system (GE Healthcare, Little Chalfont, UK). The protein concentration was determined using RotiQuant solution (Carl, Roth GmbH, Karlsruhe, Germany) according to the Bradford method32. The purity of the eluted fractions was analysed by separation on a 15% sodium dodecyl sulfate (SDS)-PAA gel.
Electrophoretic mobility shift assays
Fragments of promotor regions were amplified (for oligonucleotides, see Table S1) as decribed above, and 100 ng of DNA were mixed with increasing amounts of purified IolR-His6 in 1× Tris/borate/EDTA (EMSA) buffer to a total volume of 20 μl. After incubation for 45 min, the samples were mixed with 4 μl of 6× loading dye (MBI Fermentas) and loaded on a 12% native PAA gel prepared in EMSA buffer; the samples were then separated at 120 V and 4 °C for 2 h in the same buffer. Gel-bound DNA was stained using GelRed (Biotium, Hayward, CA, USA) and visualized by ultraviolet (UV) irradiation.
Biotinylated double-stranded (BIOT-ds) DNA fragments
BIOT-ds DNA fragments were obtained by annealing ss-DNA-oligonucleotides or via PCR. BIOT-PiolT1, BIOT-PiolR and BIOT-PreiD were amplified by PCR using the respective biotinylated DNA oligonucleotides (Sigma-Aldrich, Deisenhofen, Germany) and chromosomal DNA from strain 14028 as a template. To assemble the control promoter region PpcfA from Photorhabdus luminescens33, oligonucleotides P4568-btn_fw and P4568_rew were incubated for 5 min at 100 °C, mixed and cooled prior to annealing. BIOT-ds DNA fragments were then captured on SPR sensor chips.
SPR spectroscopy and calibration-free concentration analysis (CFCA)
SPR spectroscopy and CFCA assays were performed using a Biacore T200 device (GE Healthcare) and streptavidin-precoated Xantec SAD500-L carboxymethyl dextran sensor chips (XanTec Bioanalytics GmbH, Düsseldorf, Germany). All experiments were conducted at 25 °C with HBS-EP+ buffer [10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.05% (v/v) detergent P20] as previously described in detail17.
Assuming a globular shape, the diffusion coefficient of IolR was calculated using the Biacore diffusion constant calculator and converter webtool (https://www.biacore.com). The diffusion coefficient of IolR was determined to be D = 9.946 × 10−11 m2/s. The initial rates of the dilutions that differed by a factor of at least 1.5 were considered when calculating the concentration of IolR that interacted with the ligand. This “active” protein concentration, which was determined as 3.3 × 10−8 M (66% of the total protein concentration), was then used to calculate the binding kinetic constants and steady-state affinity.
Interaction Map (IM) analysis
IM analyses were performed as previously described17,34. SPR spectroscopy curves were determined as the sums of individual binding curves, each of which represented a monovalent interaction35 with a unique combination of the association rate ka (ON-rate) and dissociation rate kd (OFF-rate) and the consequently unique real equilibrium dissociation constant KD = kd/ka. This algorithm was used to split the experimental SPR spectroscopy data set into several theoretical monovalent binding curves and the binding curves that, when summed, best fit the experimental data. The association rate ka and dissociation rate kd were plotted within a two-dimensional distribution yield a heterogeneous binding data display in the form of a map wherein each peak corresponded to a single component that contributed to the cumulative binding curve36. To further quantify protein/DNA interactions, the OneToTwo evaluation algorithm, which is available in the Trace Drawer Software package (Ridgeview Diagnostics AB, Uppsala, Sweden), was used.
Additional Information
How to cite this article: Hellinckx, J. et al. High binding affinity of repressor IolR avoids costs of untimely induction of myo-inositol utilization by Salmonella Typhimurium. Sci. Rep. 7, 44362; doi: 10.1038/srep44362 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We acknowledge Michael Hensel for the gift of MvP101, Nicoletta Nolle for the dacB kanamycin insertion mutant, Sabine Behrend and Anna-Lena Amend for technical assistance, and Martin Haslbeck for his support of protein purification. SPR experiments were performed in the Bioanalytics Core Facility of the LMU Biocenter, Munich. This work was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) within the priority programs SPP1617 (FU 375/9-1) and SPP1617 (HE 5247/5-1).
Footnotes
The authors declare no competing financial interests.
Author Contributions Conceived and designed the experiments: J.H., R.H. and T.M.F. Performed the experiments: J.H., R.H. and A.F. Analysed the data: J.H., R.H. and T.M.F. Wrote the manuscript: J.H., R.H. and T.M.F.
References
- Staib L. & Fuchs T. M. From food to cell: nutrient exploitation strategies of enteropathogens. Microbiology 160, 1020–1039 (2014). [DOI] [PubMed] [Google Scholar]
- Kröger C. & Fuchs T. M. Characterization of the myo-inositol utilization island of Salmonella enterica serovar Typhimurium. J Bacteriol 191, 545–554 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhardt J. E., Kröger C., Broadley S. P. & Fuchs T. M. The orphan regulator ReiD of Salmonella enterica is essential for myo-inositol utilization. Mol Microbiol 94, 700–712 (2014). [DOI] [PubMed] [Google Scholar]
- Kröger C., Srikumar S., Ellwart J. & Fuchs T. M. Bistability in myo-inositol utilization by Salmonella enterica serovar Typhimurium. J Bacteriol 193, 1427–1435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellinckx J. & Fuchs T. M. Hysteresis in myo-inositol utilization by Salmonella Typhimurium. Microbiology Open accepted (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröger C., Stolz J. & Fuchs T. M. myo-Inositol transport by Salmonella enterica serovar Typhimurium. Microbiology 156, 128–138 (2010). [DOI] [PubMed] [Google Scholar]
- Yoshida K. I., Shibayama T., Aoyama D. & Fujita Y. Interaction of a repressor and its binding sites for regulation of the Bacillus subtilis iol divergon. J Mol Biol 285, 917–929 (1999). [DOI] [PubMed] [Google Scholar]
- Yoshida K. et al. myo-Inositol catabolism in Bacillus subtilis. J Biol Chem 283, 10415–10424 (2008). [DOI] [PubMed] [Google Scholar]
- Glaeser A. & Heermann R. A novel tool for stable genomic reporter gene integration to analyze heterogeneity in Photorhabdus luminescens at the single-cell level. BioTechniques 59, 74–81 (2015). [DOI] [PubMed] [Google Scholar]
- Sambrook J. & Russell D. W. Molecular cloning: a laboratory manual, 3nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y. (2001).
- Starke M. & Fuchs T. M. YmoA negatively controls the expression of insecticidal genes in Yersinia enterocolitica. Mol Microbiol 92, 287–301 (2014). [DOI] [PubMed] [Google Scholar]
- Nolle N., Felsl A., Heermann R. & Fuchs T. M. Genetic characterization of the galactitol utilization pathway of Salmonella enterica serovar Typhimurium. J Bacteriol 199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath S. C. & Kumar P. K. Biomolecular discrimination analyses by surface plasmon resonance. The Analyst 139, 2678–2682 (2014). [DOI] [PubMed] [Google Scholar]
- Katayama M., Sato T. & Kuromitsu J. Capture molecules preconditioned for kinetic analysis of high-affinity antigen-antibody complex in Biacore A100. Anal Biochem 424, 168–177 (2012). [DOI] [PubMed] [Google Scholar]
- Baquero M. R., Bouzon M., Quintela J. C., Ayala J. A. & Moreno F. dacD, an Escherichia coli gene encoding a novel penicillin-binding protein (PBP6b) with DD-carboxypeptidase activity. J Bacteriol 178, 7106–7111 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentchel K. L., Thao S., Intile P. J. & Escalante-Semerena J. C. Deciphering the Regulatory Circuitry That Controls Reversible Lysine Acetylation in Salmonella enterica. mBio 6, e00891 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr S., Heermann R. & Jung K. Insights into the DNA-binding mechanism of a LytTR-type transcription regulator. Bioscience reports 36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau D. & Losick R. Bistability in bacteria. Mol Microbiol 61, 564–572 (2006). [DOI] [PubMed] [Google Scholar]
- Geiler-Samerotte K. A. et al. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA 108, 680–685 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D. I. & Levin B. R. The biological cost of antibiotic resistance. Curr Opin Microbiol 2, 489–493 (1999). [DOI] [PubMed] [Google Scholar]
- Klumpp J. & Fuchs T. M. Identification of novel genes in genomic islands that contribute to Salmonella typhimurium replication in macrophages. Microbiology 153, 1207–1220 (2007). [DOI] [PubMed] [Google Scholar]
- Olivares J., Alvarez-Ortega C. & Martinez J. L. Metabolic compensation of fitness costs associated with overexpression of the multidrug efflux pump MexEF-OprN in Pseudomonas aeruginosa. Antimicrob Agents Chemother 58, 3904–3913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang C., Galen J. E., Kaper J. B. & Chao L. Fitness cost of the green fluorescent protein in gastrointestinal bacteria. Canadian journal of microbiology 49, 531–537 (2003). [DOI] [PubMed] [Google Scholar]
- Stoebel D. M., Dean A. M. & Dykhuizen D. E. The cost of expression of Escherichia coli lac operon proteins is in the process, not in the products. Genetics 178, 1653–1660 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eames M. & Kortemme T. Cost-benefit tradeoffs in engineered lac operons. Science 336, 911–915 (2012). [DOI] [PubMed] [Google Scholar]
- Sturm A. et al. The cost of virulence: retarded growth of Salmonella Typhimurium cells expressing type III secretion system 1. PLoS Pathog 7, e1002143 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley S. C., Plano G. V., Skrzypek E., Haddix P. L. & Fields K. A. Regulation by Ca2+ in the Yersinia low-Ca2+ response. Mol Microbiol 8, 1005–1010 (1993). [DOI] [PubMed] [Google Scholar]
- Perfeito L., Ghozzi S., Berg J., Schnetz K. & Lassig M. Nonlinear fitness landscape of a molecular pathway. PLoS Genet 7, e1002160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K. A. & Wanner B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy S., Stewart V. & Taylor R. Genetic Analysis of Pathogenic Bacteria: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbour, NY (1996).
- Bongaerts R. J., Hautefort I., Sidebotham J. M. & Hinton J. C. Green fluorescent protein as a marker for conditional gene expression in bacterial cells. Methods Enzymol 358, 43–66 (2002). [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254 (1976). [DOI] [PubMed] [Google Scholar]
- Brachmann A. O. et al. Pyrones as bacterial signaling molecules. Nature chemical biology 9, 573–578 (2013). [DOI] [PubMed] [Google Scholar]
- Schweiger R., Soll J., Jung K., Heermann R. & Schwenkert S. Quantification of interaction strengths between chaperones and tetratricopeptide repeat domain-containing membrane proteins. J Biol Chem 288, 30614–30625 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barta P., Bjorkelund H. & Andersson K. Circumventing the requirement of binding saturation for receptor quantification using interaction kinetic extrapolation. Nuclear medicine communications 32, 863–867 (2011). [DOI] [PubMed] [Google Scholar]
- Altschuh D. et al. Deciphering complex protein interaction kinetics using Interaction Map. Biochemical and biophysical research communications 428, 74–79 (2012). [DOI] [PubMed] [Google Scholar]
- Medina E. et al. Pathogenicity island 2 mutants of Salmonella typhimurium are efficient carriers for heterologous antigens and enable modulation of immune responses. Infect Immun 67, 1093–1099 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W., Rosenberg A. H., Dunn J. J. & Dubendorff J. W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol 185, 60–89 (1990). [DOI] [PubMed] [Google Scholar]
- Starke M., Richter M. & Fuchs T. M. The insecticidal toxin genes of Yersinia enterocolitica are activated by the thermolabile LTTR-like regulator TcaR2 at low temperatures Mol Microbiol 89, 596–611 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.