

Abstract
α-Pyrrolidinovalerophenone (α-PVP, 7) is an illegal synthetic stimulant that is being sold on the clandestine market as “flakka” and “gravel”. The potent pharmacological effects of α-PVP are presumably mediated by inhibition of dopamine uptake at the dopamine transporter (DAT). However, little is known about how structural modification of α-PVP influences activity at DAT. Eleven analogs of α-PVP were synthesized and examined for their ability to inhibit uptake of [3H]dopamine and [3H]serotonin in rat brain synaptosomes. None of the analogs significantly inhibited [3H]serotonin uptake when tested at 10 μM at the serotonin transporter (SERT). All of the analogs behaved as DAT reuptake inhibitors, but potencies varied over a >1500-fold range. Potency was primarily associated with the nature of the α-substituent, with the more bulky substituents imparting the highest potency. Expansion of the pyrrolidine ring to a piperidine reduced potency up to 10-fold, whereas conformational constraint in the form of an aminotetralone resulted in the least potent compound. Our study provides the first systematic and comparative structure–activity investigation on the ability of α-PVP analogs to act as inhibitors of DAT.
Keywords: Synthetic cathinones, α-PVP, flakka, α-PPP, α-PBP, dopamine transporter, serotonin transporter, DAT, SERT
Graphical abstract
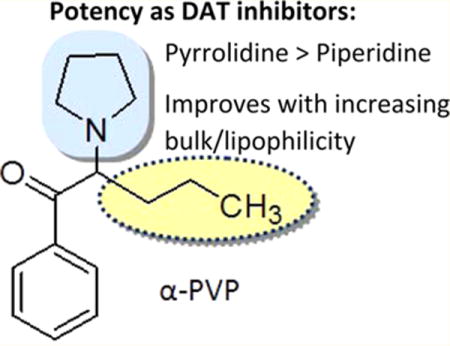
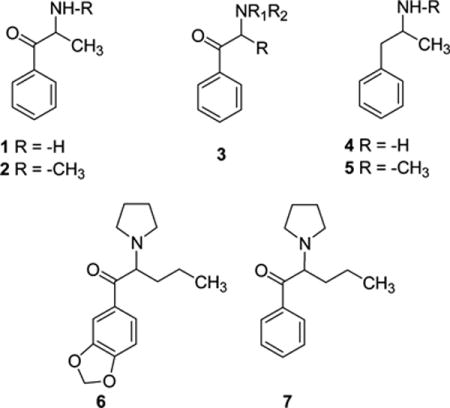
Various classes of so-called “designer drugs” have appeared on the clandestine market in recent years,1,2 and the 18-month period from July 1, 2012 to January 1, 2014 witnessed the identification of 97 new psychoactive substances (NPS) as reported by the United Nations Office of Drugs and Crime.3 One class of NPS is the “synthetic cathinones”;1 these agents are analogs of the naturally occurring substance cathinone (1) or the synthetic agent methcathinone (2)1,3 and are substituted aminophenones typified by the general chemical scaffold 3. Cathinone (1) and methcathinone (2), as well as amphetamine (4) and methamphetamine (5), act primarily as releasing agents at the dopamine transporter (DAT) (reviewed in ref 4). Some synthetic cathinone analogs, depending upon their specific pendant substituents, also are capable of releasing other neurotransmitters such as serotonin (5-HT) via the 5-HT transporter (SERT) or norepinephrine from the norepinephrine transporter (NET).4–6 One of the first synthetic cathinone analogs identified in psychoactive “bath salts” products in the United States was 3,4-methylenedioxypyrovalerone (MDPV; 6).7 MDPV (6) is structurally distinct compared with other synthetic cathinones because it contains a pyrrolidine ring, rather than a primary amine or N-methyl amine, and an extended α-carbon side chain, rather than an α-methyl group. Related to its structural peculiarities, MDPV (6) was identified as the first clandestine synthetic cathinone analog to act at DAT as a dopamine reuptake inhibitor rather than as a releasing agent.7–12 The acute effect of DAT releasing agents (i.e., substrates) and DAT inhibitors is essentially the same: increased synaptic concentrations of dopamine. Hence, whereas cathinone (1) and methcathinone (2) act analogous to amphetamine (4) and methamphetamine (5), MDPV (6) behaves more akin to cocaine.3,4
Previously, we investigated some of the structure–activity relationships (SAR) for MDPV (6) and found that a tertiary amine and an extended α-carbon side chain are optimal for these agents to act as inhibitors of DAT, with the proviso that one of these two structural elements be present.13
The presence of the MDPV (6) methylenedioxy group has little impact on DAT transporter action; that is, α-PVP (7) is a potent DAT reuptake inhibitor similar in potency to MDPV.8,15 Consistent with this action, α-PVP (7) produces locomotor stimulation in rats14 and mice,14,16 and in rats, α-PVP is self-administered,15 reduces intracranial self-stimulation (ICSS) thresholds,17 and substitutes for methamphetamine in tests of stimulus generalization.18 Taken together, preclinical data indicate that α-PVP (7) has a high risk for abuse in human users, and the agent has appeared on the clandestine market (as flakka or gravel) and was classified as a U.S. Schedule I substance in March of 2014.19
As part of a continuing SAR study on the interaction of synthetic cathinones with DAT, the present investigation focused on (a) the role of the α-PVP (7) side chain (i.e., length, bulk), (b) the impact of expansion of the pyrrolidine moiety of α-PVP to a piperidine ring, and (c) the influence of conformational constraint of the side chain. The purpose of our study was to determine the role of structural modifications of cathinones on DAT action and to provide a greater understanding of agents that have been, or might be, scheduled as controlled substances. Even though α-PVP lacks action at the serotonin transporter,15 the effect of the above structural modifications have not been examined; hence, the actions of these agents at SERT was also investigated.
RESULTS
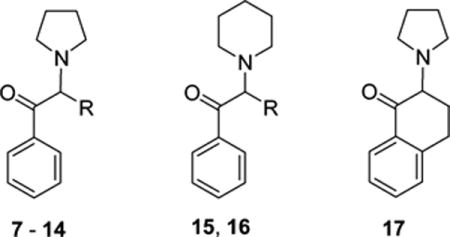
α-PVP (7), as its hydrochloride salt, was prepared as previously reported20 and was available from an earlier study.13 Compounds 1121,22 and 1523 were prepared as hydrochloride salts according to literature procedures. In general, the other compounds were prepared from their corresponding α-bromophenones by reaction with pyrrolidine or piperidine. For example, 8 was prepared from bromophenone 18, 12 from 19, 13 from 21, 14 from 22, and 16 from 23. Compound 17 was prepared in a similar manner from 2-bromo-3,4-dihydronaphthalen-1(2H)-one and pyrrolidine.
Removal of the side chain of 7 (i.e., replacement of the α-n-propyl substituent by -H) affords the simplest possible aminophenone, 8; compound 8 (IC50 = 3250 nM; Table 1 and Figure 1) was 185-fold less potent than 7 (IC50 = 17.5 nM). The α-methyl analog 9 (IC50 = 196.7 nM) displayed improved potency as did its α-ethyl homologue 10 (IC50 = 63.3 nM). The branched isopropyl analog 11 (IC50 = 92.3 nM) displayed potency similar to 10. Although the potency of the latter compounds was enhanced relative to 8, none was as potent as 7. Further homologation to the n-butyl compound (12; IC50 = 11.6 nM) resulted in a small increase in potency. The α-cyclopentyl analog was as potent as 7 (13; IC50 = 17.1 nM) whereas the potency of the corresponding cyclohexyl analog (14; IC50 = 8.3 nM) was about twice that.
Table 1.
Potency of α-PVP (7) Analogs as Inhibitors at DATa
| ||
|---|---|---|
|
| ||
| R | IC50, nM (SD) | |
| 8 | –H | 3250(±418) |
| 9 | –CH3 | 196.7b |
| 10 | –CH2 CH3 | 63.3b |
| 7 | –CH2CH2CH3 | 17.5 (±1.7) |
| 11 | –CH(CH3)2 | 92.3 (±11.6) |
| 12 | –CH2(CH2)2CH3 | 11.6 (±0.6) |
| 13 | –C5H9 | 17.1 (±2.5) |
| 14 | –C6H11 | 8.3 (±1.1) |
| 15 | –CH3 | 2490(±241) |
| 16 | –CH2CH2CH3 | 128(±13) |
| 17 | 12900(±1890) | |
Compounds had no activity at SERT at 10000 nM.
Data reported earlier.15
Figure 1.
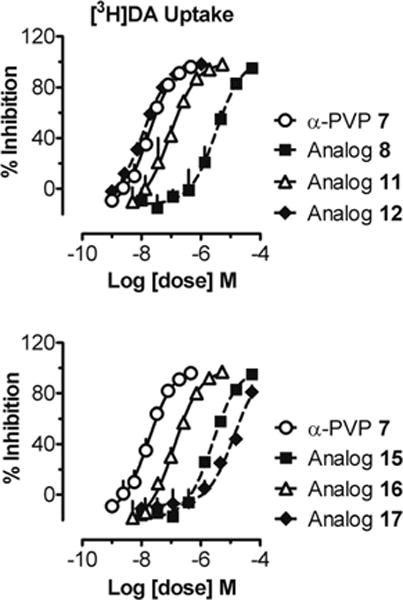
Inhibition of [3H]dopamine uptake by α-PVP analogs. The upper panel depicts effects of altering α-carbon chain length (8, 11, 12), whereas the lower panel depicts effects of ring expansion (15, 16) or conformational constraint (17). Data are mean ± SD for n = 3 experiments performed in triplicate. Uptake inhibition curves were generated by incubating rat caudate synaptosomes with compounds diluted in assay buffer as described in Methods.
Expansion of the pyrrolidine moiety to a piperidine ring resulted in decreased potency. Compounds 15 (IC50 = 2490 nM) and 16 (IC50 = 128 nM; Table 1 and Figure 1), the piperidine counterparts of 9 and 7, respectively, displayed reduced potency by as much as 10-fold. Conformational constraint of 10 as a tetralone (i.e., 17; IC50 = 12900 nM) decreased potency by 200-fold.
A cursory inspection of the data for pyrrolidine compounds 7–14 intimated a possible relationship between potency and substituent “size”. Hansch analysis of the potencies (pIC50 values for inhibition of DAT) of these eight compounds revealed correlations both with the volume (vol) of their α-substituent (r = 0.909) and with their lipophilic character (π) (r = 0.917) (Figure 2). However, for the substituents in this set, vol and π were highly intercorrelated (r = 0.997, p < 0.0001).
Figure 2.
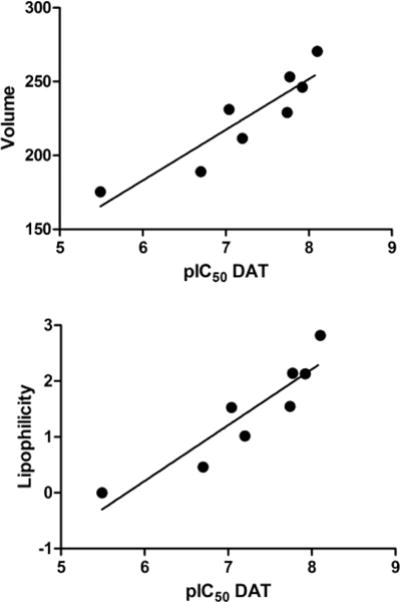
Relationship between pIC50 for inhibition of DAT versus substituent volume in Å3 (upper panel; r = 0.909, p = 0.0017) and lipophilicity (π) (lower panel; r = 0.917, p = 0.0013) for compounds 7–14.
None of the analogs significantly inhibited [3H]5-HT uptake at SERT when tested at 10 μM.
DISCUSSION
It is now appreciated that the length of the α-carbon side chain of MDPV (6) plays a role in its ability to inhibit the reuptake of dopamine.13,15 However, the precise role of various side chain features (i.e., length, bulk) has not been previously examined in a systematic fashion. In the present investigation, we used α-PVP (7), the structural parent of MDPV (6), as our reference compound. The side chain of α-PVP (7) was truncated from an n-propyl to an ethyl (i.e., 10), to a methyl group (i.e., 9), or eliminated altogether (i.e., 8). All three compounds displayed reduced potencies. Each of these compounds behaved as reuptake inhibitors at DAT but with potencies varying over a 185-fold range. Homologation of the α-n-propyl substituent of α-PVP (7) to an n-butyl group (i.e., 12) resulted in retention of potency. Branching was tolerated (comparing 11 with 10), and branching using somewhat larger substituents (comparing 13 with 7 or 12) was also tolerated or resulted in increased potency (i.e., 14).
For eight of the examined pyrrolidine analogs (7–14), there was a significant correlation between potency and both α-substituent volume (vol) and lipophilicity (π). However, due to an intercorrelation between the vol and π parameters (for the substituents that were used), additional compounds will need to be examined to determine which of the two properties is more important.
Two pairs of compounds were examined where the pyrrolidine ring was expanded to a piperidine ring (i.e., compare 15 with 9, 16 with 7); in both cases, potency was decreased. The conformationally constrained compound 17, although retaining action as a reuptake inhibitor, was only very weakly active.
To date, there has been only one investigation that examined the ability of pyrrolovalerophenones to act as inhibitors of DAT,22 and the few comparisons that can be made involve its 3,4-dichloro derivative (i.e., 3,4-dichloro-α-PVP). The results of the present investigation are generally consistent with what was previously found, although direct comparisons should be made with caution since ring substituents in the prior study might influence the overall actions of the compounds. With respect to 3,4-dichloro-α-PVP, (a) shortening the length of the side chain from an n-propyl to an ethyl group (IC50 = 43 and 55 nM, respectively) had little effect on potency as DAT reuptake inhibitors, (b) ring expansion of the pyrrolidine to a piperidine moiety (IC50 = 666 nM) decreased potency by about 15-fold, and (c) the compounds were more effective as inhibitors of DAT than of SERT.22
Overall, and in agreement with an earlier suggestion,13 an extended α-substituent is not a structural requirement for synthetic cathinones to behave as DAT inhibitors when a tertiary amine is present. For the pyrrolovalerone analogs examined here, it is evident that the length, volume, or lipophilicity of the α-substituent governs the potency of these tertiary amines in the absence of any other structural modifications. Expansion of the pyrrolidine to a piperidine ring and conformational constraint of the side chain resulted in reduced potency. Several of the compounds characterized here, including α-PVP (7), α-pyrrolidinopropiophenone (α-PPP, 9), and α-pyrrolidinobutyrophenone (α-PBP,10), have been found on the clandestine market in recent times.1 Given the predicted proliferation of stimulant NPS in the future, our findings should provide guidance as to which new agents might pose risks to public health and should be considered for future scheduling.
METHODS
Chemistry
All commercially available reagents and solvents were purchased from Sigma-Aldrich Co. (St. Louis, MO) and Platte Valley Scientific Product List (Gothenburg, NE) and used as delivered. Melting points were measured in glass capillary tubes (Thomas–Hoover melting point apparatus) and are uncorrected. 1H NMR spectra were recorded with a Bruker 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane as internal standard. Reactions and product mixtures were routinely monitored by thin-layer chromatography (TLC) on silica gel precoated F254 Merck plates.
2-(1-Pyrolidinyl)-1-acetophenone Oxalate (8)
This compound has been previously reported as its HCl salt,24 but difficulty in crystallizing the material led us to prepare an oxalate salt. Pyrrolidine (0.84 mL, 10.04 mmol) was added to a stirred solution of 2-bromoacetophenone (18; 2 g, 10.04 mmol) and NEt3 (1.02 mL, 10.04) in anhydrous benzene (15 mL) under an N2 atmosphere at room temperature. The reaction mixture was allowed to stir at room temperature for 24 h and was filtered, and the solvent was evaporated under reduced pressure to yield 1.82 g (96%) of the crude free base of which 1.00 g was dissolved in anhydrous Et2O (10 mL) and cooled to 0 °C. Oxalic acid/Et2O (15 mL) was added, and the mixture was allowed to stir at room temperature for 48 h. The precipitate was collected by filtration to yield a solid that was recrystallized from anhydrous MeOH to afford 0.35 g (24%) of the target compound as brown crystals: mp 218–220 °C. 1H NMR (DMSO-d6) δ 1.95 (s, 4H, CH2), 3.23 (m, 2H, CH2), 4.92 (s, 2H, CH2), 7.59 (s, 2H, ArH) 7.72 (s, 1H, ArH), 7.98 (s, 2H, ArH). Anal. Calcd for (C12H15NO·C2H2O4): C, 60.21; H, 6.14; N, 5.02. Found: C, 59.97; H, 6.19; N, 5.00.
1-Phenyl-2-(pyrrolidin-1-yl)hexanone Oxalate (12)
This compound has been previously reported as its HCl salt,25 but difficulty in crystallizing the material led us to prepare an oxalate salt. Pyrrolidine (3 mL) was added at 0 °C (ice-bath) to 2-bromo-1-phenylhexanone26 (19) (1.00 g, 4 mmol) under a N2 atmosphere. The reaction mixture was allowed to stir at room temperature for 22 h, quenched by the addition of H2O (25 mL), and extracted with EtOAc (3 × 15 mL). The combined organic portion was washed with H2O (4 × 25 mL) and brine (20 mL), dried (Na2SO4), and evaporated under reduced pressure to yield the free base, which was converted into an oxalate salt using oxalic acid/Et2O to give a crude solid that upon recrystallization from i-PrOH afforded 0.26 g (19%) of 12 as buff-colored crystals: mp 129–131 °C. 1H NMR (DMSO-d6) δ 0.76 (t, 3H, CH3), 1.18–1.21 (m, 4H, CH2), 1.88–1.91 (m, 6H, CH2), 3.14 (br s, 4H, CH2), 5.07 (br s, 1H, CH), 7.61 (t, 2H, Ar), 7.75 (t, 1H, Ar), 8.08 (t, 2H, Ar). Anal. Calcd for (C16H23NO·C2H2O4): C, 64.46; H, 7.51; N, 4.18. Found: C, 64.29; H, 7.28; N, 4.33.
2-Cyclopentyl-1-phenyl-2-(pyrrolidin-1-yl)ethanone Hydrochloride (13)
Pyrrolidine (0.76 mL, 10.68 mmol) was added to a solution of 21 (0.95 g, 3.56 mmol) in a mixture of anhydrous benzene (5 mL) and anhydrous Et2O (5 mL) at room temperature under a N2 atmosphere. The reaction mixture was heated at 45 °C for 24 h, diluted with Et2O (15 mL), and washed with a saturated solution of NaHCO3 (2 × 10 mL). The organic portion was extracted with HCl (1 N, 3 × 10 mL). The combined acidic portion was extracted with Et2O (10 mL), basified by addition of NaOH (5%, to pH 9–10), and re-extracted with Et2O (3 × 15 mL). The combined organic portion was washed with brine (15 mL) and dried (Na2SO4), and solvent was removed under reduced pressure. The residual brown oil was purified by flash chromatography (silica gel; hexanes/EtOAc; 95:5 to 75:25) to afford the free base as a yellow oil. The free base was dissolved in a minimal amount of anhydrous Et2O and converted to the hydrochloride salt by adding a saturated solution of HCl gas in anhydrous Et2O (ca. 10 mL). The precipitate was recrystallized first from a mixture of absolute EtOH/anhydrous Et2O and then the second time from a mixture of acetone/absolute EtOH to afford 0.26 g (25%) of the product as light-gray crystals: mp 179–180 °C. 1H NMR (DMSO-d6) δ 1.05–1.24 (m, 2H, CH2), 1.40–1.51 (m, 4H, CH2), 1.55–1.67 (m, 1H, CH2), 1.82–2.06 (m, 5H, CH2), 2.41–2.50 (m, 1H, CH2), 3.04–3.16 (m, 1H, CH2), 3.22–3.32 (m, 1H, CH2), 3.39–3.49 (m, 1H, CH2), 3.64–3.75 (m, 1H, CH2), 5.59 (t, J = 7.1 Hz, 1H, CH), 7.64 (t, J = 7.7 Hz, 2H, ArH), 7.79 (t, J = 7.4 Hz, 1H, ArH), 8.13 (d, J = 7.7 Hz, 2H, ArH), 10.40 (br s, 1H, NH+ ex with D2O). Anal. Calcd for (C17H23NO·HCl): C, 69.49; H, 8.23; N, 4.77. Found: C, 69.23; H, 8.06; N, 4.78.
2-Cyclohexyl-1-phenyl-2-(pyrrolidin-1-yl)ethanone Hydrochloride (14)
Pyrrolidine (0.42 mL, 5.01 mmol) was added to a solution of 2-bromo-2-cyclohexyl-1-phenylethanone (22; 0.47 g, 1.67 mmol) in a mixture of anhydrous benzene (3 mL) and anhydrous Et2O (3 mL) at room temperature under a N2 atmosphere. The reaction mixture was allowed to stir at room temperature for 17 h and then at reflux for 5 h. Additional pyrrolidine (0.42 mL, 5.01 mmol) was added, and the reaction mixture was heated at 45 °C for 48 h. The reaction mixture was washed with a saturated solution of NaHCO3 (2 × 10 mL) and H2O (10 mL). The organic portion was extracted with HCl (1 N, 3 × 5 mL). The combined acidic portion was basified with a solution of NaOH (5%, to pH 10) and re-extracted with Et2O (3 × 10 mL). The combined organic portion was washed with brine (10 mL) and dried (Na2SO4), and solvent was removed under reduced pressure. The obtained free base was converted to the hydrochloride salt and recrystallized twice from a mixture of absolute EtOH/anhydrous Et2O to afford 0.19 g (37%) of the product as beige crystals: mp 235–238 °C. 1H NMR (DMSO-d6) δ 0.58–0.58 (m, 1H, CH2), 0.74–0.92 (m, 2H, CH2), 1.19–1.22 (m, 2H, CH2), 1.52–1.73 (m, 4H, CH2), 1.92–2.10 (m, 6H, CH2), 3.05–3.66 (m, 4H, CH2), 5.56 (d, J = 5.9 Hz, 1H, CH), 7.63 (t, 2H, ArH), 7.79 (t, 1H, ArH), 8.11 (d, 2H, ArH), 10.30 (br s, 1H, NH+ ex with D2O). Anal. Calcd (C18H25NO·HCl): C, 70.22; H, 8.51; N, 4.55. Found: C, 69.98; H, 8.33; N, 4.45.
1-Phenyl-2-(piperidin-1-yl)pentan-1-one Hydrochloride (16)
Piperidine (1.06 g, 12.45 mmol) was added to a stirred solution of 2-bromo-1-phenylpentan-1-one (23)20 (1.00 g, 4.15 mmol) in a mixture of anhydrous benzene (5 mL) and anhydrous Et2O (5 mL), and the reaction mixture was heated at 45 °C for 20 h under an N2 atmosphere. The reaction mixture was diluted with Et2O (10 mL) and washed with H2O (2 × 10 mL). The organic portion was extracted with HCl (1 N, 3 × 10 mL). The combined acidic portion was basified with NaOH (15%, to pH 9–10) and re-extracted with Et2O (3 × 15 mL). The combined organic portion was washed with brine (15 mL) and dried (Na2SO4), and solvent was removed under reduced pressure. The residue was purified by flash chromatography (silica gel; hexane/EtOAc; 95:5) to afford a yellow oil. The oil was converted to the hydrochloride salt by dissolving the free base in anhydrous Et2O and adding a saturated solution of gaseous HCl in Et2O (ca. 10 mL). The salt was collected by filtration and recrystallized from absolute EtOH to afford 0.65 g (56%) of the product as white crystals: mp 191–193 °C (lit.27 mp 192 °C). 1H NMR (DMSO-d6) δ 0.80 (t, J = 7.2 Hz, 3H, CH3), 1.04–1.20 (m, 2H, CH2), 1.36–1.46 (m, 1H, CH2), 1.68–2.05 (m, 7H, CH2), 2.99–3.15 (m, 2H, CH2), 3.24–3.36 (m, 1H, CH2), 3.61–3.64 (m, 1H, CH2), 5.36 (dd, J = 11.2, 7.4 Hz, 1H, CH), 7.63 (t, J = 7.6 Hz, 2H, ArH), 7.78 (t, J = 7.4 Hz, 1H, ArH), 8.14 (d, J = 7.4 Hz, 2H, ArH), 10.18 (br s, 1H, NH+ ex with D2O).
2-(Pyrrolidin-1-yl)-3,4-dihydronaphthalen-1(2H)-one Oxalate (17)
A solution of 2-bromo-3,4-dihydronaphthalen-1(2H)-one28 (0.90 g, 4 mmol) in anhydrous benzene (3 mL) was added in a dropwise manner to a refluxing solution of pyrrolidine (0.85 g, 12 mmol) in anhydrous benzene (3 mL) under a N2 atmosphere. The reaction mixture was heated at reflux for 15 min. After cooling to room temperature, the reaction mixture was diluted with Et2O (15 mL) and washed with H2O (5 × 10 mL) and brine (10 mL). The organic portion was extracted with HCl (1 N, 3 × 10 mL). The combined acidic portion was washed with Et2O (15 mL), basified by addition of NaOH (15%, to pH ≈ 10), and re-extracted with Et2O (3 × 15 mL). The combined organic portion was washed with H2O (2 × 15 mL) and brine (15 mL) and dried (MgSO4), and solvent was removed under reduced pressure to afford a dark yellow oil (0.55 g). The oil was converted to its oxalate salt by dissolving the free base in absolute EtOH and adding a solution of oxalic acid in Et2O. The solution was stirred at room temperature for 2 h until the oxalate salt precipitated and solidified. The gray solid was collected by filtration and recrystallized twice from absolute EtOH to afford 0.14 g (11%) of the product as a gray powder: mp 139–143 °C. 1H NMR (DMSO-d6) δ 1.93 (m, 4H, CH2), 2.24–2.34 (m, 1H, CH2), 2.47–2.50 (m, 1H, CH2), 3.26–3.48 (m, 6H, CH2), 4.43–4.46 (m, 1H, CH), 7.40–7.44 (m, 2H, ArH), 7.64 (t, 1H, ArH), 7.92 (d, 1H, ArH). Anal. Calcd (C14H17NO·(COOH)2·0.25EtOH): C, 62.55; H, 6.52; N, 4.42. Found: C, 62.45; H, 6.33; N, 4.40.
2-Bromo-2-cyclopentyl-1-phenylethanone (21)
Bromine (0.04 mL, 0.79 mmol) was added in one portion to a stirred solution of 2-cyclopentyl-1-phenylethanone (20;29 0.70 g, 3.72 mmol) and AlCl3 (0.03 g, 0.20 mmol) in anhydrous Et2O (10 mL) at 0 °C (ice-bath) under an N2 atmosphere. The reaction mixture was allowed to warm to room temperature, and the remaining bromine (0.15 mL, 2.92 mmol) was added to the reaction solution in a dropwise manner. The reaction mixture was neutralized with a saturated solution of NaHCO3. The organic portion was separated and dried (Na2SO4), and solvent was removed under reduced pressure to afford 0.97 g (98%) of the product as a yellow oil. The compound was used without further characterization in the preparation of compound 13.
2-Bromo-2-cyclohexyl-1-phenylethanone (22)
This compound was synthesized analogously to a published procedure30 except that aluminum chloride was used as catalyst. The product was reported to be a pale-brown oil.30
Bromine (0.02 mL, 0.4 mmol) was added in one portion to a stirred solution of 2-cyclohexyl-1-phenylethanone31 (0.405 g, 2.0 mmol) and AlCl3 (0.013 g, 0.1 mmol) in anhydrous Et2O (5 mL) at 0 °C (ice-bath) under an N2 atmosphere. After 10 min, the ice-bath was removed, and the reaction mixture was allowed to warm to room temperature. The remaining bromine (0.08 mL, 1.6 mmol) was added to the reaction solution in a dropwise manner over 5 min, and then the solution was neutralized with a saturated solution of sodium NaHCO3. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic portion was dried (Na2SO4), and solvent was removed under reduced pressure to afford 0.470 g (83%) of the product as a yellow solid: mp 49–52 °C. Compound 22 was used without further purification in the preparation of 14.
Correlational Studies
Quantitative structure–activity relationship studies were performed using the α-substituent volume and lipophilicity; volume (vol) was calculated using SYBYL-X 2.1 (Tripos Inc., St. Louis, MO). Lipophilicity (π) values were obtained from the primary literature.32 Linear regressions and statistical analyses were performed between both these parameters and in vitro reuptake inhibition potency at DAT (pIC50 values) using the software Prism 5.04 (GraphPad, San Diego, CA, USA), and correlations with a p < 0.05 were considered statistically significant. pIC50 values, volume, and π values used in this investigation are shown in Table S1.
In Vitro Reuptake Assays
Subjects
Male Sprague–Dawley rats (Charles River, Wilmington, MA, USA) weighing 250–350 g were housed three per cage with free access to food and water and maintained on a 12 h light/dark cycle with lights on from 7:00 a.m. to 7:00 p.m. Animal facilities were accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, and procedures were carried out in accordance with the Institutional Animal Care and Use Committee and the National Institutes of Health guidelines on care and use of animal subjects in research (National Research Council, 2011).
Procedure
[3H]Transmitters (specific activity ranging from 30 to 50 Ci/mmol) were purchased from PerkinElmer (Shelton, CT, USA). All other chemicals and reagents were acquired from Sigma-Aldrich (St. Louis, MO, USA). Rats were euthanized by CO2 narcosis, and brains were processed to yield synaptosomes as previously described.33 Rat caudate tissue was used for DAT assays, whereas rat whole brain minus caudate and cerebellum was used for SERT assays. In uptake inhibition assays, 5 nM [3H]dopamine and 5 nM [3H]serotonin were used to assess transport activity at DAT and SERT, respectively. The selectivity of uptake assays was optimized for a single transporter by including unlabeled blockers to prevent uptake of [3H]transmitter by competing transporters. Uptake inhibition assays were initiated by adding 100 μL of tissue suspension to 900 μL Krebs–phosphate buffer (126 mM NaCl, 2.4 mM KCl, 0.83 mM CaCl2, 0.8 mM MgCl2, 0.5 mM KH2PO4, 0.5 mM Na2SO4, 11.1 mM glucose, 0.05 mM pargyline, 1 mg/mL bovine serum albumin, and 1 mg/mL ascorbic acid, pH 7.4) containing test drug and [3H]transmitter. Uptake inhibition assays were terminated by rapid vacuum filtration through Whatman GF/B filters, and retained radioactivity was quantified by liquid scintillation counting. Statistical analyses were carried out using GraphPad Prism (v. 6.0; GraphPad Scientific, San Diego, CA, USA). IC50 values for inhibition of reuptake were calculated based on nonlinear regression analysis.
Supplementary Material
Acknowledgments
This work was supported in part by Public Health Service Grant DA033930 and by the Intramural Program of the National Institute on Drug Abuse.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.5b00160.
pIC50 values, volume, and π values used in the QSAR investigation (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.UNODC. United Nations Office on Drugs and Crime. 2013. The challenge of new psychoactive substances; pp. 1–108. (World Drug Report). [Google Scholar]
- 2.USDOJ. U S Department of Justice Drug Enforcement Administration National Drug Threat Assessment Summary. 2013. pp. 1–18. (DEA-NWW-DIR-017-13). [Google Scholar]
- 3.UNODC. United Nations Office on Drugs and Crime. 2014. pp. 1–93. (World Drug Report). [Google Scholar]
- 4.Glennon RA. Bath salts, mephedrone, and methylenedioxypyrovalerone as emerging illicit drugs that will need targeted therapeutic intervention. Adv Pharmacol. 2014;69:581–620. doi: 10.1016/B978-0-12-420118-7.00015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumann MH, Partilla JS, Lehner KR. Psychoactive ‘bath salts”: Not so soothing. Eur J Pharmacol. 2013;698:1–5. doi: 10.1016/j.ejphar.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumann MH, Ayestas MA, Partilla JS, Sink JR, Shulgin AT, Daley PF, Brandt SD, Rothman RB, Ruoho AE, Cozzi NV. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37:1192–1203. doi: 10.1038/npp.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spiller HA, Ryan ML, Weston RG, Jansen J. Clinical experience with and analytical confirmation of “bath salts” and “legal highs” (synthetic cathinones) in the United States. Clin Toxicol. 2011;49:499–505. doi: 10.3109/15563650.2011.590812. [DOI] [PubMed] [Google Scholar]
- 8.Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M, Rothman RB, Goldberg SR, Lupica CR, Sitte HH, Brandt SD, Tella SR, Cozzi NV, Schindler CW. Powerful cocaine-like actions of 3,4-methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology. 2013;38:552–562. doi: 10.1038/npp.2012.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cameron K, Kolanos R, Verkariya R, De Felice L, Glennon RA. Mephedrone and methylenedioxypyrovalerone (MDPV), major constituents of “bath salts,” produce opposite effects at the human dopamine transporter. Psychopharmacology. 2013;227:493–499. doi: 10.1007/s00213-013-2967-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cameron KN, Kolanos R, Solis E, Glennon RA, De Felice LJ. Bath salts components mephedrone and methylenedioxypyrovalerone (MDPV) act synergistically at the human dopamine transporter. Br J Pharmacol. 2013;168:1750–1757. doi: 10.1111/bph.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A. Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol. 2013;85:1803–1815. doi: 10.1016/j.bcp.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME. Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol. 2013;168:458–470. doi: 10.1111/j.1476-5381.2012.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolanos R, Solis E, Sakloth F, De Felice LJ, Glennon RA. “Deconstruction” of the abused synthetic cathinone methylenedioxypyrovalerone (MDPV) and an examination of effects at the human dopamine transporter. ACS Chem Neurosci. 2013;4:1524–1529. doi: 10.1021/cn4001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aarde SM, Creehan KM, Vandewater SA, Dickerson TJ, Taffe MA. In vivo potency and efficacy of the novel cathinone α-pyrrolidinopentio- phenone and 3,4-methylenedioxypyrovalerone: Self-administration and locomotor stimulation in male rats. Psychopharmacology (Berlin) 2015;232:3045. doi: 10.1007/s00213-015-3944-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marusich JA, Antonazzo KR, Wiley JL, Blough BE, Partilla JS, Baumann MH. Pharmacology of novel synthetic stimulants structurally related to the “bath salts” constituent 3,4-methylenedioxypyrovalerone (MDPV) Neuropharmacology. 2014;87:206–213. doi: 10.1016/j.neuropharm.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaizaki A, Tanaka S, Numazawa S. New recreational drug 1-phenyl-2(-1pyrrolidinyl)-1-pentanone (alpha-PVP) activates central nervous system via dopaminergic neuron. J Toxicol Sci. 2014;39:1–6. doi: 10.2131/jts.39.1. [DOI] [PubMed] [Google Scholar]
- 17.Watterson LR, Olive MF. Synthetic cathinones and their rewarding and reinforcing effects in rodents. Adv Neurosci (Hindawi) 2014;2014:209875. doi: 10.1155/2014/209875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naylor JE, Freeman KB, Blough BE, Woolverton WL, Huskinson SL. Discriminative-stimulus effects of second generation synthetic cathinones in methamphetamine-trained rats. Drug Alcohol Depend. 2015;149:280–284. doi: 10.1016/j.drugalcdep.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Federal Register. Schedules of controlled substances: Temporary placement of 10 synthetic cathinones into Schedule I, Vol (March 7, 2014) 2014;79:12938–12943. [PubMed] [Google Scholar]
- 20.Durman J, Grayson JI, Hunt PG, Warren S. Synthesis of α-phenylthio enones and esters of α-phenylthio alkenoic acids. J Chem Soc, Perkin Trans. 1986;1:1939–1946. [Google Scholar]
- 21.Thomae K. α-Pyrrolidino ketones. 933507. UK Patent GB. 1963 Aug 8;
- 22.Meltzer PC, Butler D, Deschamps JR, Madras BK. 1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (pyrovalerone) analogues: A promising class of monoamine uptake inhibitors. J Med Chem. 2006;49:1420–1432. doi: 10.1021/jm050797a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cattaneo A, Gelmi G, Jori A, Zevio H. Some new compounds showing activity on the central nervous system. Farmaco, Ed Sci. 1962;17:308–314. [Google Scholar]
- 24.Nishimura H, Takamatsu H. Phenyl mercaptoalkylamines. I. (1964) 1-Phenyl-2(/or 3)-amino-alkanethiol derivatives. Yakugaku Zasshi. 1964;84:797–805. [PubMed] [Google Scholar]
- 25.Seeger E, Riss B. Compositions and methods for stimulating central nervous system and increasing the blood pressure. 3,287,217. US Patent. 1966 Nov 22;
- 26.Boswell GW, Musso DL, Kelley JL, Soroko FE, Cooper BR. Synthesis and anti-tetrabenazine activity of C-3 analogues of dimethyl-2-phenylmorpholines. J Heterocycl Chem. 1996;33:33–40. [Google Scholar]
- 27.Welvart Z. Modification of the reactivity of organomagnesium compounds. Orientation of their action on α-aminonitriles towards the formation of α-aminoketones. Compt Rend. 1960;250:1870–1872. [Google Scholar]
- 28.Prokopowicz M, Młynarz P, Kafarski P. Synthesis of phosphonate derivatives of 2,3-dihydroindene. Tetrahedron Lett. 2009;50:7314–7317. [Google Scholar]
- 29.Wamser CC, Wagner WR. Type II photoelimination from α-cycloalkylacetophenones and a polystyrene-bound analogue. J Am Chem Soc. 1981;103:7232–7234. [Google Scholar]
- 30.Frey B, Hufton R, Harding M, Draffan AG. Compounds for the treatment of HCV. WO2013036994 A1. International Patent. 2013 Mar 21;
- 31.Lazer ES, Miao CK, Wong H-Ch, Sorcek R, Spero DM, Gilman A, Pal K, Behnke M, Graham AG, Watrous JM, Homon CA, Nagel J, Shah A, Guindon Y, Farina PR, Adams J. Benzoxazolamines and benzothiazolamines: Potent, enantioselective inhibitors of leukotriene biosynthesis with a novel mechanism of action. J Med Chem. 1994;37:913–923. doi: 10.1021/jm00033a008. [DOI] [PubMed] [Google Scholar]
- 32.Hansch C, Leo A, Unger SH, Kim KH, Nikaitani D, Lien EJ. Aromatic” substituent constants for structure-activity correlations. J Med Chem. 1973;16:1207–1216. doi: 10.1021/jm00269a003. [DOI] [PubMed] [Google Scholar]
- 33.Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.