

Abstract
The designer stimulant methylenedioxypyrovalerone (MDPV) is a potent reuptake inhibitor at transporters for dopamine (DAT) and norepinephrine (NET) that produces a constellation of abuse-related behavioral effects. MDPV possesses a chiral center, and the abused formulation of the drug is a racemic mixture, but no data are available on the pharmacology of its isomers. Here, the individual optical isomers of MDPV were prepared and examined with respect to their neurochemical actions on neurotransmitter reuptake and behavioral effects in an assay of intracranial self-stimulation (ICSS) in rats. In assays of DAT uptake inhibition, S(+)MDPV (EC50 = 2.13 nM) was more potent than either (±)MDPV (EC50 = 4.85 nM) or R(−)MDPV (EC50 = 382.80 nM); the three drugs were less potent at NET uptake inhibition, with the same rank order of potency. Neither racemic MDPV nor its optical isomers inhibited the reuptake of serotonin at concentrations up to 10 μM. S(+)MDPV produced an abuse-related and dose-dependent facilitation of ICSS, and the potency of S(+)MDPV (significant facilitation at doses ≥ 0.1 mg/kg) was greater than that of the racemate (significant facilitation at doses ≥ 0.32 mg/kg). R(−)MDPV failed to alter ICSS at doses up to 100 times greater than the lowest effective dose of S(+)MDPV. The results indicate that abuse-related neurochemical and behavioral effects of racemic MDPV reside primarily with its S(+) isomer.
Keywords: Synthetic cathinones, MDPV, DAT, NET, reuptake inhibition, ICSS (intracranial self-stimulation), psychomotor stimulants, cocaine, drug abuse
Graphical abstract
Cathinone (1a), the active psychomotor stimulant component of the shrub Catha edulis, was first identified in 1975.1 Since then, a number of synthetic cathinone analogues have appeared on the clandestine market.2–4 Cathinone (cathinone = β-ketoamphetamine) and the synthetic cathinone analogue N-methylcathinone (methcathinone, MCAT; 1b) act, primarily, as amphetamine (2)-like substrates (i.e., releasing agents) at the dopamine transporter (DAT) in humans and rodents.2 In contrast, the synthetic cathinone analogue MDPV (i.e., methylenedioxypyrovalerone; 3), now a U.S. Schedule I substance,5 behaves as a cocaine-like reuptake inhibitor at DAT.6–10 The structural requirements of MDPV to act as a DAT reuptake inhibitor (i.e., as reflected by its hyperpolarizing action in electrophysiological studies) have been investigated; it has been established that a tertiary amine and/or an extended α-carbon side chain is required for such action.11
MDPV possesses a chiral center; hence, it was of interest to identify which optical isomer of MDPV is responsible (or more potent) for its actions as a DAT inhibitor. Here, we prepared both optical isomers and examined their actions at DAT, the norepinephrine transporter (NET), and the serotonin transporter (SERT). The isomers were also examined for their ability to produce drug abuse-related facilitation of intracranial self-stimulation12 in rats.
RESULTS
Chiral Resolution and Synthesis
A number of attempts were made to resolve racemic MDPV [(±)3] by recrystallization of its diastereomeric salts obtained upon reaction with dibenzoyl-D-tartaric acid, di-p-toluoyl-D-tartaric acid, and N-acetyl-D-2-phenylglycine. Although multiple attempts were made and various recrystallization solvents were used, conditions for satisfactory chiral resolution of the diastereomeric mixtures were not realized. In most cases, there were difficulties obtaining a crystalline form of the salts. Only in the case of di-p-toluoyl-D-tartrate salt mixtures was a small amount of crystalline material obtained and converted to an HCl salt; substantial racemization was apparent.
An attempt to stereoselectively synthesize optically pure 3S by direct alkylation of the primary amine 8S with 1,4-dibromobutane was unsuccessful and led to partial racemization of the product (Method 1, Supporting Information). Also, the attempted stereoselective synthesis of 3R, in which the starting material D-norvaline was to be converted to the product in two steps, failed and resulted in partial racemization (Method 2, Supporting Information).
Both optical isomers, 3S and 3R, were eventually prepared from the commercially available isomers of norvaline (4) by a common reaction sequence (Scheme 1). The intermediate, the appropriate isomer of N-Boc-norvaline-pivalic acid mixed anhydride, was generated in situ from 5 and pivaloyl chloride and treated with excess pyrrolidine to afford the corresponding amide 6. Pyrrolidine amide 6 was subjected to a reaction with 3,4-(methylenedioxy)phenylmagnesium bromide to afford the N-Boc protected α-aminoketone 7. Subsequent hydrolysis with a saturated solution of HCl in dioxane resulted in deprotection of the amino group. α-Aminoketone hydrochloride 8 was reduced using sodium borohydride, and the resulting α-aminoalcohol 9 was alkylated with 1,4-dibromobutane to afford the required pyrrolidine derivative 10. Finally, oxidation of the benzylic hydroxyl group of 10 with Jones reagent followed by salt formation afforded the desired enantiomer 3S or 3R.
Scheme 1a.

a(a) (Boc)2O, NaHCO3, dioxane, H2O, reflux, 24 h; (b) (i) pivaloyl chloride, Et3N, THF, 0 °C, 1 h, (ii) pyrrolidine, rt, 18 h; (c) 3,4-methylenedioxyphenylmagnesium bromide, THF, rt, 24 h; (d) HCl/dioxane, rt, 20 min; (e) (i) NaBH4, MeOH, 0 °C, 0.5 h, (ii) HCl/Et2O; (f) (i) 1,4-dibromobutane, KHCO3, CH3CN, reflux, 24 h, (ii) HCl/Et2O; (g) (i) Jones reagent, 0 °C, 1h, rt, 18 h, (ii) HCl/Et2O.
The stereochemical purity of the 3 isomers was examined using 1H NMR spectrometry. A dibenzoyl-D-tartrate salt of racemic MDPV (a diastereomeric mixture) showed two sets of triplets, one at δ = 0.79 and the other at 0.81 (CDCl3). These correspond to the ω-CH3 group of the S isomer of MDPV dibenzoyl-D-tartrate salt and R isomer of MDPV dibenzoyl-D-tartrate salt, respectively. The ratio of these two triplets was used to determine the stereochemical purity of MDPV enantiomers. The lack of a specific triplet in the 1H NMR spectra of the dibenzoyl-D-tartrate salts of the isomers prepared as shown in Scheme 1 suggested that the diastereomeric purity of the compound was >95%. An analogous method had been used previously for enantiomers of a structurally related compound, 1-(4-methylphenyl)-2-(pyrrolidin-1-yl)pentan-1-one (pyrovalerone), for which enantiomeric purity was subsequently confirmed by HPLC chiral resolution.13
Reuptake Inhibition
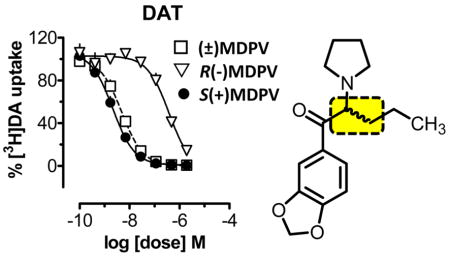
The ability of MDPV (3) to inhibit the reuptake of [3H]dopamine, [3H]norepinephrine, and [3H]-serotonin at DAT, NET, and SERT, respectively, was examined, along with (−)cocaine as a prototypical comparator compound. Dose–response data for MDPV and its isomers are depicted in Figure 1, whereas IC50 measures of potency are summarized in Table 1. Racemic MDPV [(±)3] and its isomers, 3S and 3R, were fully efficacious inhibitors of reuptake at DAT and NET. In general, potency at DAT was 2–4 times higher than potency at NET for each compound. S(+)MDPV was approximately twice as potent as the racemate at both transporters, whereas R(−)MDPV was nearly 200- and 80-fold less potent than S(+)MDPV at DAT and NET, respectively. S(+)MDPV was also 100-fold more potent than cocaine at DAT and 40-fold more potent at NET (Table 1). Neither racemic MDPV nor its optical isomers inhibited the reuptake of [3H]serotonin at SERT at concentrations up to 10 μM (data not shown).
Figure 1.

Inhibition of [3H]neurotransmitter reuptake at DAT (A) and NET (B) for MDPV (3) and its optical isomers. Data are the mean ± SEM for n = 3 experiments performed in triplicate.
Table 1.
Inhibition of [3H]Neurotransmitter Reuptake at DAT and NET for MDPV (3), Its Optical Isomers, and Cocainea
| IC50, nM
|
||
|---|---|---|
| DAT | NET | |
| (±)MDPV (3) | 4.85 ± 0.49 | 16.84 ± 1.10 |
| S(+)MDPV (3S) | 2.13 ± 0.17 | 9.86 ± 1.03 |
| R(−)MDPV (3R) | 382.80 ± 53.26 | 726.10 ± 150.10 |
| (−)cocaine | 198.80 ± 14.86 | 395.90 ± 47.80 |
Data are expressed as the mean ± SEM for n = 3 experiments performed in triplicate.
Intracranial Self-Stimulation (ICSS)
Male Sprague–Dawley rats were trained to press a response lever for pulses of electrical brain stimulation delivered to the medial forebrain bundle. After vehicle pretreatment, increasing frequencies of brain stimulation maintained increasing rates of ICSS (see vehicle data in Figure 2B,C), and the mean ± SEM number of total stimulations delivered across all brain stimulation frequencies was 228 ± 51.6.
Figure 2.

Facilitation of ICSS in rats by S(+)MDPV (0.032–1.0 mg/kg; n = 6) but not by R(−)MDPV (0.32–10 mg/kg; n = 5). (A) The abscissa shows drug dose in mg/kg (log scale; VEH = vehicle), and the ordinate shows rates of ICSS expressed as the percentage of the baseline number of stimulations delivered per component across all brain stimulation frequencies. The asterisk indicates significantly different from VEH, as determined by one-way ANOVA followed by a Dunnett’s posthoc test (p < 0.05). (B, C) The abscissa shows brain stimulation frequency in log Hz, and the ordinate shows rates of ICSS expressed as the percent of the maximum control rate (%MCR), a normalized measure of ICSS reinforcement rate. Filled points indicate significantly different from VEH, as determined by two-way ANOVA followed by the Holm–Sidak posthoc test (p < 0.05).
Figure 2A shows that S(+)MDPV (0.032–1.0 mg/kg) dose-dependently facilitated ICSS [F(4,20) = 10.99; p < 0.001], whereas R(−)MDPV (0.32–10 mg/kg) did not [F(4,16) = 2.318, p = 0.102]. Posthoc analysis indicated that doses of 0.32 and 1.0 mg/kg S(+)MDPV significantly facilitated ICSS relative to vehicle. Figure 2B,C shows effects of selected MDPV doses in more detail. Thus, Figure 2B shows that 0.32 mg/kg S(+)MDPV produced a leftward shift in the ICSS frequency–rate curve and increased rates of ICSS across a broad range of frequencies. Conversely, Figure 2C shows that a 10-fold higher dose of 3.2 mg/kg of R(−)MDPV failed to alter the ICSS frequency–rate curve relative to vehicle. Like S(+)MDPV, a dose of 10 mg/kg cocaine also significantly increased the number of stimulations per component to 170.7 ± 18.9% of baseline, and this was significantly different from effects of vehicle (t = 2.904, p = 0.044; data not shown).
DISCUSSION
Although multiple attempts at the resolution of MDPV (3) were unsuccessful, as were the initial attempts at asymmetric synthesis (see Supporting Information), eventually we were successful in preparing both optical isomers.
Previous studies have established that MDPV is a potent inhibitor of reuptake at DAT and NET in humans and in rodents.6–10 Here, we show that the actions of MDPV isomers at DAT and NET are stereoselective, with the S(+)isomer being approximately 200- and 80-fold more potent as a reuptake inhibitor than R(−)MDPV at DAT and NET, respectively. S(+)MDPV was also 100-fold more potent than (−)cocaine at DAT and 40-fold more potent at NET. However, to put this in perspective, even the less potent R(−)MDPV was still half as potent as (−)cocaine.
Drug-induced facilitation of ICSS is often interpreted as evidence of abuse liability,12 and, as reported here and previously,14 (−)cocaine facilitated ICSS. MDPV (3) produced a stereoselective facilitation of ICSS in rats. As has been earlier reported for the racemate,15 S(+)MDPV produced a dose-dependent and abuse-related facilitation of ICSS, and the potency of S(+)MDPV (significant facilitation at doses ≥ 0.1 mg/kg) was slightly greater than the potency of the racemate (significant at doses ≥ 0.32 mg/kg). Conversely, R(−)MDPV failed to significantly alter ICSS at doses up to 100-fold greater than the lowest effective dose of S(+)MDPV. Facilitation of ICSS by S(+)MDPV and (±)MDPV likely reflects their effectiveness to inhibit DAT rather than NET because previous studies have shown ICSS facilitation by other selective DAT inhibitors but not by selective NET or SERT inhibitors.12,16
These findings demonstrate that the abuse-related neurochemical and behavioral effects of MDPV are stereoselective and that S(+)MDPV is the more potent of the two optical isomers in (a) inhibiting the reuptake at DAT, (b) inhibiting reuptake at NET, and (c) producing facilitation of ICSS. Moreover, the failure of R(−)MDPV to alter ICSS suggests that even high doses of this less active isomer do not produce effects at other nontransporter targets that either enhance or oppose abuse-related effects mediated by the S(+) isomer. Given the present findings, future studies should examine the pharmacology of stereoisomers of newly emerging MDPV analogues, such as α-pyrrolidinovalerophenone (α-PVP), that are appearing in the recreational drug marketplace.
METHODS
Chemistry
All commercially available reagents and solvents were purchased from Sigma-Aldrich Co. (St. Louis, MO) and Platte Valley Scientific Product List (Gothenburg, NE) and used as delivered. Melting points were measured in glass capillary tubes (Thomas-Hoover melting point apparatus) and are uncorrected. 1H NMR spectra were recorded with a Bruker 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane as an internal standard. Mass spectra were obtained using a PerkinElmer AxION 2. Optical rotations were measured using a Jasco DIP-1000 polarimeter. Reactions and product mixtures were routinely monitored by thin-layer chromatography (TLC) on silica gel precoated F254 Merck plates.
S(+)-Methylendioxypyrovalerone Hydrochloride (3S)
Jones reagent (0.65 mL, 1.62 mmol; prepared from chromium(VI) oxide (2.50g), concentrated H2SO4 (2.50 mL), and H2O (7.50 mL)) was added in a dropwise manner to a solution of 10S (0.34 g, 1.08 mmol) in a mixture of acetone (10 mL) and H2O (2 mL) at 0 °C (ice bath). The reaction mixture was allowed to stir at 0 °C (ice bath) for 1 h and then at room temperature for 18 h. The reaction mixture was basified by adding a saturated solution of NaHCO3, and acetone was removed under reduced pressure. The aqueous solution was extracted with EtOAc (3 × 15 mL). The combined organic portion was washed with H2O (20 mL) and brine (20 mL) and dried (Na2SO4). Solvent was removed under reduced pressure. The residue was dissolved in small amount of Et2O and filtered. The filtrate was treated with a saturated solution of gaseous HCl in Et2O. The precipitate was collected by filtration and recrystallized from a mixture of absolute EtOH/Et2O to afford the product (0.19 g, 56%) as light-beige crystals. mp 232–234 °C dec. 1H NMR (DMSO-d6) δ 0.79 (t, J = 7.2 Hz, 3H, CH3), 0.99–1.12 (m, 1H, CH2), 1.15–1.28 (m, 1H, CH2), 1.82–2.03 (m, 6H, CH2), 3.95–3.05 (m, 1H, CH2), 3.15–3.25 (m, 1H, CH2), 3.38–3.49 (m, 1H, CH2), 3.57–3.63 (m, 1H, CH2), 5.43 (dd, 1H, CH), 6.20 (s, 2H, CH2), 7.15 (d, J = 8.2 Hz, 1H, ArH), 7.57 (d, J = 1.7 Hz, 1H, ArH), 7.76 (dd, J = 8.2, 1.7 Hz, 1H, ArH), 10.54 (br s, 1H, NH+ ex with D2O). MS m/z calcd, 276.1600; found, 276.1620. +17.3° (c 0.5, MeOH).
R(−)-Methylendioxypyrovalerone Hydrochloride (3R)
The compound was synthesized from 10R using the procedure described above for the synthesis of the opposite isomer. The product (81%) was obtained as light-beige crystals. mp 233–235 °C dec. 1H NMR (DMSO-d6) δ 0.79 (t, J = 7.2 Hz, 3H, CH3), 0.99–1.12 (m, 1H, CH2), 1.15–1.28 (m, 1H, CH2), 1.82–2.03 (m, 6H, CH2), 3.95–3.05 (m, 1H, CH2), 3.15–3.25 (m, 1H, CH2), 3.38–3.49 (m, 1H, CH2), 3.57–3.63 (m, 1H, CH2), 5.43 (dd, 1H, CH), 6.20 (s, 2H, CH2), 7.15 (d, J = 8.2 Hz, 1H, ArH), 7.57 (d, J = 1.7 Hz, 1H, ArH), 7.76 (dd, J = 8.2, 1.7 Hz, 1H, ArH), 10.54 (br s, 1H, NH+ ex with D2O). MS m/z calcd, 276.1600; found, 276.1598. −23.3° (c 0.5, MeOH).
N-Boc-L-norvaline (5S)
Synthesized in a manner comparable to that reported for N-Boc-D-valine;17 sodium bicarbonate (7.17 g, 85.36 mmol), di-tert-butyl dicarbonate (9.31 g, 42.68 mmol), and dioxane (60 mL) were added to a stirred suspension of L-norvaline (4S) (5.00 g, 42.68 mmol) in H2O (60 mL). The reaction mixture was heated at reflux for 24 h. Solvent was removed under reduced pressure, and the aqueous solution was washed with EtOAc (80 mL). The aqueous portion was treated with a saturated solution of KHSO4 (80 mL). The oily layer was separated, dried (Na2SO4), and filtered using EtOAc. Solvent was removed under reduced pressure to afford the product (4.85 g, 52%) as a yellow viscous oil. 1H NMR (DMSO-d6) δ 0.85 (t, J = 7.3 Hz, 3H, CH3), 1.28–1.35 (m, 2H, CH2), 1.35 (s, 9H, (CH3)3), 1.38–1.64 (m, 2H, CH2), 3.85–3.88 (m, 1H, CH), 7.02 (d, J = 8.0 Hz, 1H, NH), 12.34 (br s, 1H, COOH). −16.1° (c 1.175, MeOH), lit.18 −14° (c 1, MeOH).
N-Boc-L-norvaline (5R)
The product (63%) was obtained as a yellow oil. 1H NMR (DMSO-d6) δ 0.86 (t, J = 7.3 Hz, 3H, CH3), 1.27–1.43 (m, 2H, CH2), 1.39 (s, 9H, (CH3)3), 1.49–1.66 (m, 2H, CH2), 3.83–3.89 (m, 1H, CH), 6.99 (d, J = 8.0 Hz, 1H, NH). +13.6° (c 1.265, MeOH), lit.18 +15° (c 1, MeOH).
S-tert-Butyl(1-oxo-1-(pyrrolidin-1-yl)pentan-2-yl)carbamate (6S)
Pivaloyl chloride (2.74 g, 22.78 mmol) was added in a dropwise manner to a stirred solution of 5S (4.95 g, 22.78 mmol) and Et3N (2.31 g, 22.78 mmol) in anhydrous THF (80 mL) at 0 °C (ice bath) under an N2 atmosphere. The resulting suspension was allowed to stir at 0 °C for 1 h. Pyrrolidine (3.24 g, 45.56 mmol) was added in a dropwise manner, and the reaction mixture was allowed to stir at room temperature for 18 h. Solvent was removed under reduced pressure, and the residue was dissolved in EtOAc (50 mL) and washed with H2O (30 mL). The organic portion was dried (Na2SO4), and solvent was removed under reduced pressure. The resulting thick yellow oil was purified by Kugelrohr distillation to afford the product (5.51 g, 89%) as a yellow oil. bp 190 °C, 0.3 Torr. 1H NMR (CDCl3) δ 0.93 (t, J = 7.3 Hz, 3H, CH3), 1.36–1.45 (m, 2H, CH2), 1.43 (s, 9H, (CH3)3), 1.47–1.68 (m, 2H, CH2), 1.83–1.91 (m, 2H, CH2), 1.93–2.00 (m, 2H, CH2), 3.38–3.44 (m, 2H, CH2), 3.45–3.55 (m, 1H, CH2), 3.65–3.68 (m, 1H, CH2), 4.38–4.44 (m, 1H, CH), 5.32 (d, J = 8.4 Hz, 1H, NH). −18.6° (c 1.755, MeOH).
R-tert-Butyl(1-oxo-1-(pyrrolidin-1-yl)pentan-2-yl)carbamate (6R)
The product (81%) was obtained as a yellow oil. bp 206 °C, 0.4 Torr. 1H NMR (CDCl3) δ 0.93 (t, J = 7.3 Hz, 3H, CH3), 1.34–1.44 (m, 2H, CH2), 1.43 (s, 9H, (CH3)3), 1.49–1.68 (m, 2H, CH2), 1.82–1.90 (m, 2H, CH2), 1.93–2.00 (m, 2H, CH2), 3.38–3.45 (m, 2H, CH2), 3.49–3.55 (m, 1H, CH2), 3.62–3.68 (m, 1H, CH2), 4.38–4.44 (m, 1H, CH), 5.33 (d, J = 8.4 Hz, 1H, NH). +17.2° (c 1.545, MeOH).
S(+)-tert-Butyl(1-(benzo[d][1,3]dioxol-5-yl)-1-oxopentan-2-yl)-carbamate (7S)
3,4-(Methylenedioxy)phenylmagnesium bromide (0.5 M in THF; 76.32 mL, 38.16 mmol) was added in a dropwise manner to a stirred solution of 6S (3.47 g, 12.72 mmol) in anhydrous THF (70 mL) at 0 °C (ice bath) under an N2 atmosphere. The reaction solution was allowed to stir at room temperature for 24 h. A saturated solution of NH4Cl (170 mL) was added in a dropwise manner at 0 °C (ice bath) followed by EtOAc (90 mL). The organic layer was separated, and the aqueous portion was extracted with EtOAc (2 × 50 mL). The combined organic portion was dried (Na2SO4), and solvent was removed under reduced pressure. The residue was purified by flash chromatography (silica gel; hexanes/EtOAc 9:1) to afford the product (2.88 g, 70%) as a thick yellow oil. 1H NMR (CDCl3) δ 0.90 (t, J = 7.3 Hz, 3H, CH3), 1.33–1.60 (m, 3H, CH2), 1.44 (s, 9H, (CH3)3), 1.76–1.85 (m, 1H, CH2), 5.17–5.22 (m, 1H, CH), 5.42 (d, J = 7.8 Hz, 1H, NH), 6.06 (s, 2H, CH2), 6.87 (d, J = 8.2 Hz, 1H, ArH), 7.43 (d, J = 1.5 Hz, 1H, ArH), 7.59 (dd, J = 8.2 Hz, 1H, ArH). +17.7° (c 1.83, MeOH).
R(−)-tert-Butyl(1-(benzo[d][1,3]dioxol-5-yl)-1-oxopentan-2-yl)-carbamate (7R)
The product (78%) was obtained as a yellow oil. 1H NMR (CDCl3) δ 0.90 (t, J = 7.3 Hz, 3H, CH3), 1.26–1.53 (m, 3H, CH2), 1.44 (s, 9H, (CH3)3), 1.71–1.80 (m, 1H, CH2), 5.16–5.22 (m, 1H, CH), 5.42 (d, J = 6.7 Hz, 1H, NH), 6.06 (s, 2H, CH2O), 6.87 (d, J = 8.1 Hz, 1H, ArH), 7.44 (s, 1H, ArH), 7.59 (d, J = 7.9 Hz, 1H, ArH). −19.8° (c 1.385, MeOH).
S(+)-2-Amino-1-(benzo[d][1,3]dioxol-5-yl)pentan-1-one Hydrochloride (8S)
Compound 7S (2.75 g, 8.56 mmol) was added to a solution of HCl in dioxane (4.4 M, 90 mL), and the reaction mixture was allowed to stir at room temperature for 20 min. Solvent was removed under reduced pressure, and the resulting white solid was recrystallized from absolute EtOH to afford the product (2.02 g, 89%) as white crystals. mp 206–208 °C. 1H NMR (DMSO-d6) δ 0.82 (t, J = 7.3 Hz, 3H, CH3), 1.15–1.29 (m, 1H, CH2), 1.32–1.45 (m, 1H, CH), 1.65–1.83 (m, 2H, CH2), 5.03 (dd, J = 7.0, 4.7 Hz, 1H, CH), 6.19 (s, 2H, CH2), 7.11 (d, J = 8.2 Hz, 1H, ArH), 7.55 (d, J = 1.7 Hz, 1H, ArH), 7.71 (dd, J = 8.2, 1.7 Hz, 1H, ArH), 8.42 (br s, 3H, NH3+ ex with D2O). +48.4° (c 0.5, CH2Cl2).
R(−)-2-Amino-1-(benzo[d][1,3]dioxol-5-yl)pentan-1-one Hydrochloride (8R)
The product (90%) was obtained as a white solid. mp 206–208 °C. 1H NMR (DMSO-d6) δ 0.82 (t, J = 7.3 Hz, 3H, CH3), 1.15–1.29 (m, 1H, CH2), 1.32–1.45 (m, 1H, CH), 1.65–1.83 (m, 2H, CH2), 5.04 (m, 1H, CH), 6.19 (s, 2H, CH2O), 7.11 (d, J = 8.1 Hz, 1H, ArH), 7.56 (s, 1H, ArH), 7.72 (d, J = 8.1 Hz, 1H, ArH), 8.53 (br s, 3H, NH3+ ex with D2O). −52.9° (c 0.65, CH2Cl2).
2S(−)-2-Amino-1-(benzo[d][1,3]dioxol-5-yl)pentan-1-ol Hydrochloride (9S)
Sodium borohydride (0.52 g, 13.96 mmol) was added over a 15 min period to a solution of 8S (1.80 g, 6.98 mmol) in anhydrous MeOH (50 mL) at 0 °C (ice bath) under an N2 atmosphere. The reaction mixture was allowed to stir at 0 °C (ice bath) for 30 min, and then it was acidified by adding HCl (1N, to pH ~ 1). Solvent was removed under reduced pressure. The residue was dissolved in H2O and basified by adding NaOH (15%, to pH ~ 12). The mixture was extracted with EtOAc (3 × 30 mL). The combined organic portion was washed with H2O (30 mL) and brine (30 mL) and dried (Na2SO4), and solvent was removed under reduced pressure. The oily residue was dissolved in anhydrous Et2O and converted to the hydrochloride salt by adding a saturated solution of gaseous HCl in Et2O. Solvent was removed, and the solid was recrystallized twice from absolute EtOH to afford the product (1.52 g, 84%) as a white solid. mp 161–162 °C. 1H NMR (DMSO-d6) δ 0.77 (t, J = 6.9 Hz, 3H, CH3), 1.08–1.20 (m, 1H, CH2), 1.29–1.41 (m, 3H, CH2), 3.21–3.25 (m, 1H, CH), 4.88 (t, J = 3.6 Hz, 1H, CH), 6.00 (d, J = 4.2 Hz, 1H, CH), 6.02 (s, 2H, CH2), 6.85 (d, J = 6.0 Hz, 1H, ArH), 6.91 (d, J = 8.0 Hz, 1H, ArH), 6.96 (s, 1H, ArH), 7.98 (br s, 3H, NH3+ ex with D2O). −33.3° (c 0.64, MeOH).
2R(+)-2-Amino-1-(benzo[d][1,3]dioxol-5-yl)pentan-1-ol Hydrochloride (9R)
The product (74%) was obtained as a white solid. mp 159–161 °C. 1H NMR (DMSO-d6) δ 0.77 (t, J = 6.8 Hz, 3H, CH3), 1.07–1.20 (m, 1H, CH2), 1.26–1.41 (m, 3H, CH2), 3.21–3.25 (m, 1H, CH), 4.88 (t, J = 3.5 Hz, 1H, CH), 6.01 (d, J = 4.2 Hz, 1H, CH), 6.02 (s, 2H, CH2), 6.85 (dd, J = 8.0, 1.0 Hz, 1H, ArH), 6.91 (d, J = 8.0 Hz, 1H, ArH), 6.96 (d, J = 1.0 Hz, 1H, ArH), 8.01 (br s, 3H, NH3+ ex with D2O). +29.6° (c 0.545, MeOH).
2S(−)-1-(Benzo[d][1,3]dioxol-5-yl)-2-(pyrrolidin-1-yl)pentan-1-ol Hydrochloride (10S)
1,4-Dibromobutane (0.33 g, 1.54 mmol) was added to a mixture of 9S (0.40 g, 1.54 mmol) and potassium bicarbonate (0.77 g, 7.70 mmol) in anhydrous CH3CN (15 mL). The reaction mixture was heated at reflux for 24 h. After cooling to room temperature, it was filtered and the solid was washed with CH3CN. Solvent was removed under reduced pressure. The oily residue was dissolved in MeOH and converted to the hydrochloride salt by adding a saturated solution of gaseous HCl in Et2O. Precipitate was collected by filtration and recrystallized twice from absolute EtOH to afford the product (0.38 g, 81%) as white crystals. mp 208 °C dec. 1H NMR (DMSO-d6) δ 0.61 (t, J = 7.2 Hz, 3H, CH3), 0.71–0.81 (m, 1H, CH2), 1.04–1.13 (m, 1H, CH2), 1.31–1.35 (m, 1H, CH2), 1.63–1.72 (m, 1H, CH2), 1.89–2.10 (m, 4H, CH2), 3.05–3.16 (m, 1H, CH2), 3.27–3.37 (m, 1H, CH2), 3.50–3.58 (m, 1H, CH2), 3.62–3.68 (m, 1H, CH2), 5.18 (m, 1H, CH), 6.01 (s, 2H, CH2), 6.21 (d, J = 4.2 Hz, 1H, CH), 6.92 (m, 2H, ArH), 6.99 (s, 1H, ArH), 9.92 (br s, 1H, NH+ ex with D2O). −31.7° (c 0.535, MeOH).
2R(+)-1-(Benzo[d][1,3]dioxol-5-yl)-2-(pyrrolidin-1-yl)pentan-1-ol Hydrochloride (10R)
The product (86%) was obtained as a white solid. mp 209–210 °C dec. 1H NMR (DMSO-d6) δ 0.62 (t, J = 7.2 Hz, 3H, CH3), 0.71–0.84 (m, 1H, CH2), 1.04–1.13 (m, 1H, CH2), 1.31–1.39 (m, 1H, CH2), 1.64–1.74 (m, 1H, CH2), 1.86–2.10 (m, 4H, CH2), 3.07–3.18 (m, 1H, CH2), 3.34–3.40 (m, 1H, CH2), 3.55–3.60 (m, 1H, CH2), 3.62–3.69 (m, 1H, CH2), 5.19–5.22 (m, 1H, CH), 6.02 (s, 2H, CH2), 6.23 (d, J = 4.6 Hz, 1H, CH), 6.93 (m, 2H, ArH), 7.01 (s, 1H, ArH), 10.08 (br s, 1H, NH+ ex with D2O). +26.7° (c 0.61, MeOH).
In Vitro Reuptake Assays
Subjects
Male Sprague–Dawley rats (Charles River, Wilmington, MA) weighing 250–350 g were housed three per cage with free access to food and water and maintained on a 12 h light/dark cycle with lights on from 7:00 a.m. to 7:00 p.m. Animal facilities were accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, and procedures were carried out in accordance with the Institutional Animal Care and Use Committee and the National Institutes of Health guidelines on care and use of animal subjects in research (National Research Council, 2011).
Procedure
[3H]Transmitters (specific activity ranging from 30 to 50 Ci/mmol) were purchased from PerkinElmer (Shelton, CT). All other chemicals and reagents were acquired from Sigma-Aldrich (St. Louis, MO). Rats were euthanized by CO2 narcosis, and brains were processed to yield synaptosomes as previously described.6 Rat caudate tissue was used for DAT assays, whereas rat whole brain minus caudate and cerebellum was used for NET and SERT assays. In uptake inhibition assays, 5 nM [3H]dopamine, 10 nM [3H]norepinephrine, and 5 nM [3H]serotonin were used to assess transport activity at DAT, NET, and SERT, respectively. The selectivity of uptake assays was optimized for a single transporter by including unlabeled blockers to prevent uptake of [3H]transmitter by competing transporters. Uptake inhibition assays were initiated by adding 100 μL of tissue suspension to 900 μL of Krebs-phosphate buffer (126 mM NaCl, 2.4 mM KCl, 0.83 mM CaCl2, 0.8 mM MgCl2, 0.5 mM KH2PO4, 0.5 mM Na2SO4, 11.1 mM glucose, 0.05 mM pargyline, 1 mg/mL bovine serum albumin, and 1 mg/mL ascorbic acid, pH 7.4) containing test drug and [3H]transmitter. Uptake inhibition assays were terminated by rapid vacuum filtration through Whatman GF/B filters, and retained radioactivity was quantified by liquid scintillation counting. Statistical analyses were carried out using GraphPad Prism (v. 6.0; GraphPad Scientific, San Diego, CA). IC50 values for inhibition of reuptake were calculated based on nonlinear regression analysis.
Intracranial Self-Stimulation Procedure
Subjects
Six adult male Sprague–Dawley rats (Harlan, Frederick, MD) weighing 398–444 g at the time of surgery were individually housed and maintained on a 12 h light/dark cycle with lights on from 6:00 a.m. to 6:00 p.m. Rats had free access to food and water except during experimental sessions. Animal maintenance and research were in compliance with the National Institutes of Health guidelines for the care and use of animal subjects in research and adhered to guidelines of the Committee for Research (National Research Council, 2011). The Virginia Commonwealth University Institutional Animal Care and Use Committee approved the research protocol.
Surgery
Rats were anesthetized with isoflurane (2.5–3% in oxygen; Webster Veterinary, Phoenix, AZ) until unresponsive to toe pinch prior to implantation of stainless steel bipolar electrodes (Plastics One, Roanoke, VA). The cathode of each electrode was 0.25 mm in diameter and covered with polyamide insulation except at the flattened tip, and the anode was 0.125 mm in diameter and uninsulated. The cathode was stereotaxically implanted into the left medial forebrain bundle at the level of the lateral hypothalamus (2.8 mm posterior to the bregma, 1.7 mm lateral to midsagittal suture, 8.8 mm ventral to the skull). Three screws were placed in the skull, and the anode was wrapped around one screw to serve as the ground. The skull screws and electrode were secured to the skull with dental acrylic. Ketoprofen (5 mg/kg) was used for postoperative analgesia immediately and 24 h after surgery. Rats were allowed 7 recovery days prior to ICSS training.
Apparatus
Experimental sessions were conducted in sound-attenuating boxes containing modular acrylic and metal test chambers (29.2 × 30.5 × 24.1 cm) equipped with a response lever (4.5 cm wide, 2.0 cm deep, 3 cm off the floor), three stimulation lights (red, yellow, and green) located 7.6 cm above the lever, a 2 W house light, and an ICSS stimulator (Med Associates, St. Albans, VT). Electrodes were connected to the stimulator via a swivel commutator (Model SL2C, Plastics One, Roanoke, VA). Control of experimental events and acquisition of data were accomplished with a computer operated by Med-PC IV software and connected to test chambers by an interface system (Med Associates).
Training
Following initial shaping of lever pressing, rats were trained under a fixed-ratio 1 (FR 1) schedule of brain stimulation reinforcement using a procedure identical to that previously described.12,14,15 Each lever press resulted in the delivery of a 0.5 s train of square-wave cathodal pulses (0.1 ms per pulse) and illumination of the stimulus lights above the lever. Stimulation intensity and frequency were set at 150 μA and 2.1 log Hz, respectively, during initial 30–60 min training sessions. Stimulation intensity was then adjusted on an individual basis to the lowest value that sustained ICSS rates > 30 stimulations/min. This intensity (110–260 μA across rats) was held constant for the remainder of the study, and frequency manipulations were then introduced. Sessions involving frequency manipulations consisted of three sequential 10 min components. During each component, a descending series of 10 frequencies (2.2–1.75 log Hz in 0.05 log increments) was presented, with each frequency of stimulation available for a 60 s trial. Each frequency trial consisted of a 10 s time-out, during which five response-independent priming stimulations were delivered at the frequency of stimulation that would be available during that trial, followed by a 50 s response period, during which responding produced electrical stimulation under a FR 1 schedule. Training continued until rats responded at rates greater than 50% of the maximal control rate (see below) for the first three to six frequency trials of each component over a period of at least 3 consecutive training days.
Testing
Test sessions consisted of three sequential baseline components followed by a 30 min time-out period and then by two sequential test components. A single test drug dose was administered intraperitoneally (i.p.) at the beginning of the time-out period. Test sessions were conducted on Tuesdays and Fridays, and training sessions were conducted on all other weekdays. Dose ranges were based on previous research with (±)MDPV15 and initial empirical results. Rats were divided into two groups: one group started testing with S(+)MDPV first and the other group started testing with R(−)MDPV. After a complete dose–effect function was obtained with one isomer, rats were then tested with the other isomer. The order of testing with vehicle and drug doses was varied across subjects using a Latin-square design, with the exception that 10 mg/kg R(−)MDPV was tested after all other doses. After completion of testing with MDPV isomers, a single dose of 10 mg/kg of S(−)cocaine was tested as a positive control for comparison. One rat lost its headcap after completion of testing with S(+)MDPV and was not tested with R(−)MDPV or cocaine.
Data Analysis
The first baseline component of each test day was considered a warm-up component, and data were discarded. Data from the second and third baseline component were averaged, and data from the two test components were averaged, for further analysis. The primary dependent measure was the total number of stimulations per component. Test data were normalized to individual baseline data using the following equation: % baseline total stimulations per component = (mean total stimulations per test component)/(mean total stimulations per baseline component) × 100. Data were then averaged across rats for each dose of a given drug and analyzed by one-way ANOVA, with drug dose as the single factor. A significant ANOVA was followed by a Dunnett’s posthoc test, and the criterion for statistical significance was p < 0.05.
As a secondary measure to evaluate drug effects, the number of stimulations during each frequency trial within a component were converted to percent maximum control rate (%MCR), with maximum control rate defined as the mean of the maximal rates obtained at any frequency during the second and third baseline components for that day. Thus, %MCR values for each trial were calculated as (reinforcement rate during a frequency trial)/(MCR) × 100. For this study, the mean ± SEM MCR was 52.3 ± 5.9 stimulations per frequency trial. For each test session, data from the second and third baseline components were averaged to yield a baseline frequency–rate curve, and data from test components were averaged to generate test frequency–rate curves. Baseline and test curves were then averaged across rats to yield mean baseline and test curves for each manipulation. Group mean frequency–rate curves were analyzed by repeated measures two-way ANOVA with ICSS frequency and dose as factors. A significant ANOVA was followed by the Holm–Sidak multiple comparisons posthoc test, and the criterion for statistical significance was p < 0.05.
Supplementary Material
Acknowledgments
Funding
This work was supported in part by PHS grant R01 DA033930 and T32 DA007027.
Footnotes
Author Contributions
R.K. synthesized and characterized the target compounds. J.S.P. and M.H.B. performed the reuptake assays, and B.A.H., M.L.B., and S.S.N. conducted the ICSS assays and analyzed the data. R.A.G., M.H.B., and S.S.N. oversaw the project. R.A.G. prepared the first draft of the manuscript, and all authors contributed to the preparation of the final manuscript.
The authors declare no competing financial interest.
Two other methods explored for the synthesis of MDPV isomers that were described in the body of the article. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.United Nations Narcotic Laboratory. Studies on the chemical composition of khat. III. Investigations on the phenylalkylamine fraction. United Nations; Geneva, Switzerland: 1975. MNAR/11/75, GE.75-12624. [Google Scholar]
- 2.Glennon RA. Bath salts, mephedrone, and methyl-enedioxypyrovalerone as emerging illicit drugs that will need targeted therapeutic intervention. Adv Pharmacol. 2014;69:581–620. doi: 10.1016/B978-0-12-420118-7.00015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The challenge of new psychoactive substances. United Nations Office of Drugs and Crime; Vienna, Austria: 2013. [Google Scholar]
- 4.Baumann MH, Partilla JS, Lehner KR. Psychoactive “bath salts”: not so soothing. Eur J Pharmacol. 2013;698:1–5. doi: 10.1016/j.ejphar.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Synthetic Drug Abuse Prevention Act, Section 1152. Addition of synthetic drugs to Schedule I of the Controlled Substances Act. 2012:138–140. http://www.gpo.gov/fdsys/pkg/BILLS-112s3187enr/pdf/BILLS-112s3187enr.pdf.
- 6.Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M, Rothman RB, Goldberg SR, Lupica CR, Sitte HH, Brandt SD, Tella SR, Cozzi NV, Schindler CW. Powerful cocaine-like actions of 3,4-methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology. 2013;38:552–562. doi: 10.1038/npp.2012.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron K, Kolanos R, Verkariya R, De Felice L, Glennon RA. Mephedrone and methylenedioxypyrovalerone (MDPV), major constituents of “bath salts,” produce opposite effects at the human dopamine transporter. Psychopharmacology. 2013;227:493–499. doi: 10.1007/s00213-013-2967-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME. Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol. 2013;168:458–470. doi: 10.1111/j.1476-5381.2012.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A. Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol. 2013;85:803–815. doi: 10.1016/j.bcp.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cameron KN, Kolanos R, Solis E, Glennon RA, De Felice LJ. Bath salts components mephedrone and methylenedioxypyrovalerone (MDPV) act synergistically at the human dopamine transporter. Br J Pharmacol. 2013;68:1750–1757. doi: 10.1111/bph.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolanos R, Solis E, Sakloth F, De Felice LJ, Glennon RA. “Deconstruction” of the abused synthetic cathinone methylenedioxypyrovalerone (MDPV) and an examination of effects at the human dopamine transporter. ACS Chem Neurosci. 2013;4:1524–1529. doi: 10.1021/cn4001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Negus SS, Miller LL. Intracranial self-stimulation (ICSS) to evaluate abuse potential of drugs. Pharmacol Rev. 2014;66:869–917. doi: 10.1124/pr.112.007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meltzer PC, Butler D, Deschamps JR, Madras BK. 1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (pyrovalerone) analogues: a promising class of monoamine uptake inhibitors. J Med Chem. 2006;49:1420–1432. doi: 10.1021/jm050797a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonano JS, Runyon SP, Hassler C, Glennon RA, Negus SS. Effects of the neuropeptide S receptor antagonist RTI-118 on abuse-related facilitation of intracranial self-stimulation produced by cocaine and methylenedioxypyrovalerone (MDPV) in rats. Eur J Pharmacol. 2014;743:98–105. doi: 10.1016/j.ejphar.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonano JS, Glennon RA, De Felice LJ, Banks ML, Negus SS. Abuse-related and abuse-limiting effects of methcathinone and the synthetic “bath salts” cathinone analogs methylenedioxypyrovalerone (MDPV), methylone and mephedrone on intracranial self-stimulation in rats. Psychopharmacology. 2014;231:199–207. doi: 10.1007/s00213-013-3223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg MB, Carroll FI, Negus SS. Effects of monoamine reuptake inhibitors in assays of acute pain-stimulated and pain-depressed behavior in rats. J Pain. 2013;14:246–259. doi: 10.1016/j.jpain.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nájera C, Abellán T, Sansano JM. Asymmetric synthesis of α-methyl α-amino acids through diastereoselective alkylation under mild reaction conditions of an alanine template with a 1,2,3,6-tetrahydro-2-pyrazinone structure. Eur J Org Chem. 2000;15:2809–2820. [Google Scholar]
- 18.Sasaki NA, Hashimoto Ch, Potier P. A novel approach to the synthesis of optically pure non protein α-amino acids in both L and D configurations from L-serine. Tetrahedron Lett. 1987;28:6069–6072. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.