

Late endosomes are a focal point of cholesterol transport. NPC and LAMP proteins, which are required for cholesterol export from late endosomes, are necessary for effective Plasmodium berghei development.
Abstract
While lysosomes are degradative compartments and one of the defenses against invading pathogens, they are also hubs of metabolic activity. Late endocytic compartments accumulate around Plasmodium berghei liver-stage parasites during development, and whether this is a host defense strategy or active recruitment by the parasites is unknown. In support of the latter hypothesis, we observed that the recruitment of host late endosomes (LEs) and lysosomes is reduced in uis4− parasites, which lack a parasitophorous vacuole membrane protein and arrest during liver-stage development. Analysis of parasite development in host cells deficient for late endosomal or lysosomal proteins revealed that the Niemann–Pick type C (NPC) proteins, which are involved in cholesterol export from LEs, and the lysosome-associated membrane proteins (LAMP) 1 and 2 are important for robust liver-stage P. berghei growth. Using the compound U18666A, which leads to cholesterol sequestration in LEs similar to that seen in NPC- and LAMP-deficient cells, we show that the restriction of parasite growth depends on cholesterol sequestration and that targeting this process can reduce parasite burden in vivo. Taken together, these data reveal that proper LE and lysosome function positively contributes to liver-stage Plasmodium development.
INTRODUCTION
Although many intracellular pathogens encounter, or even exploit, the host endocytic pathway upon invasion, apicomplexan parasites avoid the endocytic pathway upon entry through their active invasion of cells (Sibley, 2011). Recent studies of Plasmodium berghei parasites, however, found that late endosomes (LEs) and lysosomes of the host progressively accumulate around the parasitophorous vacuole (PV) and associated tubovesicular network (TVN) during the first 24 h of parasite liver-stage development (Lopes da Silva et al., 2012; Grützke et al., 2014). The mechanisms and consequences of this recruitment are thus far undefined.
After development within hepatocytes, Plasmodium parasites infect red blood cells and cause malaria. Although red blood cells have minimal resources available to the parasites, blood-stage Plasmodium are well adapted to take advantage of their host environment, for example, through the metabolism of host hemoglobin (Sigala and Goldberg, 2014). Hepatocytes as host cells could offer more opportunities for parasites to usurp host processes but could also present a greater chance of encountering cellular defense pathways. Fusion of the liver-stage PV with host lysosomes can act as one of these defense pathways and lead to parasite killing (Prado et al., 2015; Boonhok et al., 2016); however, for the majority of liver-stage parasites, any fusion of the host late endocytic compartments with the PV membrane (PVM) is minimal because markers of LEs and lysosomes such as CD63 and lysosome-associated membrane protein 1 (LAMP1) do not appear to label the membrane of the PV, and the PV does not have the acidity of lysosomes (Lopes da Silva et al., 2012; Grützke et al., 2014; Prado et al., 2015).
The PV interacts directly with the host autophagy pathway, as seen by the recruitment of the autophagy protein LC3 to the developing parasite (Grützke et al., 2014; Prado et al., 2015; Thieleke-Matos et al., 2016). Typically, autophagosome maturation results in fusion with lysosomes, and some of the lysosomes associated with the PV also possess the autophagy marker LC3; however, the PV of properly developing parasites does not appear to mature as an autophagosome beyond LC3 recruitment, and further maturation seems detrimental to the parasites (Prado et al., 2015). Some of the late endocytic vesicles surrounding the PV do possess both LC3 and lysosomal markers, indicating that some of these associated vesicles are maturing amphisomes (Thieleke-Matos et al., 2016). Parasite-encoded proteins within the liver-stage PVM and TVN are at the interface between the parasite and host and likely play a critical role in the interaction with the host cell and its organelles. For instance, the liver stage–specific protein up-regulated in infective sporozoites 4 (UIS4) delineates the PVM, and reverse genetics established the necessity of this PVM protein for parasite maturation inside the liver (Mueller et al., 2005), but the underlying molecular mechanism that leads to growth restriction in the absence of UIS4 remains enigmatic.
To elucidate the function and mechanism of late endocytic organelle recruitment to liver stage parasites, we assessed this recruitment upon infection with parasites deficient for specific parasite PVM proteins and investigated the influence of late endosomal host proteins on parasite development. We analyzed parasite development within cells deficient for the late endosomal/lysosomal proteins Niemann–Pick type C (NPC) 1 and 2, LAMP1, and LAMP2. The prominent phenotypes of cells deficient for these proteins include defects in autophagic flux and the accumulation of cholesterol in LEs and lysosomes due to inefficient export of free cholesterol from these vesicles (Eskelinen et al., 2004; Chang et al., 2005; Schneede et al., 2011). In the present model, endocytosed cholesteryl esters are hydrolyzed upon reaching LEs, and the resulting unesterified cholesterol in the lysosomal lumen is initially bound by NPC2, and subsequently transferred to NPC1 in the limiting LE/lysosomal membrane (Wang et al., 2010). Cells deficient for LAMP1 and LAMP2 phenocopy the defects observed in cells deficient for either NPC1 or NPC2, indicating that these two type I transmembrane glycoproteins are also required for proper export of cholesterol out of LEs and lysosomes (Schneede et al., 2011).
Plasmodium is unable to synthesize cholesterol de novo, but the parasite PVM is enriched with cholesterol (Bano et al., 2007). Although Plasmodium can access cholesterol from the host via either the endogenous or exogenous pathways (Labaied et al., 2011), neither pathway has been shown to be necessary for parasite growth, and the significance of this cholesterol acquisition for Plasmodium liver-stage development is undetermined.
RESULTS
Efficient recruitment of host late endocytic compartments requires UIS4
The striking recruitment of host LEs and lysosomes to developing P. berghei liver-stage parasites (Lopes da Silva et al., 2012; Supplemental Figure S1A) and to the liver-stage TVN (Grützke et al., 2014) prompted us to investigate whether parasite PVM/TVN proteins play a role in recruitment of these vesicles. We tested recombinant parasite lines deficient for the proteins UIS4 (Mueller et al., 2005) and IBIS1 (Ingmundson et al., 2012) for their capacity to recruit late endocytic compartments (Figure 1, A and B). Both UIS4 and IBIS1 are transmembrane P. berghei-encoded proteins that localize to the liver-stage PVM/TVN. Although absence of IBIS1 does not appear to affect liver-stage growth (Ingmundson et al., 2012; Supplemental Figure S2A), infection with uis4− parasites results in reduced parasite burden in the liver and liver-stage arrest (Mueller et al., 2005; Supplemental Figure S2A). In vitro, uis4− parasites are less likely to successfully develop into liver-stage forms (Mueller et al., 2005; Supplemental Figure S2B), although those that do develop are approximately the same size as wild-type parasites (Supplemental Figure S2C). We confirmed the absence of UIS4 in these parasites with antibody staining of infected cells (Supplemental Figure S2D).
FIGURE 1:

UIS4 is required for maximal host LE and cholesterol recruitment to developing liver-stage P. berghei parasites. (A–C) Huh7 cells infected with GFP-expressing wild-type (WT), uis4−, or ibis1− parasites were fixed 24 h postinfection and stained with filipin (blue) and antibodies against CD63 (red) and GFP (green). Scale bars, 10 μm. (D) Huh7 cells were infected with P. berghei WT or mutant sporozoites, and infection was left to proceed until the indicated time points. Samples were stained with anti-GFP (parasite) and either anti-CD63 (LE/lysosomes; (top), anti-LAMP2 (LE/lysosomes; middle), or anti-LC3 (autophagic bodies; bottom). (E) WT and Atg5−/− MEFs infected with P. berghei sporozoites were fixed 24 h postinfection and stained with antibodies against PbHsp70 (parasite) and LAMP2 (LE/lysosomes). At least 50 parasites per well were scored for host marker recruitment. Results are shown as a mean percentage of total parasites analyzed per well (±SD). One representative experiment of three performed is shown. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001 (Fisher's exact test).
Although ibis1− parasites recruited LEs at a frequency comparable to wild-type parasites, noticeably fewer uis4− parasites were surrounded by CD63-positive vacuoles 24 h after infection. By assessing LE recruitment to parasites over time from soon after host cell invasion until the beginning of merosome development, we saw that the percentage of uis4− parasites that recruit LEs was at least 34% lower than that of wild-type parasites throughout infection (Figure 1C). The same pattern was observed in uis4−-infected cells stained with the LE and lysosome marker LAMP2 (Figure 1C).
Other known host components that have been shown to be associated with the liver-stage PVM are cholesterol and the autophagy protein LC3 (Labaied et al., 2011; Grützke et al., 2014; Prado et al., 2015). As previously reported, membranes surrounding liver-stage parasites are enriched with cholesterol, as detected by filipin staining (Labaied et al., 2011). When cells are infected with uis4− parasites, this enrichment of the PVM with cholesterol is detected less frequently (Figure 1, A and C), and when it is detected, the intensity of filipin staining surrounding parasites is typically less than around wild-type parasites (Supplemental Figure S2, E and F).
Furthermore, UIS4-deficient parasites recruit LC3 less efficiently than wild-type parasites when LC3 localization is examined over the course of infection (Figure 1C and Supplemental Figure S2G). Autophagosome maturation is associated with lysosome fusion. Therefore we assessed whether recruitment of lysosomes to the PVM depends on LC3 recruitment by examining recruitment of LAMP2-positive vesicles to liver-stage parasites in Atg5−/− cells, which are not capable of LC3 processing and lipidation (Kuma et al., 2004) and do not allow LC3 recruitment to developing parasites (Prado et al., 2015). Lysosome accumulation around liver-stage parasites does not depend on LC3 recruitment: no significant difference in LAMP2-vesicle association to parasites was seen in Atg5−/− cells compared with wild-type cells (Figure 1D). Therefore absence of LC3 around uis4− parasites is not directly responsible for the lack of recruited late endocytic vesicles. These results demonstrate that the absence of UIS4 on the PVM negatively affects the recruitment of host lysosomes and host cholesterol during liver-stage development. Although this result might be an indirect consequence of incomplete vacuole development of uis4− parasites rather than a direct interaction of UIS4 with the host endocytic pathway, the correlation between arrested growth and reduced LE and lysosome recruitment in uis4− parasites indicates that the recruitment of host late endocytic compartments signifies proper liver-stage development and might be of benefit to developing parasites.
Cells deficient for LAMP or NPC proteins fail to support efficient liver-stage parasite development
To begin to ascertain which host lyososomal functions could contribute to liver-stage parasite development, we infected embryonic fibroblasts (MEFs) derived from knockout mouse lines lacking specific lysosomal proteins (Scimeca et al., 2000; Eskelinen et al., 2004; Huang et al., 2014; Kissing et al., 2015). Because mouse embryonic fibroblasts are not a natural target cell for Plasmodium sporozoite infection, we wanted to ensure that these cells support normal parasite development. A comparison of parasite growth in hepatoma (Huh7) and wild-type MEFs showed that, although parasite numbers are in general lower in MEFs (unpublished data), once sporozoites establish infection, they develop into similarly sized liver-stage forms as in Huh7 cells (Supplemental Figure S3A). These data, together with successful merosome production (see Figure 3B later in this article), suggest that MEFs are a suitable model cell line for Plasmodium infection and development. MEFs deficient for the vacuolar ATPase subunit V0A3 (Scimeca et al., 2000) or V-ATPase accessory protein ATP6AP2 (Kissing et al., 2015) support P. berghei liver-stage growth as well as the comparable wild-type cell lines (Figure 2, A and B). These findings indicate that these proteins are dispensable for liver-stage maturation. In contrast, parasites developing within cells deficient for the lysosomal glycoproteins LAMP1 and LAMP2 were significantly smaller 24 h after sporozoite infection than those growing in wild-type cells (Figure 2C). Parasite growth was similarly restricted in cells lacking the NPC1 and NPC2; Figure 2D).
FIGURE 3:

Host LAMP1 and LAMP2 are required for efficient P. berghei liver-stage development. (A, B) WT and Lamp1−/−Lamp2−/− MEFs were infected with P. berghei sporozoites. Parasite size was assessed 48 h postinfection (A; right, representative images of mean-sized parasites), and the total number of merosomes was counted 70 h postinfection (B). (C) Protein levels of LAMP1 or LAMP2 in Huh7 cells transfected with siRNAs targeting either Lamp1 or Lamp2 were evaluated by Western blotting 48 h after transfection. The effect of three independent siRNAs (A–C) targeting Lamp1 or Lamp2 were compared with a nonspecific negative control siRNA (neg) using antibodies against LAMP1 or LAMP2, respectively. (D) siRNA-transfected cells were infected with P. berghei sporozoites 48 h posttransfection, and parasite size was evaluated 48 h postinfection. Parasites were visualized with anti-PbHsp70 antibody. One representative of at least three independent experiments is shown. ns, not significant; *p < 0.05; **p < 0.01 (Student's unpaired t test, n = 3).
FIGURE 2:

Host proteins involved in lipid export from late endocytic vacuoles are important for P. berghei development. (A–D) MEF cell lines deficient for late endocytic/lysosomal host proteins and corresponding WT MEFs were infected with P. berghei sporozoites, and parasite size was assessed by fluorescence microscopy 24 h postinfection. In all experiments, parasites were visualized by staining with anti-PbHsp70 antibody. One representative experiment of at least three performed is shown. ns, not significant; **p < 0.01; ***p < 0.001 (Student's unpaired t test, n = 3).
One phenotype shared by both cell lines in which reduced parasite growth was observed is the accumulation of cholesterol in LEs (Liscum, 2000; Eskelinen et al., 2004). One important function of late endocytic organelles lies in the processing and sorting of exogenously derived cholesterol, and deletion of Npc1, Npc2, or Lamp1 and Lamp2 blocks the export of free cholesterol from LEs (Brasaemle and Attie, 1990; Liscum, 2000; Eskelinen et al., 2004).
To determine the effect of cholesterol retention in LEs on infection outcome, we followed infection of Lamp1−/−Lamp2−/− cells until parasite egress. At 48 h after infection, the difference in parasite size between control and knockout cell lines was even more severe than at 24 h (Figure 3A). Parasites in control cells increased in size by more than sevenfold from 24 to 48 h, whereas parasites in LAMP1/LAMP2-deficient cells increased by only approximately fourfold. Because MEF cell lines are derived from individual embyronic day 12.5 embryos, they are prone to clonal differences. To further support our findings, we tested P. berghei growth in a second, independently generated LAMP1/LAMP2-deficient cell line. Parasite growth was similarly restricted in this second clone in comparison to the congenic wild-type line (Supplemental Figure S3, B and C).
Of note, the percentage of infected cells differed between wild-type and knockout MEF in the two LAMP1/LAMP2-deficient cell lines. Lamp1−/−Lamp2−/− cells of clone 1, but not clone 2, harbored more parasites after infection than wild-type control cell lines (unpublished data), indicating that infection rates can differ between MEF lines but cannot be attributed to loss of LAMP1 and LAMP2.
The impaired parasite growth in Lamp1−/− Lamp2−/− cells led to reduced production of merosomes (Figure 3B), the final stage of liver development, in which parasites are released (Sturm et al., 2006). Merosome numbers were reduced by >60% in knockout cells compared with wild-type MEFs. Because of the higher number of infected cells in Lamp1−/−Lamp2−/− cells, these results indicate that formation of first-generation merozoites is severely impaired in cells lacking LAMP1 and LAMP2.
To confirm independently the data we generated in the Lamp1−/−Lamp2−/− MEFs and address whether both LAMP proteins are similarly important for parasite development, we used small interfering RNA (siRNA) to target LAMP1 and LAMP2 expression individually. Gene expression was targeted with at least three distinct siRNA sequences (A–C) against either Lamp1 or Lamp2, and each siRNA was transfected individually into hepatoma cells. Using antibodies against LAMP1 or LAMP2, we confirmed efficient knockdown for all three siRNAs targeting Lamp1 and for siRNA A and B but not siRNA C targeting Lamp2 (Figure 3C). When the cells were infected, only siRNAs that reduced protein expression of LAMP2 caused significant reductions in parasite size (Figure 3D). Despite efficient knockdown of LAMP1, no effect on parasite development was observed. When LAMP2 knockdown was not achieved (siRNA C), parasite size was comparable to that for infected control cells. Combining LAMP1 knockdown with LAMP2 knockdown did not further enhance the phenotype (unpublished data). We conclude that LAMP2 expression is crucial for efficient parasite liver-stage development.
Sequestration of cholesterol in late endocytic vesicles restricts liver-stage growth
To validate the parasite growth phenotype observed in NPC- and LAMP-deficient cells, we used the amphipathic steroid 3-b-[2-(diethylamine)eth-oxy]androst-5-en-17-one (U18666A), which is well established to mimic the cellular phenotype caused by Npc mutations (Cenedella, 2009; Lu et al., 2015). U18666A blocks the exit of low-density lipoprotein (LDL)–derived free cholesterol from LEs and causes its accumulation in these compartments (Liscum and Faust, 1989; Roff et al., 1991). P. berghei growing in U18666A-treated Huh7 cells reached ∼50% the size of parasites growing in control cells at 24 h postinfection (Figure 4A). This phenotype was confirmed in primary murine hepatocytes treated with U18666A (Figure 4A). To test potential activity of the drug against the parasite, we pretreated sporozoites with U18666A under multiple conditions before infection (Supplemental Figure S4A). This treatment caused no difference in liver-stage development compared with development of phosphate-buffered saline (PBS)–treated control sporozoites, which is consistent with the assumption that the effect of the drug on infection is due to disruption specifically of the host cell physiology.
FIGURE 4:

P. berghei development is impaired when host cell cholesterol export from late endocytic vacuoles is blocked. (A) Hepatoma cells and mouse primary hepatocytes or (B) WT and LAMP1/LAMP2-deficient MEFs were treated with 3 μM U18666A 12 h before and throughout infection with P. berghei sporozoites. Parasite size was assessed 24 h postinfection by fluorescence microscopy. Parasites were visualized by staining with anti-PbHsp70 antibody. One representative of at least three independent experiments is shown, with the exception of the experiment in primary hepatocytes, in which the combined results of two experiments are shown. ns, not significant; *p < 0.05, **p < 0.01 (Student's unpaired t test, n = 3).
We predict that the defects in parasite development in the LAMP1/LAMP2-deficient cells and the U18666A-treated cells is due to inhibition of the same host cellular pathways in both conditions. If this assumption is true, combining these two conditions through treatment of cells deficient in LAMP1 and LAMP2 with U18666A should not have an additive effect and lead to further reduced parasite growth. When this was tested, indeed, U18666A treatment of Lamp1−/−Lamp2−/− cells resulted in parasite growth that was similar to that seen in untreated Lamp1−/−Lamp2−/− cells or U18666A-treated wild-type cells (Figure 4B). These results are consistent with the hypothesis that LAMP deficiency and U18666A treatment disrupt a common cellular pathway important for parasite development, which further confirms that the effect of U18666A on infection is due to modulation of the host cell.
If the defect in parasite development upon U18666A treatment is due to the sequestration of cholesterol in LEs and lysosomes, we could predict, on the basis of previous studies (Rosenbaum et al., 2010), that low concentrations of methyl-β-cyclodextrin (MβCD) should release the accumulated cholesterol and therefore restore parasite growth. Cyclodextrins are water-soluble compounds that can release cholesterol from membranes (Yancey et al., 1996; Atger et al., 1997), and MβCD efficiently reduces cholesterol accumulation in late endocytic compartments of cells with Npc mutations (Camargo et al., 2001; Lope-Piedrafita et al., 2008; Liu et al., 2009). In cells treated with U18666A alone, LAMP2-positive compartments are enlarged (Supplemental Figure S4B), and cholesterol, as detected by filipin, accumulates in enlarged intracellular vacuoles (Figure 5A and Supplemental Figure S4C). With the addition of 1 mM MβCD, lysosomal morphology was restored, and filipin labeled the plasma membrane, similar to control cells. Addition of MβCD also rescued the effect on parasite size caused by U18666A (Figure 5B). Parasites developing in cells treated with both U18666A and MβCD were significantly larger than those growing in cells treated with U18666A alone. MβCD treatment alone does not alter parasite growth, indicating that the drug at this concentration has no effect on parasite development.
FIGURE 5:
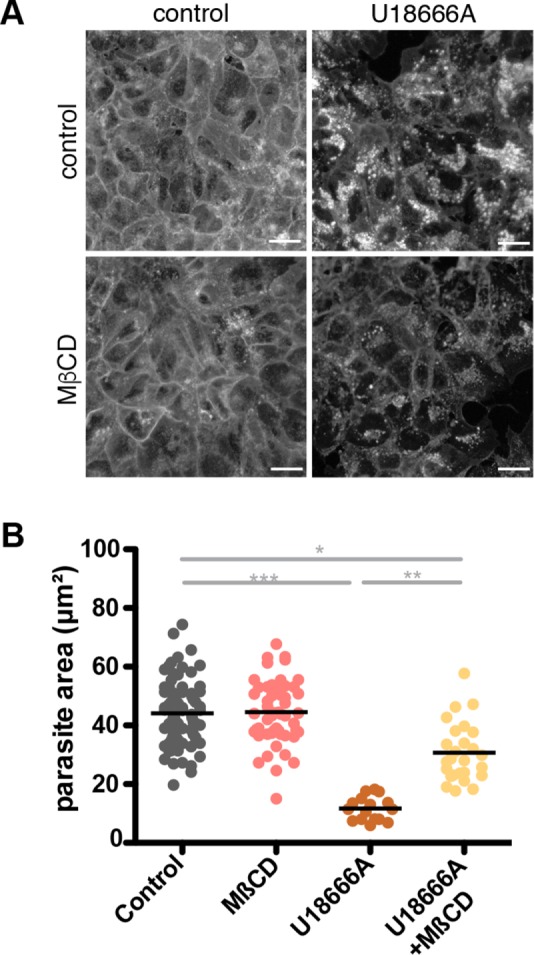
Release of cholesterol from LE/lysosomes rescues P. berghei development in U18666A-treated cells. Huh7 cells were treated with 3 μM U18666A or 1 mM MβCD 36 h before infection. Where indicated, MβCD was added to U18666A-treated cells 12 h before infection. Cells were either fixed and stained with filipin (A; scale bars, 30 μm) or infected with P. berghei sporozoites and kept under continual presence of the drug until fixation 24 h postinfection (B). Parasites were visualized by staining with an anti-PbHsp70 antibody. Size was assessed, and the area of each measured parasite was plotted individually. One representative of three independent experiments is shown. *p < 0.05; **p < 0.01; ***p < 0.001 (Student's unpaired t test, n = 3).
Together these data demonstrate that cholesterol accumulation in host LEs and lysosomes impairs parasite growth and indicate that the export of cholesterol from the late endocytic compartments is needed for normal P. berghei development.
The defect in autophagic flux in U18666A-treated or LAMP1/LAMP2-deficient cells is not likely responsible for restricted parasite development
In addition to their effect on cholesterol export from LEs and lysosomes, Npc mutations, U18666A treatment, or LAMP1/LAMP2-deficiencies cause an increase in the steady-state number of LC3-II–positive autophagosomes due to a defect in amphisome formation (Tanaka et al., 2000; Eskelinen et al., 2004; Ishibashi et al., 2009; Sarkar et al., 2013; Maetzel et al., 2014). A similar accumulation of LC3-positive membranes can be observed in cells treated with 1 mM MβCD (Cheng et al., 2006; Supplemental Figure S4B). P. berghei liver-stage parasites growing in cells treated with MβCD showed no defect in development, suggesting that accumulation of autophagic bodies alone does not disturb parasite growth. Recently it was shown that LC3 is recruited to developing liver-stage parasites (Grützke et al., 2014; Prado et al., 2015; Boonhok et al., 2016; Thieleke-Matos et al., 2016). Published data also indicated that early recruitment of autophagy markers promotes successful liver-stage development (Prado et al., 2015). To examine further whether the impaired autophagic flux seen in cells with impaired cholesterol transport could influence parasite growth, we monitored the recruitment of LC3 to developing liver-stage parasites over time in cells treated with U18666A. We saw no change in the recruitment of LC3 to liver-stage parasitophorous vacuoles in U18666A-treated Huh7 cells and only a very small difference in LAMP1/LAMP2-deficient MEFs compared with control cells (Supplemental Figure S4, D–F). Thus we have no evidence that the accumulation of autophagic bodies in the knockout and drug-treated cells alters the manner in which the parasite vacuole interacts with the host autophagy pathway or is responsible for the observed impairment of parasite growth.
Block of cholesterol export from late endocytic organelles affects liver-stage development and time to blood-stage patency in vivo
We sought to validate the in vitro data indicating the detrimental effect of cholesterol sequestration in LEs and lysosomes on P. berghei infection in vivo. To this end, we administered U18666A to C57BL/6 mice daily for 15 d, and after drug treatment on day 15, we intravenously infected the mice with P. berghei sporozoites (Figure 6A). Blood-stage parasites were detected 3 d postinfection in most control animals. However, U18666A-treated mice displayed a 2- to 3-d delay in patency, with blood-stage infection becoming apparent in most animals 5–6 d postinfection. This delay is unlikely to be due to inhibition of blood-stage parasite replication because subsequent blood-stage replication proceeded normally in both groups (Supplemental Figure S5). To ensure that the delay in patency was due to liver-stage development, we assessed the parasite burden in the livers of infected animals with a quantitative-PCR–based assay 42 h after infection (Figure 6B). Thus administration of U18666A resulted in a significant reduction of liver-stage parasite load and increased time to the appearance of P. berghei in the blood.
FIGURE 6:

Blocking cholesterol export from late endocytic organelles delays time to blood-stage patency and reduces liver-stage parasite burden in vivo. Mice were treated daily with 10 mg/kg U18666A (intraperitoneally) for 15 d before receiving an intravenous dose of 10,000 P. berghei sporozoites. (A) The prepatent period was followed by microscopic examination of Giemsa-stained blood smears. (B) Parasite burden in infected mouse livers at 42 h postinfection was quantified by quantitative PCR. Relative expression levels of the Pb18S gene were normalized to expression levels of the mouse GAPDH gene. Data from two biological replicates (n = 3 and 5) are displayed. *p < 0,05 (Student's unpaired t test; n = 8). (C) After treatment with U18666A for 15 d, mouse livers were removed and immediately frozen in liquid nitrogen. Sections (7 μm) were Formalin fixed, stained with filipin (0.05 mg/ml), and imaged under identical conditions.
To confirm that U18666A influenced cholesterol transport in livers of treated mice, we visualized cholesterol in liver sections by filipin staining and fluorescence microscopy (Figure 6C). Whereas in control livers, filipin intensities were weak and only a few areas with cholesterol accumulation could be detected, liver sections from drug-treated animals showed a strong filipin signal and a clear increase in cholesterol accumulation in liver tissue. These data demonstrate that functional cholesterol transport in host cells is required for efficient liver-stage growth of P. berghei also in vivo.
DISCUSSION
LEs and lysosomes fulfill many different functions in the endocytic network. The highly acidic vesicles are part of the cells' antimicrobial defense mechanism, and their fusion with the PVM can lead to parasite death (Prado et al., 2015, Boonhok et al., 2016). However, late endocytic vesicles are also a distribution point of substances taken up by the cells and could therefore serve as a nutrient source. During their development, liver-stage Plasmodium parasites undergo a striking size increase, for which they require large amounts of nutrients, which they likely acquire from the nutrient-rich environment in the hepatocyte; however, our knowledge of the mechanisms by which the parasites acquire these resources from host cells is limited.
When we began our studies, the potential benefit of lysosomal recruitment to liver-stage parasites as a source of nutrients had been proposed (Lopes da Silva et al., 2012). However, this suggestion was based on the use of chemical agents that deacidify the lysosomes of not only the host cell, but also of the parasite, making interpretation of these data difficult. Recently it was shown that generation of phosphatidylinositol 3,5-bisphosphate by PIKfyve at endosomal membranes is required for efficient liver-stage parasite growth, which demonstrated that functions occurring at the host late endocytic compartments are important for parasite growth (Thieleke-Matos et al., 2014). Here we found that LEs/lysosomes are not efficiently recruited to liver-stage-arrested uis4− parasites and that perturbing host LE/lysosome function reduced parasite burden. Together these results further support the hypothesis that LE/lysosome recruitment indicates proper PV maturation and may support parasite development.
What drives the association between late endocytic compartments and the liver-stage PV is unknown. The presence of the autophagy protein LC3 in the PVM may contribute to lysosomal recruitment; however, because we showed that lysosomes are still recruited to the PV in ATG5-deficient cells, the interaction with the host autophagic pathway cannot be the sole factor responsible for lysosomal recruitment. Reduced recruitment of LEs/lysosomes to PVs containing uis4− parasites could be due to direct interaction between UIS4 and LE/lysosome membranes or proteins, or the cause could be indirect and instead due to improper maturation of the vacuole in the absence of UIS4. In either case, poor liver-stage growth, which has been demonstrated for uis4− parasites (Mueller et al., 2005), coincides with reduced recruitment of host LC3, LEs and lysosomes, and cholesterol. Although several PVM proteins have been shown to be important for parasite development, to our knowledge, this is the first example in which the absence of a parasite protein is shown to alter the interaction between the PV and host cell. These results demonstrate that perturbing the protein composition of the PVM negatively affects the recruitment of host LEs during liver-stage development and suggest that accumulation of these organelles is a normal part of PV maturation.
To investigate whether host lysosomal functions contribute to liver-stage Plasmodium development, we assessed parasite growth in MEFs deficient for specific LE/lysosome proteins. Whereas ATP6V0A3 and ATP6AP2 are associated with the vacuolar-ATPase, which is responsible for lysosomal acidification, lysosomes remain acidified in both of these knockout lines (Kissing et al., 2015). Therefore, on the basis of our observation that cells deficient for ATP6V0A3 or ATP6AP2 support efficient parasite growth, we cannot claim that acidification of host lysosomes is dispensable for parasite development, but only that these proteins are not required for parasite development. In contrast, cells deficient for NPC1, NPC2, LAMP1, or LAMP2 did not support proficient P. berghei growth in comparison to wild-type cells.
NPC2 is a small soluble protein in the LE/lysosome lumen, and NPC1 is a large membrane glycoprotein in the outer LE/lysosome membrane (Higgins et al., 1999; Naureckiene et al., 2000). The NPC proteins actively participate in cholesterol transport (Vanier and Millat, 2004), and mutation in either of these two genes leads to the lysosomal storage disease Niemann–Pick disease type C (Sokol et al., 1988). Other lipids, for example, sphingolipids, also accumulate in LEs/lysosomes of cells containing Npc mutations, but this is likely a consequence of the cholesterol sequestration (reviewed in Vance and Karten, 2014). Further, whereas sphingolipid levels are elevated in cells with Npc mutations (Chang et al., 2005), sphingolipid levels in LAMP1/LAMP2 doubly deficient cells appear comparable to those in wild-type cells (Eskelinen et al., 2004).
In infection studies, host NPC function has been found to be crucial for the filoviruses Marburg and Ebola (Carette et al., 2011), as well as for HIV-1 infectivity and production (Tang et al., 2009; Coleman et al., 2012). Furthermore, the replication rate of Toxoplasma gondii is reduced in NPC-deficient cells (Coppens et al., 2000).
LAMP1 and LAMP2 are the most abundant proteins in mammalian lysosomes. The exact function of these proteins is unknown, but characterization of cells and animals deficient for these proteins reveals their necessity for proper autophagosome maturation (Tanaka et al., 2000), phagosome maturation (Huynh et al., 2007), and cholesterol egress from late endocytic compartments (Eskelinen et al., 2004). These studies found that deletion of Lamp1 alone is compensated for by increased expression of LAMP2 and therefore results in no severe phenotypes. In contrast, LAMP1 is not able to fully compensate functionally for the loss of LAMP2. Accordingly, knockdown of Lamp1 did not affect Plasmodium infection, whereas knockdown of Lamp2 resulted in smaller developing parasites.
Previous infection studies in cells deficient for the LAMPs used pathogens that reside within LAMP-positive compartments during their development. LAMP1/LAMP2-deficient cells fail to kill Neisseria gonorrhoeae (Binker et al., 2007), harbor faster-maturing, smaller Coxiella burnetii–containing phagosomes (Schulze-Luehrmann et al., 2016), and are less permissive to Trypanosoma cruzi invasion (Albertti et al., 2010).
Autophagic flux is altered in cells deficient for the LAMPs or NPC proteins and in cells treated with U18666A (Tanaka et al., 2000; Eskelinen et al., 2004; Ishibashi et al., 2009; Sarkar et al., 2013; Maetzel et al., 2014). These cells display a defect in amphisome formation, which increases the number of steady-state autophagic bodies. Because the liver-stage PV is known to interact with the autophagy pathway (Grützke et al., 2014; Prado et al., 2015; Thieleke-Matos et al., 2016), we initially hypothesized that defects in parasite growth in these conditions relate to changes in host autophagosome maturation. However, parasites grow robustly in cells treated with 1 mM MβCD, which leads to a similar accumulation of autophagic bodies as seen in the aforementioned conditions. Furthermore, LC3 recruitment to liver-stage parasites is unchanged in U18666A-treated cells and barely reduced in LAMP-deficient cells. Thus, although we cannot rule out the possibility that alterations in autophagic maturation contribute to the parasite growth phenotypes we describe here, we have no evidence that autophagy is responsible for the P. berghei growth phenotypes.
The other shared phenotype caused by Npc mutations, Lamp mutations, and U18666A treatment is the sequestration of cholesterol in late endocytic compartments. Cells can acquire cholesterol via two pathways: by uptake of LDL, which is hydrolyzed along the endocytic pathway and released from LEs to allow transport to other membranes in the cell, or via synthesis by the mevalonate pathway in the endoplasmic reticulum. In any of the conditions we tested in which cholesterol export from lysosomes is blocked, parasite growth was inhibited. We were able to validate these results in vivo by infecting mice treated with U18666A. Of note, we could rescue the in vitro phenotype with MβCD. Cyclodextrins bind cholesterol and can extract it from membranes (Christian et al., 1997), and MβCD reduces cholesterol accumulation in late endocytic compartments of NPC-deficient cells and U18666A-treated cells and rescues the NPC-associated phenotype (Camargo et al., 2001; Lope-Piedrafita et al., 2008; Liu et al., 2009). These rescue experiments are performed using 1 mM MβCD, which is 5–10 times less than the concentration typically used to deplete cholesterol from membranes. MβCD rescues the U18666A-induced phenotype by acting from within LEs (Abi-Mosleh et al., 2009; Rosenbaum et al., 2010), and an increase in esterified cholesterol has been detected in this context, indicating that this concentration of MβCD does not just remove cholesterol but makes it available for transport to the ER, the site of cholesterol esterification (Rosenbaum et al., 2010). It is therefore tempting to speculate that liver-stage Plasmodium grows poorly when cholesterol is sequestered in LEs because the parasite has reduced access to exogenously acquired cholesterol. Hepatocytes can compensate for deficits in cholesterol uptake through cholesterol synthesis (Rudney and Sexton, 1986), but it is not sufficient to rescue the parasite growth phenotype.
Previous studies showed that host cholesterol labels the liver-stage PVM and that both exogenous and endogenous sources contribute to the cholesterol in these membranes (Labaied et al., 2011). Whereas cholesterol levels at the parasite PVM were shown to decrease when progesterone treatment blocked exogenous cholesterol trafficking, parasite development was not investigated in this context (Labaied et al., 2011). Here we see that preventing release of late endocytic cholesterol has a negative effect on parasite development. These results could be due to blocked access to cholesterol by the parasite, which would indicate that blocking transfer of cholesterol out of late endosomes is more problematic for the parasite than temporarily depleting the source of exogenous cholesterol. Alterations in access to other late endocytic lipids could also contribute to the observed phenotypes. Cholesterol is not the only lipid species obtained by the parasite from the host cell. Acquisition of both lipoic acid (Deschermeier et al., 2012) and phosphatidylcholine (Itoe et al., 2014) from the host environment is needed for robust liver-stage parasite growth. The latter is present in LE membranes but also in all other cellular membranes, so it seems unlikely that LEs and lysosomes would be the exclusive source of this lipid. However, transfer of this or other lipid species from LEs and lysosomes to the parasite or PVM could be altered when the cholesterol content of LE and lysosomes is increased.
Taken together, our results show that specific targeting of host LE/lysosomal proteins can restrict liver-stage parasite development. Because interfering with the lipid homeostasis in LEs interferes with parasite development, we conclude that proper host LE function actively contributes to successful parasite growth. Although fusion of lysosomes with the PVM can be a successful host defense against the parasite, we and others have provided evidence that these host late endocytic compartments positively contribute to parasite growth and that their close association with the PV and potential fusion with the liver-stage TVN could provide a source of lipids for the parasite during liver-stage development. Successful liver-stage parasites appear to maintain a balance between exploiting these degradative host compartments and avoiding their antiparasitic potential.
MATERIALS AND METHODS
Experimental animals, cells, and parasite lines
Female C57BL/6 and NMRI mice were obtained from Charles River Laboratories and were 6–12 wk of age at the time of experiments. All animal work was conducted in accordance with the German Tierschutzgesetz in der Fassung vom 18. Mai 2006 (BGBl. I S. 1207), which implements the Directive 86/609/EEC from the European Union and the European Convention for the protection of vertebrate animals used for experimental and other scientific purposes. All appropriate measures were taken to reduce the pain or discomfort of the animals, and the protocol was approved by the ethics committee of the Max Planck Institute for Infection Biology and the Berlin state authorities (LAGeSo Reg# G0469/09 and G0294/15). The hepatoma cells used in this study were Huh7 cells cultured in RPMI supplemented with 10% fetal calf serum (FCS), 100 U penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 1× nonessential amino acids (Life Technologies). MEFs including wild type, Npc−/− (Huang et al., 2014; kindly provided by Peter Lobel, Rutgers University, New Brunswick, NJ), Lamp1/2−/− (Eskelinen et al., 2004), Atg5−/− (Kuma et al., 2004; kindly provided by Ingo Schmitz, Helmholz Centre for Infection Research, Braunschweig, Germany), Atp6v0a3−/− (derived from the TCIRG1oc/oc line; Scimeca et al., 2000; Kissing et al., 2015); and Atp6ap2−/− (derived from the Atp6ap2 conditional deletion line; Kissing et al., 2015) were maintained in DMEM supplemented with 10% FCS, 100 U penicillin, and 100 μg/ml streptomycin. Primary murine hepatocytes were isolated from NMRI mice as described (Renia et al., 1990) using liver perfusion, liver digest, and hepatocyte wash medium (Life Technologies) and plated on collagen-coated dishes in William's E Medium supplemented with 10% fetal bovine serum and penicillin-streptomycin (Life Technologies). Wild-type P. berghei were either ANKA clone 507 (Janse et al., 2006) or ANKA Bergreen (Kooij et al., 2012), both of which express green fluorescent protein (GFP). The ibis1−P. berghei parasite line was described previously (Ingmundson et al., 2012). The uis4− ANKA P. berghei was generated in clone 507 as described (Mueller et al., 2005). Anopheles stephensi mosquitoes were raised under a 14-h light/10-h dark cycle at 28°C and 80% humidity and were fed daily on 10% sucrose.
In vitro infections with P. berghei
Huh7 cells, MEFs, and primary hepatocytes seeded in collagen-coated 96-well µclear plates (Greiner) were infected with P. berghei sporozoites as previously described (Silvie et al., 2008). When required, cells were treated as indicated in the figure legends with 3 μM U18666A (Enzo Life Sciences; Liscum and Faust, 1989) and 1 μM MβCD (Sigma-Aldrich). Because the drugs are reversible (Härmälä et al., 1994; Rodal et al., 1999), they remained in the medium throughout infection. For merosome production, infection proceeded for 70 h; merosomes in the culture medium were harvested and counted in a Neubauer chamber.
Indirect immunofluorescence analysis
At the indicated time points, samples were fixed with 4% paraformaldehyde in PBS and permeabilized with either 100% ice-cold methanol (for the LC3 antibody) or 0.3% Triton X-100 (all other antibodies) and labeled with the following primary antibodies: polyclonal chicken anti-GFP (1:1000; ab13970; Abcam), monoclonal mouse anti-PbHsp70 (Potocnjak et al., 1980), polyclonal rabbit anti-UIS4 (Müller et al., 2011), monoclonal mouse anti-LC3 (1:100; 5F10; Nanotools), polyclonal rabbit anti-EEA1 (1:500; ab2900; Abcam), monoclonal mouse anti-CD63 (1:100; CBL553; Merck Millipore), and mouse anti-LAMP2 (1:500; H4B4; Developmental Studies Hybridoma Bank) followed by Alexa Fluor–conjugated secondary antibodies (Invitrogen) and Hoechst 33342 (Invitrogen). When required, filipin staining was carried out before permeabilization using 0.025 mg/ml of filipin III (Sigma-Aldrich) in PBS for 1 h at room temperature.
Fluorescence microscopy, parasite quantification, and quantification of LE/lysosome recruitment
Fluorescent samples were analyzed using a Zeiss Axio Observer.Z1 microscope (Zeiss) equipped with 10, 40, and 63×/1.2 oil immersion lens, and pictures were collected with an AxioCam MRm (Zeiss) and Zeiss AxioVision software (Zeiss). Images were processed in Fiji (Schindelin et al., 2012) or Adobe Photoshop.
The P. berghei liver-stage size (in micrometers squared) was measured in Fiji by identifying and measuring areas of fluorescence signal using defined threshold levels. The identified fluorescent areas were used to measure parasite area. Quantification of LE/lysosome recruitment was performed visually on stained samples. Parasites were considered to recruit LEs/lysosomes when a continuous portion of the parasite periphery comprising at least 30% of the parasite circumference had closely associated LAMP2- or CD63-positive vesicles. If <30% of the parasite was covered with these signals, the parasite was considered to not recruit these vesicles.
siRNA knockdown
Huh7 cells were reverse transfected with target-specific or control siRNAs (Silencer Select; Life Technologies) according to the manufacturer's instructions. In brief, 24 h after cell seeding, siRNAs diluted in Opti-MEM (Life Technologies) and Lipofectamine RNAiMAX transfection solution (Life Technologies) were added to cells. At 48 h after siRNA transfection, either knockdown efficiency was analyzed by Western blot using antibodies against LAMP1 (Abcam) and LAMP2 (Developmental Studies Hybridoma Bank) or cells were infected with P. berghei sporozoites.
Quantification of liver parasite load by quantitative PCR
Quantification of parasite burden in mouse liver after sporozoite infection into U18666A- or PBS-treated animals was performed as described (Friesen et al., 2010). Briefly, C57Bl/6 mice were injected intraperitoneally with 10 mg/kg U18666A in PBS or PBS alone daily for 15 d. On day 15, animals were injected with 104 sporozoites intravenously. At 42 h after infection, livers of infected animals were harvested and homogenized in TRIzol (Life Technologies), total RNA was extracted, and cDNA was prepared using the RETROscript reverse transcription kit (Life Technologies). Relative parasite levels were determined with quantitative PCR by comparing the mean Ct value of the P. berghei 18s ribosomal subunit to the mean Ct value of the Mus musculus GAPDH. To assess time to patency and parasitemia, Giemsa-stained blood smears were examined microscopically daily starting at day 3 after infection.
Staining of liver tissue sections
Liver tissue sections were generated from U18666A-treated and control animals. At 42 h postinfection, mice were killed and perfused with PBS. Livers were removed and frozen in liquid nitrogen immediately. Thin liver sections (7 μm) were stained with filipin as described (Vanier and Latour, 2015). Briefly, after fixing for 15 min in Formalin, sections were washed with PBS, and 0.05 mg/ml filipin was added to the sections for 1 h. Slides were mounted using ProLong Gold Antifade Mountant (Life Technologies) and examined using an Olympus BX50 microscope. Images were processed in Fiji (Schindelin et al., 2012).
Statistical analysis
Statistics for parasite size analysis are based on the average values of three wells, not on the sizes of the individual parasites. All experiments were carried out at least three times, with the exception of those in primary hepatocytes and in mice, which were both performed twice. In each figure, one representative experiment is shown. All statistical analyses were performed using Prism 5.0 for Mac OS X (GraphPad Software, San Diego, CA). p < 0.05 was considered to indicate statistical significance.
Supplementary Material
Acknowledgments
We thank Peter Lobel, Noboru Mizushima, Ingo Schmitz, Meryem Senkara, and Sandra Kissing for providing cell lines and reagents and Özlem Günay-Esiyok, Nishith Gupta, and Katja Müller for experimental assistance and advice. We also thank Carolin Rauch, Manuel Rauch, and Petra Matylewski for technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (SPP 1580; IN 182/1-1) and in part by the Max Planck Society and the European Commission through the EviMalaR network (#34).
Abbreviations used:
- LAMP
lysosome-associated membrane protein
- LE
late endosome
- MβCD
methyl-β-cyclodextrin
- NPC
Niemann–Pick type C
- PV
parasitophorous vacuole
- PVM
parasitophorous vacuole membrane
- TVN
tubovesicular network
- UIS4
up-regulated in infective sporozoites 4.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0531) on January 25, 2017.
REFERENCES
- Abi-Mosleh L, Infante RE, Radhakrishnan A, Goldstein JL, Brown MS. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc Natl Acad Sci USA. 2009;106:19316–19321. doi: 10.1073/pnas.0910916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertti LA, Macedo AM, Chiari E, Andrews NW, Andrade LO. Role of host lysosomal associated membrane protein (LAMP) in Trypanosoma cruzi invasion and intracellular development. Microbes Infect. 2010;12:784–789. doi: 10.1016/j.micinf.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atger VM, De La Llera Moya M, Stoudt GW, Rodrigueza WV, Phillips MC, Rothblat GH. Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. J Clin Invest. 1997;99:773–780. doi: 10.1172/JCI119223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano N, Romano JD, Jayabalasingham B, Coppens I. Cellular interactions of Plasmodium liver stage with its host mammalian cell. Int J Parasitol. 2007;37:1329–1341. doi: 10.1016/j.ijpara.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Binker MG, Cosen-Binker LI, Terebiznik MR, Mallo GV, Mccaw SE, Eskelinen EL, Willenborg M, Brumell JH, Saftig P, Grinstein S, et al. Arrested maturation of Neisseria-containing phagosomes in the absence of the lysosome-associated membrane proteins, LAMP-1 and LAMP-2. Cell Microbiol. 2007;9:2153–2166. doi: 10.1111/j.1462-5822.2007.00946.x. [DOI] [PubMed] [Google Scholar]
- Boonhok R, Rachaphaew N, Duangmanee A, Chobson P, Pattaradilokrat S, Utaisincharoen P, Sattabongkot J, Ponpuak M. LAP-like process as an immune mechanism downstream of IFN-gamma in control of the human malaria Plasmodium vivax liver stage. Proc Natl Acad Sci USA. 2016;113:E3519–E3528. doi: 10.1073/pnas.1525606113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasaemle DL, Attie AD. Rapid intracellular transport of LDL-derived cholesterol to the plasma membrane in cultured fibroblasts. J Lipid Res. 1990;31:103–112. [PubMed] [Google Scholar]
- Camargo F, Erickson RP, Garver WS, Hossain GS, Carbone PN, Heidenreich RA, Blanchard J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001;70:131–142. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenedella RJ. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids. 2009;44:477–787. doi: 10.1007/s11745-009-3305-7. [DOI] [PubMed] [Google Scholar]
- Chang TY, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CC. Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005;280:20917–20920. doi: 10.1074/jbc.R400040200. [DOI] [PubMed] [Google Scholar]
- Cheng J, Ohsaki Y, Tauchi-Sato K, Fujita A, Fujimoto T. Cholesterol depletion induces autophagy. Biochem Biophys Res Commun. 2006;351:246–252. doi: 10.1016/j.bbrc.2006.10.042. [DOI] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Coleman EM, Walker TN, Hildreth JE. Loss of Niemann Pick type C proteins 1 and 2 greatly enhances HIV infectivity and is associated with accumulation of HIV Gag and cholesterol in late endosomes/lysosomes. Virol J. 2012;9:31. doi: 10.1186/1743-422X-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppens I, Sinai AP, Joiner KA. Toxoplasma gondii exploits host low-density lipoprotein receptor-mediated endocytosis for cholesterol acquisition. J Cell Biol. 2000;149:167–180. doi: 10.1083/jcb.149.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschermeier C, Hecht LS, Bach F, Rutzel K, Stanway RR, Nagel A, Seeber F, Heussler VT. Mitochondrial lipoic acid scavenging is essential for Plasmodium berghei liver stage development. Cell Microbiol. 2012;14:416–430. doi: 10.1111/j.1462-5822.2011.01729.x. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, Tanaka Y, Lullmann-Rauch R, Hartmann D, Heeren J, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell. 2004;15:3132–3145. doi: 10.1091/mbc.E04-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen J, Silvie O, Putrianti ED, Hafalla JC, Matuschewski K, Borrmann S. Natural immunization against malaria: causal prophylaxis with antibiotics. Sci Transl Med. 2010;2:40ra49. doi: 10.1126/scitranslmed.3001058. [DOI] [PubMed] [Google Scholar]
- Grützke J, Rindte K, Goosmann C, Silvie O, Rauch C, Heuer D, Lehmann MJ, Mueller AK, Brinkmann V, Matuschewski K, Ingmundson A. The spatiotemporal dynamics and membranous features of the Plasmodium liver stage tubovesicular network. Traffic. 2014;15:362–382. doi: 10.1111/tra.12151. [DOI] [PubMed] [Google Scholar]
- Härmälä AS, Porn MI, Mattjus P, Slotte JP. Cholesterol transport from plasma membranes to intracellular membranes is inhibited by 3 beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. Biochim Biophys Acta. 1994;1211:317–325. doi: 10.1016/0005-2760(94)90156-2. [DOI] [PubMed] [Google Scholar]
- Higgins ME, Davies JP, Chen FW, Ioannou YA. Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab. 1999;68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- Huang L, Pike D, Sleat DE, Nanda V, Lobel P. Potential pitfalls and solutions for use of fluorescent fusion proteins to study the lysosome. PLoS One. 2014;9:e88893. doi: 10.1371/journal.pone.0088893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26:313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingmundson A, Nahar C, Brinkmann V, Lehmann MJ, Matuschewski K. The exported Plasmodium berghei protein IBIS1 delineates membranous structures in infected red blood cells. Mol Microbiol. 2012;83:1229–1243. doi: 10.1111/j.1365-2958.2012.08004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Yamazaki T, Okamoto K. Association of autophagy with cholesterol-accumulated compartments in Niemann-Pick disease type C cells. J Clin Neurosci. 2009;16:954–959. doi: 10.1016/j.jocn.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Itoe MA, et al. Host cell phosphatidylcholine is a key mediator of malaria parasite survival during liver stage infection. Cell Host Microbe. 2014;16:778–786. doi: 10.1016/j.chom.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse CJ, Franke-Fayard B, Mair GR, Ramesar J, Thiel C, Engelmann S, Matuschewski K, Van Gemert GJ, Sauerwein RW, Waters AP. High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol Biochem Parasitol. 2006;145:60–70. doi: 10.1016/j.molbiopara.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kissing S, Hermsen C, Repnik U, Nesset CK, Von Bargen K, Griffiths G, Ichihara A, Lee BS, Schwake M, De Brabander J, et al. Vacuolar ATPase in phagosome-lysosome fusion. J Biol Chem. 2015;290:14166–14180. doi: 10.1074/jbc.M114.628891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooij TW, Rauch MM, Matuschewski K. Expansion of experimental genetics approaches for Plasmodium berghei with versatile transfection vectors. Mol Biochem Parasitol. 2012;185:19–26. doi: 10.1016/j.molbiopara.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Labaied M, Jayabalasingham B, Bano N, Cha SJ, Sandoval J, Guan G, Coppens I. Plasmodium salvages cholesterol internalized by LDL and synthesized de novo in the liver. Cell Microbiol. 2011;13:569–586. doi: 10.1111/j.1462-5822.2010.01555.x. [DOI] [PubMed] [Google Scholar]
- Liscum L. Niemann-Pick type C mutations cause lipid traffic jam. Traffic. 2000;1:218–225. doi: 10.1034/j.1600-0854.2000.010304.x. [DOI] [PubMed] [Google Scholar]
- Liscum L, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J Biol Chem. 1989;264:11796–11806. [PubMed] [Google Scholar]
- Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc Natl Acad Sci USA. 2009;106:2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lope-Piedrafita S, Totenhagen JW, Hicks CM, Erickson RP, Trouard TP. MRI detects therapeutic effects in weanling Niemann-Pick type C mice. J Neurosci Res. 2008;86:2802–2807. doi: 10.1002/jnr.21707. [DOI] [PubMed] [Google Scholar]
- Lopes Da Silva M, Thieleke-Matos C, Cabrita-Santos L, Ramalho JS, Wavre-Shapton ST, Futter CE, Barral DC, Seabra MC. The host endocytic pathway is essential for Plasmodium berghei late liver stage development. Traffic. 2012;13:1351–1363. doi: 10.1111/j.1600-0854.2012.01398.x. [DOI] [PubMed] [Google Scholar]
- Lu F, Liang Q, Abi-Mosleh L, Das A, De Brabander JK, Goldstein JL, Brown MS. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife. 2015;4:e12177. doi: 10.7554/eLife.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maetzel D, Sarkar S, Wang H, Abi-Mosleh L, Xu P, Cheng AW, Gao Q, Mitalipova M, Jaenisch R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep. 2014;2:866–880. doi: 10.1016/j.stemcr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AK, Camargo N, Kaiser K, Andorfer C, Frevert U, Matuschewski K, Kappe SH. Plasmodium liver stage developmental arrest by depletion of a protein at the parasite-host interface. Proc Natl Acad Sci USA. 2005;102:3022–3027. doi: 10.1073/pnas.0408442102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller K, Matuschewski K, Silvie O. The Puf-family RNA-binding protein Puf2 controls sporozoite conversion to liver stages in the malaria parasite. PLoS One. 2011;6:e19860. doi: 10.1371/journal.pone.0019860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Potocnjak P, Yoshida N, Nussenzweig RS, Nussenzweig V. Monovalent fragments (Fab) of monoclonal antibodies to a sporozoite surface antigen (Pb44) protect mice against malarial infection. J Exp Med. 1980;151:1504–1513. doi: 10.1084/jem.151.6.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado M, Eickel N, De Niz M, Heitmann A, Agop-Nersesian C, Wacker R, Schmuckli-Maurer J, Caldelari R, Janse CJ, Khan SM, et al. Long-term live imaging reveals cytosolic immune responses of host hepatocytes against Plasmodium infection and parasite escape mechanisms. Autophagy. 2015;11:1561–1579. doi: 10.1080/15548627.2015.1067361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renia L, Mattei D, Goma J, Pied S, Dubois P, Miltgen F, Nussler A, Matile H, Menegaux F, Gentilini M, et al. A malaria heat-shock-like determinant expressed on the infected hepatocyte surface is the target of antibody-dependent cell-mediated cytotoxic mechanisms by nonparenchymal liver cells. Eur J Immunol. 1990;20:1445–1449. doi: 10.1002/eji.1830200706. [DOI] [PubMed] [Google Scholar]
- Rodal SK, Skretting G, Garred O, Vilhardt F, Van Deurs B, Sandvig K. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol Biol Cell. 1999;10:961–974. doi: 10.1091/mbc.10.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roff CF, Goldin E, Comly ME, Cooney A, Brown A, Vanier MT, Miller SP, Brady RO, Pentchev PG. Type C Niemann-Pick disease: use of hydrophobic amines to study defective cholesterol transport. Dev Neurosci. 1991;13:315–319. doi: 10.1159/000112179. [DOI] [PubMed] [Google Scholar]
- Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci USA. 2010;107:5477–5482. doi: 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudney H, Sexton RC. Regulation of cholesterol biosynthesis. Annu Rev Nutr. 1986;6:245–272. doi: 10.1146/annurev.nu.06.070186.001333. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, Cohen MA, Chakraborty S, Wang H, Spooner E, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013;5:1302–1315. doi: 10.1016/j.celrep.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneede A, Schmidt CK, Holtta-Vuori M, Heeren J, Willenborg M, Blanz J, Domanskyy M, Breiden B, Brodesser S, Landgrebe J, et al. Role for LAMP-2 in endosomal cholesterol transport. J Cell Mol Med. 2011;15:280–295. doi: 10.1111/j.1582-4934.2009.00973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Luehrmann J, Eckart RA, Olke M, Saftig P, Liebler-Tenorio E, Lührmann A. LAMP proteins account for the maturation delay during the establishment of the Coxiella burnetii-containing vacuole. Cell Microbiol. 2016;18:181–194. doi: 10.1111/cmi.12494. [DOI] [PubMed] [Google Scholar]
- Scimeca JC, Franchi A, Trojani C, Parrinello H, Grosgeorge J, Robert C, Jaillon O, Poirier C, Gaudray P, Carle GF. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone. 2000;26:207–213. doi: 10.1016/s8756-3282(99)00278-1. [DOI] [PubMed] [Google Scholar]
- Sibley LD. Invasion and intracellular survival by protozoan parasites. Immunol Rev. 2011;240:72–91. doi: 10.1111/j.1600-065X.2010.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigala PA, Goldberg DE. The peculiarities and paradoxes of Plasmodium heme metabolism. Annu Rev Microbiol. 2014;68:259–278. doi: 10.1146/annurev-micro-091313-103537. [DOI] [PubMed] [Google Scholar]
- Silvie O, Goetz K, Matuschewski K. A sporozoite asparagine-rich protein controls initiation of Plasmodium liver stage development. PLoS Pathog. 2008;4:e1000086. doi: 10.1371/journal.ppat.1000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol J, Blanchette-Mackie J, Kruth HS, Dwyer NK, Amende LM, Butler JD, Robinson E, Patel S, Brady RO, Comly ME, et al. Type C Niemann-Pick disease. Lysosomal accumulation and defective intracellular mobilization of low density lipoprotein cholesterol. J Biol Chem. 1988;263:3411–3417. [PubMed] [Google Scholar]
- Sturm A, Amino R, Van De Sand C, Regen T, Retzlaff S, Rennenberg A, Krueger A, Pollok JM, Menard R, Heussler VT. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science. 2006;313:1287–1290. doi: 10.1126/science.1129720. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, Von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- Tang Y, Leao IC, Coleman EM, Broughton RS, Hildreth JE. Deficiency of niemann-pick type C-1 protein impairs release of human immunodeficiency virus type 1 and results in Gag accumulation in late endosomal/lysosomal compartments. J Virol. 2009;83:7982–7985. doi: 10.1128/JVI.00259-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieleke-Matos C, da Silva ML, Cabrita-Santos L, Pires CF, Ramalho JS, Ikonomov O, Seixas E, Shisheva A, Seabra MC, Barral DC. Host PI(3,5)P2 activity is required for Plasmodium berghei growth during liver stage infection. Traffic. 2014;15:1066–1082. doi: 10.1111/tra.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieleke-Matos C, Lopes Da Silva M, Cabrita-Santos L, Portal MD, Rodrigues IP, Zuzarte-Luis V, Ramalho JS, Futter CE, Mota MM, Barral DC, et al. Host cell autophagy contributes to Plasmodium liver development. Cell Microbiol. 2016;18:437–450. doi: 10.1111/cmi.12524. [DOI] [PubMed] [Google Scholar]
- Vance JE, Karten B. Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J Lipid Res. 2014;55:1609–1621. doi: 10.1194/jlr.R047837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier MT, Latour P. Laboratory diagnosis of Niemann-Pick disease type C: the filipin staining test. Methods Cell Biol. 2015;126:357–375. doi: 10.1016/bs.mcb.2014.10.028. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Millat G. Structure and function of the NPC2 protein. Biochim Biophys Acta. 2004;1685:14–21. doi: 10.1016/j.bbalip.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Wang ML, Motamed M, Infante RE, Abi-Mosleh L, Kwon HJ, Brown MS, Goldstein JL. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010;12:166–173. doi: 10.1016/j.cmet.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey PG, Rodrigueza WV, Kilsdonk EP, Stoudt GW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.