

Knockout of the centriolar linker C-NAP1 increased centriole separation and dispersed centriolar satellites. Primary cilia formed normally, even though the ciliary rootlet was mislocalized, and mechanosensory ciliary signaling was intact. Reduced levels of centrosome amplification after DNA damage indicate the importance of centriole proximity.
Abstract
Duplication of the centrosomes is a tightly regulated process. Abnormal centrosome numbers can impair cell division and cause changes in how cells migrate. Duplicated centrosomes are held together by a proteinaceous linker made up of rootletin filaments anchored to the centrioles by C-NAP1. This linker is removed in a NEK2A kinase-dependent manner as mitosis begins. To explore C-NAP1 activities in regulating centrosome activities, we used genome editing to ablate it. C-NAP1–null cells were viable and had an increased frequency of premature centriole separation, accompanied by reduced density of the centriolar satellites, with reexpression of C-NAP1 rescuing both phenotypes. We found that the primary cilium, a signaling structure that arises from the mother centriole docked to the cell membrane, was intact in the absence of C-NAP1, although components of the ciliary rootlet were aberrantly localized away from the base of the cilium. C-NAP1–deficient cells were capable of signaling through the cilium, as determined by gene expression analysis after fluid flow–induced shear stress and the relocalization of components of the Hedgehog pathway. Centrosome amplification induced by DNA damage or by PLK4 or CDK2 overexpression was markedly reduced in the absence of C-NAP1. We conclude that centriole splitting reduces the local density of key centriolar precursors to impede overduplication.
INTRODUCTION
Each of the centrosomes at the poles of the mitotic spindle consists of two orthogonally arranged microtubule barrels—the centrioles— surrounded by the pericentriolar material (PCM; Conduit et al., 2015). At the end of mitosis, this orthogonal arrangement is lost through the activity of Polo-like kinase 1 (PLK1) and separase (Nigg and Stearns, 2011; Firat-Karalar and Stearns, 2014), and a proteinaceous linker is established between the proximal ends of the two centrioles of each daughter cell (Mayor et al., 2000; Agircan et al., 2014). Both of these centrioles can now initiate procentriole duplication, with the initial site of procentriole formation during late G1/S phase specifying an orthogonal arrangement of the new “daughter” centriole with respect to the preexisting “mother.” By the onset of mitosis, cells possess two mother–daughter pairs of centrioles, each within their own PCM. The linker is removed, and the centrosomes can move apart to provide the spindle poles.
The proteinaceous linker, which has also been termed a G1-G2 tether (Nigg and Stearns, 2011), is composed of a number of large coil-coiled proteins (Paintrand et al., 1992). Rootletin, the major component of the ciliary rootlet structure seen in ciliated cells (Yang et al., 2002), and CEP68 form filaments that span the intercentriolar space. The docking proteins, centrosomal NEK2-associated protein 1 (C-NAP1) and centlein, anchor rootletin and CEP68 filaments, respectively, to the base of the centriole (Mayor et al., 2000; Bahe et al., 2005; Yang et al., 2006; Graser et al., 2007b; Fang et al., 2014). Loss or disruption of any of these protein results in impaired centrosome cohesion. Other linker-associated components, including β-catenin, LRRC45, and CEP215 (CDK5RAP2), also contribute to maintenance of the centrosomal linker (Bahmanyar et al., 2008; He et al., 2013; Pagan et al., 2015). Key to the removal of the linker is the PLK1-mediated activation of the kinase, Never in mitosis A (NIMA)–related kinase 2A (NEK2A). Active NEK2A phosphorylates linker components, triggering their removal and the dissolution of centrosome cohesion at the onset of mitosis (Fry et al., 1998; Bahe et al., 2005; Fang et al., 2014).
Primary cilia are antenna-like structures that extend from the surface of cells to regulate signaling pathways such as Hedgehog (Hh) and Wnt in response to changes in both the extracellular biochemical and biophysical environment. A range of developmental abnormalities are caused by ciliary dysfunction and are termed ciliopathies (Veland et al., 2009; Oh and Katsanis, 2012). The mother centriole acts as the basal body for primary cilium formation. Ciliogenesis involves the docking of the mother centriole at an intracellular ciliary vesicle, followed by its migration to the cell membrane and the formation of the ciliary axoneme (Ye et al., 2014; Lu et al., 2015). Primary cilia can form in mammalian cells that lack the ciliary rootlet or the proteinaceous linker (Yang et al., 2005; Panic et al., 2015). However, long-term cilium stability in specialized photoreceptor cells of the retina is reduced in rootletin-deficient mice, suggesting a role for the ciliary rootlet in ciliary structural integrity (Yang et al., 2005). In Drosophila, rootletin deficiency ablates the ciliary rootlet in sensory neurons and impairs their mechanosensing function (Styczynska-Soczka and Jarman, 2015).
Scattered in the immediate surrounding of the centrosome are the pericentriolar satellites. These are electron-dense granules that contain a range of proteins that regulate centriole and cilium formation, which are shuttled to and from the centrosome via dynein-mediated transport along the centrosomal microtubule network (Barenz et al., 2011; Tollenaere et al., 2015a). During ciliogenesis, centriolar satellite distribution is altered, as is their behavior after exposure to cellular stresses such as irradiation and heat shock (Loffler et al., 2013; Villumsen et al., 2013; Tollenaere et al., 2015b).
Here, we describe the effect of C-NAP1 ablation on centriole structure and duplication, cilium formation and function, and centriolar satellite distribution in hTERT-RPE1 cells. We find that C-NAP1–deficient cells undergo premature centriole cohesion loss, accompanied by loss of the linker and cytoplasmic aggregation of rootletin. Cilia formed efficiently in the absence of C-NAP1 and the ciliary rootlet, and C-NAP1–null cells are capable of responding to ciliary signals such as Hh stimulation and mechanical stimulation such as fluid shear. The centriolar satellites were less densely assembled around C-NAP1–null centrioles, and DNA damage– or regulatory kinase overexpression–induced centriole amplification was reduced in C-NAP1–deficient cells. These findings indicate the roles of C-NAP1 in regulating specific interphase centriole activities.
RESULTS
To examine how centrosome cohesion affects primary ciliation and centriolar satellite behavior, we used clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology to disrupt the CEP250 (C-NAP1) locus in the immortalized hTERT-RPE1 cell line, with guides designed to target exon 8 (protein-coding exon 5). We screened 15 candidate clones using a new monoclonal antibody (mAb) to C-NAP1, 6F2C8. As shown in Supplemental Figure S1A, 6F2C8 recognized a major band at 250 kDa in immunoblot experiments, which disappeared upon treatment of cells with small interfering RNA (siRNA) against C-NAP1. Similarly, a centrosomal signal detected with 6F2C8 was lost upon siRNA knockdown of C-NAP1 (Supplemental Figure S1B). From these results, we concluded that 6F2C8 is specific for C-NAP1. From our screen, we isolated eight clones that lacked detectable C-NAP1 signal by immunoblot and then confirmed the mutation of the CEP250 locus by genomic PCR and DNA sequencing of three of these clones (Figure 1, A and B). Stable integration of a construct that expressed full-length C-NAP1 was used to obtain a rescue clone (Figure 1A). Proliferative analysis confirmed that C-NAP1 loss did not affect cell doubling times, indicating that C-NAP1 deficiency did not have a major effect on cell cycle progression (Figure 1C).
FIGURE 1:

Generation of C-NAP1 null hTERT-RPE1 cells and preliminary phenotypic analysis. (A) Immunoblot of wild-type, C-NAP1–knockout (KO) clone 1, and C-NAP1 rescue (R1) cells using anti–C-NAP1 monoclonal 6F2C8. α-Tubulin was used as a loading control. (B) Sequence analysis of C-NAP1–deficient clones. PCR was performed on genomic DNA from the candidate clones and both total (shown in the traces) and cloned, individual PCR products were cloned and sequenced (five per clone). All sequences for clones 1 and 2 were identical. In sequencing clone 3 products, deletion of a thymine occurred in three of the five samples, with incorporation of a guanine in the other two. (C) Proliferative analysis of cells of the indicated C-NAP1 genotype. We plated 2 × 105 cells in 2 ml at 0 h and at 24, 48, 72, and 96 h. The cells were counted and the culture split 1:2. Data points show mean ± SD of three independent experiments. No significant difference was observed between wild type (WT) and C-NAP1 nulls at any time point. (D–F) Immunofluorescence microscopy of the indicated centrosomal markers in cells of the indicated C-NAP1 genotype. Scale bar, 5 μm.
We next examined the effect of C-NAP1 loss on centrosome structure. The loss of C-NAP1 was confirmed in microscopy experiments with the 6F2C8 mAb (Figure 1D). C-NAP1 nulls showed intact centriole structure, as detected by CEP135 and centrin localization, along with apparently normal pericentriolar material, as determined by staining for γ-tubulin (Figure 1, D–F). We noted a loss of ninein signal in one of the separated C-NAP1 centrioles (Figure 1F), which we attribute to the loss of ninein from the centriolar proximal ends while it was being retained at the subdistal appendages, as recently described in another C-NAP1–knockout line (Mazo et al., 2016). However, although we concluded from these observations that the overall structure of individual centrioles was unaffected by C-NAP1 deficiency, we noted some alterations in the distribution of the centriolar satellite protein PCM1 in the absence of C-NAP1 (Figure 1E), which we analyze in more detail later. We observed notable changes in the composition of the centriolar linker in the absence of C-NAP1, as described in previous experiments in which loss or inhibition of C-NAP1 resulted in a marked loss of cohesion between the centrioles during interphase (Mayor et al., 2000; Bahe et al., 2005; Yang et al., 2006; Fang et al., 2014; Panic et al., 2015). Consistent with observations knockouts generated with zinc finger nuclease (ZFN)–mediated targeting of C-NAP1 exon 14 (Panic et al., 2015) and with a CRISPR-mediated disruption of C-NAP1 exon 20 (Mazo et al., 2016), 30% of our C-NAP1 nulls showed a distance of ≥2 μm between G1-phase centrioles compared with 5% in wild-type cells (Figure 2, A and B). Of importance, the percentage of prematurely separated centrioles was restored to wild-type levels when C-NAP1 was reexpressed in the null cells (Figure 2, A and B). Immunofluorescence microscopy showed the absence of the linker components rootletin and Nek2 from the proximal end of the centrioles, confirming the loss of the intercentriolar tether (Figure 2, A and C). We saw no change in the levels of rootletin or Nek2 protein (Figure 2, D and E), from which we concluded that the loss of centriole cohesion was due to C-NAP1 deficiency preventing the recruitment of its binding partners to the proximal ends of the centrioles. These observations are consistent with the loss of linker components in other experiments that used reverse genetics to ablate C-NAP1 (Bahe et al., 2005; Graser et al., 2007b; Conroy et al., 2012; He et al., 2013; Fang et al., 2014; Panic et al., 2015; Mazo et al., 2016).
FIGURE 2:

Loss of the centriole linker causes centriole separation in C-NAP1–deficient cells. (A, C) Immunofluorescence microscopy of the indicated centriolar and linker proteins in cells of the indicated C-NAP1 genotype. Scale bar, 5 μm. (B) Quantitation of centriolar separation in the absence of C-NAP1. The percentage of G1 cells that exhibited a distance of >2 μm between centrioles was calculated based on the result of three individual experiments analyzing 200 cells in each case. (D, E) Immunoblot of rootletin (D) and Nek2 (E) levels in wild-type, C-NAP1 knockout (KO) clone 1, and C-NAP1 rescue (R1) cells. Ponceau S staining of the membrane after protein transfer was used as a loading control. ***p < 0.001 by unpaired t test.
We next investigated the effect of C-NAP1 deletion on primary cilium formation and functioning. We saw no change in cilium frequency in C-NAP1–deficient cells (Figure 3A). This contrasts with a reduction we observed in a previous knockdown experiment (Conroy et al., 2012). We found that treating our C-NAP1–nulls with same siRNA and serum starvation used in that study also led to a reduction in ciliation frequency (Supplemental Figure S2), indicating an off-target effect. From this result, we conclude that C-NAP1 is not required for primary cilium formation, consistent with recent knockout studies of C-NAP1 function (Panic et al., 2015; Mazo et al., 2016) and a previous siRNA analysis (Graser et al., 2007a). However, we observed a moderate C-NAP1–dependent increase in cilium length (Figure 3B) and a notable alteration in rootletin distribution, with large, filamentous rootletin structures being distributed in the cytoplasm in 80% of C-NAP1–null cells after serum starvation (Figure 3, C and D). These structures resembled the ciliary rootlet, a polymer of rootletin (Yang et al., 2002), except for their displacement from the proximal end of the basal body. To test whether the loss of centriole cohesion and the ciliary rootlet impaired ciliary function, we used the sonic hedgehog (SHh)-dependent localization to cilia of Smoothened (Smo) as a readout for ciliary signaling (Kiprilov et al., 2008). As shown in Figure 3, E and F, we found that Smo Agonist (SAG) treatment of cells caused the same level of cilia to show Smo localization in C-NAP1–null cells as in wild-type controls, demonstrating intact SHh signaling in the absence of C-NAP1 and the ciliary rootlet.
FIGURE 3:

C-NAP1 nulls show normal ciliation without attached ciliary rootlets. (A) Bar chart showing ciliation frequency in wild-type, C-NAP1 knockout (KO) clone 1, and C-NAP1 rescue (R1) cells. Ciliation percentages were determined using microscopy for detyrosinated tubulin in three separate experiments in which 100 cells were counted. (B) Length of cilia in cells of the indicated genotype. Maximum intensity projections of cilia were captured and measured in Volocity using the Line tool. Thirty ciliated cells were counted in three individual experiments. (C) Immunofluorescence microscopy of the ciliary rootlet in cells of the indicated genotype. Acet. tub, acetylated tubulin; Root, rootletin. Scale bar, 5 μm (main image), 0.5 μm (inset). (D) Bar chart showing the frequency of cells that show mislocalized rootletin, that is, an aggregate not localized to the base of the cilium. One hundred ciliated cells were counted in three separate experiments. (E) Immunofluorescence microscopy of Smoothened (Smo) in the absence and presence of SAG in serum-starved (SS) cells of the indicated C-NAP1 genotype. Scale bar, 5 μm. (F) Quantitation of the frequency with which Smo was detected at cilia in the absence and presence of SAG. One hundred ciliated cells were counted in three separate experiments. ***p < 0.001; *p < 0.05 by unpaired t test.
Analysis of rootletin-knockout mice revealed no major defects in sensory or motile cilium behavior, although certain ciliated cells showed signs of premature degeneration, suggestive of a long-term loss of cytoskeletal stability (Yang et al., 2005). However, recent work in Drosophila indicates that the loss of the ciliary rootlet impairs neuronal sensory responses to mechanical stimuli (Chen et al., 2015; Styczynska-Soczka and Jarman, 2015). We tested how mechanosensitive cilia-associated signaling was affected by the loss of C-NAP1 by monitoring the expression of genes that have been described as responsive to ciliary signaling of shear stress. Specifically, we looked at PTGS2, VEGFA, the Wnt pathway gene AXIN2, and the Hh target GLI1, which have been described as responding to shear stress in different cell types (Thi et al., 2007; Hoey et al., 2012; Cha et al., 2016). To do this, we exposed cells to oscillatory fluid flow–induced shear stress and measured the expression levels of genes of interest by quantitative reverse transcription PCR (qRT-PCR). We found that GLI1 and PTGS2 showed flow-dependent increases and AXIN2 a decline in expression in both wild type and C-NAP1 nulls, with VEGFA showing no significant changes in either genotype. The basal expression levels of GLI1 and VEGFA were unaffected by C-NAP1 deficiency, although a notable decline in PTGS2 expression and an increase in AXIN2 expression were seen in the C-NAP1 knockouts (Figure 4, A and B). From these data, we conclude that ciliary signaling pathways that respond to mechanical stress are intact and responsive in the absence of centriole cohesion or an attached rootlet.
FIGURE 4:
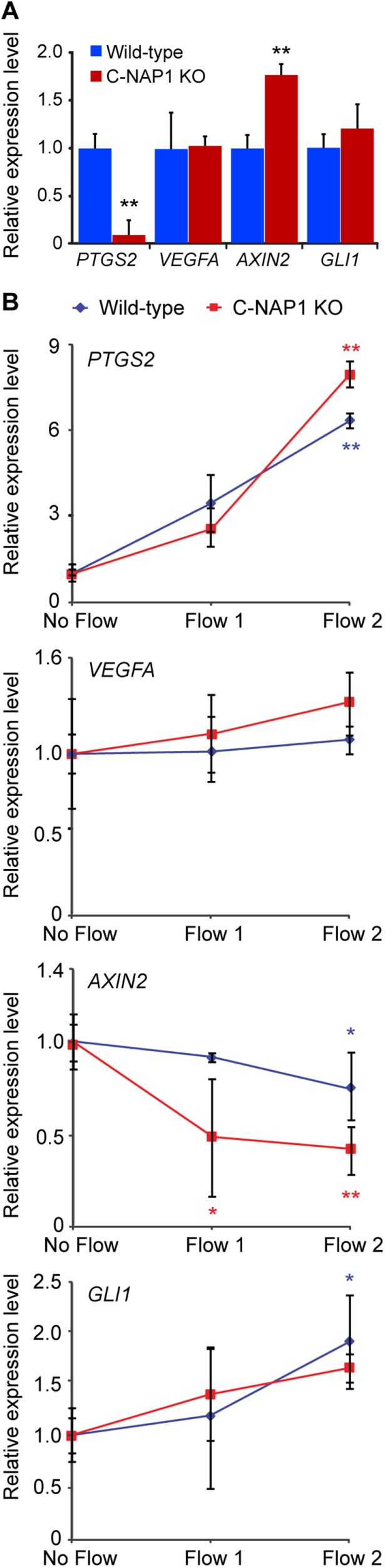
Intact mechanosensory responses in C-NAP1 nulls. (A) Basal expression levels of the genes of interest. (B) Responses of the various genes to indicated flow stimuli. Flows 1 and 2 correspond to a shear stress magnitude of 1 and 2 Pa, respectively. To determine whether the effect of flow elicited a response, an unpaired t test with Welch's correction was executed for each flow magnitude. The statistical difference of the level of mRNA expression between WT and KO was tested using the same approach. *p < 0.05; **p < 0.01.
As noted earlier, we observed an altered distribution of the centriolar satellite marker PCM1 in the absence of C-NAP1 (Figure 1E). We measured the total fluorescence intensity of the signal from PCM1 and another satellite marker, OFD1, in a standard volume around the centrioles. We observed a reduction in the centriolar satellite levels around C-NAP1–null centrioles in comparison to wild-type controls, as determined by the intensity of the signal seen with antibodies to the centriolar satellite components PCM1 and OFD1 (Figure 5, A and B). The reexpression of C-NAP1 restored centriole satellites to wild-type levels. The reduction in signal intensity appears to be due to altered distribution of the satellites in the absence of C-NAP1, as immunoblot analysis revealed no difference in the cellular levels of PCM1, OFD1, or CEP72 (Figure 5C).
FIGURE 5:

Reduced centriolar satellite density in C-NAP–knockout cells. (A) Immunofluorescence microscopy of the indicated satellite markers in cells of the indicated C-NAP1 genotype. Scale bar, 5 μm. (B) Quantitation of the centriolar satellite density in cells of the indicated genotype. Maximum intensity projections of 10 centrosomes from G1 cells were analyzed in three separate experiments. Graphs show the sum of PCM1 or OFD1 fluorescence intensities in a 25-µm2 circle around the two centrioles in each cell in arbitrary fluorescence units (A.U.). (C) Immunoblot of centriolar satellite proteins in cells of the indicated genotype. Ponceau S staining of the membrane after protein transfer was used as a loading control. ***p < 0.001 by unpaired t test.
Previous studies suggest that centriolar satellite densities control centrosome duplication (Prosser et al., 2009; Loffler et al., 2013; Kodani et al., 2015). We quantitated centrosome amplification after ionizing radiation–induced DNA damage (IR) and hydroxyurea (HU) treatment. C-NAP1 nulls exhibited significantly lower levels of centrosome amplification 48 h after 5-Gy IR than wild-type controls, with a partial rescue of this phenotype seen when C-NAP1 was reexpressed in the knockout cells (Figure 6, A and B). IR-induced activation of CHK1 and cell cycle delay were indistinguishable between wild-type and C-NAP1–null cells, showing that DNA damage response signaling was unaffected by the absence of C-NAP1 and excluding this possible explanation for the reduced centrosome amplification we saw (Figure 6C and Supplemental Figure S3). Similarly, HU treatment led to a lower level of centrosome amplification in C-NAP1–deficient cells than in controls (Figure 6D). We next tested whether centrosome amplification induced by the overexpression of PLK4 (Kleylein-Sohn et al., 2007) or CDK2 (Matsumoto et al., 1999; Meraldi et al., 1999) was affected by the loss of C-NAP1. As shown in Figure 7, A–C, loss of C-NAP1 strongly reduced centrosome amplification in both cases, showing that C-NAP1 is required for the centrosome overduplication induced by a number of stimuli.
FIGURE 6:
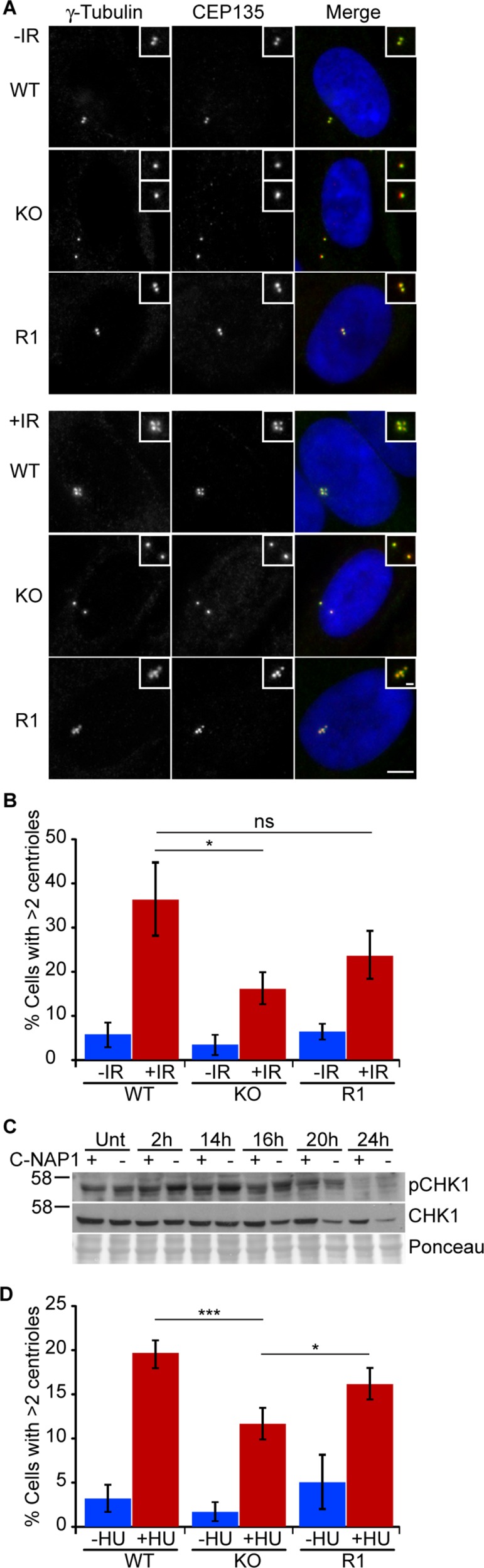
Reduced DNA damage–induced centrosome amplification in the absence of C-NAP1. (A) Immunofluorescence microscopy showing γ-tubulin (green) and CEP135 (red) in cells of the indicated C-NAP1 genotype at 48 h after 5-Gy IR treatment. Scale bar, 5 μm. (B) Centrosome quantitation in cells of the indicated genotype and treatment 48 h after 5-Gy IR. Centrosomes were quantitated using antibodies for CEP135, and bar graph indicates mean ± SD of three separate experiments in which at least 100 cells were counted. (C) Immunoblot of CHK1 activation in wild-type and C-NAP1–knockout cells in untreated cells and in cells at the indicated times after exposure to 5-Gy IR. Ponceau S staining of the membrane after protein transfer was used as a loading control. Size markers at left are in kilodaltons. (D) Cells of the indicated genotype were treated with 4 mM HU for 48 h before fixation. Centrosomes were quantitated by staining with antibodies to glutamylated tubulin and γ-tubulin. Bar graph indicates mean ± SD of three separate experiments in which at least 200 cells were counted. ***p < 0.001; *p < 0.05; ns, not significant by unpaired t test.
FIGURE 7:
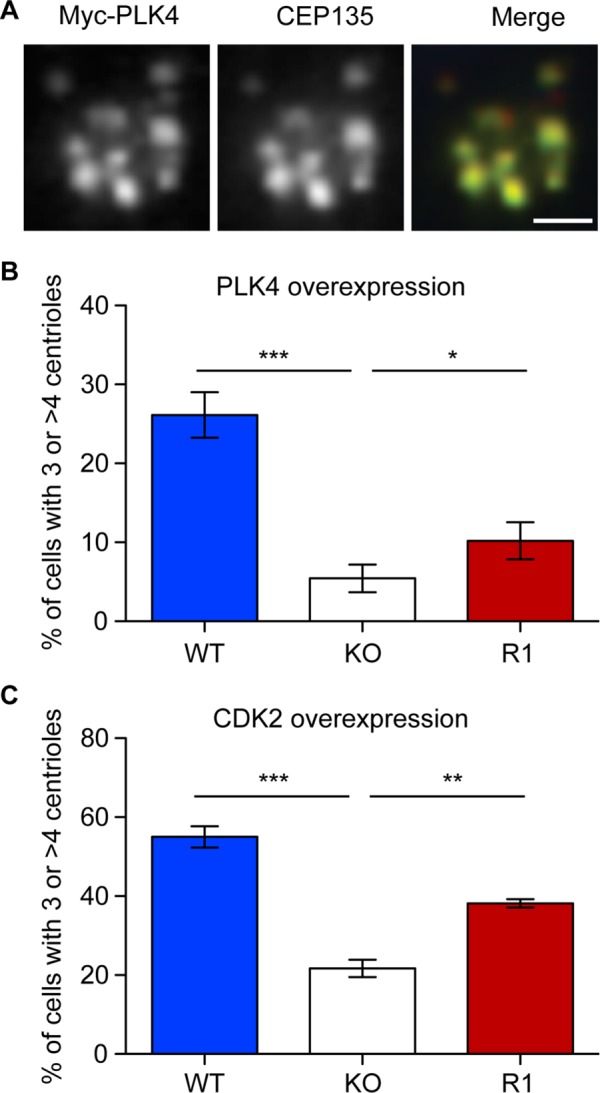
Reduced centrosome amplification induced by PLK4 and CDK2 overexpression in C-NAP1–deficient cells. (A) Immunofluorescence microscopy of a centriole rosette visualized with antibodies to myc (green) and CEP135 (red) 72 h after transfection of wild-type hTERT-RPE1 cells with a myc-PLK4 overexpression construct. Scale bar, 2 μm. (B, C) Centrosome quantitation in cells of the indicated genotype 72 h after transfection with constructs encoding (B) myc-PLK4 or (C) HA-CDK2. Centrioles were scored by staining with antibodies against CEP135 or centrin and transfected cells identified using antibodies to myc or CDK2. Bar graphs indicate mean ± SD of three separate experiments in which at least 50 transfected cells were counted. ***p < 0.001, **p < 0.01, and *p < 0.05 by unpaired t test.
DISCUSSION
We show here that CRISPR/Cas9-mediated ablation of C-NAP1 caused the loss of centriolar linker components from the proximal end of the centriole and increased centriole separation. The centrioles appeared otherwise normal and supported proliferation rates that were indistinguishable from those of wild-type hTERT-RPE1 cells. These data are consistent with previous reverse genetic analyses of C-NAP1 function that used siRNA (Bahe et al., 2005; Graser et al., 2007b; Conroy et al., 2012), ZFN-mediated gene targeting (Panic et al., 2015), or CRISPR targeting of a different exon (Mazo et al., 2016) to remove C-NAP1. A truncating mutation in CEP250 gives rise to caprine-like generalized hypoplasia syndrome in Montbéliarde cattle. (Floriot et al., 2015), in which a very similar splitting phenomenon is seen with otherwise normal centrioles.
The relatively moderate effect of C-NAP1 deficiency prompted us to examine another centriolar function, that of providing the basal body for the primary cilium. We found that C-NAP1 deficiency was compatible with normal levels of primary cilium formation after serum starvation. Individual cilia arose from single basal bodies, with no evidence for abnormality deriving from the separated centrioles. Ciliation was previously reported to occur normally in C-NAP1–knockout hTERT-RPE1s (Panic et al., 2015; Mazo et al., 2016), and neither C-NAP1 nor rootletin (CROCC) have been identified as candidate ciliary genes in siRNA screens or interaction analyses (Gupta et al., 2015; Roosing et al., 2015; Wheway et al., 2015). The slight increase in mean ciliary length that we observed in our C-NAP1–deficient cells was not seen in the other CRISPR knockout study, in which there was a moderate decline in mean cilium length (Mazo et al., 2016). Comparing these data, it seems likely that the alteration in cilium length was an effect of clonal variation.
Recent data show that the combined loss of C-NAP1 and centriolar subdistal appendage proteins causes detachment of the ciliated centriole from the Golgi apparatus and an alteration in cilium positioning from being “submerged” in a deep membrane invagination to a position at the apical cell surface (Mazo et al., 2016). Ciliary “surfacing” allowed increased ciliary motion and the activation of Hh signaling even without SAG treatment, indicating that ciliary position can regulate signaling through the cilium. Our analysis of the submerged cilia induced by serum starvation in C-NAP1–null cells showed that they were responsive to chemical and mechanical stimuli, as determined by the ciliary localization of Smo upon stimulation with its agonist (Kiprilov et al., 2008) and by the response to fluid flow–mediated shear stress of a series of known cilium-regulated genes. These data suggest that the loss of the ciliary rootlet did not block ciliary signaling. However, although rootletin is dispensable for development in the mouse, its absence leads to mechanical instability of sensory cilia in specialized cells (Yang et al., 2005), and findings in in Drosophila demonstrate that the absence of the ciliary rootlet impairs neuronal mechanosensory responses (Chen et al., 2015; Styczynska-Soczka and Jarman, 2015). Our analysis of C-NAP1–deficient cilia indicates that the short-term stability and signaling capability of these submerged cilia are independent of C-NAP1 and of the ciliary rootlet in retinal pigmented epithelial cells, although our data do not exclude the possibility of an altered signaling response.
In wild-type cells, the splitting of centrosomes into individual centrioles is seen soon after cells are exposed to DNA damage (Saladino et al., 2009; Inanc et al., 2010). Centriole amplification occurs later after DNA damage, when the DNA replication and centrosome duplication cycles become disconnected (Balczon et al., 1995; Dodson et al., 2004; Bourke et al., 2007; Nigg, 2007). Our initial speculation when performing these analyses was that C-NAP1 deficiency–induced loss of centriole cohesion might stimulate centriole duplication. However, we observed a reduction in DNA damage–induced centriole amplification in the absence of C-NAP1. Furthermore, overexpression of key regulators of centriole duplication, PLK4 and CDK2, also led to reduced levels of centriole amplification in C-NAP1 nulls. Previous analyses showed that neither C-NAP1 nor rootletin levels are affected after irradiation (Conroy et al., 2012), so the mechanism by which C-NAP1 loss reduces centrosome amplification is not related to a change in the cellular levels of centriole cohesion proteins. It is unclear how centriole splitting arises so frequently after DNA damage, a condition that drives centriole overduplication.
Centriole splitting can affect various aspects of cell behavior, altering migratory activities or Golgi organization (Godinho et al., 2014; Kushner et al., 2014; Panic et al., 2015). Our data suggest that centriole amplification after DNA damage may also be affected. Several studies implicated centriolar satellite densities in the control of centrosome overduplication (Prosser et al., 2009; Loffler et al., 2013; Kodani et al., 2015). Recent data highlight the importance of local concentration in the activation of PLK4 (Lopes et al., 2015), providing a model for how specific regions of the cytoplasm around a centriole may determine the capacity of that centriole to duplicate during the normal cell cycle. Dispersion of the satellites around centrioles that have split due to the absence of C-NAP1 may impede centriole duplication by reducing the available centriolar precursor concentrations, consistent with the reduced centriole amplification seen in C-NAP1 nulls after overexpression of PLK4 or CDK2. The ATM-stimulated inhibition of NEK2 by PLK1 to block premature centrosome separation (Fletcher et al., 2004; Zhang et al., 2005; Mardin et al., 2011) may thus contribute to PLK1's positive role in centriole amplification (Inanc et al., 2010; Douthwright and Sluder, 2014) through a novel mechanism. Our data suggest that centriole splitting may impede, rather than potentiate, centrosome amplification.
MATERIALS AND METHODS
Cell culture
hTERT-RPE1 cells were obtained from the American Type Culture Collection and grown in DMEM-F12 with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a humidified 5% CO2 atmosphere at 37°C. Mycoplasma testing was performed every 2 mo. Hydroxyurea at 4 mM (Sigma-Aldrich) was added for 48 h. Irradiations were performed using a 137Cs source at 9.5 Gy/min (Mainance Engineering). For flow cytometry, cells were fixed in 70% ethanol at −20°C overnight and then resuspended in phosphate-buffered saline (PBS) containing 200 μg/ml RNase A and 20 μg/ml propidium iodide and incubated for 30 min. Cell cycle analysis was performed on an Accuri C6 Sampler (BD Biosciences). To deplete cells of serum, 0.6 × 106 cells were washed with unsupplemented DMEM-F12 before addition of DMEM-F12 with 0.1% FBS for 48 h. For serum starvation after siRNA, 0.2% FBS in DMEM-F12 was added for 24 h. For treatment with SAG, cells were serum starved for 24 h and incubated for 4 h with 100 nM SAG (EMD Millipore).
For mechanical stimulation, cells were seeded on collagen I (0.15 mg/ml in 0.2 M acetic acid)–coated glass slides at a seeding density of 7000 cells/cm2 and cultured for 24 h in standard growth medium, followed by 48 h of serum starvation in 0.5% FBS. Cells were exposed to oscillatory fluid flow–induced shear stress using a custom-designed parallel-plate flow chamber (Hoey et al., 2012). Fluid flow was achieved by applying a pressure-driven flow via a syringe pump (Alladin 1660). The volumetric rate of flow (Q) necessary for a given shear stress was calculated using the equation
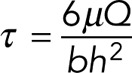 |
where τ is the shear stress, μ is the fluid viscosity, b is the width of the channel (38 mm), and h is the height of the channel (300 μm). Slides were placed within the chamber, incubated for 15 min, and then exposed to 1-Pa (45 ml/min) or 2-Pa (90 ml/min) shear stress at frequency 1 Hz in serum starvation medium for 1 h. No-flow control slides were placed within the chamber but not exposed to fluid flow.
Cloning and genome editing
Primers targeting C-NAP1 exon 8 (Mali et al., 2013) were cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 (plasmid 43330; Addgene; Cong et al., 2013): 5′-CACCGACATTCCGACGCCACTTCC-3′ and 5′-AAACGGAAGTGGCGTCGGAATGTC-3′. hTERT-RPE1 cells at 80% confluency were transfected using Lipofectamine (Thermo Fisher) as per the manufacturer's instructions with 3 μg of pX330-Ex8 and 2 μg of pLOX-Neo (Arakawa et al., 2001) for 24 h at 37°C. Cells were then trypsinized, and serial dilutions were performed into medium containing 1 mg/ml G418 (Invitrogen). Cells were placed under selection for 48 h, after which the medium was replaced with normal growth medium and then incubated at 37°C for 10 d. Colonies were lifted using cloning disks (Sigma-Aldrich) and expanded. Full-length C-NAP1 was assembled into pcDNA3.1-BSR from five fragments (A–E) that were cloned by RT-PCR using the following primers: A, 5′-GAGGCTCTTAAGATGGAGACAAGAAGCCCT-3′ and 5′-AGTAGTCGACCTGCAAAGCATTTCTCGCCT-3′; B, 5′-AGTAGTCGACCTGGCGGAGGCAGAGAAGAG-3′ and 5′-AGTAAAGCTTGTGGAGGGCAGATGCTACTG-3′; C, 5′-AGTAAAGCTTCATCAAGACCTGTGGAAGAC-3′ and 5′-GATACCATGGGCAGCTGCTCTAAAACAGAC-3′; D, 5′-GATACCATGGCCGTCCAGGAGCGAGAGCAG-3′ and 5′-GATACATATGGGCTTGCTCCAGAGCTCCCT-3′; and E, 5′-AGGACATATGACACTGAAGGAGCGTCATGG-3′ and 5′-GCGGCCGCCTACCTGGAGGCGGCTTG-3′. A C-NAP1 rescue cell line was generated using 9 μl of Lipofectamine combined with 4 μg of linearized plasmid. DNA was transfected for 48 h to 0.5 × 106 cells in a 35-mm dish. The medium was then replaced with a 1:1 mix of fresh to conditioned medium containing 10 µg/ml of blasticidin S (Sigma-Aldrich). Selection was carried out for 10–14 d with medium replacement every 3 d. Cells were expanded and screened by Western blot and immunofluorescence microscopy, followed by genomic PCR and clonal DNA sequencing for C-NAP1–disrupted clones (Source Bioscience). For transient overexpression experiments, cells were transfected with 2 μg of plasmids that encode Myc-PLK4 (pCMV-3Tag2-PLK4; Agilent) or HA-CDK2 (pCMV-HA-Cdk2; 1883; Addgene) using Lipofectamine 2000.
RNA-mediated interference
hTERT-RPE1 cells were transfected with an ON-TARGETplus SMART pool of RNA duplexes inhibitory to C-NAP1 (L-012364-00-0005; Dharmacon), Silencer Select siRNA oligonucleotides specific to GAPDH (s5573; Ambion), or an ON-TARGETplus Non-targeting Control Pool (D-001810-10-05; Dharmacon) using Oligofectamine (Invitrogen). A 50- or 100-nmol amount of siRNA was complexed with Oligofectamine in serum-free OptiMEM (Gibco) and added to cells at 30–40% confluency. Serum was added 5 h after transfection and fresh medium 24 h after transfection. Cells were analyzed 24 and 48 h after transfection. Where indicated, cells were serum starved 24 h after transfection.
mAb generation
A fragment of the human C-NAP1 cDNA encoding amino acids 1513–1750 was cloned into pGEX4T2 and expressed in bacteria as a glutathione S-transferase fusion protein. The C-NAP1 fragment was purified from a glutathione column by thrombin cleavage and used for hybridoma preparation (Dundee Cell Products). The best-performing clone 6F2 was expanded and subcloned to give 6F2C8, which produces IgG1κ.
Immunoblotting
Primary monoclonal mouse antibodies were used in immunoblot analyses as follow: α-tubulin (1:5000; B512, Sigma-Aldrich), glutamylated tubulin (1:750; GT335; Adipogen), centrin (20H5; 1:500; Millipore), Chk1 (1:1000; DCS-310; Sigma-Aldrich), pChk1 (1:1000; 2348S 133D3; Cell Signaling), myc (9E10, produced in-house from the hybridoma; 1:100), NEK2 (1:250; BD Transduction), and C-NAP-1 (1:2; 6F2C8 hybridoma supernatant; this study). A monoclonal rabbit antibody to glyceraldehyde-3-phosphate dehydrogenase (2118; Cell Signaling) was used at 1:5000. Polyclonal rabbit antibodies used were against CEP72 (1:500; A301-297A; Bethyl), CDK2 (1:500; M2/sc-163, Santa Cruz Biotechnology), CEP135 (1:1000; Ab75005; Abcam), OFD1 (1:500; 32843; Novus), PCM1 (1:10,000; 817; Dammermann and Merdes, 2002), and rootletin (1:1000; 80820, Novus).
Microscopy
hTERT-RPE1 cells were grown on glass coverslips and fixed in methanol/5 mM ethylene glycol tetraacetic acid at −20°C for 10 min. Before fixation and staining with acetylated or detyrosinated tubulin, cells were incubated on ice for 30 min to depolymerize the microtubules unless otherwise indicated. Cells were blocked in 1% bovine serum albumin before 1 h of incubation with primary antibodies and 45 min of incubation with Alexa 488- or 594–labeled secondary antibodies (Jackson). Coverslips were mounted in 80% (vol/vol) glycerol in PBS containing 3% (wt/vol) N-propyl-gallate and 4′,6-diamidino-2-phenylindole. Monoclonal antibodies were used as follows: γ-tubulin (1:500; GTU88; Sigma-Aldrich), acetylated tubulin (1:2000; T6793; Sigma-Aldrich), NEK2 (1:250; BD Transduction Laboratories), and C-NAP1 (1:2; this study). Polyclonal rabbit antibodies used were against γ-tubulin (1:1000; T3559; Sigma-Aldrich), pericentrin (1:2000; ab4448; Abcam), detyrosinated tubulin (1:500; ab48389; Abcam), ninein (1:200; ab4447; Abcam), Arl13b (1:500; 17711-1-AP; Proteintech), rootletin (1:750; NBP1-80820; Novus), PCM1 (1:10,000; Dammermann and Merdes, 2002; a gift from A. Merdes, University of Toulouse), Smoothened (1:500; ab38686; Abcam), and Cep135 (1:1000; Bird and Hyman, 2008; a gift from A. Bird, Max Planck Institute of Molecular Physiology, Dortmund). Imaging was performed with an Olympus IX81 microscope (Hamamatsu C4742-80-12AG camera), 100× objective, numerical aperture 1.35, using Volocity software (PerkinElmer). Images were saved as Adobe Photoshop CS2 files (version 9.0). Satellite intensity was measured by determining the total fluorescence intensity of the area surrounding each centriole. Deconvolved maximum intensity projections were used for analyzing satellite intensity, with the three-dimensional volumes collapsed into two-dimensional images for calculations. Fluorescence intensity within a 25-µm2 circle around each centriole was determined using the Measurement tool of Volocity.
qRT-PCR
Immediately after flow treatment, total RNA was isolated using TriReagent (Sigma-Aldrich). A 1-µg amount of RNA was reverse transcribed into cDNA using a High Capacity cDNA kit (Life Technologies). qPCR was performed using SYBR Select Mastermix with ROX passive dye (ThermoFisher). The expression of AXIN2, COX2, GAPDH, GLI1, and VEGFA was quantified using the following primers (Sigma-Aldrich): AXIN2 (Tm = 60°C, 400 nM), 5′-AAAGAGAGGAGGTTCAGATG-3′ and 3′-CTGAGTCTGGGAATTTTTCTTC-5′; PTGS2 (Tm = 60°C, 150 nM), 5′-AAGCAGGCTAATACTGATAGG-3′ and 3′-TGTTGAAAAGTAGTTCTGGG-5′; GAPDH (Tm = 60°C, 300 nM), 5′-ACAGTTGCCATGTAGACC-3′ and 3′-TTTTTGGTTGAGCACAGG-5′; GLI1 (Tm = 61°C, 300 nM), 5′-CTCGTAGCTTTCATCAACTC-3′ and 3′-TTTTTGGTGATTCATCTGGG-5′; and VEGFA (Tm = 60°C, 400 nM), 5′-AATGTGAATGCAGACCAAAG-3′ and 3′-GACTTATACCGGGATTTCTTG-5′. The amplification of the target product was executed with an ABI7500 Fast Real Time PCR machine, and melt curve analysis was implemented as a control for primer dimer formation. Each sample was normalized to reference gene GAPDH and static control.
Statistical analysis
Statistical analyses were performed with Prism, version 5.0 (GraphPad).
Supplementary Material
Acknowledgments
We acknowledge the National Biophotonics and Imaging Platform Ireland and the National Centre for Biomedical Engineering Science Flow Cytometry Core Facility, which were supported by Irish Government Programme for Research in Third-Level Institutions Cycles 4 and 5. This work was funded by Science Foundation Ireland Principal Investigator Award 10/IN.1/B2972 and European Commission SEC-2009-4.3-02, Project 242361 ‘BOOSTER’ (to C.G.M.), an Irish Research Council Postgraduate Scholarship (to S.B.), and European Research Council Grant 336882 and SFI ERC Support Grant SFI 13/ERC/L2864 (to D.A.H.).
Abbreviations used:
- C-NAP1
centrosomal NEK2-associated protein
- CRISPR
clustered regularly interspaced short palindromic repeats
- FBS
fetal bovine serum
- Hh
Hedgehog
- hTERT-RPE1
human telomerase reverse transcriptase-immortalized retinal pigmented epithelium cells
- HU
hydroxyurea
- IR
ionizing radiation
- mAb
monoclonal antibody
- NEK2A
Never in mitosis A (NIMA)-related kinase 2A
- PCM
pericentrolar material
- PLK
polo-like kinase
- qRT-PCR
quantitative reverse transcription PCR
- SAG
smoothened agonist
- SHh
sonic hedgehog
- siRNA
small interfering RNA
- Smo
smoothened
- ZFN
zinc finger nuclease.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0325) on January 18, 2017.
REFERENCES
- Agircan FG, Schiebel E, Mardin BR. Separate to operate: control of centrosome positioning and separation. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130461. doi: 10.1098/rstb.2013.0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahe S, Stierhof YD, Wilkinson CJ, Leiss F, Nigg EA. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J Cell Biol. 2005;171:27–33. doi: 10.1083/jcb.200504107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahmanyar S, Kaplan DD, Deluca JG, Giddings TH, Jr, O'Toole ET, Winey M, Salmon ED, Casey PJ, Nelson WJ, Barth AI. beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008;22:91–105. doi: 10.1101/gad.1596308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR. Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol. 1995;130:105–115. doi: 10.1083/jcb.130.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barenz F, Mayilo D, Gruss OJ. Centriolar satellites: busy orbits around the centrosome. Eur J Cell Biol. 2011;90:983–989. doi: 10.1016/j.ejcb.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bird AW, Hyman AA. Building a spindle of the correct length in human cells requires the interaction between TPX2 and Aurora A. J Cell Biol. 2008;182:289–300. doi: 10.1083/jcb.200802005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke E, Dodson H, Merdes A, Cuffe L, Zachos G, Walker M, Gillespie D, Morrison CG. DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep. 2007;8:603–609. doi: 10.1038/sj.embor.7400962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B, Geng X, Mahamud MR, Fu J, Mukherjee A, Kim Y, Jho EH, Kim TH, Kahn ML, Xia L, et al. Mechanotransduction activates canonical Wnt/beta-catenin signaling to promote lymphatic vascular patterning and the development of lymphatic and lymphovenous valves. Genes Dev. 2016;30:1454–1469. doi: 10.1101/gad.282400.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JV, Kao LR, Jana SC, Sivan-Loukianova E, Mendonca S, Cabrera OA, Singh P, Cabernard C, Eberl DF, Bettencourt-Dias M, Megraw TL. Rootletin organizes the ciliary rootlet to achieve neuron sensory function in Drosophila. J Cell Biol. 2015;211:435–453. doi: 10.1083/jcb.201502032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16:611–624. doi: 10.1038/nrm4062. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy PC, Saladino C, Dantas TJ, Lalor P, Dockery P, Morrison CG. C-NAP1 and rootletin restrain DNA damage-induced centriole splitting and facilitate ciliogenesis. Cell Cycle. 2012;11:3769–3778. doi: 10.4161/cc.21986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammermann A, Merdes A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J Cell Biol. 2002;159:255–266. doi: 10.1083/jcb.200204023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson H, Bourke E, Jeffers LJ, Vagnarelli P, Sonoda E, Takeda S, Earnshaw WC, Merdes A, Morrison C. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 2004;23:3864–3873. doi: 10.1038/sj.emboj.7600393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douthwright S, Sluder G. Link between DNA damage and centriole disengagement/reduplication in untransformed human cells. J Cell Physiol. 2014;229:1427–1436. doi: 10.1002/jcp.24579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Zhang D, Yin H, Zheng L, Bi X, Yuan L. Centlein mediates an interaction between C-Nap1 and Cep68 to maintain centrosome cohesion. J Cell Sci. 2014;127:1631–1639. doi: 10.1242/jcs.139451. [DOI] [PubMed] [Google Scholar]
- Firat-Karalar EN, Stearns T. The centriole duplication cycle. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130460. doi: 10.1098/rstb.2013.0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher L, Cerniglia GJ, Nigg EA, Yend TJ, Muschel RJ. Inhibition of centrosome separation after DNA damage: a role for Nek2. Radiat Res. 2004;162:128–135. doi: 10.1667/rr3211. [DOI] [PubMed] [Google Scholar]
- Floriot S, Vesque C, Rodriguez S, Bourgain-Guglielmetti F, Karaiskou A, Gautier M, Duchesne A, Barbey S, Fritz S, Vasilescu A, et al. C-Nap1 mutation affects centriole cohesion and is associated with a Seckel-like syndrome in cattle. Nat Commun. 2015;6:6894. doi: 10.1038/ncomms7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry AM, Mayor T, Meraldi P, Stierhof YD, Tanaka K, Nigg EA. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J Cell Biol. 1998;141:1563–1574. doi: 10.1083/jcb.141.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D. Oncogene-like induction of cellular invasion from centrosome amplification. Nature. 2014;510:167–171. doi: 10.1038/nature13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA. Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol. 2007a;179:321–330. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graser S, Stierhof YD, Nigg EA. Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J Cell Sci. 2007b;120:4321–4331. doi: 10.1242/jcs.020248. [DOI] [PubMed] [Google Scholar]
- Gupta GD, Coyaud E, Goncalves J, Mojarad BA, Liu Y, Wu Q, Gheiratmand L, Comartin D, Tkach JM, Cheung SW, et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015;163:1484–1499. doi: 10.1016/j.cell.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Huang N, Bao Y, Zhou H, Teng J, Chen J. LRRC45 is a centrosome linker component required for centrosome cohesion. Cell Rep. 2013;4:1100–1107. doi: 10.1016/j.celrep.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Hoey DA, Tormey S, Ramcharan S, O'Brien FJ, Jacobs CR. Primary cilia-mediated mechanotransduction in human mesenchymal stem cells. Stem Cells. 2012;30:2561–2570. doi: 10.1002/stem.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanc B, Dodson H, Morrison CG. A centrosome-autonomous signal that involves centriole disengagement permits centrosome duplication in G2 phase after DNA damage. Mol Biol Cell. 2010;21:3866–3877. doi: 10.1091/mbc.E10-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiprilov EN, Awan A, Desprat R, Velho M, Clement CA, Byskov AG, Andersen CY, Satir P, Bouhassira EE, Christensen ST, Hirsch RE. Human embryonic stem cells in culture possess primary cilia with hedgehog signaling machinery. J Cell Biol. 2008;180:897–904. doi: 10.1083/jcb.200706028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. Plk4-induced centriole biogenesis in human cells. Dev Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Kodani A, Yu TW, Johnson JR, Jayaraman D, Johnson TL, Al-Gazali L, Sztriha L, Partlow JN, Kim H, Krup AL, et al. Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. Elife. 2015;4:e07519. doi: 10.7554/eLife.07519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner EJ, Ferro LS, Liu JY, Durrant JR, Rogers SL, Dudley AC, Bautch VL. Excess centrosomes disrupt endothelial cell migration via centrosome scattering. J Cell Biol. 2014;206:257–272. doi: 10.1083/jcb.201311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffler H, Fechter A, Liu FY, Poppelreuther S, Kramer A. DNA damage-induced centrosome amplification occurs via excessive formation of centriolar satellites. Oncogene. 2013;32:2963–2972. doi: 10.1038/onc.2012.310. [DOI] [PubMed] [Google Scholar]
- Lopes CA, Jana SC, Cunha-Ferreira I, Zitouni S, Bento I, Duarte P, Gilberto S, Freixo F, Guerrero A, Francia M, et al. PLK4 trans-autoactivation controls centriole biogenesis in space. Dev Cell. 2015;35:222–235. doi: 10.1016/j.devcel.2015.09.020. [DOI] [PubMed] [Google Scholar]
- Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS, et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat Cell Biol. 2015;17:228–240. doi: 10.1038/ncb3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardin BR, Agircan FG, Lange C, Schiebel E. Plk1 controls the Nek2A-PP1gamma antagonism in centrosome disjunction. Curr Biol. 2011;21:1145–1151. doi: 10.1016/j.cub.2011.05.047. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Hayashi K, Nishida E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol. 1999;9:429–432. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- Mayor T, Stierhof YD, Tanaka K, Fry AM, Nigg EA. The centrosomal protein C-Nap1 is required for cell cycle-regulated centrosome cohesion. J Cell Biol. 2000;151:837–846. doi: 10.1083/jcb.151.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazo G, Soplop N, Wang WJ, Uryu K, Tsou MB. Spatial control of primary ciliogenesis by subdistal appendages alters sensation-associated properties of cilia. Dev Cell. 2016;39:424–437. doi: 10.1016/j.devcel.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17:215–221. doi: 10.1016/j.tcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Stearns T. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154–1160. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh EC, Katsanis N. Cilia in vertebrate development and disease. Development. 2012;139:443–448. doi: 10.1242/dev.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan JK, Marzio A, Jones MJ, Saraf A, Jallepalli PV, Florens L, Washburn MP, Pagano M. Degradation of Cep68 and PCNT cleavage mediate Cep215 removal from the PCM to allow centriole separation, disengagement and licensing. Nat Cell Biol. 2015;17:31–43. doi: 10.1038/ncb3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintrand M, Moudjou M, Delacroix H, Bornens M. Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol. 1992;108:107–128. doi: 10.1016/1047-8477(92)90011-x. [DOI] [PubMed] [Google Scholar]
- Panic M, Hata S, Neuner A, Schiebel E. The centrosomal linker and microtubules provide dual levels of spatial coordination of centrosomes. PLoS Genet. 2015;11:e1005243. doi: 10.1371/journal.pgen.1005243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser SL, Straatman KR, Fry AM. Molecular dissection of the centrosome overduplication pathway in S-phase-arrested cells. Mol Cell Biol. 2009;29:1760–1773. doi: 10.1128/MCB.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosing S, Hofree M, Kim S, Scott E, Copeland B, Romani M, Silhavy JL, Rosti RO, Schroth J, Mazza T, et al. Functional genome-wide siRNA screen identifies KIAA0586 as mutated in Joubert syndrome. Elife. 2015;4:e06602. doi: 10.7554/eLife.06602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saladino C, Bourke E, Conroy PC, Morrison CG. Centriole separation in DNA damage-induced centrosome amplification. Environ Mol Mutagen. 2009;50:725–732. doi: 10.1002/em.20477. [DOI] [PubMed] [Google Scholar]
- Styczynska-Soczka K, Jarman AP. The Drosophila homologue of Rootletin is required for mechanosensory function and ciliary rootlet formation in chordotonal sensory neurons. Cilia. 2015;4:9. doi: 10.1186/s13630-015-0018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi MM, Iacobas DA, Iacobas S, Spray DC. Fluid shear stress upregulates vascular endothelial growth factor gene expression in osteoblasts. Ann NY Acad Sci. 2007;1117:73–81. doi: 10.1196/annals.1402.020. [DOI] [PubMed] [Google Scholar]
- Tollenaere MA, Mailand N, Bekker-Jensen S. Centriolar satellites: key mediators of centrosome functions. Cell Mol Life Sci. 2015a;72:11–23. doi: 10.1007/s00018-014-1711-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollenaere MA, Villumsen BH, Blasius M, Nielsen JC, Wagner SA, Bartek J, Beli P, Mailand N, Bekker-Jensen S. p38- and MK2-dependent signalling promotes stress-induced centriolar satellite remodelling via 14-3-3-dependent sequestration of CEP131/AZI1. Nat Commun. 2015b;6:10075. doi: 10.1038/ncomms10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009;111:39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villumsen BH, Danielsen JR, Povlsen L, Sylvestersen KB, Merdes A, Beli P, Yang YG, Choudhary C, Nielsen ML, Mailand N, Bekker-Jensen S. A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. EMBO J. 2013;32:3029–3040. doi: 10.1038/emboj.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen TM, Racher H, Phelps IG, Toedt G, Kennedy J, Wunderlich KA, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat Cell Biol. 2015;17:1074–1087. doi: 10.1038/ncb3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Adamian M, Li T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol Biol Cell. 2006;17:1033–1040. doi: 10.1091/mbc.E05-10-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Gao J, Adamian M, Wen XH, Pawlyk B, Zhang L, Sanderson MJ, Zuo J, Makino CL, Li T. The ciliary rootlet maintains long-term stability of sensory cilia. Mol Cell Biol. 2005;25:4129–4137. doi: 10.1128/MCB.25.10.4129-4137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Yue G, Adamian M, Bulgakov O, Li T. Rootletin, a novel coiled-coil protein, is a structural component of the ciliary rootlet. J Cell Biol. 2002;159:431–440. doi: 10.1083/jcb.200207153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Zeng H, Ning G, Reiter JF, Liu A. C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc Natl Acad Sci USA. 2014;111:2164–2169. doi: 10.1073/pnas.1318737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Fletcher L, Muschel RJ. The role of Polo-like kinase 1 in the inhibition of centrosome separation after ionizing radiation. J Biol Chem. 2005;280:42994–42999. doi: 10.1074/jbc.M505450200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.