

Abstract
Ischemic stroke results in excessive release of glutamate, which contributes to neuronal cell death. Here, we test the hypothesis that otherwise neurotoxic glutamate can be productively metabolized by glutamate oxaloacetate transaminase (GOT) to maintain cellular energetics and protect the brain from ischemic stroke injury. The GOT-dependent metabolism of glutamate was studied in primary neural cells and in stroke-affected C57-BL6 mice using magnetic resonance spectroscopy and GC-MS. Extracellular Glu sustained cell viability under hypoglycemic conditions and increased GOT-mediated metabolism in vitro. Correction of stroke-induced hypoxia using supplemental oxygen in vivo lowered Glu levels as measured by 1H magnetic resonance spectroscopy. GOT knockdown abrogated this effect and caused ATP loss in the stroke-affected brain. GOT overexpression increased anaplerotic refilling of tricarboxylic acid cycle intermediates in mouse brain during ischemic stroke. Furthermore, GOT overexpression not only reduced ischemic stroke lesion volume but also attenuated neurodegeneration and improved poststroke sensorimotor function. Taken together, our results support a new paradigm that GOT enables metabolism of otherwise neurotoxic extracellular Glu through a truncated tricarboxylic acid cycle under hypoglycemic conditions.—Rink, C., Gnyawali, S., Stewart, R., Teplitsky, S., Harris, H., Roy, S., Sen, C. K., Khanna, S. Glutamate oxaloacetate transaminase enables anaplerotic refilling of TCA cycle intermediates in stroke-affected brain.
Keywords: anaplerosis, hypoglycemia, ischemia, oxygen
Ischemic stroke challenges the brain with energetic failure (ATP deficiency) due to the focal arrest of cerebral blood flow, which prevents delivery of glucose to brain tissue. Glutamatergic neurons rely on the liberal production of ATP from glucose via tricarboxylic acid cycle (TCA) in order to maintain energy-expensive ion pumps that regulate neurotransmitter release and uptake (1). To that end, the disruption of glucose supply due to ischemic stroke incurs a rapid neurotoxic release of excitatory neurotransmitter glutamate at the synapse that contributes to neural cell death (2).
We have previously reported that correction of hypoxia by supplemental oxygen (SO) therapy during cerebral ischemia induces expression of the glutamate-metabolizing enzyme glutamate oxaloacetate transaminase (GOT) and rescues brain tissue from stroke-induced injury (2–4). GOT-mediated metabolism of glutamate in brain tissue was first characterized by Krebs in 1935 (5). Krebs originally identified a metabolic role for glutamate in the brain prior to the elucidation of a neurotransmitter function by Hayashi in the 1950s (6). Krebs observed that glutamate was the only amino acid that increased cellular respiration in the cortex of rabbit brain under hypoglycemic conditions (5). Given the pathophysiological role of glutamate-mediated neurotoxicity and cell death in ischemic stroke injury (7) and in light of our observation that SO-mediated induction of GOT protects against ischemic stroke injury (2–4), we hypothesize that GOT can utilize otherwise neurotoxic glutamate to support cell survival in the face of ischemia-induced hypoglycemia. The current study is designed to test the significance of GOT-mediated glutamate metabolism in neural cell culture and the hypoglycemic stroke-affected brain. A small but growing body of literature recognizes the therapeutic potential of GOT during ischemic stroke. These efforts have primarily focused on the glutamate grabbing properties of circulating GOT to lower neurotoxic glutamate in brain (8, 9). The current work addresses glutamate clearance from the stroke site and seeks to test whether glutamate is productively metabolized by GOT to generate TCA cycle intermediates in energy-starved, stroke-affected brain tissue.
MATERIALS AND METHODS
All animal studies were performed in accordance with protocols approved by the Institutional Laboratory Animal Care and Use Committee of The Ohio State University. l-Glutamic acid monosodium salt, DMSO, sodium propionate 13C3, and propidium iodide were obtained from Sigma-Aldrich (St. Louis, MO, USA); calcein AM was obtained from Thermo Fisher Scientific (Waltham, MA, USA), and U-13C glutamate was obtained from Cambridge Isotopes (Andover, MA, USA). For cell culture, minimum essential medium, Neurobasal-A Medium, no d-glucose, l-aspartic acid, l-glutamic acid, l-glutamine, or sodium pyruvate, B-27 Supplement Minus AO (50X), d-glucose, l-glutamine, sodium pyruvate, fetal calf serum, and antibiotics (100 μg/ml streptomycin, 100 U/ml penicillin, and 0.25 μg/ml amphotericin) were purchased from Thermo Fisher Scientific. Culture dishes were obtained from Nunc (Roskilde, Denmark).
Primary cortical neurons
Neurons were isolated from the cerebral cortex of rat fetuses (Sprague-Dawley, d 17 of gestation) (Harlan, Indianapolis, IN, USA) as described previously (3, 10–14). Cells were cultured in minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum, 40 μM cystine, and antibiotics (100 μg/ml streptomycin, 100 U/ml penicillin, and 0.25 μg/ml amphotericin) and maintained at 37°C in 5% CO2 and 95% air in a humidified incubator. Unless otherwise stated, all experiments were carried out 24 h after plating. After 24 h of cell seeding, culture medium was replaced with Neurobasal-A Medium with glucose (25 or 1 mM) or without glucose. For hypoxia, cells were placed in a Billups-Rothenburg chamber (Billups-Rothenburg, San Diego, CA, USA) with 95% N2 and 5% CO2. Glutamate treatment was performed as previously described (3). Glutamate was added to the medium as an aqueous solution (3, 15, 16). No change in the medium pH was observed in response to the addition of glutamate.
Determination of cell viability
Cell viability was measured using a Vybrant MTT Cell Proliferation Assay Kit (Thermo Fisher Scientific) per the manufacturer’s instructions. At 24 h after treatment, cells were incubated in fresh Neurobasal culture medium containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for 4–6 h at 37°C with 5% CO2. After MTT treatment, medium was removed, and DMSO was added (10–20 min at 37°C with 5% CO2) to solubilize formazan produced as a result of MTT metabolism. DMSO extract from each well (100 μl) was collected in a 96-well plate, and formazan content was determined by reading absorbance at 540 nm (17). Cell viability was also measured by Calcein-AM and propidium iodide solutions as described previously (3, 10). Cells were incubated with Calcein-AM (3 μM) and propidium iodide (2.5 μM) in PBS for 15 min in cell culture incubators. After incubation, digital images were collected using a Zeiss Observer.Z1 microscope and Zen 2012 (blue edition) (Carl Zeiss AG, Oberkochen, Germany) suited for imaging cells growing in regular culture plates (3, 10, 16).
Glutathione assay
To measure glutathione (GSH) content, primary cortical neurons (0.5 × 106 cells/well) were seeded in 96 well flat, clear-bottom, white polystyrene TC–treated microplates (Corning, Corning, NY, USA). GSH concentrations were measured in cells using a GSH-Glo Glutathione assay kit (Promega, Madison, WI, USA) per the manufacturer’s instructions as previously described (18, 19).
Glutamate labeling and sample preparation
Primary cortical neurons were plated in a 60 mm plate (15–17 × 106 cells/plate). After 24 h of cell plating, medium was changed, and cells were incubated with [U-13C]glutamate (0.25 mM) plus unlabeled glutamate (0.25 mM) in the presence or absence of glucose for 2 h, after which cell extracts were prepared as described in Bakken et al. (20). Briefly, cells were washed with ice-cold PBS, frozen on liquid nitrogen, and extracted with 1 ml of perchloric acid (7% w/v). Acid extracts were centrifuged at 4000 g for 10 min. Supernatants were titrated with KOH (9 M) to pH 7.5, and the precipitate (KClO4) was removed by centrifugation (4000 g for 10 min). The supernatants were lyophilized and stored at −80°C until NMR analysis. Pellets were dissolved in 1 M KOH for 30 min at 37°C, and protein concentrations were determined using bicinchoninic acid protein reagents. Each sample was prepared by pooling cells from 6 plates.
GOT siRNA knockdown
For GOT-knockdown studies, 3 pregnant rats (∼45 fetuses, 350 × 106 cells primary cortical neurons) were used. For mRNA expression, cells were plated in 12-well plates (2.5 × 106 cells/well) or in 60 mm plates (15 × 106 cell/plate) for glutamate labeling 24 h before transfection. DharmaFect 1 transfection reagent was used to transfect cells with GOT siRNA pool for 96 h (GE Dharmacon, Lafayette, CO, USA) per the manufacturer’s instructions and as described (10, 14). A total of 200 nM GOT siRNA per well for 12-well plates and 2000 nM GOT siRNA per 60-mm plate were used. siControl nontargeting siRNA pool (GE Dharmacon) was used for control transfections. Samples were collected after 96 h of siRNA transfection for quantification of mRNA expression or labeled with glutamate as described in the glutamate labeling and sample preparation above. For glutamate labeling experiments in transfected cells (120 h in culture), 10 × 60-mm plates were pooled. The abundance of mRNA for GOT was quantified by real-time PCR using SYBR green-I. The following primer sets were used: rGOT, (forward) 5′-CGGAAAAGAGCATGACAGCG-3′; rGOT, (reverse) 5′-AGGGGGATTGGACCAGGTG-3′; 18s_rRNA, (forward) 5′-GTAACCCGTTGAACCCCATT-3′; and 18s_rRNA, (reverse) 5′- CCATCCAATCGGTAGTAGCG-3′.
Mouse stroke model
Focal cerebral ischemia was induced in C57BL/6 male mice (8–10 wk old) (The Jackson Laboratory, Bar Harbor, ME, USA) by the intraluminal suture method of middle cerebral artery occlusion (MCAO) under room air (RA) conditions or with supplemental oxygen (SO; 100% O2 inhaled) as described previously (3, 4). Successful MCAO was validated by Laser Doppler Flowmetry (3, 4). For magnetic resonance spectroscopy experiments, MCAO was permanent, and spectroscopy was acquired 30 min after occlusion. For anaplerosis experiments, mice were killed after 30 min of MCAO while ischemia persisted. For lentiviral GOT overexpression experiments, mice were subjected to 90 min of transient MCAO as previously described (3, 4).
NMR spectroscopy
In vitro
High-resolution NMR spectroscopy of cell extracts was performed at 400 MHz on a wide-bore spectrometer (Avance III400; Bruker, Billerica, MA, USA). Each lyophilized cell extract was dissolved in 0.3 ml of D2O with 0.05% dioxanne. The pH was adjusted to 7, and the solution was placed in a 5-mm Shigemi tube (Shigema, Allison Park, PA, USA). Proton decoupled carbon spectra (Waltz-16) were obtained at 100.62 MHz using a 5-mm Bruker BBO probe during an overnight acquisition with the following conditions: 30° pulse, repetition time 1.6 s, spectral width 24 kHz, data point 64 k, and 256 dummy scans. A Lorentz to Gauss transformation (LB = −1 Hz; GB = 0.1) was applied to the raw data before Fourier transformation. Under these conditions, protonated carbons were fully relaxed, and nuclear Overhauser effects were assumed to be maximum for CHs and CH2s. Using the dioxanne signal, we were able to measure the number of nanomoles per milligram protein of enriched glutamate and aspartate. After 13C spectroscopy, the solution was diluted to 0.6 ml by adding 0.3 ml of D2O and introduced in a 5-mm NMR tube. An external standard made of a sealed capillary containing a solution of trimethylsilylpropionic acid in D2O was introduced in the NMR tube and used as chemical shift reference and quantitation standard. Fully relaxed water-suppressed proton spectra were acquired with the same Bruker probe. Standard acquisition conditions were as follows: 45° pulse, repetition time 8.8 s, water saturation during the relaxation delay of 4 s, 6775 Hz pectral width, data point 64 k, AQ 4.8 s, 4 dummy scans, and 64 scans.
In vivo
Magnetic resonance spectroscopy (MRS) imaging was performed 30 min after onset of MCAO while ischemia persisted. MRS was performed using a 9.4-T magnet. A transceiver mouse brain coil (internal diameter, 20 mm) was used to transmit and receive the radio frequency signal. Two groups of mice (n = 6) were used for study under RA and SO conditions. Before MRS, T2-weighted anatomic imaging using RARE sequence was performed as well as localization with shimming and Fastmap scans. Mouse brain was localized for each MRS measurement with multivoxel 2-dimensional chemical shift imaging. Chemical shift imaging was performed using chemical shift imaging spin-echo sequence and the Point Resolved SpectroScopy (PRESS) protocol with the following parameters: TR/TE = 1 s/35 ms, slice thickness 2 mm, total number of averages 1800 with scan time of 30 min per scan, matrix size 16 × 16. For analyses, rectangular phase encoding was performed over a field of view of 128 × 128 mm2. Voxels from the stroke-affected S1 cortex were selected for quantification. Spectra were Fourier transformed in the spectral domain using gaussian line-shapes, and the ratios of Glu/total creatinine resonance areas (area under peak) were calculated using 3DiCSI (Qi Zhao, Columbia University, New York, NY, USA).
MRI
T2-weighted imaging was performed separately on stroke-affected mice receiving lentiviral delivery at 48 h after MCAO using a 11.7T (500 MHz) MR system comprised of a vertical bore magnet (Bruker Biospin; Bruker) as previously described (3, 10, 14, 16).
In vivo anaplerosis
For in vivo anaplerosis, succinate content was measured in mouse brain from the following groups: RA, RA with plasmid lentivirus (pLV)-control, RA with pLV-GOT, SO, SO with pLV-control, or SO with pLV-GOT. Sodium propionate 13C3 (2 mM) was prepared in ultrapure water and administered via jugular catheterization for 30 min at 0.1 mmol/g/min before MCAO to achieve steady-state delivery. After 30 min MCAO and while propionate infusion continued, animals were killed, and stroke-affected and contralateral control tissues were quickly snap frozen in liquid nitrogen within 15 s of decapitation. Succinate content (nmol/mg wet weight) was analyzed from tissue extracts using GC-MS on a mass spectrometer (Agilent 5973; Agilent, Santa Clara, CA, USA) equipped with an Agilent 6890 gas chromatograph and an HP-5MS 5% phenyl methyl siloxane fused silica capillary column (60 m, 250 mm inner diameter, 0.25-mm film thickness). Sample preparation and GC-MS procedures were performed as previously described (21, 22).
In vivo GOT lentiviral delivery in the mouse brain
To knockdown GOT in mouse brain, iLenti-GFP scramble siRNA, and iLenti-GFP GOT-1 siRNA and for overexpression of GOT-1 pLenti-GFP (pLV control) or pLenti-GOT-1-GFP (pLV GOT-1), lentiviral vector (8 μl of 1.0 × 108) (Applied Biologic Materials, Inc., Richmond, BC, Canada) was delivered to the S1 cortex of C57BL/6 male mice (8–10 wk old) using stereotaxic injection as previously described (3). This approach was effective in the knockdown and overexpression of GOT protein in the brain 7 d after lentiviral delivery (3). Stroke-affected S1 cortex was specifically targeted using the following coordinates: −0.1 mm bregma, +2.0 mm lateral, −1.0 mm dorsal. Subsequent experiments were performed 1 wk after delivery.
Determination of brain ATP during stroke
GOT-knockdown mice and scramble controls (n = 6) underwent MCAO under SO. Mice were killed after 30 min and while stroke-induced ischemia persisted. Stroke-affected (ipsilateral) and contralateral brain tissues were collected within 20 s of death and snap frozen in liquid nitrogen for downstream processing at the same time. Changes in brain ATP were measured using a bioluminescent assay (EnzyLight ATP Assay Kit; BioAssay Systems, Hayward, CA, USA) as previously described (3). The ATP content in each sample was normalized to the protein concentration that was determined using the bicinchoninic acid assay.
Sensorimotor assessment
At 24 h before MCAO (baseline) and 48 h after stroke, sensorimotor assessment was performed as described previously (10). In brief, mice were placed in the center of a 1- × 1-m open field and allowed to freely move for 5 min while being recorded overhead using AnyMaze video tracking software (Stoelting, v4.5; Kiel, WI, USA). The software calculated distance, mean speed, and time mobile for baseline and 48 h poststroke open-field tests.
RNA isolation and real-time PCR from brain tissue
Brain tissue frozen in optimal cutting temperature (OCT) compound (Sakura, Tokyo, Japan) was cut into 12-μm coronal sections and mounted onto polyethylene naphthalate–membrane slides for laser capture microdissection as described (3, 10). RNA was isolated from laser-captured contralateral control and stroke-affected S1 cortex (1 × 106 μm2) using a PicoPure RNA Isolation kit (Applied Biosystems, Foster City, CA, USA) (3). GOT mRNA expression was quantified by real-time PCR as described in Rink et al. (3).
Histology
OCT-embedded frozen brains of 48-h poststroke mice were sectioned (12 μm) and mounted on slides. Brain sections were stained with rabbit monoclonal antibody to GOT (1:100) (Abcam, Cambridge, MA, USA) and 0.0001% Fluoro-Jade C (Millipore, Temecula, CA, USA) (10, 23).
Statistical analyses
All data are reported as means ± sd. Before experimentation, mice were randomized to treatment groups and coded for blinded data analyses. Sample size for each group was estimated on the basis of experience with the MCAO model (3, 4, 10, 23, 24). The number of per group for each experiment is reported in the figure legends. Differences in means were tested using Student’s t test or 1-way ANOVA followed by Scheffé’s post hoc test, where P < 0.05 was considered statistically significant.
RESULTS
Extracellular Glu supports neuronal cell survival under hypoglycemic conditions and is metabolized by GOT
Cell viability was measured in response to extracellular Glu (10 mM) challenge in primary neurons cultured under normoglycemic (25 mM glucose), hypoglycemic (1 mM), and aglycemic (no glucose) conditions. As expected, after 24 h, extracellular Glu caused 90% lower cell viability under normoglycemic conditions (Fig. 1A, B). Neural cell viability was lower under hypoglycemic (72.2%) and aglycemic conditions (89.4%) as compared with normoglycemic conditions (no glutamate). Exposure of hypoglycemic neurons to extracellular Glu rescued cell viability. Cell viability was 6-fold higher under hypoglycemic conditions and 1.5-fold higher under aglycemic conditions when compared with normoglycemic conditions in the presence of extracellular glutamate. Importantly, Glu rescue of cell viability under hypoglycemic conditions was independent of cellular GSH levels (Supplemental Fig. 1). Under conditions of hypoxia, no cell viability was observed irrespective of glucose or glutamate conditions (Fig. 1A). High-resolution NMR spectroscopy was used to determine the metabolic fate of uniformly 13C-labeled extracellular glutamate ([U-13C]Glu) in primary neurons under normoglycemic, hypoglycemic, and aglycemic culture conditions (Fig. 1C, D). The generation of [U-13C]Asp and [1,2,3-13C]Glu indicated increased anaplerotic refilling of TCA cycle intermediates by GOT-mediated metabolism of U-13C Glu into α-ketoglutatarate (Fig. 1C) (20). After 2-h incubation, [U-13C]Glu uptake was highest in primary neurons cultured under hypoglycemic conditions (Fig. 1D). The detection of resynthesized glutamate ([1,2,3-13C]Glu) requires the presence of 12C atoms from acetyl Co-A (25, 26). Under hypoglycemic conditions, [1,2,3-13C]Glu levels were 70% higher as compared with normoglycemic conditions, whereas resynthesized glutamate was not detectable under aglycemic conditions (Fig. 1D). The loss of [1,2,3-13C]Glu resynthesis under aglycemic conditions is explained by insufficient acetyl Co-A substrate in the absence of glucose and glycolysis. Significantly higher levels of [U-13C]Asp were detected in primary neurons under hypoglycemic and aglycemic conditions as compared with normoglycemia (Fig. 1D). [U-13C]Asp levels under hypoglycemia and aglycemia were 6.6- and 24.8-fold higher than normoglycemia culture conditions. Next, primary neurons were cultured to test MRS outcomes with GOT knockdown. Because of MRS sensitivity in knockdown cells, 10 plates had to be pooled. Results are shown in Fig. 1E.
Figure 1.
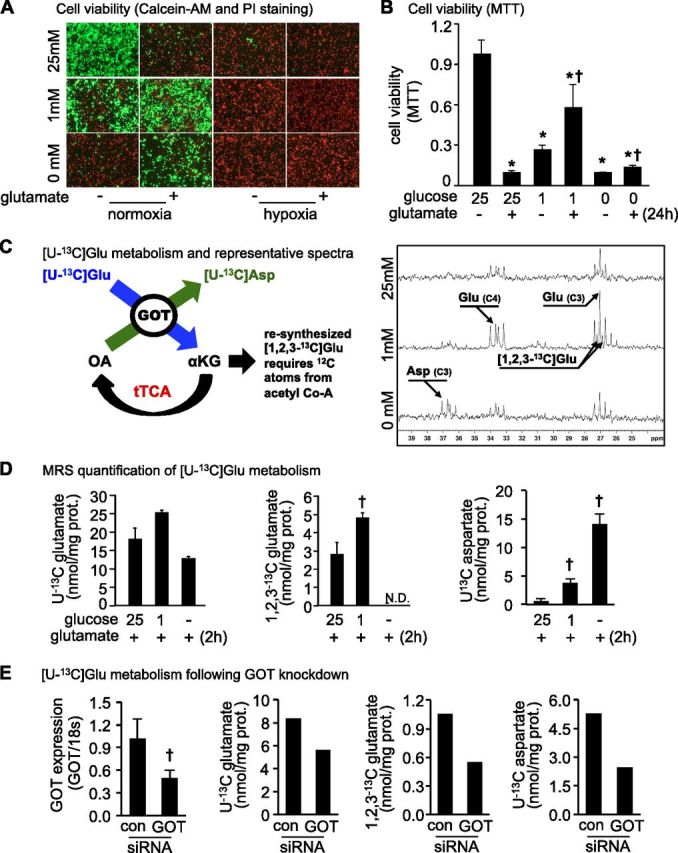
Extracellular Glu supports neuronal cell survival under hypoglycemic conditions by glutamate metabolism via a truncated TCA (tTCA) cycle. Under hypoglycemic conditions, glutamate (10 mM) protected against neural cell death. A, B) Cell viability was determined after 24 h of glutamate challenge by using Calcein-AM (green, live cells) and propidium iodide (red, dead cells) staining (A) or MTT assay (B). *P < 0.05 vs. 25 mM glucose/−glutamate. †P < 0.05 vs. 25 mM glucose/+glutamate. C) Glutamate metabolism was assessed by NMR spectroscopy. The presence of [U-13C]Asp marks metabolism of glutamate to α-ketoglutarate by GOT and anaplerotic refilling to enable a truncated TCA cycle. After 24 h of cell plating, medium was changed, and cells were incubated with [U-13C]glutamate in the presence or absence of glucose for 2 h. After 2 h, samples were collected for NMR analysis. Representative NMR spectra under normoglycemic (25 mM glucose), hypoglycemic (1 mM), and aglycemic (0 mM) conditions. D) [U-13C]Glu and [1,2,3-13C]Glu levels in neuronal cells were highest in hypoglycemic conditions (1 mM). [U-13C]Asp levels were significantly higher in neuronal cells under hypoglycemic and aglycemic conditions compared with normoglycemia. Data are means ± sd. N.D., not detected. †P < 0.05 vs. 25 mM glucose/+glutamate. E) Effect of GOT knockdown on [U-13C]Glu uptake, [1,2,3-13C]Glu, and [U-13C]Asp levels in neuronal cells under aglycemic conditions. GOT mRNA expression data (n = 3). GOT MRS outcomes are pooled from 10 plates per group due to the sensitivity of MRS in knockdown cells. Con, control. †P < 0.05.
GOT reduces cortical Glu and protects against stroke-induced loss of ATP
Glu levels were measured in mouse brain using 1H MRS obtained by chemical shift imaging. Acquisition of spectra from stroke-affected brain began 30 min after the onset of MCAO under RA, SO, or SO with GOT knockdown in a separate cohort (Fig. 2A, B). Here, SO (100% oxygen inhaled) was tested as a condition in which GOT activity is known to increase during ischemic stroke (3). After 60 min of MCAO, Glu levels in the stroke-affected (ipsilateral) cortex under SO conditions were significantly lower than the ipsilateral cortex under RA conditions (Fig. 2C, D). As hypothesized, the effect of SO in lowering Glu levels in the ipsilateral hemisphere were lost under GOT knockdown conditions (Fig. 2C, D). To test the significance of GOT on brain ATP levels after MCAO, we used a lentiviral knockdown approach (3). In mice receiving scramble siRNA (control), SO protected against the loss of brain ATP in the stroke-affected ipsilateral hemisphere (Fig. 2E), as reported previously in naive rats (3). This protection was lost in mice with lentiviral GOT knockdown (Fig. 2E).
Figure 2.

GOT enables reduction of cortical Glu in stroke-affected brain. A) Experimental protocol. B) 1H MRS chemical shift images were acquired 30 min after the onset of MCAO under RA, SO, or SO + GOT-knockdown conditions. Spectra were acquired from contralateral (c) and stroke-affected ispsilateral (i) regions in the S1 cortex. C) Representative RA (red), SO (blue), and SO + GOT-knockdown (black) spectra from voxel in ipsilateral cortex. Zoomed inset depicts Glu peaks under each condition. D) Percentage Glu decrease in ipsilateral cortex compared with contralateral cortex under RA (red bar), SO (blue bar), and SO + GOT knockdown (black bar) conditions. Data are means ± sd (n = 6). *P < 0.05 vs. RA. †P < 0.05 vs. SO + GOT knockdown. E) Percentage change in brain ATP (ipsilateral vs. contralateral) from GOT-knockdown and scramble control mice under SO conditions after 30 min of MCAO. Data are means ± sd (n = 6). *P < 0.05 vs. contralateral (con) hemisphere within group. †P < 0.05 vs. ipsilateral (ipsi) scramble.
Oxygen- and GOT-sensitive changes in stroke-affected brain anaplerosis
Metabolism of glutamate by GOT can enable anaplerotic refilling of TCA cycle intermediates that are otherwise depleted in the absence of glucose delivery to neurons during ischemic stroke. To determine oxygen and GOT-dependent changes in anaplerosis, we quantified [U-13C] propionate metabolism into propionyl CoA and TCA cycle intermediate succinyl CoA in stroke-affected brain (Fig. 3A–C) (21). To test the significance of GOT on anaplerosis in stroke-affected brain, we used SO (100% O2 inhaled) during ischemia, which is known to increase GOT activity (3), and targeted lentiviral overexpression of GOT in S1 cortex (3). Delivery of pLV control lentivirus did not affect anaplerosis in stroke-affected brain tissue under RA conditions (Fig. 3D). When GOT was overexpressed under RA conditions, anaplerosis was significantly higher (57% increase) in the stroke-affected brain as compared with RA controls. Likewise, under SO in which GOT activity increased, anaplerosis in stroke-affected brain tissue was 50% higher as compared with RA conditions. Whereas GOT overexpression combined with SO therapy did not increase anaplerosis beyond what was already enabled by targeted overexpression of GOT under RA conditions or SO therapy alone, SO and GOT combined increased anaplerosis by 89% as compared with RA controls (Fig. 3D).
Figure 3.
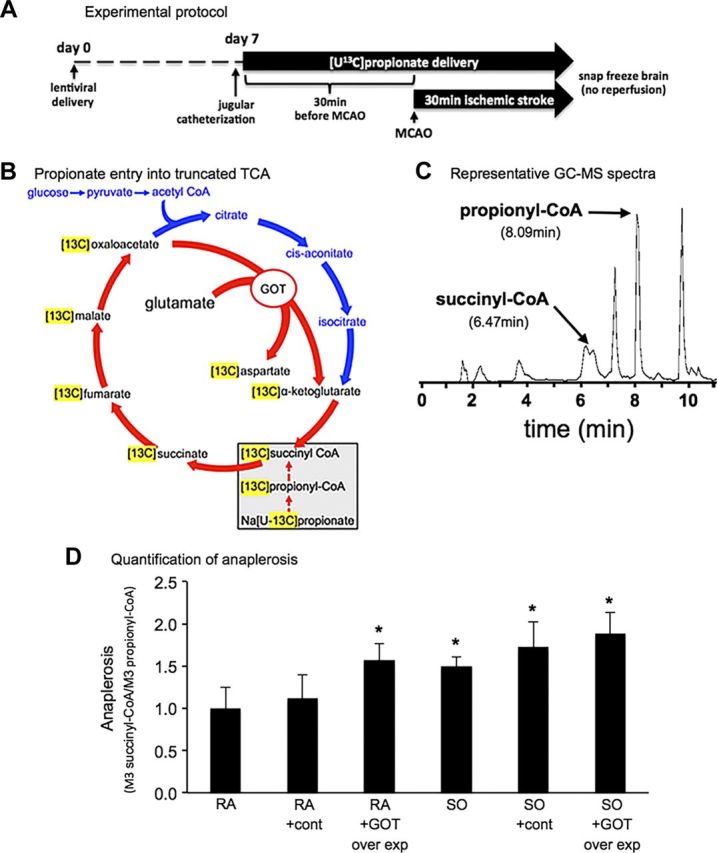
Oxygen- and GOT-sensitive changes in anaplerosis. A) Experimental protocol. B) [U-13C] propionate enters the TCA cycle at the level of succinyl CoA. C) Representative GC-MS spectra of control (cont) under RA. D) Quantification of anaplerosis under all conditions. Data are mean ± sd fold change compared with RA (n/group: RA = 4, RA pLV con = 3, RA pLV GOT = 7, SO = 8, SO pLV con = 4, SO pLV GOT = 4). *P < 0.05 vs. RA.
Functional significance of GOT against stroke in vivo
Because targeted lentiviral overexpression of GOT was as effective as SO in increasing anaplerosis in stroke-affected brain, we sought to test the pathophysiological significance of GOT overexpression as it relates to stroke outcomes. Stereotaxic delivery of lentiviral particles was performed 7 d before stroke surgery to allow sufficient time for GOT overexpression (Fig. 4A). The gene delivery was successful in elevating GOT mRNA (Fig. 4B) and GOT protein expression (Fig. 4C, D). Stroke lesion volume quantified from T2-weighted MRI revealed that the delivery of GOT decreased the stroke-induced brain lesion by 46% (Fig. 5A, B). This observation was consistent with our previous study (3). Additional experiments conducted here examined the effects of GOT overexpression on poststroke neurodegeneration and functional outcomes. Stroke-induced glutamate release contributes to neurodegeneration and cell death (7). Immunohistochemical localization of neurodegeneration using Fluoro-Jade staining demonstrated that GOT gene delivery significantly attenuated stroke-induced neurodegeneration as compared with controls (Fig. 5C, D). Furthermore, sensorimotor function as quantified in an open-field test was markedly improved when GOT was overexpressed (Fig. 6), where mice moved further, faster, and for a longer duration as compared with controls.
Figure 4.

A) Experimental protocol. B) GOT mRNA expression in contralateral (contra) and stroke-affected S1 cortex laser captured from mice receiving pLV scramble (control) or GOT-overexpressing lentiviral vector (n = 5). C, D) Representative micrographs of GOT immunostaining (C) and quantification of GOT immunostaining (D) in stroke-affected S1 cortex from control and GOT-overexpressing mice (n = 5). *P < 0.05 vs. contra control. †P < 0.05 vs. contra GOT-overexpressing. #P < 0.05 vs. stroke control.
Figure 5.

GOT overexpression attenuates stroke-induced lesion volume and neurodegeneration. A) Representative T2-weighted coronal MRI images at 48 h after MCAO in mice transfected with pLV scramble control or GOT-overexpressing lentiviral vector. B) Percentage hemisphere lesion volume calculated from T2-weighted MRI at 48 h after MCAO. *P < 0.05. C) Micrographs depicting Fluoro-Jade staining of neurodegeneration in contralateral control and stroke-affected S1 cortex of control and GOT-overexpressing mice (green, Fluoro-Jade; blue, DAPI-counterstained nuclei). Scale bars, 50 μm. D) Floro-Jade quantification. Data are means ± sd (n = 5). N.D., not detected. *P < 0.05 vs. contralateral. †P < 0.05 vs. pLV control.
Figure 6.

GOT overexpression improves poststroke sensorimotor function. A, B) Representative heat maps (A) and movement maps (B) of mice in open-field test at baseline and 48 h after MCAO in control and GOT-overexpressing mice. C) Distance (m), speed (m/s), and time mobile (s) were calculated by AnyMaze software for control and GOT-overexpressing mice at baseline (BL) and 48 h after MCAO. Data are means ± sd (n = 5). *P < 0.05 vs. BL within group. †P < 0.05 vs. 48 h control.
DISCUSSION
Acute ischemic stroke limits the supply of key blood-borne survival factors to the affected areas of the brain. Hypoxia and hypoglycemia represent two such major insults that affect the stroke site. Our prior efforts (3, 4) to understand the specific significance of the hypoxic component of ischemic insult during stroke uncovered a set of novel mechanisms that shed light on how to minimize injury to the brain site challenged by hypoglycemia and flooded with glutamate as a consequence of focal brain ischemia. We previously reported that SO protects brain from ischemic stroke injury as measured by T2-weighted MRI acquired 48 h after MCAO (4). Although SO intervention addressed stroke-induced hypoxia, it did not account for the threat of stroke-induced hypoglycemia. Indeed, hypoglycemia is known to contribute to glutamate release and neurotoxicity (27–29). In the context of ischemic stroke, elevated extracellular glutamate is neurotoxic by mechanisms including excitotoxicity (30) and oxytosis (31). In previously published work, we demonstrated that SO during cerebral ischemia induces expression of the glutamate-metabolizing enzyme GOT (3). This work led to new hypotheses tested here that GOT could mitigate neurotoxicity by metabolism of glutamate. The two most significant findings in the current work are that GOT enables metabolism of otherwise neurotoxic extracellular Glu through a truncated TCA cycle under hypoglycemic conditions, and that GOT is required for anaplerotic refilling of TCA cycle intermediates in the stroke-affected brain. Importantly, GOT overexpression was observed not only to attenuate stroke lesion volume but also to decrease neurodegeneration and to improve functional outcomes after cerebral ischemia.
In the absence of acetyl co-A from the glycolytic metabolism of glucose, and without glycogen stores, neurons challenged by stroke-induced hypoglycemia must turn to alternative energy substrates to sustain their high metabolic demands. Glutamate is positioned to serve as an ideal metabolic substitute given its sequestration in neurons and the need to mitigate neurotoxic accumulation under pathophysiological conditions such as ischemic stroke. As a transaminase, GOT catalyzes the transfer of the amino group from glutamate to the 4-carbon TCA cycle intermediate oxaloacetate to generate aspartate and the 5-carbon TCA cycle intermediate α-ketoglutarate (2). This ability enables the anaplerotic flux of GOT-metabolized glutamate into a truncated TCA cycle. In the current work, we observed that the same amount of extracellular glutamate that induces neuronal cell death in vitro (11, 12, 32–35) loses its toxic potency as the concentration of glucose in medium decreases. Furthermore, hypoglycemic neural cells treated with U-13C glutamate generated more 13C-aspartate, suggesting increased GOT-mediated metabolism of glutamate through a truncated TCA cycle to sustain cell viability. In vivo, glutamate levels were significantly lower in the stroke-affected hemisphere under SO and when GOT was overexpressed. Even under normoxic (RA) conditions, glutamate levels appeared lower in the stroke-affected hemisphere as compared with the contralateral control. This likely represents the basal physiologic response to stroke injury because GOT protein is constitutively expressed under resting conditions in brain tissue (3).
Taken together, the findings of this study pave the way for a new paradigm suggesting that the functional significance of glutamate at the stroke site is switched by GOT from being a neurotoxic inducer of cell death to metabolic fuel that sustains neural tissue in the face of stroke-induced hypoglycemia. Importantly, this paradigm recognizes the therapeutic potential to target GOT for improved neurotoxic glutamate clearance and anaplerotic flux of TCA cycle intermediates in the hypoglycemic, stroke-affected brain.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
This research was supported by U.S. National Institutes of Health, National Institute of Neurological Disorders and Stroke Grant R01NS085272 (to C.R. and S.K.), and by American Heart Association Grants 12SDG11780023 (to C.R.) and NS42617 (to C.K.S.). The authors thank Jessica Weist (The Ohio State University Wexner Medical Center) for technical support with in vivo experiments.
Glossary
- GOT
glutamate oxaloacetate transaminase
- GSH
glutathione
- MCAO
middle cerebral artery occlusion
- MRS
magnetic resonance spectroscopy
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- pLV
plasmid lentivirus
- RA
room air
- SO
supplemental oxygen
- TCA
tricarboxylic acid
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
C. Rink, C. K. Sen, and S. Khanna conceived and designed the studies; C. Rink, S. Gnyawali, R. Stewart, S. Teplitsky, H. Harris, S. Roy, and S. Khanna acquired, analyzed, and interpreted data; C. Rink, C. K. Sen, and S. Khanna drafted the manuscript; and C. Rink, S. Gnyawali, S. Roy, C. K. Sen, and S. Khanna provided critical revision of the manuscript for important intellectual content.
REFERENCES
- 1.Hofmeijer J., van Putten M. J. (2012) Ischemic cerebral damage: an appraisal of synaptic failure. Stroke , 607–615 [DOI] [PubMed] [Google Scholar]
- 2.Khanna S., Briggs Z., Rink C. (2015) Inducible glutamate oxaloacetate transaminase as a therapeutic target against ischemic stroke. Antioxid. Redox Signal. , 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rink C., Gnyawali S., Peterson L., Khanna S. (2011) Oxygen-inducible glutamate oxaloacetate transaminase as protective switch transforming neurotoxic glutamate to metabolic fuel during acute ischemic stroke. Antioxid. Redox Signal. , 1777–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rink C., Roy S., Khan M., Ananth P., Kuppusamy P., Sen C. K., Khanna S. (2010) Oxygen-sensitive outcomes and gene expression in acute ischemic stroke. J. Cereb. Blood Flow Metab. , 1275–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krebs H. A. (1935) Metabolism of amino-acids: deamination of amino-acids. Biochem. J. , 1620–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayashi T. (1954) Effects of sodium glutamate on the nervous system. Keio J. Med. , 192–193 [Google Scholar]
- 7.Lai T. W., Zhang S., Wang Y. T. (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog. Neurobiol. , 157–188 [DOI] [PubMed] [Google Scholar]
- 8.Pérez-Mato M., Ramos-Cabrer P., Sobrino T., Blanco M., Ruban A., Mirelman D., Menendez P., Castillo J., Campos F. (2014) Human recombinant glutamate oxaloacetate transaminase 1 (GOT1) supplemented with oxaloacetate induces a protective effect after cerebral ischemia. Cell Death Dis. , e992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campos F., Sobrino T., Ramos-Cabrer P., Argibay B., Agulla J., Pérez-Mato M., Rodríguez-González R., Brea D., Castillo J. (2011) Neuroprotection by glutamate oxaloacetate transaminase in ischemic stroke: an experimental study. J. Cereb. Blood Flow Metab. , 1378–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khanna S., Rink C., Ghoorkhanian R., Gnyawali S., Heigel M., Wijesinghe D. S., Chalfant C. E., Chan Y. C., Banerjee J., Huang Y., Roy S., Sen C. K. (2013) Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. J. Cereb. Blood Flow Metab. , 1197–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khanna S., Roy S., Park H. A., Sen C. K. (2007) Regulation of c-Src activity in glutamate-induced neurodegeneration. J. Biol. Chem. , 23482–23490 [DOI] [PubMed] [Google Scholar]
- 12.Khanna S., Roy S., Ryu H., Bahadduri P., Swaan P. W., Ratan R. R., Sen C. K. (2003) Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J. Biol. Chem. , 43508–43515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy T. H., Schnaar R. L., Coyle J. T. (1990) Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. , 1624–1633 [PubMed] [Google Scholar]
- 14.Park H. A., Kubicki N., Gnyawali S., Chan Y. C., Roy S., Khanna S., Sen C. K. (2011) Natural vitamin E α-tocotrienol protects against ischemic stroke by induction of multidrug resistance-associated protein 1. Stroke , 2308–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khanna S., Venojarvi M., Roy S., Sen C. K. (2002) Glutamate-induced c-Src activation in neuronal cells. Methods Enzymol. , 191–198 [DOI] [PubMed] [Google Scholar]
- 16.Park H. A., Khanna S., Rink C., Gnyawali S., Roy S., Sen C. K. (2009) Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ. , 1167–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherwood T. W., Lee K. G., Gormley M. G., Askwith C. C. (2011) Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J. Neurosci. , 9723–9734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jamaluddin M. S., Wang X., Wang H., Rafael C., Yao Q., Chen C. (2009) Eotaxin increases monolayer permeability of human coronary artery endothelial cells. Arterioscler. Thromb. Vasc. Biol. , 2146–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang Y., Scheef E. A., Wang S., Sorenson C. M., Marcus C. B., Jefcoate C. R., Sheibani N. (2009) CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood , 744–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakken I. J., White L. R., Unsgård G., Aasly J., Sonnewald U. (1998) [U-13C]glutamate metabolism in astrocytes during hypoglycemia and hypoxia. J. Neurosci. Res. , 636–645 [DOI] [PubMed] [Google Scholar]
- 21.Puchowicz M. A., Zechel J. L., Valerio J., Emancipator D. S., Xu K., Pundik S., LaManna J. C., Lust W. D. (2008) Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J. Cereb. Blood Flow Metab. , 1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L., Kasumov T., Yu L., Jobbins K. A., David F., Previs S. F., Kelleher J. K., Brunengraber H. (2006) Metabolomic assays of the concentration and mass isotopomer distribution of gluconeogenic and citric acid cycle intermediates. Metabolomics , 85–94 [Google Scholar]
- 23.Khanna S., Roy S., Slivka A., Craft T. K., Chaki S., Rink C., Notestine M. A., DeVries A. C., Parinandi N. L., Sen C. K. (2005) Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke , 2258–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khanna S., Heigel M., Weist J., Gnyawali S., Teplitsky S., Roy S., Sen C. K., Rink C. (2015) Excessive α-tocopherol exacerbates microglial activation and brain injury caused by acute ischemic stroke. FASEB J. , 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonnewald U., White L. R., Odegård E., Westergaard N., Bakken I. J., Aasly J., Unsgård G., Schousboe A. (1996) MRS study of glutamate metabolism in cultured neurons/glia. Neurochem. Res. , 987–993 [DOI] [PubMed] [Google Scholar]
- 26.Yang L., Kasumov T., Kombu R. S., Zhu S. H., Cendrowski A. V., David F., Anderson V. E., Kelleher J. K., Brunengraber H. (2008) Metabolomic and mass isotopomer analysis of liver gluconeogenesis and citric acid cycle: II. Heterogeneity of metabolite labeling pattern. J. Biol. Chem. , 21988–21996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butcher S. P., Jacobson I., Sandberg M., Hagberg H., Hamberger A. (1987) 2-Amino-5-phosphonovalerate attenuates the severe hypoglycemia-induced loss of perforant path-evoked field potentials in the rat hippocampus. Neurosci. Lett. , 296–300 [DOI] [PubMed] [Google Scholar]
- 28.Engelsen B., Westerberg E., Fonnum F., Wieloch T. (1986) Effect of insulin-induced hypoglycemia on the concentrations of glutamate and related amino acids and energy metabolites in the intact and decorticated rat neostriatum. J. Neurochem. , 1634–1641 [DOI] [PubMed] [Google Scholar]
- 29.Wieloch T. (1985) Hypoglycemia-induced neuronal damage prevented by an N-methyl-D-aspartate antagonist. Science , 681–683 [DOI] [PubMed] [Google Scholar]
- 30.Olney J. W. (1986) Inciting excitotoxic cytocide among central neurons. Adv. Exp. Med. Biol. , 631–645 [DOI] [PubMed] [Google Scholar]
- 31.Tan S., Schubert D., Maher P. (2001) Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. , 497–506 [DOI] [PubMed] [Google Scholar]
- 32.Fukui M., Song J. H., Choi J., Choi H. J., Zhu B. T. (2009) Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. , 1–11 [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto M., Murphy T. H., Schnaar R. L., Coyle J. T. (1989) Antioxidants protect against glutamate-induced cytotoxicity in a neuronal cell line. J. Pharmacol. Exp. Ther. , 1132–1140 [PubMed] [Google Scholar]
- 34.Murphy T. H., Miyamoto M., Sastre A., Schnaar R. L., Coyle J. T. (1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron , 1547–1558 [DOI] [PubMed] [Google Scholar]
- 35.Zhu M. Y., Piletz J. E., Halaris A., Regunathan S. (2003) Effect of agmatine against cell death induced by NMDA and glutamate in neurons and PC12 cells. Cell. Mol. Neurobiol. , 865–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.