

Introduction
Pain is deemed chronic if it lasts more than 12 weeks and can sometimes last a lifetime. While some clinical pain syndromes are traced back to specific causes such as traumatic or metabolic nerve injuries the majority of cases have no clear causative event/injury [11; 106]. Pain can even be perceived to arise from an amputated, and hence an absent, limb [93; 121]. Genetic and functional findings have linked voltage-gated sodium channels that are expressed in peripheral sensory neurons to human pain disorders, and support targeting these channels for development of new analgesics.
Nine genes (SCN1A-SCN5A and SCN8A-SCN11A) encode diverse pore-forming sodium channel α-subunits (Nav1.1-Nav1.9) which manifest distinct expression patterns and biophysical and pharmacological properties [24]. Sodium channels are composed of 1700–2000 amino acids which fold into four domains (DI-DIV) with each domain consisting of six transmembrane segments, linked by three intracellular loops, and cytoplasmic N- and C-termini [23]. Adult peripheral sensory neurons can express channels Nav1.1, Nav1.6 and Nav1.7, which are blocked by nanomolar concentrations of the neurotoxin tetrodotoxin (TTX-S), and channels Nav1.8 and Nav1.9, which are resistant to micromolar concentrations of TTX (TTX-R). The TTX-S channel Nav1.3 is predominantly expressed in embryonic sensory neurons, [116] but is up-regulated following traumatic or metabolic nerve injury in rodent DRG neurons [35; 116]. Functional studies of these channels within neurons in which they are normally expressed [38] as well as studies in knock-out mice have yielded important information about the contribution of individual sodium channels to electrogenesis within these neurons [42; 47; 97](Figure 1).
Figure 1. Contribution of sodium channel isoforms to action potential.
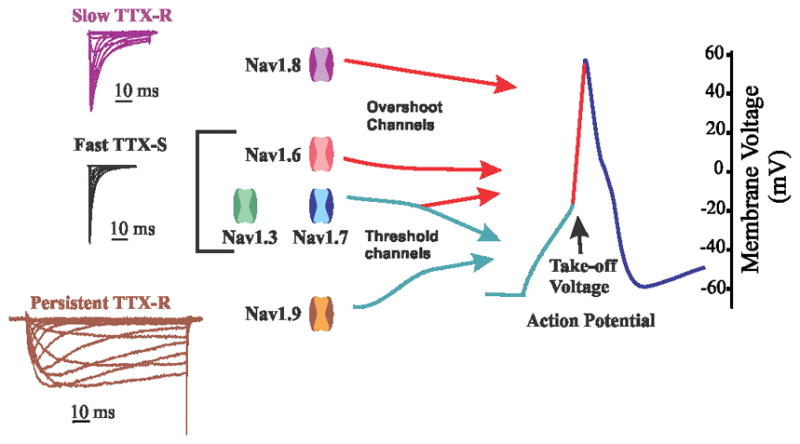
Based on their ability to boost subthreshold stimuli -- TTX-S channels Nav1.3 and Nav1.7 -- or hyperpolarized and persistent current -- TTX-R channel Nav1.9 -- these channels have been considered threshold channels for action potential firing. TTX-S channels Nav1.3, Nav1.6 (especially at nodes of Ranvier), and TTX-R channel Nav1.8 (in the neurons in which it is expressed), contribute most of the current for the action potential. Adapted with permission from [42]
Three of the channels that are expressed in sensory neurons — Nav1.7, Nav1.8 and Nav1.9 — are preferentially expressed in peripheral neurons, whereas Nav1.1 and Nav1.6 are also widely expressed in the CNS. Nav1.7 and Nav1.8 channels have garnered intense interest because of the accumulating evidence for their role in human pain disorders and relative ease of studying these channels in isolation in heterologous expression systems or in native neurons [43; 57]. Evidence for mutations in Nav1.9 [60; 61; 64; 72; 73; 87; 118; 127] and Nav1.6 [108] in human pain conditions has started to appear in the past few years. Recent comprehensive reviews have covered this topic in detail [15; 36; 37; 40; 43; 57]. We provide in this article an overview of the more recent developments that further our understanding of the contribution of peripheral sodium channels to a spectrum of human pain disorders, and the clinical testing of both new and existing sodium channel blockers for treatment of patients carrying mutations in Nav1.7.
Nav1.6 in trigeminal neuralgia
Nav1.6 is the major sodium channel at axon initial segments (AIS) and mature nodes of Ranvier in myelinated fibers of the central and peripheral nervous systems [21; 100], and contributes to the C-wave of compound action potential of the sciatic nerve, consistent with its expression in small DRG neurons [17]. Nav1.6 produces a TTX-S current characterized by fast activation, inactivation and repriming, and produces persistent current –incomplete inactivation of the channel– and resurgent current – channel reopening upon hyperpolarization following strong depolarization – in DRG neurons [32; 98]. These properties poise Nav1.6 to contribute to neuronal repetitive firing.
A recent study reported a Nav1.6 mutation in a patient with trigeminal neuralgia [108], adding to evidence from animal studies [34; 105; 122; 123] supporting a role for this channel in pain. Trigeminal neuralgia (TN) is characterized by paroxysmal facial neuropathic pain along one or more branches of the trigeminal nerve, often triggered by innocuous stimuli, such as light touch, eating, shaving, or applying makeup [30]. Although neurovascular compression of the trigeminal nerve is prevalent in patients with TN, symptoms can occur in the absence of nerve compression or only in a subpopulation of patients with nerve compression [5; 68; 69; 77], which suggests the contribution of other factors in disease manifestation. The sodium channel blockers carbamazepine (CBZ) and oxcarbazepine are the first-line treatment for classical TN [30], which supports involvement of sodium channels in the pathophysiology of TN. We have recently reported the first mutation located in transmembrane segment 1 of domain I in Nav1.6 in a patient with trigeminal neuralgia and no other comorbidity The Met136Val mutation in Nav1.6, which was not previously reported in any of the sequence databases, confers gain-of-function attributes on the channel – increased transient and resurgent current density – and enhances excitability of trigeminal neurons –reduced threshold for action potential and increased repetitive firing– that express this mutant channel [108]. Although this mutation by itself is not enough to cause TN, it may act to predispose the carrier to the manifestation of TN symptoms. This study provides the first link of voltage-gated sodium channel NaV1.6 to a human pain disorder.
Nav1.8 in human peripheral painful neuropathy
Nav1.8 is preferentially expressed in peripheral sensory neurons, and is present in >90% of small DRG neurons and in about 40% of cutaneous afferents [103]. Nav1.8 is characterized by depolarized voltage dependence of activation and inactivation, compared to other sodium channels, and a slow rate of inactivation and rapid recovery from inactivation [2; 3], and a slow resurgent current [107]. Nav1.8 contributes most the current underlying the upstroke of the action potential in small DRG neurons [18; 96]. As a result of these properties, Nav1.8 supports repetitive firing in response to sustained depolarization. Recently, we have shown species-specific differences in Nav1.8 (Figure 2), for example a larger persistent current is produced by human Nav1.8, compared to rodent Nav1.8 channels, which is correlated by wider action potentials in both human DRG neurons or when the human Nav1.8 channel is expressed in rodent DRG neurons [57]. The presence or absence of Nav1.8 in a particular cell is a major contributor to the cell background effect, which is manifested when the same mutations that cause depolarization of resting membrane potential will lead to hypoexcitabile neurons if Nav1.8 is absent and hyperexcitable neurons if Nav1.8 is present [58; 97].
Figure 2. Species-specific differences in Nav1.8 and action potential properties.

(A, B) Representative Nav1.8 current family traces recorded from rat (A) or human DRG neuron (B). Human DRG neurons produce a Nav1.8 current with slower kinetics of inactivation, and larger persistent current compared to the Nav1.8 current in rat DRG neurons. (C, D) Representative action potential traces recorded from rat (C) or human (D) DRG neuron. Human DRG neurons produce broader action potentials than rat neurons. (E) Comparison of the half-width of action potential between rat and human DRG neurons. The mean ± standard error of the half-width is represented by the two larger symbols. Adapted with permission from [57].
De novo gain-of-function mutations of Nav1.8 that produce DRG neuron hyperexcitability have been found in 4% of a group of 393 consecutive patients diagnosed with small fiber neuropathy (SFN) at one medical center, including familial episodic pain syndrome (FEPS2, OMIM#61551), establishing a role for this channel in pathogenesis [19; 59], and a single nucleotide polymorphism biases experimental pain in healthy human subjects [46]. The gain-of-function attributes that these mutations confer on Nav1.8 include hyperpolarized voltage-dependence of activation, accelerated recovery from inactivation, enhanced ramp current and impaired inactivation. Nav1.8 selective blockers have been developed and showed efficacy in animal models [65; 88], providing proof-of-principle that it is possible to target Nav1.8 in vivo. The new links of Nav1.8 in human pain disorders and success in pre-clinical studies using Nav1.8 selective blockers suggest new therapeutic strategies when clinically-relevant blockers become available.
Nav1.9 in human painful and painless disorders
Nav1.9 is preferentially expressed in small-diameter (<30 μm diameter) DRG neurons, trigeminal ganglion neurons, including functionally-identified nociceptors, and intrinsic myenteric neurons [37]. Nav1.9 channels produce a TTX-R current which characteristically activates at hyperpolarized voltages, compared to other neuronal sodium channels, and inactivates with unusually ‘ultra-slow’ kinetics causing the persistence of the sodium current after activation [31] [41]. The large overlap between activation and inactivation of Nav1.9 results in a big window current within the physiological voltage domain close to resting membrane potential of DRG neurons (-70 mV to -40 mV), permitting these channels to depolarize the membrane potential to reach threshold for firing the all-or-none action potential. These features have led to the designation of Nav1.9 as a threshold channel. Although studies on Nav1.9 have been hampered by poor expression in heterologous expression systems, recently, expression of Nav1.9 in the DRG-derived cell line ND7/23 [111] and HEK293 cells [75] have been reported which may accelerate studies of this channel and development of isoform-selective blockers.
A spectrum of human pain disorders has been linked to dominant gain-of-function mutations in Nav1.9, including painful (FEPS3, OMIM#615552) and painless channelopathies (HSAN VII, OMIM#615548) [37]. Mutations in Nav1.9 have been identified in several families with pain in distal extremities [61; 87; 127] and in one family with cold-aggravated pain [72]. Sporadic mutations of Nav1.9 have been identified in patients with painful small fiber neuropathy [60; 64]. Unexpectedly a gain-of-function mutation in Nav1.9 has been reported in patients with insensitivity to pain [73; 118], and another mutation was identified in a family with early-onset insensitivity to pain associated with chronic diarrhea, but functional assessment of this mutation has not been reported thus far [90]. The gain-of-function attributes that these mutations confer on Nav1.9 include massive hyperpolarizing shift in activation, increased amplitude of ramp current and slower deactivation. The expression of Nav1.9 in nociceptors and myenteric neurons is consistent with the symptoms of distal extremity pain and gastrointestinal disturbances reported by patients carrying mutations in this channel [61; 64; 72; 87; 127]. However, a mechanistic basis for the loss of pain sensation in patients with gain-of-function mutations in Nav1.9 remains to be determined [73; 118].
Nav1.7 in human pain disorders
Arguably, we know more about the contribution of Nav1.7 to pain in humans than any other sodium channel. Nav1.7 is preferentially expressed in peripheral somatic and visceral sensory neurons – including the vast majority of functionally-identified nociceptors – olfactory sensory neurons, and sympathetic ganglion neurons [43]. Nav1.7 accumulates both at nerve fiber endings in the periphery and presynaptic terminals in the dorsal horn of the spinal cord [43]. Nav1.7 produces a TTX-S current that rapidly activates and inactivates but slowly recovers from inactivation (reprimes) [67], and is also characterized by slow closed-state inactivation, allowing the channel to produce a substantial current in response to small, slow “ramp” depolarizations [33; 63]. This ability of Nav1.7 to boost subthreshold stimuli increases the probability of neurons reaching their threshold for action potential firing [42; 47; 97]. The implementation of dynamic-clamp recordings in native rodent DRG neurons using physiologically-relevant levels of Nav1.7 currents with native biophysical properties, showed a linear correlation between the level of Nav1.7 conductance and current threshold in these cells [112]. Pharmacological block of Nav1.7 increases threshold and reduces amplitude of action potential in DRG neurons and reduces neurotransmitter release from both peripheral and central terminals of primary afferents [4]. Thus, both anatomical and functional evidence supports a role for Nav1.7 as a threshold channel.
Many mutations in Nav1.7 have been found in patients with heritable pain disorders that follow a dominant Mendelian inheritance pattern and confer gain-of-function attributes on the channel, supporting the conclusion that these mutations are pathogenic [117] and confirming a definitive role for Nav1.7 in human pain signaling. Dominantly inherited gain-of-function missense mutations in SCN9A, the gene encoding Nav1.7, are found in patients with inherited erythromelalgia (IEM, also known as primary EM, OMIM #133020)[125] and paroxysmal extreme pain disorder (PEPD, previously known as familial rectal pain, OMIM #167400)[49]. By contrast, recessively inherited loss-of-function mutations in SCN9A are linked to congenital insensitivity (indifference) to pain (CIP, OMIM #243000) characterized by sensory loss limited to pain sensation and anosmia, and without autonomic deficits [29], and hereditary sensory and autonomic neuropathy IID (HSANIID, OMIM#243000) which is characterized by adolescent or congenital onset with loss of pain and temperature sensation, autonomic nervous dysfunctions, hearing loss, and hyposmia [126]. These data provided the biological basis to investigate the contribution of this channel to more common pain disorders and led to the identification of gain-of-function variants of Nav1.7 in roughly 30% of patients with idiopathic small fiber neuropathy and biopsy-confirmed loss of intra-epidermal nerve fibers [48]. These studies have solidified the status of Nav1.7 as a major contributor to human pain disorders.
Although patients with IEM and PEPD present with distinct clinical symptoms, they both carry gain-of-function mutations in Nav1.7. Patients with PEPD report severe perirectal pain starting typically in infancy and is normally triggered by bowel movement or probing of the rectal or perineal areas, which is accompanied by tonic posturing and immediately followed by flushing in a uni- or bi-lateral fashion [50], and sometimes in a harlequin pattern which can alternate between the left and right sides of the body during different pain episodes [26]. Ocular and mandibular pain, sometimes triggered by cold or irritants, becomes the more prominent complaints in older patients. Patients with IEM start to exhibit symptoms as early as 1 year of age and as late as late-teens, and episodes of burning pain are triggered by mild warmth or exercise together with erythema and mild swelling in the hands and feet, with partial relief of symptoms by cooling affected extremities [45]. Despite the apparent uniformity of these symptoms, one recent study that looked in more details into the natural history of IEM in a cohort of 13 patients from four families demonstrated substantial variability in symptom presentation and triggers among members of the same family, including, for example, patients who reported that cooling evokes pain rather than relieves pain [82], while another study on two patients (parent/child) showed large variability in the severity of ongoing pain and in the number of nightly awakening due to pain [55]. The distinct patterns of affected body regions in IEM and PEPD, and the substantial variability of pain symptoms among members of the same family with the same mutation in IEM, suggest additional factors that regulate the severity of the symptoms and which parts of the body are affected.
Functional characterization of the mutations in Nav1.7 has shed light on the pathophysiological basis for nociceptor excitability in these disorders, establishing a mechanistic link to pain. At the channel level, these mutations confer multiple gain-of-function attributes on mutant channels. These include hyperpolarizing shift in activation, increased amplitude of ramp current and slower deactivation, impaired inactivation and increased persistent current, and enhancement of resurgent current. At the cellular levels, expression of these mutant channels in sensory neurons lead to increased excitability of DRG neurons [43], which reflects a cellular correlate of pain that these patients experience.
Studies of mutant Nav1.7 channels have also unmasked the important observation that the neuronal background governs the cellular response to the expression of a mutant Nav1.7 channel. Experimental evidence shows that the same mutation can induce hyperexcitability of sensory neurons while causing hypoexcitability of sympathetic neurons, and that this dichotomy is caused by the presence of Nav1.8 in sensory neurons and its absence in sympathetic neurons [58; 97]. This divergent effect of the same mutation in different neuronal backgrounds has been linked to the mutation-induced depolarization of the resting potential of the neurons. The depolarization of resting membrane potential of sensory neurons facilitates the activation of the Nav1.8 channels that have a depolarized voltage-dependence of activation and inactivation compared to the other sodium channels and can support firing action potentials. The depolarization of resting membrane potential of sympathetic neurons leads to resting inactivation of the sodium channels leading to reduced probability of firing an action potential. While hyperexcitability of sensory neurons is intuitively linked to pain symptoms, hypoexcitability of sympathetic neurons may lead to reduced sympathetic vasoconstriction tone, contributing to skin flushing that is observed in these patients.
The majority of the functional studies discussed above have been carried out in an overexpression system in which the wild-type or mutant channels are expressed at supraphysiological levels in rodent DRG neurons; however, additional lines of evidence from assays under more physiological settings produced results that are in agreement with these data. Implementation of dynamic-clamp recordings in which physiologically-relevant levels of a wild-type or mutant Nav1.7 channel could be injected into DRG neurons using a computer-controlled amplifier, showed that the Nav1.7 Leu858His mutant current caused a 27-fold amplification of net sodium influx during subthreshold depolarizations and even greater amplification during interspike intervals [112]. These findings contribute to the mechanistic basis for reduced current threshold and enhanced action potential firing probability in sensory neurons. Another independent line of evidence is provided by studies using sensory neurons that are differentiated from induced pleuripotent stem cells from patients with IEM. These patient-derived neurons have a lower threshold and increased firing frequency [22]. Taken together, the genetic and functional studies show that Nav1.7 channels play as a major role in pain signaling in humans.
There are no published animal models for a gain-of-function mutation in Nav1.7, thus it has not been possible to assess the effect of mutant channels in vivo. By contrast, global, or conditional knockout of Nav1.7 in sensory and sympathetic ganglia, have been reported to recapitulate CIP and anosmia in the mouse [56; 85]. Importantly, these animal studies have shown no haploinsufficiency due to the loss of one Scn9a allele, just as is the case in humans [29]. Whether a mouse with a knock-in mutation in Nav1.7 recapitulates the IEM or PEPD phenotypes remains to be seen.
Nav1.7 as a validated target for development of pain therapeutics
Despite the clear role of sodium channels in the pathogenesis of pain in individuals carrying mutations in Nav1.7, treatment with sodium channel inhibitors which bind to the local anesthetics (LA) site of sodium channels has produced mixed outcomes [43]. However, the genetic and functional validation of Nav1.7 as a major contributor to human pain spurred the development of isoform-specific blockers. Several small-molecule inhibitors of Nav1.7 have been recently described [10]. A new class of sulfonamide-based molecules with selectivity for Nav1.7 have been developed which bind to a novel site in the voltage-sensing domain of domain IV of Nav1.7 [1; 81]. An orally-bioavailable arylsulfonamide blocker, PF-05089771, with notable selectivity for Nav1.7 over other voltage-gated sodium channel isoforms (by 10–900-fold), and 1000-fold selectivity over potassium and calcium channels, binds preferentially to the slow-inactivated state of the channel in a use-dependent manner [4]. Other sulfonamide compounds have also been reported with comparable selectivity for Nav1.7 over other sodium channels [1; 44]. These molecules combine molecular selectivity and functional selectivity for hyperactive channels, and hold the promise for improved therapeutic potential.
Patients with well-defined gain-of-function mutations in Nav1.7 present an opportunity to validate therapeutic efficacy of existing and novel small molecule sodium channel blockers because of the near certainty that the target — Nav1.7 — plays a major role in pain. Capitalizing on the availability of a cohort of patients carrying mutations in Nav1.7 that have been functionally tested and shown to produce DRG neuron hypersensitivity, and the development of an orally bioavailable Nav1.7 selective inhibitor, a phase II proof-of-principle randomized, double-blind placebo-controlled cross-over clinical trial (NCT01769274) was conducted to evaluate safety and efficacy of a single dose PF-05089771 (1600 mg) in these patients following a thermal challenge to the foot that was previously shown to evoke a pain episode [82]. There were 5 individuals who met inclusion/exclusion criteria and carried the following mutations: Ser241Thr (1 subject), Val400Meth (2 subjects), Ile848Thr (I subject) and Phe1449Val (1 subject). The clinical data show variability in the response of the subjects with different mutations, differential responses of subjects carrying the same mutation, and the differential response in the two testing sessions. The primary endpoint for the trial, the average pain score on the numeric rating scale (NRS) at 0–4 hrs postdose, was not met, possibly due to lower than expected plasma concentrations of the PF05089771 which reached TMax at 4–6 hours post dosing. However, compared to placebo, four of the patients showed a reduction in the maximum pain score of at least 2 points on the 10-point NRS in at least one session and three patients responded in both sessions; improvement in pain scores occurred more than 4 hrs post-dose [22]. Interestingly, the subject with the Ser241Thr mutation was the only subject who did not respond to treatment in either session. Despite the limitations of this study especially the small number of subjects and the single dose design, it is encouraging that individual subjects achieved a substantial level of pain relief, which supports further evaluation of Nav1.7 blockers in larger cohorts with multiple dosing that might produce more favorable pharmacokinetics, or with an improved generation of Nav1.7 selective compounds.
Subjects who were enrolled in the clinical trial gave additional consent to generate induced pluripotent cells, which were used to differentiate patient-specific sensory neurons that were evaluated for firing properties in response to a physiologically-relevant thermal stimulus, and for their response to incubation with clinically-relevant concentration of PF-05089771. The data show that the patient-specific sensory neurons with the mutations Val400Meth, Ile848Thr and Phe1449Val recapitulated the clinical phenotype in that they fired more action potentials compared to sensory neurons that were differentiated from control subjects; there was attenuation of excitability of these neurons upon incubation with PF-05089771. Sensory neurons from the subject with Ser241Thr showed a rheobase within the range of control neurons, and unlike the neurons from the other IEM subjects, did not manifest a reduction in the rheobase upon exposure to a thermal stimulus of 40°C. Moreover, the reversal of elevated heat sensitivity seen with the other IEM-derived sensory neurons following exposure to an analogue of PF-05089771 was not seen with the Ser241Thr sensory neurons [22]. This in vitro pharmacological data parallels that clinical data showing the subject with the Ser241Thr mutation did not respond to the single dose treatment with PF-05089771. The reason for the lack of efficacy of the selective Nav1.7 blocker on cells with the Ser241Thr mutation and in the subject carrying this mutation is not known at this time.
Although the majority of patients with IEM due to mutations in Nav1.7 do not respond to treatment with sodium channel blockers in clinical use, a few cases of IEM patients and the PEPD patients respond to treatment with these drugs, indicating that monotherapy could be effective when there is adequate target engagement. Pharmacological studies have shown that carbamazepine inhibits gain-of-function attributes of PEPD Nav1.7 mutations, namely reducing the persistent current and shifting fast-inactivation in a hyperpolarizing direction, consistent with the effective response of these patients to treatment with carbamazepine [14; 49]. The lack of efficacy of sodium channel blockers in patients with Nav1.7 IEM mutations is not well understood, albeit in one case it has been linked to a reduced affinity of the mutant channel, Asn395Lys, to sodium channel blockers that bind at the local anesthetic binding site [102]. However, there are case reports of effective treatment with mexiletine in a patient carrying the Val872Gly [27], and CBZ in patients carrying the mutations Val400Met [52] and Ser241Thr [55]. The efficacy of mexiletine in the patient carrying the Val872Gly mutation has been linked to a stronger use-dependent fall-off in the current [27]. By contrast, CBZ was shown to shift the Val400Met mutant channel activation in a depolarizing direction, bringing it closer to that of wild-type channels [52].
In the era of affordable individualized genomic screening, we implemented a multi-disciplinary approach to predict the effect of treatment of a novel channel variant based on the response of a “seed” variant, thus potentially moving away from a trial and error approach to a more predictive approach for choice of pain therapeutic. Using atomic-level modeling and thermodynamic analysis, we were able to show that clinically relevant concentration of CBZ attenuates firing of DRG neurons expressing the Ser241Thr mutant channels (Figure 3), thus predicting CBZ-responsiveness of the Ser241Thr mutation and shown that clinically-relevant concentrations [124], and this pharmacogenomic guided therapy was successfully implemented in a placebo-controlled double-blind clinical trial in two patients carrying this mutation [55]. This represents a case study that illustrates the power of structural modeling and pharmacogenomic approaches for the treatment of pain, moving us closer to the promise of personalized medicine.
Figure 3. Carbamazepine reduces excitability of DRG neurons expressing the Nav1.7-S241T mutation and is accompanied by a shift in brain activity toward a pattern associated with decreased pain.

Schematic represents burning pain pathway in patients with inherited erythromelalgia. Typically, pain attacks which can be reproducibly and predictably triggered by warming distal limbs, are accompanied by skin flushing and swelling of affected limb which also becomes hot. Primary afferents innervating the foot consist of unmyelinated C fibers (black), lightly myelinated Aδ fibers (brown) and heavily myelinated Aβ fibers (magenta). Primary afferents form their first synapse in the spinal cord, and the signal is transmitted to higher brain centers. DRG neurons that express the Nav1.7-S241T mutation fire repetitively, and the firing frequency is markedly attenuated when neurons are treated with a clinically-relevant concentration of carbamazepine (CBZ). Functional brain imaging in a patient treated with placebo shows that pain, after termination of the thermal stimulus, is accompanied by increased activity in valuation areas posterior cingulate cortex (PCC) and anterior cingulate cortex (ACC), and nucleus accumbans (NAc). Treatment with CBZ is accompanied by a shift in brain activity to the primary somatosensory cortex (SI) and parietal attention areas. Adapted with permission from [55].
Functional brain imaging in pain studies
Given that pain reports are subjective and rely on patients’ ratings it follows therefore that consciousness, and hence activity within cortical circuitry, are a pre-requisite for the experience of pain [12; 53; 93; 94]. More than half-a-century of animal research in the setting of a peripheral injury has now extensively described the neural nociceptive machinery that transmits information about potentially painful stimuli cephalad and demonstrates its plasticity in animal models of pain [119; 120]. However, conditions such as fibromyalgia can defy modeling with animal research because the causative injury, if any, is unclear [106]. The advent of functional brain imaging (fMRI) has opened a window into studying the subjective experience of pain by allowing the simultaneous measurement of subjective pain ratings and brain activity.
In the past 25 years hundreds of positron emission tomography and functional brain imaging studies have reported brain responses to acute painful thermal, mechanical, chemical and visceral stimuli [7; 20; 79]. These studies have described activations in the thalamus, primary (SI) and secondary somatosensory area (SII), insula, and anterior cingulate cortex (ACC), areas collectively referred to as the pain matrix [109], with variable reports of activation in the cerebellum, striatum, amygdala, medial and dorso-lateral prefrontal cortices. Pain intensity ratings collected during some of these experiments covaried with activity in the insula and ACC [28]. Such findings were consistent across hundreds of studies in healthy participants, providing support for the suggestion that the pain matrix might be a mediator of the conscious perception of pain or provide a brain signature of pain [109; 114]. Nevertheless, brain activity associated with chronic clinical pain such as low-back pain (CBP), fibromyalgia, migraine or neuropathic pain demonstrated a very different picture, engaging mainly limbic brain areas such as amygdala, striatum (dorsal and ventral), and medial prefrontal cortex, mPFC [6; 7; 12; 78; 104]. These studies provided evidence that areas mediating emotional decision-making, learning and valuation [62; 66; 92] correlate more strongly with chronic pain than areas mediating accurate sensations [84] and attention or salience [83]. These findings also challenged the concept of the pain matrix [7]. Behavioral studies describing deficits in hedonic perception in CBP [54] and emotional decision making tasks [9; 16; 115] in CBP and fibromyalgia patients are consistent with the idea that chronic pain taxes the limbic brain.
Recent findings provide evidence that altered structure and function of areas in the limbic brain are critical factors in determining the risk of transition from acute clinical pain to the chronic condition [13; 110]. Baliki et al., [13] showed that the magnitude of cortico-striatal functional connectivity between the nucleus accumbens (NAc, part of the ventral striatum) and the mPFC predicts the odds that an episode of sub-acute back pain (period between 6 to 12 weeks) would persist or remit one year later. The same group provided evidence that the volume of amygdala and hippocampus and their functional and structural connectivity to the medio-dorsal prefrontal cortex predict the magnitude of the risk of transitioning to chronic low-back pain at 1 and 3 years follow-up, and mediate the effect of genetic risk factors for that transition [110]. Both the amygdala and hippocampus are crucial brain areas for learning and memory formation [91; 92] implying that pain persistence presupposes new learning, most likely driven by peripheral nociceptive input. In agreement with these conclusions, a recent study demonstrated that adult hippocampal neurogenesis, which is a hallmark of new learning, is necessary for the development of neuropathic pain in rodents [8]. Collectively, these results show that the brain correlates of adaptive (i.e. protective) acute pain are different from those of chronic (i.e. maladaptive) pain and point to a causal role of the limbic circuitry and limbic plasticity in clinical pain perception and the transition to the chronic state.
Functional brain imaging in pain studies: Lessons from IEM
As discussed in previous sections, IEM is a rare chronic pain condition caused by gain-of-function mutations in the Nav1.7 channel present on peripheral nociceptors. Patients with IEM suffer from severe pain attacks when they are exposed to thermal stimulus that would not otherwise elicit pain in individuals with normal Nav1.7 channels. These attacks can be relieved by cooling of the affected body part [39]. Therefore, if chronic pain is conceived, at least in part, as a persistent peripheral barrage from nociceptors [113], IEM offers a unique opportunity to examine this model of hyperactive nociceptors in the context of brain imaging because peripheral input can be manipulated via the control of skin temperature (Figure 3). Seghredahl et al. [101] measured cerebral blood flow (CBF) in a patient with IEM to assess differences between acute thermal heating and cooling using arterial spin labeling. Acute thermal heating triggered a pain attack while cooling induced analgesia. Compared to cooling, the pain attacks were accompanied by increased CBF in the thalamus, striatum, SI, insula, inferior frontal gyrus, posterior, mid- and anterior cingulate cortex. Activation in these areas has been shown to predict thermal heat pain intensity [114], and is reminiscent of activity in the areas of the pain matrix [109]. One disadvantage of presenting an outside painful stimulation during brain imaging, however, is the difficulty of controlling for the activation elicited by the saliency of the stimulus itself [71]. Interestingly, activation of the pain matrix can be elicited by stimuli from other modalities (i.e. visual, auditory, tactile) [86], and occurs in the absence of pain in patients with a loss-of-function mutation of the Nav1.7 channel [99], raising issues about the specificity of activation in these brain areas to pain perception.
In keeping with the learning model of chronic pain described above, we therefore hypothesized that examining IEM patients’ stimulus-free pain, an approach used with multiple other chronic pain conditions [6], would uncover the shift of neural correlate of this chronic pain syndrome towards the limbic brain. We studied two patients suffering from IEM due to the same gain-of-function mutation Nav1.7-S241T using fMRI blood oxygen level dependent (BOLD) signal. BOLD signal was collected simultaneously with continuous pain intensity ratings under two different conditions: (1) during an attack of IEM pain elicited by a thermal heating boot wrapped around the foot; and (2) after the termination of the stimulus, while patients reported their ongoing stimulus-free pain during attacks that persisted for more than an hour. The neural correlates of stimulus-free pain engaged mostly the limbic brain including the prefrontal cortex, dorsal and ventral striatum and showed a minimal overlap (~ 2%) with areas of the pain matrix in the insula. In contrast, the neural correlates of the acute thermal pain stimulation showed a larger overlap with areas of the pain matrix (~11%) and resembled more the results reported by Segherdahl et al [101]. More importantly, we assessed the treatment effects of CBZ vs. placebo on IEM pain and brain activity. Our choice of CBZ was genomically guided by in vitro work pointing to its specific effect on the Nav1.7-S241T mutation carried by both patients [55; 124]. Clinical pain improvement was observed after two weeks of treatment with CBZ but not placebo and was accompanied by a shift in brain activity from ventral striatum, rostral ACC and posterior cingulate cortex towards primary sensori-motor and attention areas [55]. This shift in brain activity suggests that chronic IEM pain treatment is associated with increased activity in areas mediating accurate sensation and attention and decreased activity in areas mediating emotional learning (Figure 3).
Half a century ago Wilder Penfield described the brain as a “passive observer” of peripheral nociceptive input based on the absence of evidence for specific cortical neuronal responses supporting the presence of a “pain cortex” [89]. More recent cortical stimulation studies confirmed that painful responses to cortical stimulation are very rare and confined to the posterior insula and adjacent parietal operculum only [80]. However, brain imaging findings described above as well as a large literature on the psychological modulation of pain [20; 51] support the role of the limbic brain as mechanistically involved in the perception of pain and the “chronification” of clinical conditions. Two different rodent studies of nerve injury induced pain [70; 95] to-date have been able to control pain behavior via the modulation of activity in neuronal projections from the pre-limbic PFC (the rodent equivalent of the human mPFC) into the ventral striatum. In agreement with these observations, imaging of IEM patients suggests that although the peripheral source of afferent barrage is well defined and can be readily manipulated, pain perception and pain relief involve the limbic brain, in particular the valuation circuitry [55; 66; 74]. Taken together, evidence from animal and human research, support the hypothesis therefore that this circuitry, particularly the ventral striatum-medial prefrontal axis, may function as a gate-keeper controlling which nociceptive input emerges into consciousness as pain [12].
However promising the findings of brain imaging in clinical pain conditions including IEM are, many limitations persist. For example, it is still unclear how conscious perception of pain arises from the activation of nociceptors. Some advance in predicting pain intensity using fMRI data has been recently demonstrated [25; 114]. While IEM pain can be reproducibly triggered in study subjects and imaging data can thus be correlated with well-defined pain trigger, the causative chain of events in the overwhelming majority of clinical pain conditions that are studied by fMRI remain unknown. Nevertheless, studies using fMRI data within the framework of a longitudinal design have proven crucial in understanding the mechanism of, and predicting the transition from early to chronic pain syndromes [13; 110]. Finally, fMRI measures brain activity based on the BOLD signal. BOLD is a slow signal relative to the frequency of neuronal firing [76]. Combined electroencephalogram-fMRI studies may help on this front.
Conclusions
Elucidation of the role of individual sodium channels in action potential firing has informed our understanding of pathophysiological mechanisms underlying painful channelopathies. The parallel outcome of attenuation of firing of DRG neurons expressing the Ser241Thr mutation and pain relief of the subjects carrying this mutation, and the lack of efficacy of treatment with PF-05089771 in the in vitro pharmacological assay and the clinical outcome in the subject with this mutation, suggests that in vitro pharmacological assays using patient-specific differentiated neurons might be predictive of clinical outcomes of subjects receiving this treatment. These are examples of a complicated pharmacogenomic picture that may underlie the diverse response that is seen in the clinic. The variability in the response of individual patient to existing drugs or to new ones like PF-05089771 is to be expected, but we’ll have to wait for additional clinical trials with larger cohorts of subjects to make more definitive conclusions. The promise of the new generation of sodium channel blockers is that their isoform selectivity will reduce the potential adverse effects that limit their effective clinical utility.
References
- 1.Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, Shriver S, Young C, Lin S, Johnson JP, Jr, Wu P, Li J, Coons M, Tam C, Brillantes B, Sampang H, Mortara K, Bowman KK, Clark KR, Estevez A, Xie Z, Verschoof H, Grimwood M, Dehnhardt C, Andrez JC, Focken T, Sutherlin DP, Safina BS, Starovasnik MA, Ortwine DF, Franke Y, Cohen CJ, Hackos DH, Koth CM, Payandeh J. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science. 2015;350(6267):aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- 2.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379(6562):257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 3.Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2(6):541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- 4.Alexandrou AJ, Brown AR, Chapman ML, Estacion M, Turner J, Mis MA, Wilbrey A, Payne EC, Gutteridge A, Cox PJ, Doyle R, Printzenhoff D, Lin Z, Marron BE, West C, Swain NA, Storer RI, Stupple PA, Castle NA, Hounshell JA, Rivara M, Randall A, Dib-Hajj SD, Krafte D, Waxman SG, Patel MK, Butt RP, Stevens EB. Subtype-Selective Small Molecule Inhibitors Reveal a Fundamental Role for Nav1. 7 in Nociceptor Electrogenesis, Axonal Conduction and Presynaptic Release. PLoS One. 2016;11(4):e0152405. doi: 10.1371/journal.pone.0152405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonini G, Di Pasquale A, Cruccu G, Truini A, Morino S, Saltelli G, Romano A, Trasimeni G, Vanacore N, Bozzao A. Magnetic resonance imaging contribution for diagnosing symptomatic neurovascular contact in classical trigeminal neuralgia: a blinded case-control study and meta-analysis. Pain. 2014;155(8):1464–1471. doi: 10.1016/j.pain.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87(2):81–97. doi: 10.1016/j.pneurobio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9(4):463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Apkarian AV, Mutso AA, Centeno MV, Kan L, Wu M, Levinstein M, Banisadr G, Gobeske KT, Miller RJ, Radulovic J, Hen R, Kessler JA. Role of adult hippocampal neurogenesis in persistent pain. Pain. 2016;157(2):418–428. doi: 10.1097/j.pain.0000000000000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN, Chialvo DR. Chronic pain patients are impaired on an emotional decision-making task. Pain. 2004;108(1–2):129–136. doi: 10.1016/j.pain.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 10.Bagal SK, Chapman ML, Marron BE, Prime R, Storer RI, Swain NA. Recent progress in sodium channel modulators for pain. Bioorg Med Chem Lett. 2014;24(16):3690–3699. doi: 10.1016/j.bmcl.2014.06.038. [DOI] [PubMed] [Google Scholar]
- 11.Balague F, Mannion AF, Pellise F, Cedraschi C. Non-specific low back pain. Lancet. 2012;379(9814):482–491. doi: 10.1016/S0140-6736(11)60610-7. [DOI] [PubMed] [Google Scholar]
- 12.Baliki MN, Apkarian AV. Nociception, Pain, Negative Moods, and Behavior Selection. Neuron. 2015;87(3):474–491. doi: 10.1016/j.neuron.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV. Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci. 2012;15(8):1117–1119. doi: 10.1038/nn.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bednarek N, Arbues AS, Motte J, Sabouraud P, Plouin P, Morville P. Familial rectal pain: a familial autonomic disorder as a cause of paroxysmal attacks in the newborn baby. Epileptic Disord. 2005;7(4):360–362. [PubMed] [Google Scholar]
- 15.Bennett DL, Woods CG. Painful and painless channelopathies. Lancet Neurol. 2014;13(6):587–599. doi: 10.1016/S1474-4422(14)70024-9. [DOI] [PubMed] [Google Scholar]
- 16.Berger SE, Baria AT, Baliki MN, Mansour A, Herrmann KM, Torbey S, Huang L, Parks EL, Schnizter TJ, Apkarian AV. Risky monetary behavior in chronic back pain is associated with altered modular connectivity of the nucleus accumbens. BMC Res Notes. 2014;7:739. doi: 10.1186/1756-0500-7-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Black JA, Renganathan M, Waxman SG. Sodium channel Nav1. 6 is expressed along nonmyelinated axons and it contributes to conduction. Mol Brain Res. 2002;105(1–2):19–28. doi: 10.1016/s0169-328x(02)00385-6. [DOI] [PubMed] [Google Scholar]
- 18.Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22(23):10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst. 2014;19(2):53–65. doi: 10.1111/jns5.12071. [DOI] [PubMed] [Google Scholar]
- 20.Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14(7):502–511. doi: 10.1038/nrn3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Nav1. 6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97(10):5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao L, McDonnell A, Nitzsche A, Alexandrou A, Saintot PP, Loucif AJ, Brown AR, Young G, Mis M, Randall A, Waxman SG, Stanley P, Kirby S, Tarabar S, Gutteridge A, Butt R, McKernan RM, Whiting P, Ali Z, Bilsland J, Stevens EB. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med. 2016;8(335):335ra356. doi: 10.1126/scitranslmed.aad7653. [DOI] [PubMed] [Google Scholar]
- 23.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 24.Catterall WA, Goldin AL, Waxman SG International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol Rev. 2005;57(4):397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 25.Cecchi GA, Huang L, Hashmi JA, Baliki M, Centeno MV, Rish I, Apkarian AV. Predictive dynamics of human pain perception. PLoS Comput Biol. 2012;8(10):e1002719. doi: 10.1371/journal.pcbi.1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi JS, Boralevi F, Brissaud O, Sanchez-Martin J, Te Morsche RH, Dib-Hajj SD, Drenth JP, Waxman SG. Paroxysmal extreme pain disorder: a molecular lesion of peripheral neurons. Nat Rev Neurol. 2011;7(1):51–55. doi: 10.1038/nrneurol.2010.162. [DOI] [PubMed] [Google Scholar]
- 27.Choi JS, Zhang L, Dib-Hajj SD, Han C, Tyrrell L, Lin Z, Wang X, Yang Y, Waxman SG. Mexiletine-responsive erythromelalgia due to a new Nav1. 7 mutation showing use-dependent current fall-off. Exp Neurol. 2009;216(2):383–389. doi: 10.1016/j.expneurol.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 28.Coghill RC, Sang CN, Maisog JM, Iadarola MJ. Pain intensity processing within the human brain: a bilateral, distributed mechanism. J Neurophysiol. 1999;82(4):1934–1943. doi: 10.1152/jn.1999.82.4.1934. [DOI] [PubMed] [Google Scholar]
- 29.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cruccu G, Bonamico LH, Zakrzewska JM. Cranial neuralgias. Handb Clin Neurol. 2010;97:663–678. doi: 10.1016/S0072-9752(10)97056-5. [DOI] [PubMed] [Google Scholar]
- 31.Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current In SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19(24):RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. Nav1. 6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005;579(10):2166–2170. doi: 10.1016/j.febslet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18(23):9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deuis JR, Zimmermann K, Romanovsky AA, Possani LD, Cabot PJ, Lewis RJ, Vetter I. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Na1. 6 in peripheral pain pathways. Pain. 2013;154(9):1749–1757. doi: 10.1016/j.pain.2013.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dib-Hajj S, Black JA, Felts P, Waxman SG. Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci U S A. 1996;93(25):14950–14954. doi: 10.1073/pnas.93.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 2009;10(7):1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- 37.Dib-Hajj SD, Black JA, Waxman SG. NaV1. 9: a sodium channel linked to human pain. Nat Rev Neurosci. 2015;16(9):511–519. doi: 10.1038/nrn3977. [DOI] [PubMed] [Google Scholar]
- 38.Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, Waxman SG. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009;4(8):1118–1126. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- 39.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Nav1. 7 and human pain disorders. Trends Neurosci. 2007;30(11):555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 40.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium Channels in Normal and Pathological Pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 41.Dib-Hajj SD, Tyrrell L, Cummins TR, Black JA, Wood PM, Waxman SG. Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Lett. 1999;462(1–2):117–120. doi: 10.1016/s0014-5793(99)01519-7. [DOI] [PubMed] [Google Scholar]
- 42.Dib-Hajj SD, Waxman SG. Diversity of composition and function of sodium channels in peripheral sensory neurons. Pain. 2015;156(12):2406–2407. doi: 10.1097/j.pain.0000000000000353. [DOI] [PubMed] [Google Scholar]
- 43.Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The NaV1. 7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14(1):49–62. doi: 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- 44.DiMauro EF, Altmann S, Berry LM, Bregman H, Chakka N, Chu-Moyer M, Bojic EF, Foti RS, Fremeau R, Gao H, Gunaydin H, Guzman-Perez A, Hall BE, Huang H, Jarosh M, Kornecook T, Lee J, Ligutti J, Liu D, Moyer BD, Ortuno D, Rose PE, Schenkel LB, Taborn K, Wang J, Wang Y, Yu V, Weiss MM. Application of a Parallel Synthetic Strategy in the Discovery of Biaryl Acyl Sulfonamides as Efficient and Selective NaV1. 7 Inhibitors. J Med Chem. 2016;59(17):7818–7839. doi: 10.1021/acs.jmedchem.6b00425. [DOI] [PubMed] [Google Scholar]
- 45.Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603–3609. doi: 10.1172/JCI33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duan G, Han C, Wang Q, Guo S, Zhang Y, Ying Y, Huang P, Zhang L, Macala L, Shah P, Zhang M, Li N, Dib-Hajj SD, Waxman SG, Zhang X. A SCN10A SNP biases human pain sensitivity. Mol Pain. 2016:12. doi: 10.1177/1744806916666083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eijkelkamp N, Linley JE, Baker MD, Minett MS, Cregg R, Werdehausen R, Rugiero F, Wood JN. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135(Pt 9):2585–2612. doi: 10.1093/brain/aws225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JP, Waxman SG, Merkies IS. Gain of function NaV1. 7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012a;71(1):26–39. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- 49.Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg-Olofsson O, Elmslie FV, Griesemer DA, Goutieres F, Kirkpatrick M, Malmros IN, Pollitzer M, Rossiter M, Roulet-Perez E, Schubert R, Smith VV, Testard H, Wong V, Stephenson JB. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome) Neurology. 2007;69(6):586–595. doi: 10.1212/01.wnl.0000268065.16865.5f. [DOI] [PubMed] [Google Scholar]
- 51.Fields HL. Understanding how opioids contribute to reward and analgesia. Reg Anesth Pain Med. 2007;32(3):242–246. doi: 10.1016/j.rapm.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Fischer TZ, Gilmore ES, Estacion M, Eastman E, Taylor S, Melanson M, Dib-Hajj SD, Waxman SG. A novel Nav1. 7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol. 2009;65(6):733–741. doi: 10.1002/ana.21678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gatchel RJ, Peng YB, Peters ML, Fuchs PN, Turk DC. The biopsychosocial approach to chronic pain: scientific advances and future directions. Psychol Bull. 2007;133(4):581–624. doi: 10.1037/0033-2909.133.4.581. [DOI] [PubMed] [Google Scholar]
- 54.Geha P, Dearaujo I, Green B, Small DM. Decreased food pleasure and disrupted satiety signals in chronic low back pain. Pain. 2014;155(4):712–722. doi: 10.1016/j.pain.2013.12.027. [DOI] [PubMed] [Google Scholar]
- 55.Geha P, Yang Y, Estacion M, Schulman BR, Tokuno H, Apkarian AV, Dib-Hajj SD, Waxman SG. Pharmacotherapy for Pain in a Family With Inherited Erythromelalgia Guided by Genomic Analysis and Functional Profiling. JAMA Neurol. 2016;73(6):659–667. doi: 10.1001/jamaneurol.2016.0389. [DOI] [PubMed] [Google Scholar]
- 56.Gingras J, Smith S, Matson DJ, Johnson D, Nye K, Couture L, Feric E, Yin R, Moyer BD, Peterson ML, Rottman JB, Beiler RJ, Malmberg AB, McDonough SI. Global nav1. 7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One. 2014;9(9):e105895. doi: 10.1371/journal.pone.0105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han C, Estacion M, Huang J, Vasylyev DV, Zhao P, Dib-Hajj S, Waxman SG. Human Nav1. 8: enhanced persistent and ramp currents contribute to distinct firing properties of human DRG neurons. J Neurophysiol. 2015;113(9):3172–3185. doi: 10.1152/jn.00113.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han C, Hoeijmakers JG, Liu S, Gerrits MM, Te Morsche RH, Lauria G, Dib-Hajj SD, Drenth JP, Faber CG, Merkies IS, Waxman SG. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain. 2012b;135(Pt 9):2613–2628. doi: 10.1093/brain/aws187. [DOI] [PubMed] [Google Scholar]
- 59.Han C, Huang J, Waxman SG. Sodium channel Nav1.8: Emerging links to human disease. Neurology. 2016;86(5):473–483. doi: 10.1212/WNL.0000000000002333. [DOI] [PubMed] [Google Scholar]
- 60.Han C, Yang Y, de Greef BT, Hoeijmakers JG, Gerrits MM, Verhamme C, Qu J, Lauria G, Merkies IS, Faber CG, Dib-Hajj SD, Waxman SG. The Domain II S4–S5 Linker in Nav1. 9: A Missense Mutation Enhances Activation, Impairs Fast Inactivation, and Produces Human Painful Neuropathy. Neuromolecular Med. 2015;17(2):158–169. doi: 10.1007/s12017-015-8347-9. [DOI] [PubMed] [Google Scholar]
- 61.Han C, Yang Y, Te Morsche RH, Drenth JP, Politei JM, Waxman SG, Dib-Hajj SD. Familial gain-of-function Nav1.9 mutation in a painful channelopathy. Journal of neurology, neurosurgery, and psychiatry. 2016 doi: 10.1136/jnnp-2016-313804. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 62.Hart G, Leung BK, Balleine BW. Dorsal and ventral streams: the distinct role of striatal subregions in the acquisition and performance of goal-directed actions. Neurobiology of learning and memory. 2014;108:104–118. doi: 10.1016/j.nlm.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG. Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1. 7 sodium channels in mouse spinal sensory neurons. J Physiol (Lond) 2003;551(Pt 3):741–750. doi: 10.1113/jphysiol.2003.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM, Tyrrell L, Lauria G, Faber CG, Dib-Hajj SD, Merkies IS, Waxman SG. Gain-of-function mutations in sodium channel Nav1. 9 in painful neuropathy. Brain. 2014;137(Pt 6):1627–1642. doi: 10.1093/brain/awu079. [DOI] [PubMed] [Google Scholar]
- 65.Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, Gauvin D, Wade C, Zhu C, Pai M, Scanio M, Shi L, Drizin I, Gregg R, Matulenko M, Hakeem A, Gross M, Johnson M, Marsh K, Wagoner PK, Sullivan JP, Faltynek CR, Krafte DS. A-803467, a potent and selective Nav1. 8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci (USA) 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kable JW, Glimcher PW. The neural correlates of subjective value during intertemporal choice. Nat Neurosci. 2007;10(12):1625–1633. doi: 10.1038/nn2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14(6):1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ko AL, Lee A, Raslan AM, Ozpinar A, McCartney S, Burchiel KJ. Trigeminal neuralgia without neurovascular compression presents earlier than trigeminal neuralgia with neurovascular compression. J Neurosurg. 2015;123(6):1519–1527. doi: 10.3171/2014.11.JNS141741. [DOI] [PubMed] [Google Scholar]
- 69.Lee A, McCartney S, Burbidge C, Raslan AM, Burchiel KJ. Trigeminal neuralgia occurs and recurs in the absence of neurovascular compression. J Neurosurg. 2014;120(5):1048–1054. doi: 10.3171/2014.1.JNS131410. [DOI] [PubMed] [Google Scholar]
- 70.Lee M, Manders TR, Eberle SE, Su C, D’Amour J, Yang R, Lin HY, Deisseroth K, Froemke RC, Wang J. Activation of corticostriatal circuitry relieves chronic neuropathic pain. J Neurosci. 2015;35(13):5247–5259. doi: 10.1523/JNEUROSCI.3494-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Legrain V, Iannetti GD, Plaghki L, Mouraux A. The pain matrix reloaded: a salience detection system for the body. Prog Neurobiol. 2011;93(1):111–124. doi: 10.1016/j.pneurobio.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 72.Leipold E, Hanson-Kahn A, Frick M, Gong P, Bernstein JA, Voigt M, Katona I, Oliver Goral R, Altmuller J, Nurnberg P, Weis J, Hubner CA, Heinemann SH, Kurth I. Cold-aggravated pain in humans caused by a hyperactive NaV1. 9 channel mutant. Nat Commun. 2015;6:10049. doi: 10.1038/ncomms10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stodberg T, Hennings JC, Bergmann M, Altmuller J, Thiele H, Wetzel A, Nurnberg P, Timmerman V, De Jonghe P, Blum R, Schaible HG, Weis J, Heinemann SH, Hubner CA, Kurth I. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet. 2013;45(11):1399–1404. doi: 10.1038/ng.2767. [DOI] [PubMed] [Google Scholar]
- 74.Levy DJ, Glimcher PW. The root of all value: a neural common currency for choice. Curr Opin Neurobiol. 2012;22(6):1027–1038. doi: 10.1016/j.conb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin Z, Santos S, Padilla K, Printzenhoff D, Castle NA. Biophysical and Pharmacological Characterization of Nav1. 9 Voltage Dependent Sodium Channels Stably Expressed in HEK-293 Cells. PLoS One. 2016;11(8):e0161450. doi: 10.1371/journal.pone.0161450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Logothetis NK. The neural basis of the blood-oxygen-level-dependent functional magnetic resonance imaging signal. Philos Trans R Soc Lond B Biol Sci. 2002;357(1424):1003–1037. doi: 10.1098/rstb.2002.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maarbjerg S, Wolfram F, Gozalov A, Olesen J, Bendtsen L. Significance of neurovascular contact in classical trigeminal neuralgia. Brain. 2015;138(Pt 2):311–319. doi: 10.1093/brain/awu349. [DOI] [PubMed] [Google Scholar]
- 78.Maleki N, Becerra L, Brawn J, McEwen B, Burstein R, Borsook D. Common hippocampal structural and functional changes in migraine. Brain Struct Funct. 2013;218(4):903–912. doi: 10.1007/s00429-012-0437-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martucci KT, Mackey SC. Imaging Pain. Anesthesiol Clin. 2016;34(2):255–269. doi: 10.1016/j.anclin.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mazzola L, Isnard J, Peyron R, Mauguiere F. Stimulation of the human cortex and the experience of pain: Wilder Penfield’s observations revisited. Brain. 2012;135(Pt 2):631–640. doi: 10.1093/brain/awr265. [DOI] [PubMed] [Google Scholar]
- 81.McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D, Padilla KM, Lin Z, Wagoner PK, Swain NA, Stupple PA, de Groot M, Butt RP, Castle NA. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci U S A. 2013;110(29):E2724–2732. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDonnell A, Schulman B, Ali Z, Dib-Hajj SD, Brock F, Cobain S, Mainka T, Vollert J, Tarabar S, Waxman SG. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain. 2016;139(Pt 4):1052–1065. doi: 10.1093/brain/aww007. [DOI] [PubMed] [Google Scholar]
- 83.Menon V. Large-scale brain networks and psychopathology: a unifying triple network model. Trends Cogn Sci. 2011;15(10):483–506. doi: 10.1016/j.tics.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 84.Mesulam MM. From sensation to cognition. Brain. 1998;121(Pt 6):1013–1052. doi: 10.1093/brain/121.6.1013. [DOI] [PubMed] [Google Scholar]
- 85.Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, Malcangio M, Wood JN. Distinct Nav1. 7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. 2012;3:791. doi: 10.1038/ncomms1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mouraux A, Diukova A, Lee MC, Wise RG, Iannetti GD. A multisensory investigation of the functional significance of the “pain matrix”. Neuroimage. 2011;54(3):2237–2249. doi: 10.1016/j.neuroimage.2010.09.084. [DOI] [PubMed] [Google Scholar]
- 87.Okuda H, Noguchi A, Kobayashi H, Kondo D, Harada KH, Youssefian S, Shioi H, Kabata R, Domon Y, Kubota K, Kitano Y, Takayama Y, Hitomi T, Ohno K, Saito Y, Asano T, Tominaga M, Takahashi T, Koizumi A. Infantile Pain Episodes Associated with Novel Nav1. 9 Mutations in Familial Episodic Pain Syndrome in Japanese Families. PLoS One. 2016;11(5):e0154827. doi: 10.1371/journal.pone.0154827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Payne CE, Brown AR, Theile JW, Loucif AJ, Alexandrou AJ, Fuller MD, Mahoney JH, Antonio BM, Gerlach AC, Printzenhoff DM, Prime RL, Stockbridge G, Kirkup AJ, Bannon AW, England S, Chapman ML, Bagal S, Roeloffs R, Anand U, Anand P, Bungay PJ, Kemp M, Butt RP, Stevens EB. A novel selective and orally bioavailable Na 1.8 blocker PF-01247324 attenuates nociception and sensory neuron excitability. Br J Pharmacol. 2015;172(10):2654–2670. doi: 10.1111/bph.13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Penfield W. Temporal lobe epilepsy. Br J Surg. 1954;41(168):337–343. doi: 10.1002/bjs.18004116802. [DOI] [PubMed] [Google Scholar]
- 90.Phatarakyijnirund V, Mumm S, McAlister WH, Novack D, Wenkert D, Clements KL, Whyte MP. Congenital insensitivity to pain: Fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1. 9. Bone. 2016;84:289–298. doi: 10.1016/j.bone.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43(6):897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 92.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48(2):175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 93.Ramachandran VS. Consciousness and body image: lessons from phantom limbs, Capgras syndrome and pain asymbolia. Philos Trans R Soc Lond B Biol Sci. 1998;353(1377):1851–1859. doi: 10.1098/rstb.1998.0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ramachandran VS, Hirstein W. The perception of phantom limbs. The D. O. Hebb lecture. Brain. 1998;121(Pt 9):1603–1630. doi: 10.1093/brain/121.9.1603. [DOI] [PubMed] [Google Scholar]
- 95.Ren W, Centeno MV, Berger S, Wu Y, Na X, Liu X, Kondapalli J, Apkarian AV, Martina M, Surmeier DJ. The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nat Neurosci. 2016;19(2):220–222. doi: 10.1038/nn.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Renganathan M, Cummins TR, Waxman SG. Contribution of Nav1. 8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86(2):629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- 97.Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103(21):8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1. 6, expressed in mouse spinal sensory neurons. J Physiol (Lond) 2005;564(3):803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Salomons TV, Iannetti GD, Liang M, Wood JN. The “Pain Matrix” in Pain-Free Individuals. JAMA Neurol. 2016;73(6):755–756. doi: 10.1001/jamaneurol.2016.0653. [DOI] [PubMed] [Google Scholar]
- 100.Schafer DP, Custer AW, Shrager P, Rasband MN. Early events in node of Ranvier formation during myelination and remyelination in the PNS. Neuron Glia Biol. 2006;2(2):69–79. doi: 10.1017/S1740925X06000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Segerdahl AR, Xie J, Paterson K, Ramirez JD, Tracey I, Bennett DL. Imaging the neural correlates of neuropathic pain and pleasurable relief associated with inherited erythromelalgia in a single subject with quantitative arterial spin labelling. Pain. 2012;153(5):1122–1127. doi: 10.1016/j.pain.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sheets PL, Jackson JO, II, Waxman SG, Dib-Hajj S, Cummins TR. A Nav1. 7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol (Lond) 2007;581:1019–1031. doi: 10.1113/jphysiol.2006.127027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD. NaV1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153(10):2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 104.Simons LE, Moulton EA, Linnman C, Carpino E, Becerra L, Borsook D. The human amygdala and pain: evidence from neuroimaging. Hum Brain Mapp. 2014;35(2):527–538. doi: 10.1002/hbm.22199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, Alzheimer C, Grafe P, Carr RW. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype NaV1.6-resurgent and persistent current. Proc Natl Acad Sci U S A. 2012;109(17):6704–6709. doi: 10.1073/pnas.1118058109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sluka KA, Clauw DJ. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience. 2016;338:114–129. doi: 10.1016/j.neuroscience.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tan ZY, Piekarz AD, Priest BT, Knopp KL, Krajewski JL, McDermott JS, Nisenbaum ES, Cummins TR. Tetrodotoxin-resistant sodium channels in sensory neurons generate slow resurgent currents that are enhanced by inflammatory mediators. J Neurosci. 2014;34(21):7190–7197. doi: 10.1523/JNEUROSCI.5011-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tanaka BS, Zhao P, Dib-Hajj FB, Morisset V, Tate S, Waxman SG, Dib-Hajj SD. A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol Med. 2016:22. doi: 10.2119/molmed.2016.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tracey I, Mantyh PW. The cerebral signature for pain perception and its modulation. Neuron. 2007;55(3):377–391. doi: 10.1016/j.neuron.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 110.Vachon-Presseau E, Tetreault P, Petre B, Huang L, Berger SE, Torbey S, Baria AT, Mansour AR, Hashmi JA, Griffith JW, Comasco E, Schnitzer TJ, Baliki MN, Apkarian AV. Corticolimbic anatomical characteristics predetermine risk for chronic pain. Brain. 2016;139(Pt 7):1958–1970. doi: 10.1093/brain/aww100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vanoye CG, Kunic JD, Ehring GR, George AL., Jr Mechanism of sodium channel NaV1. 9 potentiation by G-protein signaling. J Gen Physiol. 2013;141(2):193–202. doi: 10.1085/jgp.201210919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vasylyev DV, Han C, Zhao P, Dib-Hajj S, Waxman SG. Dynamic-clamp Analysis of Wild-type hNaV1. 7 and Erythromelalgia Mutant Channel L858H. J Neurophysiol. 2014;111(7):1429–1443. doi: 10.1152/jn.00763.2013. [DOI] [PubMed] [Google Scholar]
- 113.von Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73(4):638–652. doi: 10.1016/j.neuron.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wager TD, Atlas LY, Lindquist MA, Roy M, Woo CW, Kross E. An fMRI-based neurologic signature of physical pain. N Engl J Med. 2013;368(15):1388–1397. doi: 10.1056/NEJMoa1204471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Walteros C, Sanchez-Navarro JP, Munoz MA, Martinez-Selva JM, Chialvo D, Montoya P. Altered associative learning and emotional decision making in fibromyalgia. J Psychosom Res. 2011;70(3):294–301. doi: 10.1016/j.jpsychores.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 116.Waxman SG, Kocsis JD, Black JA. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol. 1994;72(1):466–470. doi: 10.1152/jn.1994.72.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Waxman SG, Merkies IS, Gerrits MM, Dib-Hajj SD, Lauria G, Cox JJ, Wood JN, Woods CG, Drenth JP, Faber CG. Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol. 2014;13(11):1152–1160. doi: 10.1016/S1474-4422(14)70150-4. [DOI] [PubMed] [Google Scholar]
- 118.Woods CG, Babiker MO, Horrocks I, Tolmie J, Kurth I. The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p. L811P. Eur J Hum Genet. 2015;23(5):561–563. doi: 10.1038/ejhg.2014.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 120.Woolf CJ, Salter MW. Plasticity and pain: role of the dorsal horn. In: McMahon SB, Koltzenburg M, editors. Textbook of Pain. New York: Churchill-Livingstone; 2006. pp. 91–105. [Google Scholar]
- 121.Wu CL, Tella P, Staats PS, Vaslav R, Kazim DA, Wesselmann U, Raja SN. Analgesic effects of intravenous lidocaine and morphine on postamputation pain: a randomized double-blind, active placebo-controlled, crossover trial. Anesthesiology. 2002;96(4):841–848. doi: 10.1097/00000542-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 122.Xie W, Strong JA, Ye L, Mao JX, Zhang JM. Knockdown of sodium channel Na1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain. 2013;154(8):1170–1180. doi: 10.1016/j.pain.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xie W, Strong JA, Zhang JM. Local knockdown of the Na1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience. 2015;291:317–30. doi: 10.1016/j.neuroscience.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang Y, Dib-Hajj SD, Zhang J, Zhang Y, Tyrrell L, Estacion M, Waxman SG. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Nav1. 7 mutant channel. Nat Commun. 2012;3:1186. doi: 10.1038/ncomms2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yuan J, Matsuura E, Higuchi Y, Hashiguchi A, Nakamura T, Nozuma S, Sakiyama Y, Yoshimura A, Izumo S, Takashima H. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology. 2013;80(18):1641–1649. doi: 10.1212/WNL.0b013e3182904fdd. [DOI] [PubMed] [Google Scholar]
- 127.Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, Wang T, Ran K, Li Y, Li X, Xu M, Luo J, Feng S, Ma X, Ma H, Chai Z, Zhou Z, Yao J, Zhang X, Liu JY. Gain-of-Function mutations in SCN11A cause familial episodic pain. Am J Hum Genet. 2013;93(5):957–966. doi: 10.1016/j.ajhg.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]