

Abstract
Transient outward potassium current (Ito) in the heart underlies phase 1 repolarization of cardiac action potentials and thereby affects excitation–contraction coupling. Small molecule activators of Ito may therefore offer novel treatments for cardiac dysfunction, including heart failure and atrial fibrillation. NS5806 has been identified as a prototypic activator of canine Ito. This study investigated, for the first time, actions of NS5806 on rabbit atrial and ventricular Ito. Whole cell patch‐clamp recordings of Ito and action potentials were made at physiological temperature from rabbit ventricular and atrial myocytes. 10 μmol/L NS5806 increased ventricular Ito with a leftward shift in Ito activation and accelerated restitution. At higher concentrations, stimulation of Ito was followed by inhibition. The EC 50 for stimulation was 1.6 μmol/L and inhibition had an IC 50 of 40.7 μmol/L. NS5806 only inhibited atrial Ito (IC 50 of 18 μmol/L) and produced a modest leftward shifts in Ito activation and inactivation, without an effect on restitution. 10 μmol/L NS5806 shortened ventricular action potential duration (APD) at APD 20‐APD 90 but prolonged atrial APD. NS5806 also reduced atrial AP upstroke and amplitude, consistent with an additional atrio‐selective effect on Na+ channels. In contrast to NS5806, flecainide, which discriminates between Kv1.4 and 4.x channels, produced similar levels of inhibition of ventricular and atrial Ito. NS5806 discriminates between rabbit ventricular and atrial Ito, with mixed activator and inhibitor actions on the former and inhibitor actions against the later. NS5806 may be of significant value for pharmacological interrogation of regional differences in native cardiac Ito.
Keywords: action potential; flecainide; Ito; Kv4.3, Kv 4.2, Kv1.4; NS5806; potassium channels; transient outward current
Introduction
Genetically distinct potassium (K+) ion channel currents are responsible for the repolarization of cardiac action potentials (APs) (Tamargo et al. 2004). The rapid and slow delayed rectifier K+ currents (IKr and IKs) contribute to ventricular AP repolarization over plateau voltages, while the inward rectifier K+ current (IK1) plays key roles in both terminal repolarization and in setting the resting membrane potential of nonpacemaker myocytes (Nerbonne 2000; Tamargo et al. 2004). In many species, including (for example) man, dog, ferret, rabbit, and rodent, initial rapid repolarization (phase 1) takes place before the AP plateau (phase 2). This arises from a combination of rapid inactivation of fast Na+ current (INa) and from the activation of a voltage‐dependent transient outward K+ current (Ito) and, in the atria, of ultrarapid delayed rectifier K+ current (IKur) (Nerbonne 2000; Tamargo et al. 2004). The pore‐forming subunits of channels that underlie Ito are derived from KCND3 (Kv4.3), KCND2 (Kv4.2), and KCNA4 (Kv1.4) genes (Nerbonne and Kass 2005; Niwa and Nerbonne 2010). Kv4.2 and 4.3 are believed to underlie an Ito that exhibits fast recovery kinetics (Ito,f), whilst Kv1.4 is responsible for Ito with slower kinetics (Ito,s) (Nerbonne and Kass 2005; Niwa and Nerbonne 2010). Regional and species differences in Ito are likely to result from the relative balance between these Ito subtypes (Niwa and Nerbonne 2010). Native Ito,f channels require interactions between Kv4.x and K+ Channel interacting Protein 2 (KChIP2), while other proteins (Kvβ, DPP6 and members of the KCNE family) may also modulate the current (Radicke et al. 2006; Niwa and Nerbonne 2010).
Ito contributes to phase 1 repolarization, but can also affect both the plateau (phase 2) and repolarization (phase 3) of the AP, due to the time‐ and voltage‐dependent behavior of IKr, IKs, and L‐type Ca2+ current (ICa,L) (Nerbonne 2000; Niwa and Nerbonne 2010). Reductions in Ito are seen in heart failure (HF) and human atrial fibrillation, and abnormal Ito regulation may also contribute to Brugada syndrome (Brandt et al. 2000; Antzelevitch 2006; Niwa and Nerbonne 2010). Indeed, pharmacological modification of Ito coupled with ICa,L block has recently been utilized as a way of studying electrogram fractionation in Brugada syndrome (Szel and Antzelevitch 2014; Patocskai et al. 2016). On the other hand, action potential clamping has shown that a loss of Ito in human APs directly leads to reduced and dyssynchronous Ca2+ release, raising the possibility that pharmacological Ito activation may have therapeutic value in HF (Cooper et al. 2010) (see also (Sah et al. 2003)). Consistent with this idea, data obtained from a canine HF model, using a single NS5806 concentration of 10 μmol/L to stimulate Ito, suggest that ventricular Ito stimulation may be able to mitigate electrophysiological changes in HF (Cordeiro et al. 2012).
In experiments on recombinant Kv4.x and Kv1.4 channels, the response of Kv4.3 to NS5806 has been shown to be modulated by co‐expression with KChIP2, while current carried by recombinant Kv1.4 channels was inhibited rather than activated by the compound (Lundby et al. 2010). It follows that the net effect of NS5806 on native Ito may vary both with the levels of Kv4.x/1.4 isoforms expressed as well as their possible association with KChIP2. To our knowledge, all studies to date of NS5806 effects on native Ito have used the dog (Calloe et al. 2009, 2010, 2011; Cordeiro et al. 2012) and the effect of NS5806 on native human ventricular Ito has yet to be ascertained. Some differences between canine and human Ito have been reported (Akar et al. 2004; Jost et al. 2013). Rabbits are widely used in studies of cardiac electrophysiology and can provide a cost‐effective alternative to larger species such as dog, while possessing ventricular action potentials closer to human than those from rats or mice (Milani‐Nejad and Janssen 2014; Camacho et al. 2016). Normal rabbit atrial and ventricular tissue each express Kv1.4, 4.2 and 4.3 (Wang et al. 1999; Bosch et al. 2003; Rose et al. 2005), and Kv1.4, Kv4.2, and Kv4.3 have all been detected by RT PCR in human ventricle (Kaab et al. 1998; Gaborit et al. 2007) although only the presence of Kv1.4 and Kv4.3 have been confirmed by Western blotting (Akar et al. 2004). While rabbit Ito is known to be slower than human Ito to recover from inactivation (e.g., Fermini et al. 1992; Mitcheson and Hancox 1999), it is nevertheless instructive to determine the effects of NS5806 on Ito from this species both to further knowledge of modulation of Ito from a widely used model species and for comparison with available information on canine Ito. The aim of this paper, therefore, was to study the modulatory effects of NS5806 on rabbit ventricular and atrial Ito. The results reveal distinct responses of rabbit atrial and ventricular Ito to NS5806.
Materials and Methods
Rabbit ventricular and atrial myocyte isolation and storage
Myocytes were isolated from the right ventricle and left atrium of hearts of male New Zealand White rabbits (2–3 kg). All procedures were in accordance with the UK Home Office Animals (Scientific Procedures) Act, 1986 and had institutional ethical approval. Ventricular and atrial myocytes were isolated by enzymatic and mechanical dispersion, using previously described methods (Hancox et al. 1993; Howarth et al. 1996). Cells were temporarily stored in a Kraft‐Brühe solution (Isenberg and Klockner 1982) at 4°C prior to electrophysiological recording.
Electrophysiological recording
Myocytes were placed in an experimental chamber mounted on an inverted microscope (Nikon Eclipse TE2000‐U) and superfused with a standard ‘normal’ Tyrode's solution containing (in mmol/L): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 5 HEPES (pH 7.4 with NaOH). This solution was used in all experiments to obtain the whole‐cell recording mode and was also used as superfusate for action potential measurements. For Ito measurements, the above solution was modified as previously described (Mitcheson and Hancox 1999): N‐methyl‐D‐glucamine (NMDG) chloride was substituted for NaCl and 20 μmol/L nifedipine was used to inhibit ICa,L. During experimental recordings, the superfusates were applied to the cell, using a home‐built device capable of exchanging solution bathing the cell in <1 sec (Levi et al. 1996). Borosilicate patch pipettes (A‐M Systems Inc, Sequim, WA) were pulled, using a Narishige vertical puller and fire‐polished (PP‐830 and MF83, Narishige Japan) to a resistance of 2–3 MΩ. For Ito recording, pipettes were filled with a solution containing (in mmol/L): 113 KCl, 10 HEPES, 0.4 MgCl2, 5 glucose, 5 K2ATP, 5 K4BAPTA (pH 7.2 with KOH). For AP recording, the pipette solution contained (in mmol/L): 110 KCl, 10 NaCl, 0.4 MgCl2, 10 HEPES, 5 glucose, 5 K2ATP, 0.5 GTP‐Tris (pH 7.1 with KOH). Series resistance values (typically 4–7 MΩ) were compensated by >70%. All recordings were made at 35–37°C. NS5806 (1‐[3,5‐bis(trifluoromethyl)phenyl]‐3‐[2,4‐dibromo‐6‐(2H‐tetrazol‐5‐yl)phenyl]urea) was obtained from Tocris (Bristol, UK) and dissolved in DMSO to produce stock solutions between 1 and 100 mmol/L (stored at −20°C). Stock solutions were diluted with the external solutions to obtain the final concentrations as given in the Results, with a final DMSO concentration in the superfusate of 1 in 1000 vv. Higher concentrations of stock solution in DMSO showed poor solubility in our hands, limiting the maximum concentration tested in the experimental solutions to 100 μmol/L. Flecainide was obtained from Sigma‐Aldrich (UK), and dissolved in distilled water to produce stock solutions between 1 and 100 mmol/L.
Data analysis
Data are presented as mean ± SEM, except for EC50/IC50 values derived from concentration‐response plots, for which 95% confidence intervals are given. Statistical analyses were performed, using Microsoft Excel (Microsoft) and Prism (GraphPad Software Inc.) and fits to particular datasets were made using either Prism or the Clampfit module of pClamp 10 (Axon Instruments, Molecular Devices). Statistical comparisons employed paired or unpaired t‐test, 1 or 2‐way ANOVA (with Bonferroni post‐test) as appropriate (P < 0.05 was taken as statistically significant).
Results
Concentration‐dependent effects of NS5806 on ventricular and atrial Ito
Prior canine studies have employed a single NS5806 concentration of 10 μmol/L for Ito experiments. Here, a wide range of concentrations (10 nmol/L to 100 μmol/L) was investigated against ventricular Ito. An exemplar ventricular Ito activated by depolarization from −80 mV to +40 mV in control solution, in the presence of 10 μmol/L NS5806 and following washout is shown in Figure 1A. The marked augmentation of Ito amplitude by NS5806 is apparent; this effect was largely reversible on drug washout. Current remaining after the initial time‐dependent, inactivating component was not altered by NS5806 at this concentration. Figure 1B shows the mean time course for augmentation of time‐dependent (peak minus end‐pulse) ventricular Ito at +40 mV by 10 μmol/L NS5806 (n = 26): the maximal response was seen within 1 min of drug application. The increase in Ito amplitude was accompanied by acceleration of Ito inactivation time course (mean inactivation thalf = 30.3 ± 2.4 msec in control and 21.5 ± 1.2 msec in 10 μmol/L NS5806; P < 0.01, n = 26). Despite this modest acceleration, the integral of the inactivating current was increased to 150.7 ± 10.5% of control (P < 0.01). Four additional concentrations of NS5806 were tested. At 1 μmol/L and 10 nmol/L, qualitatively similar but smaller responses to that with 10 μmol/L were seen. However, at higher concentrations (30 and 100 μmol/L), the response of peak Ito to NS5806 became biphasic with an initial increase in peak Ito followed by a decrease. Figure 1C shows representative traces for the effects of 100 μmol/L NS5806. The initial peak Ito (trace at 5 sec) showed a rapid increase in amplitude compared to control, but then declined to a level below that in control solution (trace at 2 min); this effect was poorly reversible. An additional effect of this concentration was a progressive increase in outward current following the initially inactivating current component. This secondary effect was partially reversible on washout. Figure 1D shows the time course of the biphasic effect of 100 μmol/L NS5806 on peak minus end‐pulse current amplitude (n = 10). In order to quantify the concentration‐dependence of NS5806 action, two concentration‐response relations were constructed: Figure 1E shows the relationship for the maximal stimulatory effect of the compound, whilst Figure 1F shows the relationship at steady‐state effect. The derived EC50 for augmentation of peak minus end‐pulse Ito (Fig. 1E) was 1.6 μmol/L (LogEC50 mean ± SEM: −5.80 ± 0.12; 95% C.I: 0.6–3.9 μmol/L), with a Hill slope of 0.55 ± 0.08. For a similar plot for augmentation of the peak current amplitude (not shown), the derived EC50 was also 1.6 μmol/L (LogEC50 mean±SEM: −5.81 ± 0.21; 95% C.I: 0.3–7.1 μmol/L), with a Hill slope of 0.58 ± 0.14. The peak minus end‐pulse data in Figure 1F could not be described by a single Hill equation, but could be fitted by two site model in which the EC50 describing augmentation of Ito was fixed to the value obtained from Figure 1E (1.6 μmol/L), whilst the IC50 value derived for the descending phase of the relationship was 40.7 μmol/L (LogIC50 mean ± SEM: −4.39 ± 0.13; 95% C.I: 11.7–112.2 μmol/L), with a Hill slope of −1.15 ± 0.22. A similar analysis of the biphasic effect of NS5806 on the peak current amplitude (not shown), again utilizing an EC50 of 1.6 μmol/L for augmentation of Ito, yielded an IC50 for the descending phase of the relationship of 21.2 μmol/L (LogIC50 mean ± SEM: −4.67 ± 0.30; 95% C.I: 10 μmol/L to 143 mmol/L), with minimum of 74% of control and a Hill slope of −1.09 ± 0.50.
Figure 1.
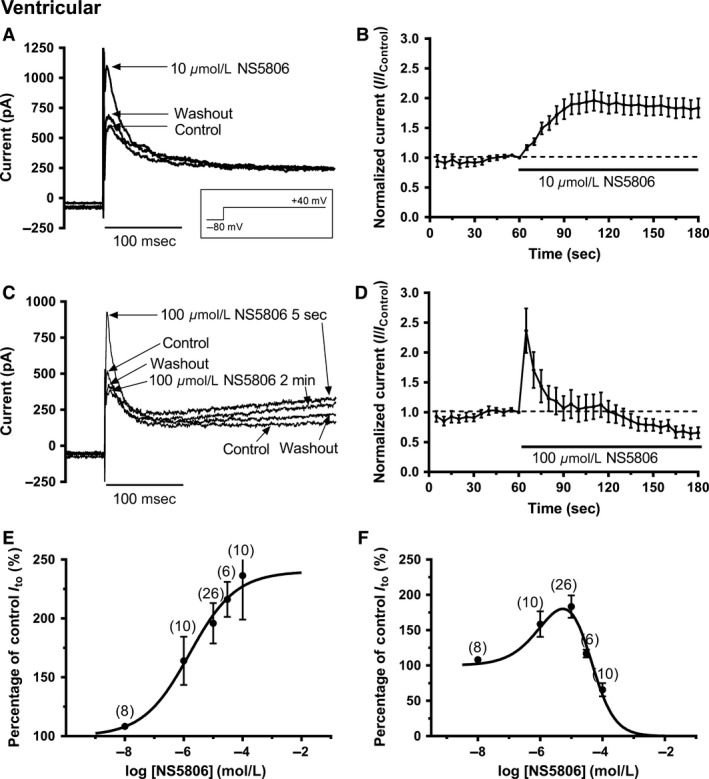
NS5806 modulation of I to from rabbit ventricular cells. (A) Representative current records in control, after application of 10 μmol/L NS5806 and after washout with control solution. The voltage protocol is shown as an inset. (B) Mean (± SEM, n = 26) time course of response to 10 μmol/L NS5806 of Ito (measured as peak minus end‐pulse current), using protocol shown in A. (C) Representative current records in control, after application of 100 μmol/L NS5806 and after washout with control solution. The voltage protocol is the same as that used for panel A. (D) Mean (± SEM, n = 10) time course of response of Ito (peak minus end‐pulse current) to 100 μmol/L NS5806, using protocol shown in A. Note the initial stimulation followed by inhibition. (E) Concentration‐dependence of the maximal agonist effect of NS5806 on Ito. Data (for peak minus end‐pulse current effects) were fitted with a standard Hill‐equation to get the EC 50 and nH values given in the ‘Results’ text. Values in parentheses denote number of independent replicates at each concentration. (F) Concentration–response relation for the steady‐state effect of NS5806 on Ito. For each cell, the effect of NS5806 at 120 sec was recorded and current amplitude expressed as a % of control. Data (for peak minus end‐pulse current effects) were fitted with a two site (agonist and antagonist) Hill‐equation to get the EC 50/IC 50 and nH values. Values in parentheses denote number of independent replicates at each concentration.
Figure 2A shows representative traces of atrial Ito activated by depolarization from ‐80 mV to +40 mV in control solution, in the presence of 10 μmol/L NS5806 and following washout. In contrast to the effects seen on ventricular Ito, NS5806 reduced atrial Ito amplitude and this was accompanied by a modest slowing of Ito inactivation time course (inactivation thalf in control of 13.5 ± 0.9 msec and in 10 μmol/L NS5806 of 17.3 ± 1.0 msec; P < 0.01, n = 21). The current remaining after the initial time‐dependent inactivating current was little affected by this concentration of NS5806. The integral of inactivating current in 10 μmol/L NS5806 for atrial cells decreased to 70.9 ± 6.6% of control (P < 0.01) and inhibitory effects of this NS5806 concentration did not fully reverse on washout. Figure 2B shows the mean time course of action of 10 μmol/L NS5806 (n = 21) on peak minus end‐pulse Ito. Figure 2C contains representative traces showing the effect of 100 μmol/L NS5806. The rapidly activating peak Ito was strongly suppressed at this concentration of NS5806. Residual current was somewhat elevated but no progressively activating outward current was seen at this concentration, in contrast to the effect seen in ventricular cells (compare Fig. 1C and 2C). Figure 2D shows the mean time course of action of 100 μmol/L NS5806 (n = 9) on peak minus end‐pulse Ito. Figure 2E shows mean concentration‐response data for 1, 10 and 100 μmol/L on peak minus end‐pulse current. A fit to these data with a one site Hill equation yielded an IC50 of 18.2 μmol/L (LogIC50 mean ± SEM: −4.74 ± 0.05; 95% C.I: 4.2–80.0 μmol/L; Hill coefficient: −0.74 ± 0.06). Analysis of peak current inhibition gave an IC50 of 34.7 μmol/L (LogIC50 mean ± SEM: −4.46 ± 0.04; 95% C.I: 11.9–101.8 μmol/L; Hill coefficient: −0.54 ± 0.03).
Figure 2.
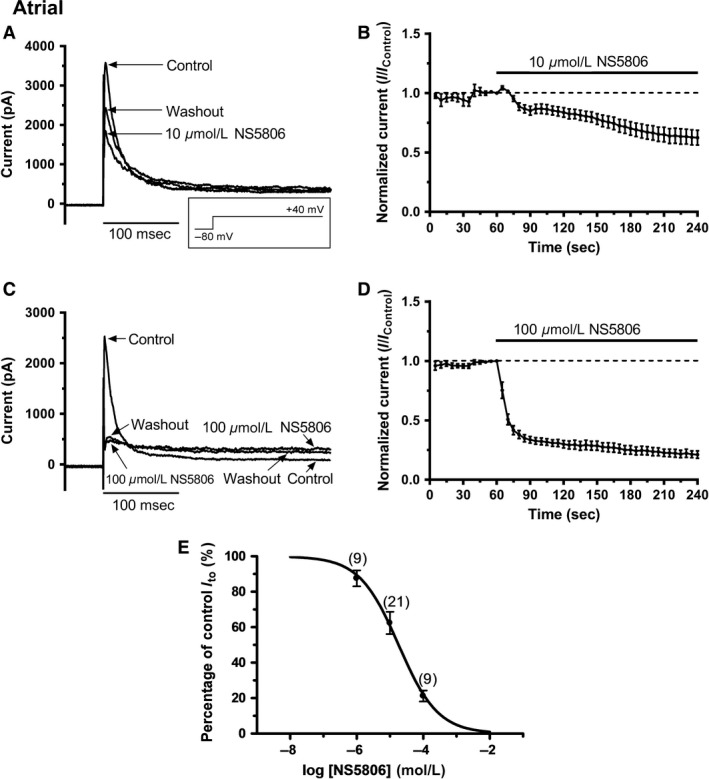
NS5806 inhibition of I to from rabbit atrial cells. A: Representative current records in control, 10 μmol/L NS5806 and washout with control solution elicited by the protocol shown as an inset (same protocol as Figure 1). B: Mean (± SEM, n = 21) time course of response to 10 μmol/L NS5806 of Ito (measured as peak minus end‐pulse current, using protocol shown in A. C: Representative current records in control, after application of 100 μmol/L NS5806 and after washout with control solution. The voltage protocol is the same as that used for panel A. D: Mean (± SEM, n = 9) time course of response of Ito (peak minus end‐pulse current) to 100 μmol/L NS5806, using protocol shown in A. E: Isochronal concentration–response relation for the inhibition of atrial Ito by NS5806. For each cell, the effect of NS5806 at 180 s (on peak minus end‐pulse currents) was recorded and current amplitude expressed as a % of control. Data were fitted with a Hill‐equation to get the IC 50 and nH values given in the ‘Results’ text.
Effects of NS5809 on voltage‐dependent activation and inactivation of Ito
The voltage dependence of activation and inactivation of Ito were determined, using a classical Hodgkin‐Huxley protocol ((Mitcheson and Hancox 1999); see Figure 3 and Figure 4 legends for details). Figures 3Ai and Aii show families of ventricular Ito elicited by depolarization to a range of membrane potentials both in the absence and presence of 10 μmol/L NS5806. Peak Ito was increased by 10 μmol/L NS5806 (n = 7) at all potentials greater than 0 mV, as shown the current‐voltage (I‐V) plots in Figure 3Aiii (data normalized to cell capacitance). No significant difference in mean end‐pulse current was seen between control and 10 μmol/L NS5806 between −60 and +50 mV (P > 0.05). Figure 3Aiv shows the voltage dependence of Ito activation derived from normalized conductance voltage (G‐V) plots, with Boltzmann fits used to derive half‐maximal activation voltage (V0.5) and slope factor (k a). In control solution, ventricular Ito activation V0.5 was +25.3 ± 2.6 mV (k a=25.4 ± 3.1 mV), whilst in 10 μmol/L NS5806 V0.5 was −3.4 ± 2.7 mV (P < 0.01 versus control; k a = 16.7 ± 1.1 mV, also P < 0.01 versus control).
Figure 3.
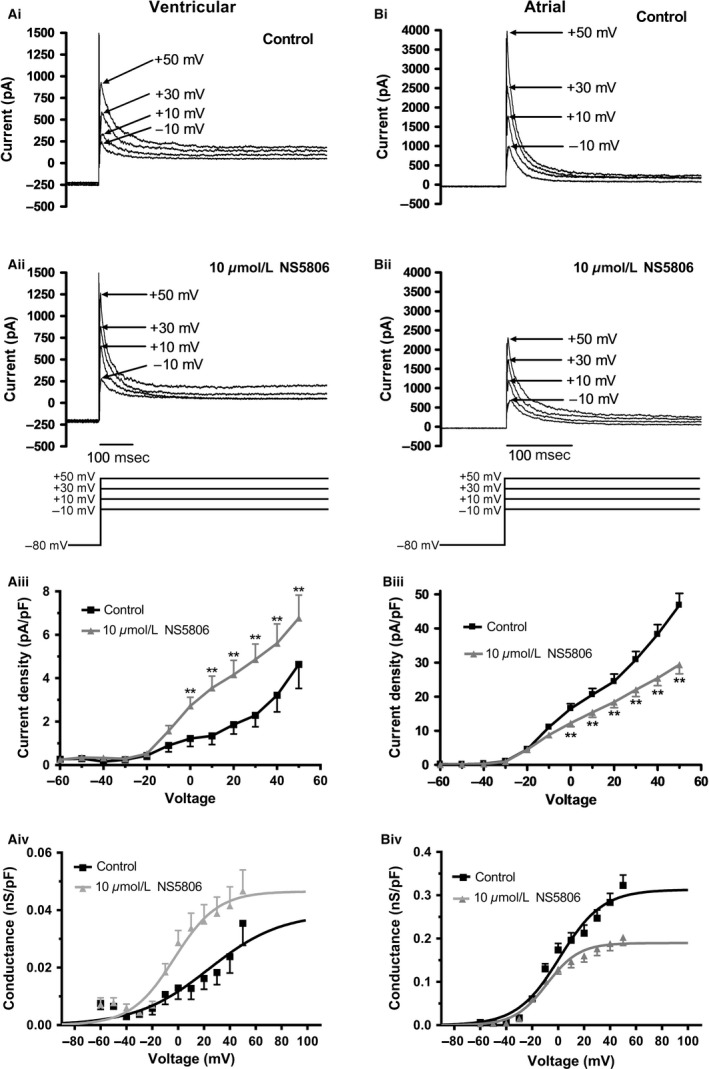
Effects of NS5806 on voltage dependence of I to activation. Ai‐Aii: Representative ventricular current traces with control solution (Ai) and 10 μmol/L NS5806 (Aii) at the potentials indicated (protocol shown as lower panel of Aii). From the holding potential of ‐80 mV, an initial 1‐second duration ‘conditioning’ step was applied to potentials between −90 and +50 mV in 10 mV increments. The conditioning step both enabled activation of Ito (on depolarization) and also enabled subsequent inactivation during the maintained depolarization. A second 500 msec ‘test’ step to +40 mV was applied to determine how availability (inactivation) of Ito was influenced by the conditioning pulse. A brief (3‐msec) step at −80 mV was included between the first and second steps to ensure that any residual capacitance artefacts that occurred during the test depolarization were not influenced by differing conditioning voltages. Interpulse interval was 5 sec. Aiii: Mean I‐V relations (normalized to cell capacitance) for ventricular Ito elicited by the initial 1s step of protocol described above, in control and in 10 μmol/L NS5806 (same protocol as Ai,Aii; n = 7). Control data are shown in black; NS5806 data are shown in gray (error bars indicate SEM). ** denotes significant difference at P < 0.01. Aiv: Voltage‐dependence of conductance for ventricular Ito (same experiments as shown in Aiii). Data were fitted with a Boltzmann equation of the form: G/Gmax=1/[1 + exp[(V0.5‐V)/ka]], where G=conductance at test voltage V, Gmax= maximal conductance, V0.5=half‐maximal activation voltage, and ka=activation slope factor. V0.5 and ka values are given in the ‘Results’ text. Bi‐Bii: Representative atrial current traces with control solution (Bi) and 10 μmol/L NS5806 (Bii) at the potentials indicated. The protocol was the same as for ventricular cells as shown in Ai. Biii: Mean I‐V relations (normalized to cell capacitance) for atrial Ito in control and in the presence of 10 μmol/L NS5806 (same protocol as Bi,Bii; n = 8). Control data are shown in black (and with +SEM bars); NS5806 data are shown in gray (and with ‐SEM bars). ** denotes significant difference between control and NS5806 at P < 0.01. Biv: Voltage‐dependent activation curves for atrial Ito (data from same experiments as Biii). For each experiment and each recording condition (control and NS5806) macroscopic conductance values were calculated at each voltage, normalized to maximal conductance during the protocol and pooled data fitted with the Boltzmann equation as described above.
Figure 4.
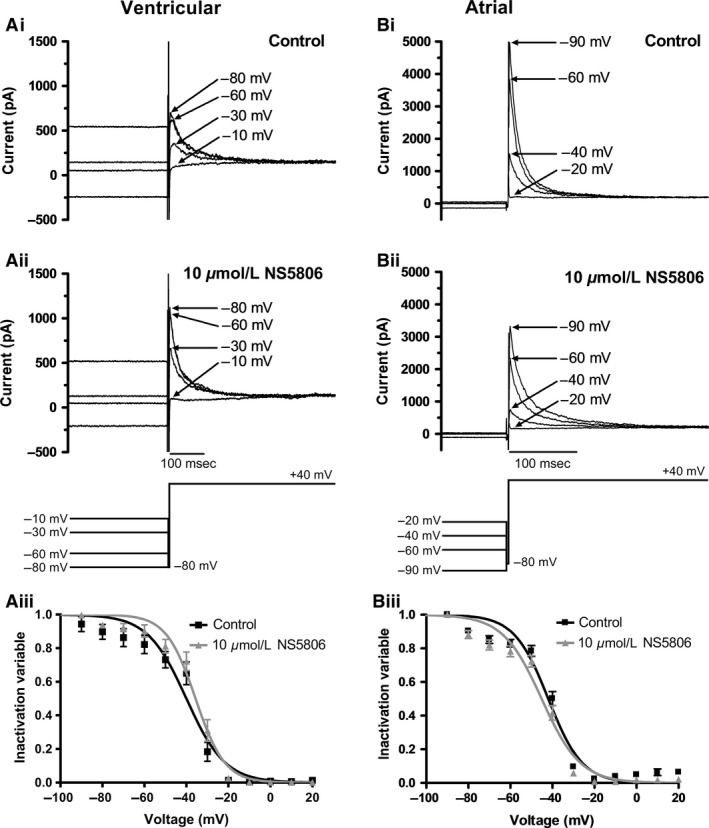
Effect of NS5806 on voltage‐dependent inactivation of I to. Ai–Aii: Representative ventricular current traces with control solution (Ai) and 10 μmol/L NS5806 (Aii) elicited by protocol shown as lower panel of Aii. Full protocol contained 1 sec conditioning steps in 10 mV increments between −90 mV and +20 mV, followed by a 500 msec test pulse to +40 mV. Conditioning and test steps were separated by a brief (3 msec) period at −80 mV. The figure focuses on currents elicited by the test step following conditioning steps to the voltages indicated. Currents at selected voltages are shown for clarity of display. Aiii: Mean (± SEM) plots of inactivation variables against conditioning voltage in control and in the presence of 10 μmol/L NS5806 (n = 7). For each experiment and each condition, currents during each test command were normalized to the maximal test current observed during the protocol, pooled and plotted against conditioning voltage. Data were fitted by a Boltzmann function: I/Imax=1‐[1/[1 + exp((V0.5‐V)/ki)]], where I=current during the test pulse (+40 mV), V= conditioning voltage, Imax= maximal test current, V0.5=half‐maximal inactivation voltage, and ki=inactivation slope factor. V0.5 and ki values are given in the Results text. Bi–Bii: Representative atrial current traces with control solution (Bi) and 10 μmol/L NS5806 (Bii) elicited by protocol shown as lower panel of Bii. Voltage protocol as described for ‘A’. The figure shows currents elicited by the test step after selected conditioning steps (voltages indicated on traces). Biii: Mean (± SEM) plots of atrial inactivation variables against conditioning voltage in control and NS5806 (n = 8). Data were fitted by the Boltzmann equation described in ‘A’. V0.5 and k i values are given in the Results text.
Figures 3Bi and Bii show families of atrial Ito during depolarizations to a range of voltages and demonstrate that, in marked contrast to ventricular myocytes, peak Ito was decreased by 10 μmol/L NS5806 over the range of potentials tested. Mean I‐V relations in control and after application of 10 μmol/L NS5806 (n = 8; normalized to cell capacitance) are shown in Figure 3Biii and NS5806 significantly reduced Ito amplitude at all voltages greater than 0 mV. No significant difference in mean end‐pulse current was seen between control and 10 μmol/L NS5806 between −60 and +50 mV (P > 0.05). Figure 3Biv shows normalized G‐V plots of atrial Ito fitted with a Boltzmann function to derive activation parameters. The activation V0.5 for atrial Ito in control was 2.8 ± 2.5 mV (k a=16.7 ± 1.3 mV), whilst in NS5806 it was ‐8.6 ± 1.5 mV (P < 0.01 versus control; k a = 12.3 ± 0.9 mV, also P < 0.01 vs. control).
Thus, NS5806 produced a leftward shift and decrease in slope in the voltage dependence of activation of Ito in both cell types, though the magnitude of this effect was much greater in ventricular than atrial myocytes.
Figures 4Ai and Aii show families of Ito elicited by the test depolarization in ventricular myocytes following different conditioning steps in both control (Fig. 4Ai) and after adding 10 μmol/L NS5806 (Fig. 4Aii). Under both conditions, Ito was greater at more negative conditioning voltages. After normalizing the test pulse Ito to the maximal test Ito observed following the different conditioning pulses protocol and fitting a Boltzmann function (Fig. 4Aiii; n = 7) the half‐maximal inactivation voltage (V0.5) and slope factor (k i) values were not significantly changed by NS5806 (Control: V0.5 of ‐40.9 ± 2.7 mV; k i = 9.1 ± 1.7 mV and with NS5806: V0.5 of −36.2 ± 2.0 mV; k i = 6.9 ± 1.1 mV; P > 0.1 for both).
Figure 4Bi and Bii shows equivalent data for atrial Ito in control and NS5806 and Figure 4Biii shows mean data and Boltzmann fits. In eight experiments the mean atrial Ito inactivation V0.5 in control was ‐42.3 ± 1.4 mV which was shifted to −45.6 ± 1.6 mV in 10 μmol/L NS5806 (P < 0.01). The slope factors appeared unchanged; in control k i was 8.0 ± 1.0 mV and in NS5806 k i was 9.1 ± 1.1 mV (P > 0.05 vs. control). Thus, NS5806 produced a modest but significant leftward shift in voltage‐dependent inactivation of atrial Ito, with no significant shift in inactivation of ventricular Ito.
Effects of NS5806 on Ito restitution
In order to measure restitution of ventricular Ito (recovery from inactivation) a paired‐pulse protocol (shown schematically in the insets to Fig. 5A and B) was used (Mitcheson and Hancox 1999). Figure 5A shows mean data from six experiments in which restitution of Ito from ventricular cells was measured in control solution and following exposure to 10 μmol/L NS5806. In both control and NS5806, Ito restitution followed a single exponential time course, with time constants of 2417 ± 117 msec and 1814 ± 82 msec in control and NS5806, respectively (P < 0.01, n = 6). Restitution of Ito from atrial cells (Fig. 5B) was best described by a bi‐exponential time course: the fast component had time constants of 452 ± 146 msec and 521 ± 287 msec in control and with NS5806, respectively, while for the slow component the corresponding values were 3023 ± 241 msec and 3045 ± 400 msec, respectively. The fraction of fast atrial Ito restitution was 21.2 ± 6.0% in control and 20.0 ± 10.8% in NS5806. None of these values differed significantly between control and NS5806 (n = 7). When restitution of atrial cell Ito was additionally fitted with monoexponential function to facilitate comparison with ventricular Ito, this yielded time constants in control and NS5806, respectively of 2147 ± 57 msec and 2253 ± 71 msec (n = 7; P > 0.05). Taken together, these data indicate that NS5806 significantly accelerated restitution of Ito from rabbit ventricular cells, but did not significantly affect restitution of Ito from atrial cells.
Figure 5.
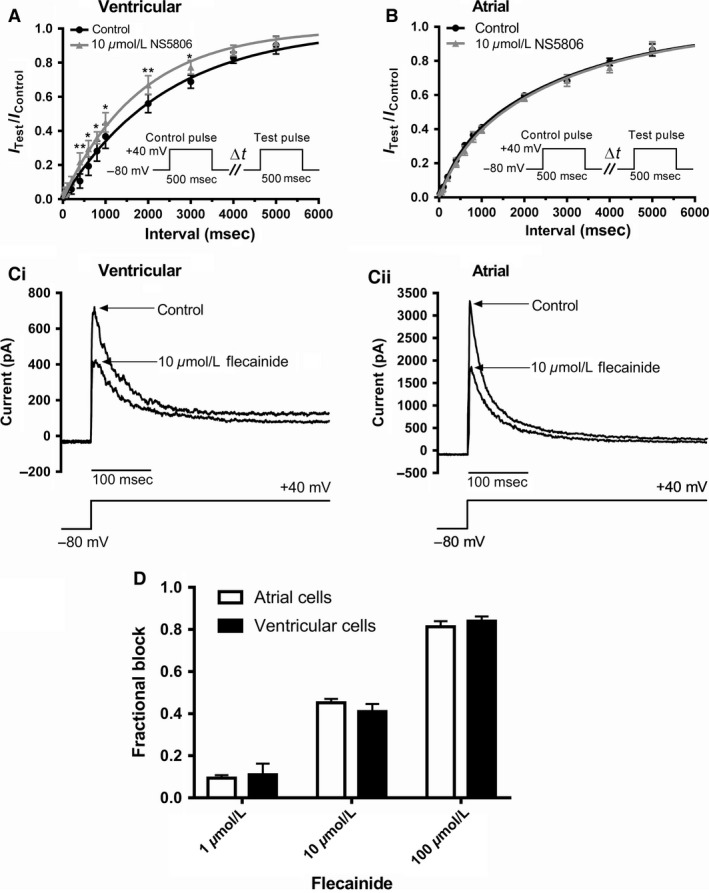
Effect of NS5806 on recovery of I to from inactivation (“restitution”) in atrial and ventricular myocytes and response to flecainide. The protocol for studying restitution is illustrated in the insets to panels A and B: an initial 500 msec depolarizing step from −80 mV to +40 mV was followed by varying intervals (Δt, 20 msec to 5000 ms) at −80 mV followed by a ‘test’ depolarization to +40 mV. Each pair of pulses was separated by 10 sec. For each pulse‐pair, the magnitude of Ito elicited by the second command (IT est) was expressed as the fraction of that elicited by the first (IC ontrol). A: Mean (± SEM) data (n = 6) for recovery of Ito from inactivation for ventricular myocytes, in control (black) and 10 μmol/L NS5806 (gray). Data were fitted by a single exponential function to get time constant values given in the Results. *difference between Control and NS5806 at P < 0.05; ** P < 0.01. B: Mean (± SEM) data (n = 7) for recovery of Ito from inactivation for atrial myocytes, in control (black) and 10 μmol/L NS5806 (gray). Data were fitted by a bi‐exponential function to get time constant values given in the Results text. C: Representative traces of Ito in control and following exposure to 10 μmol/L flecainide (same protocol as used in Figures 1 and 2) for ventricular (Ci) and atrial (Cii) myocytes. D: Bar chart plots for flecainide inhibition of ventricular and atrial Ito (n = 6–7 cells for each concentration for both cell types). 2‐way ANOVA with Bonferroni's post‐test confirmed that for each cell type the concentration dependence of the inhibitory effect was significant (P < 0.05), whilst at no concentration did the extent of inhibition differ significantly between atrial and ventricular cells.
Effect of flecainide on ventricular and atrial Ito
Since Ito arises from multiple channel isoforms the atrial‐ventricular differences in response to NS5806 might reflect different functional Kv1.4 and Kv4.x tissue expression. Flecainide has been reported to discriminate between Kv4.x and 1.4 channels, with the latter exhibiting lower sensitivity to inhibition by the drug (Yeola and Snyders 1997; Singarayar et al. 2003; Herrera et al. 2005). Effects of flecainide on rabbit ventricular and atrial Ito were therefore examined to probe the functional expression of these channel subunits. Figure 5Ci shows representative ventricular Ito traces in the absence and presence of 10 μmol/L flecainide, whilst Figure 5Cii shows comparable data for atrial Ito. The bar charts in Figure 5D show mean fractional block for Ito from the two cell types with 1, 10 and 100 μmol/L flecainide (n ≥ 6 for each concentration). At no concentration did the inhibitory effect of flecainide differ significantly between atrial and ventricular cells. When the data for each cell type were fitted to standard concentration‐response relations to estimate IC50 values (constraining minimal and maximal possible fractional block values to 0 and 1, respectively; plot not shown) the derived value for ventricular Ito was 14.7 μmol/L, (LogIC50 mean ± SEM = −4.83 ± 0.03, 95% C.I = 5.6–39.0 μmol/L; nH = 0.83 ± 0.05), whilst for atrial Ito, the derived IC50 was 13.8 μmol/L (LogIC50 mean ± SEM = −4.86 ± 0.05, 95% C.I = 3.2 to 60.3 μmol/L; nH of 0.79 ± 0.07). Thus, in contrast to their distinct responses to NS5806, ventricular and atrial Ito exhibited similar sensitivity to inhibition by flecainide.
Effects of NS5806 on ventricular and atrial APs
In a final set of experiments, the action of 10 μmol/L NS5806 on ventricular and atrial AP profiles was compared. For both cell types, APs were elicited in membrane potential (current clamp) recording mode, by brief (5–7 msec) duration suprathreshold depolarizing current pulses (0.6‐1 nA for ventricular myocytes and 0.4–0.5 nA for atrial myocytes) at a stimulation frequency of 0.5 Hz. Figure 6A shows representative ventricular APs in control and following application of 10 μmol/L NS5806. The compound had no significant effect on the AP upstroke or initial overshoot (see Table 1); however, AP duration (APD) was abbreviated in the presence of the drug. We evaluated AP shortening at 20%, 50% and 90% repolarization (APD20, APD50, APD90), respectively. NS5806 shortened APD20 by 36.5 ± 5.0%, APD50 by 31.2 ± 3.3% and APD90 by 24.7 ± 3.0% (n = 7 for all; see Table 1 for absolute APD values). Figure 6B shows representative atrial APs in control solution and following application of 10 μmol/L NS5806. In contrast to the AP shortening seen for ventricular APs, atrial APD was prolonged by the drug, particularly during early repolarization. APD20, APD50 and APD90 were prolonged by 90.9 ± 14.7%, 88.6 ± 18.8% and 30.7 ± 12.0%, respectively (n = 7 for all; see Table 1). In addition, and in contrast to ventricular myocytes, atrial AP overshoot and upstroke were also affected (Table 1), with a marked (77.4 ± 3.8%) reduction in upstroke velocity in accord with dog atrial data in a previous report (Calloe et al. 2011). Further experiments with a higher concentration (100 μmol/L) of NS5806 were not attempted, because the likely lack of selectivity of this concentration for ventricular Ito (Fig. 1C) would have make its effects on APs difficult to interpret.
Figure 6.
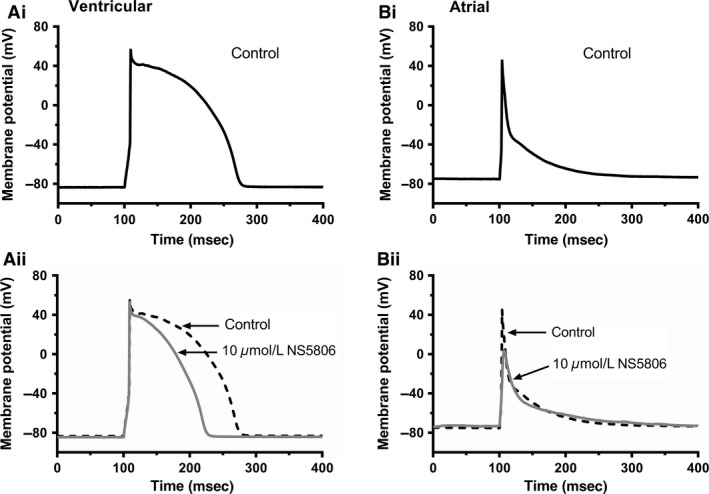
Effect of 10 μmol/L NS5806 on ventricular and atrial action potentials Ai–Aii: Representative ventricular action potentials in control (Ai, black) and in 10 μmol/L NS5806 (Aii, gray, with control action potential superimposed as dashed black line). Bi–Bii: Representative atrial action potentials in control (Bi, black) and in 10 μmol/L NS5806 (Bii, gray, with control action potential superimposed as dashed black line). For A and B, depolarizing stimuli were applied at 2 sec intervals. Mean ventricular cell resting potential of −81.5 ± 0.7 mV was obtained with zero current injection. Atrial cell resting membrane potential was somewhat depolarized (~−50 to −40 mV) with zero current and so a small hyperpolarizing (−50 pA) current was injected to give the mean resting potential of −79.9 ± 1.9 mV. Mean action potentials parameters for both cell types in Control and NS5806 are given in Table 1.
Table 1.
Effect of 10 μmol/L NS5806 on action potentials from rabbit ventricular and atrial myocytes
| Ventricular cells (n = 7) | Atrial cells (n = 7) | |||
|---|---|---|---|---|
| Control | 10 μmol/L NS5806 | Control | 10 μmol/L NS5806 | |
| Overshoot (mV) | 56.0 ± 2.0 | 54.8 ± 2.0 | 43.5 ± 3.2 | −2.3 ± 4.4 b |
| AP amplitude (mV) | 137.5 ± 1.9 | 137.0 ± 2.0 | 123.2 ± 2.8 | 73.9 ± 3.8 b |
| Upstroke velocity (mV/msec) (percentage change,%) | 128.4 ± 8.6 | 134.3 ± 7.0 | 120.9 ± 8.3 | 27.4 ± 4.9 b(−77.4 ± 3.8) |
| APD20 (msec) (percentage change,%) | 73.6 ± 8.0 | 46.0 ± 4.9 b (−36.5 ± 5.0) | 8.2 ± 1.1 | 16.0 ± 2.8 b(90.9 ± 14.7) |
| APD50 (msec) (percentage change,%) | 133.8 ± 8.4 | 91.6 ± 6.4 b (−31.2 ± 3.3) | 18.0 ± 2.8 | 32.9 ± 5.8 b(88.6 ± 18.8) |
| APD90 (msec) (percentage change,%) | 167.4 ± 9.7 | 125.9 ± 8.8 b (−24.7 ± 3.0) | 101.8 ± 7.3 | 128.6 ± 5.9 a(30.7 ± 12.0) |
P < 0.05.
P < 0.01, paired t‐test.
Discussion
Comparison with prior canine ventricular and atrial Ito data
To our knowledge, this is the first study to investigate the concentration‐dependent effects of NS5806 on native cardiac Ito. Previous work in dogs has shown that NS5806 increases the depth of phase 1 repolarization in both left and right ventricles in a concentration‐dependent fashion between 5 and 15 μmol/L and, when phase 1 repolarization became very pronounced, could lead to AP collapse (Calloe et al. 2009). A lack of concentration‐response data on canine ventricular Ito for NS5806 means that direct comparison with our data is limited to the typical 10 μmol/L concentration used in most prior dog studies (Calloe et al. 2010, 2011; Cordeiro et al. 2012). Table 2 compares the effects of NS5806 on rabbit and normal canine ventricular Ito. The agonist effect of NS5806 at 10 μmol/L is similar between the two species. However, concentration‐response data are not available for canine ventricular Ito to determine whether or not the biphasic concentration response relation we obtained at steady‐state is shared by the two species. Voltage‐dependent activation data are also lacking for dog Ito, precluding comparison with the marked leftward shift in activation V0.5 found here for rabbit Ito. Differences between the dog and rabbit Ito response to NS5806 are: (1) an apparent acceleration, not slowing of rabbit ventricular Ito inactivation time course with the compound (either as a result of direct inactivation modulation or some modest open channel block during the inactivating phase of the current); (2) no significant shift in voltage‐dependent inactivation V0.5 with NS5806 was seen in rabbit.
Table 2.
Comparison of effects of NS5806 on normal rabbit and dog ventricular Ito
| Ito property | Rabbit | Source | Dog | Reference |
|---|---|---|---|---|
| Ventricle | ||||
| Current amplitude |
Initial: ↑ EC50 1.6 μmol/L Steady state: “bell‐shaped” EC50 1.6 μmol/L; IC50 40 μmol/L |
This study |
↑ at 10 μmol/L ↑ at 10 μmol/L (Epi by 80%, Mid by 82% Endo by 16%) |
Calloe et al. 2009
Calloe et al. 2010, 2011 |
| Voltage dependence of activation | Negative shift in V0.5 of ~ −29 mV | This study | No data | |
| Time course of inactivation | Accelerated: thalf at +40 mV decreased from 30.3 msec to 21.5 msec (by 29%) | This study |
Slowed: Tau at +40 mV increased from 12.6 to 20.3 ms (by 61%) Ito integral increased to 227%, 192% and 83% of control in EPI, MID and EPI |
Calloe et al. 2009
Calloe et al. 2010; |
| Voltage dependence of inactivation | No statistical difference | This study | Negative shift in V0.5 of −6 mV EPI, −5 mV MID, −3.4 ENDO | Calloe et al. 2010 Calloe et al. 2011 |
| Restitution | Accelerated: tau from 2417 msec to 1814 msec (by ~25%) | This study | Accelerated EPI and MID and biexponential to single exponential time course |
Calloe et al. 2009
Calloe et al. 2010 |
The effects of NS5806 on rabbit atrial Ito differed significantly both from those seen in rabbit ventricular myocytes in this study and in canine atrial cells (summarized in Table 3). We observed a concentration‐dependent inhibition of atrial Ito amplitude (Fig. 2), accompanied by a ~−11 mV shift in voltage‐dependent activation (Fig. 3), a ~−3 mV shift in voltage‐dependent inactivation (Fig. 4), slowed inactivation time course, but unchanged restitution (Fig. 5). Canine atrial Ito was modestly increased (25%) by NS5806, and its restitution was accelerated – effects that differ markedly from those seen here in rabbit. No canine data are available on effects on voltage dependence of atrial Ito activation, whilst effects on Ito inactivation time course and voltage dependence are similar between the two species. The marked inhibitory effect of NS5806 on Ito accounts for atrial AP prolongation seen in our experiments (Fig. 6, Table 1). 10 μmol/L NS5806 was reported to not alter phase 1 repolarisation in perfused dog atrial preparations, but shortened the APD90 (Calloe et al. 2011).
Table 3.
Comparison of effects of NS5806 on normal rabbit and dog atrial Ito
| Ito property | Rabbit | Source | Dog | Reference |
|---|---|---|---|---|
| Atrium | ||||
| Current amplitude | ↓ IC50 18.2 μmol/L | This study | ↑ at 10 μmol/L (25%) | Calloe et al. 2011 |
| Voltage dependence of activation | Negative shift in V0.5 of ~ 11 mV | This study | No data | |
| Time course of inactivation | Slowed: thalf at +40 mV increased from 13.5 msec to 17.3 msec (by 28%) | This study | Slowed: Tau at +50 mV increased from 20 to 26.5 ms (by 32.5%) | Calloe et al. 2011 |
| Voltage dependence of inactivation | Negative shift in V0.5 of ~ ‐3.3 mV | This study | Negative shift in V0.5 of ‐7.3 mV | Calloe et al. 2011 |
| Restitution | No significant change | This study | Accelerated and biexponential changed to single exponential time course | Calloe et al. 2011 |
In atrial, but not ventricular myocytes, NS5806 produced a substantial slowing of AP upstroke velocity and amplitude (Fig. 6, Table 1), consistent with a selective reduction in atrial INa. Such ‘off target’ actions of NS5806 on AP upstroke velocity were noted in dog atrial tissue and found to correlate with intrinsic atrial‐ventricular differences in INa inactivation kinetics that may favor atrial INa inhibition by the compound (Calloe et al. 2011). Thus, our own observations in respect of effects of NS5806 on atrial AP upstroke velocity are consistent with previously reported atrial‐ventricular differences in INa and atrio‐selectivity of drug INa modulation (Burashnikov et al. 2007; Calloe et al. 2011; Suzuki et al. 2013).
On the mechanism of NS5806 action
The decrease in the slope factor for voltage‐dependent activation of ventricular Ito suggests that NS5806 either effectively alters the membrane field sensed by the Ito voltage sensor or decreases the net effective charge of the voltage sensor. The positive residues in the S4 region play a key role in forming the voltage sensor of Kv channels (for review see Swartz 2004), and since NS5806 should be negatively charged at pH7.2, it could decrease the slope of the activation curve by binding near the voltage sensor. However, NS5806 may also bind and exert effects outside the immediate S4 region. Consistent with this, NS5806 has been reported to produce an agonist action on Kv4.3/KChIP2/DPP6 channels expressed in mammalian CHO‐K1 cells and a smaller agonist effect on Kv4.3/DPP6 in Xenopus oocytes, whilst peak current carried by Kv4.3 alone was reduced by NS5806 (Lundby et al. 2010). The effects of NS5806 on inactivation (and recovery from inactivation) of Kv4.3 also seem to be sensitive to the interaction of NS5806 with KChIP2 (Lundby et al. 2010). Moreover, in canine ventricular myocytes, variation in response to NS5806 across the ventricular wall correlated with varying transmural KChIP2 expression levels in the presence of similar transmural levels of Kv4.3 (Calloe et al. 2010). Thus, to stimulate Ito, it seems likely that NS5806 either interacts directly with the Kv4.3‐KChIP2 accessory subunit complex, or the interaction between Kv4.3 and KChIP2 exposes an interaction site for NS5806 on the Kv4.3 protein. In this regard, it is notable that a recent study investigating effects of NS5806 on the interaction between Kv4.3 and the KChIP2 relative KChIP3 has provided evidence that NS5806 binds at a hydrophobic site on the C terminus of KChIP3 and increases the affinity between KChIP3 and the N terminus of Kv4.3 (Gonzalez et al. 2014). Significantly, alignment of KChIP3 and KChiP2 (Uniprot Q9Y2W7 and Q9NS61, respectively) indicates that hydrophobic amino acid residues in KChiP3 (Tyr‐174 and Phe‐218) identified to be important for NS5806 binding (Gonzalez et al. 2014) are present in analogous positions in KChIP2, making it likely that the two interact similarly with NS5806.
Our data on ventricular Ito showed a biphasic concentration response relation to NS5806, with higher concentrations producing an initial stimulation followed by inhibition. In prior investigation of recombinant Kv channels, the response of Kv4.3/KChIP2/DPP6 to 100 μmol/L NS5806 was smaller than that at 10 μmol/L (see Fig. 2B in Lundby et al. 2010 at 100 μmol/L –although this data‐point was excluded from the concentration‐response fit). In the same study, for Kv4.3/KChIP2 and Kv4.3/KChIP2/DPP6, concentrations up to 10 μmol/L increased current amplitude and 30 μmol/L produced some reduction (Fig. 3 in Lundby et al. 2010). In a different study directed toward the molecular pharmacology of hippocampal A‐current (based on Kv4.2 rather than Kv4.3), NS5806 increased Kv4.2/KChIP2 current amplitude at concentrations up to 20–60 μmol/L, with an EC50 of 5.6 μmol/L, but was inhibited at 200 μmol/L (Witzel et al. 2012). Importantly, when Kv4.2/DPP6S or Kv4.2/KChiP3/DPP6a were co‐expressed, NS5806 produced a low‐affinity monophasic inhibition of the A current. These results support the idea that NS5806 interacts at more than one site to affect Kv4.x channels, with a lower affinity site, possibly on accessory subunits, mediating the inhibitory action. However, as Kv1.4 is inhibited by NS5806 (Lundby et al. 2010), an additional factor to be considered is contribution of Kv1.4 to the overall macroscopic rabbit Ito. As shown in Fig. 5 (and discussed in more detail below), the similar sensitivity of ventricular and atrial Ito to flecainide argue against the differential effect of NS5806 on atria and ventricles being solely due to the presence of Kv1.4 in atria. Instead, it seems more likely that stimulation and inhibition combine, so that NS5806 acts as both an agonist and antagonist for ventricular Ito on the same channel complex(es). An additional unexpected feature of the response of ventricular cells to 100 μmol/L NS5806 was the induction of a time‐dependent increase in outward current following initial inactivation of Ito (Fig. 1C). In principle, this could result from: (1) induction of an additional low NS5806 affinity gating mode of Ito or (2) some other off target effect (such as effects on the membrane or another current). The overall profile of the current in 100 μmol/L NS5806 makes (1) unlikely; it seems improbable that Ito would inactivate then reactivate slowly during a test pulse to a fixed voltage. Off target membrane effects also seem less likely because 100 μmol/L NS5806 did not produce a similar slow outward current in atrial cells (Fig. 2C). In addition, we tested for membrane effects in a limited number of additional experiments with a structurally closely related compound NS11021 (N′‐[3,5‐Bis(trifluoromethyl)phenyl]‐N‐[4‐bromo‐2‐(2H‐tetrazol‐5‐yl)phenyl]‐thiourea), which would be expected to have similar interactions with the cell membrane to NS5806. At 100 μmol/L this compound did not produce a comparable slowly activating current to that with NS5806 in ventricular cells. Thus, it seems most likely that 100 μmol/L NS5086 both affected ventricular Ito with biphasic time dependence (an increase followed by subsequent decrease in amplitude) and had an additional nonselective effect of activating another (unidentified) current. This secondary effect mitigates against the use of high concentrations of NS5806 for the selective enhancement of ventricular Ito.
Our data on ventricular Ito inactivation and its modification by NS5806 have some notable similarities to those reported for Kv4.3 + KChIP2 expression in CHO cells by Calloe et al. (Calloe et al. 2010) in terms of V0.5 and k. However, the recovery from inactivation was slowed by NS5806 in that expression system unlike the acceleration seen both here and in dog (Calloe et al. 2009, 2010). This difference might be explained by heteromultimeric channel assembly (Po et al. 1993; Wang et al. 1999) which is encountered in many Kv channel families (for review see (Birnbaum et al. 2004)). In connection with this, heterologous expression produced by adding Kv1.4 subunits to an amphibian Kv4.3 expression system resulted in NS5806 speeding the recovery from inactivation (Lundby et al. 2010). However, in that expression system NS5806 had little effect on Ito amplitude which makes any simple translation of those results to the behavior of mammalian native cardiac Ito problematic.
The previously reported inhibitory effect of NS5806 on Kv1.4 (Lundby et al. 2010) together with our atrial Ito data might suggest a dominant role for Kv1.4 in rabbit atrial Ito as Kv4.3, 4.2 and 1.4 are all expressed in atria (e.g., Rose et al. 2005; Abd Allah et al. 2012)). However, antisense oligodeoxynucleotide probes show a slightly larger effect when directed against Kv4.3 than Kv4.2 and 1.4 (Wang et al. 1999; Bosch et al. 2003; Rose et al. 2005), so one would not expect a purely inhibitory effect of NS5806 in atria. In some preliminary experiments (not shown), 3 μmol/L CP‐339,818, a compound which exerts preferential inhibition of Kv1.4 over 4.2 channels (Nguyen et al. 1996), partially inhibited both atrial and ventricular Ito. Furthermore, the similar inhibitory potency of flecainide (as a probe to differentiate between Kv1.4 and Kv4.x channels) on ventricular and atrial Ito (Fig. 5) is not consistent with a more dominant role for Kv1.4 in atria as a lower atrial potency (compared to ventricle) should then occur (Yeola and Snyders 1997; Singarayar et al. 2003; Herrera et al. 2005) and this was not seen. An alternative explanation for the monophasic inhibitory effect of NS5806 on atrial Ito would be that KChIP2 has a weaker association with the Kv4.3 isoform in atria, a possibility that could be tested by examining the effect of NS5806 in KChIP2 native knockdown/overexpression systems in a future study.
Although rabbit ventricular Ito restitution time course is slower than reported for dog (Akar et al. 2004; Jost et al. 2013), the accelerated restitution of rabbit ventricular Ito produced by NS5806 (Fig. 5) is qualitatively similar to that seen in prior canine studies (Calloe et al. 2009, 2010). In contrast, the compound did not affect rabbit atrial restitution. These results differ markedly from the slowing of restitution by NS5806 seen for recombinant Kv4.3, even when KChIP2 is co‐expressed (Calloe et al. 2010; Lundby et al. 2010). This difference underscores present uncertainty as to the precise molecular makeup of native Ito, and that caution is needed in the extrapolation of data obtained in expression systems to actual tissues.
Functional relevance?
In our ventricular AP experiments, APs showed rapid initial repolarisation, but lacked an inscribed notch. Rabbit ventricular APs lacking a pronounced notch have also been seen in other studies (e.g., Giles and Imaizumi 1988; Kelly et al. 2013; Meedech et al. 2015). In our experiments, 10 μmol/L NS5806 produced significant AP shortening at APD20‐APD90, an effect distinct from phase 1 repolarization (Fig. 6, Table 1). Incorporation of baseline rabbit Ito kinetics and of the Ito effects of NS5806 into a human ventricular AP model (O'Hara et al. 2011) qualitatively reproduced the experimentally observed AP shortening reported here (data not shown). In some respects, the ability of NS5806 to increase phase 1 and shorten phase 2 would oppose some of the deleterious changes seen in APs from failing human hearts. The restoration of the phase 1 notch in human should increase Ca2+ release synchrony (Cooper et al. 2010), whilst shortening of the AP should also reduce the duration of the Ca2+ transient (Cannell et al. 1987), via suppression of late release events (Cooper et al. 2010) as well stimulation of Ca2+ extrusion via sodium‐calcium exchange (Crespo et al. 1990). Consistent with this notion, recent data have shown that a dual Ito and IKr activator, NS3623, restores both the AP notch and protects against early after‐depolarisations in ventricular myocytes with reduced repolarisation reserve (Calloe et al. 2016).
Conclusions
This study has demonstrated a biphasic concentration‐dependent modulation of rabbit ventricular Ito by NS5806 and a monophasic inhibitory effect of the compound on atrial Ito. As both prior canine data and the present rabbit study indicate that NS5806 acts as a ventricular Ito agonist at the lower end of the μmol/L range, it seems likely that at such concentrations the compound would also stimulate human native ventricular Ito. However, at the same concentration as used in prior canine studies (Calloe et al. 2009, 2010, 2011; Cordeiro et al. 2012), NS5806 produced unexpected opposite effects on rabbit ventricular and atrial APD. Our ventricular data indicate that the consequences of Ito stimulation on ventricular repolarization can vary between species, depending on underlying Ito kinetics. The discordance between our rabbit atrial Ito data and prior canine atrial Ito data complicates extrapolation of these results to human atrial Ito. With that caveat, whilst ventricular Ito activation might be anticipated to be beneficial in heart failure, concomitant atrial Ito inhibition could in principle promote initiation of re‐entrant arrhythmia in healthy atrial tissue if it promoted dispersion of atrial APD (Aslanidi and Hancox 2015). On the other hand, in a setting of electrically remodeled atria the APD lengthening effect of NS5806 could be beneficial and protect against sustained re‐entry (Aslanidi and Hancox 2015). Our data support the previously proposed notion that NS5806 additionally exerts atrio‐selective Na+ channel inhibitory effects (Calloe et al. 2011) and effects of combined atrial Ito and INa inhibition may well differ from those of Ito inhibition alone. Concomitant atrial INa inhibition by a ventricular Ito agonist may not be desirable unless abnormal atrial excitability is also present, and should be considered carefully during future design/development of such agents. Finally, the uncertainty as to the precise composition of native Ito channels means that the underlying basis of action of NS5806 and related molecules may best be further elucidated by the study of native rather than recombinant Ito, combined with genetic modification of Kv and KChiP isoform expression.
Conflicts of Interest
None.
Acknowledgments
We thank Hanne Gadeberg and Stephanie Choisy for assistance with rabbit myocyte isolations, Professor David Jane for helpful chemistry discussion and Professor András Varró and Dr László Virág for helpful discussion of dog Ito.
Cheng H., Cannell M. B., Hancox J. C.. Differential responses of rabbit ventricular and atrial transient outward current (Ito) to the Ito modulator NS5806, Physiol Rep, 5 (5), 2017, e13172, doi: 10.14814/phy2.13172
Funding information
We thank the British Heart Foundation for financial support (PG/11/24; PG/14/21; PG/14/42; PG/16/55). MBC was also supported by a Royal Society Wolfson award.
Contributor Information
Mark B. Cannell, Email: mark.cannell@bristol.ac.uk
Jules C. Hancox, Email: jules.hancox@bristol.ac.uk
References
- Abd Allah, E. S. , Aslanidi O. V., Tellez J. O., Yanni J., Billeter R., Zhang H., et al. 2012. Postnatal development of transmural gradients in expression of ion channels and Ca2+‐handling proteins in the ventricle. J. Mol. Cell. Cardiol. 53:145–155. [DOI] [PubMed] [Google Scholar]
- Akar, F. G. , Wu R. C., Deschenes I., Armoundas A. A., Piacentino V. III, Houser S. R., et al. 2004. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: insights into molecular composition of ventricular Ito . Am. J. Physiol. Heart Circ. Physiol. 286:H602–H609. [DOI] [PubMed] [Google Scholar]
- Antzelevitch, C. 2006. Brugada syndrome. Pacing Clin. Electrophysiol. 29:1130–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidi, O. , and Hancox J. C.. 2015. Initiation and sustenance of reentry are promoted by two different mechanisms. Heart Rhythm 12:e2. doi:10.1016/j.hrthm.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum, S. G. , Varga A. W., Yuan L. L., Anderson A. E., Sweatt J. D., and Schrader L. A.. 2004. Structure and function of Kv4‐family transient potassium channels. Physiol. Rev. 84:803–833. [DOI] [PubMed] [Google Scholar]
- Bosch, R. F. , Scherer C. R., Rub N., Wohrl S., Steinmeyer K., Haase H., et al. 2003. Molecular mechanisms of early electrical remodeling: transcriptional downregulation of ion channel subunits reduces ICaL and Ito in rapid atrial pacing in rabbits. J. Am. Coll. Cardiol. 41:858–869. [DOI] [PubMed] [Google Scholar]
- Brandt, M. C. , Priebe L., Bohle T., Sudkamp M., and Beuckelmann D. J.. 2000. The ultrarapid and the transient outward K+ current in human atrial fibrillation. Their possible role in postoperative atrial fibrillation. J. Mol. Cell. Cardiol. 32:1885–1896. [DOI] [PubMed] [Google Scholar]
- Burashnikov, A. , Di Diego J. M., Zygmunt A. C., Belardinelli L., and Antzelevitch C.. 2007. Atrium‐selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloe, K. , Cordeiro J. M., Di Diego J. M., Hansen R. S., Grunnet M., Olesen S. P., et al. 2009. A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc. Res. 81:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloe, K. , Soltysinska E., Jespersen T., Lundby A., Antzelevitch C., Olesen S. P., et al. 2010. Differential effects of the transient outward K+ current activator NS5806 in the canine left ventricle. J. Mol. Cell. Cardiol. 48:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloe, K. , Nof E., Jespersen T., Di Diego J. M., Chlus N., Olesen S. P., et al. 2011. Comparison of the effects of a transient outward potassium channel activator on currents recorded from atrial and ventricular cardiomyocytes. J. Cardiovasc. Electrophysiol. 22:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloe, K. , Di Diego J. M., Hansen R. S., Nagle S. A., Treat J. A., and Cordeiro J. M.. 2016. A dual potassium channel activator improves repolarization reserve and normalizes ventricular action potentials. Biochem. Pharmacol. 108:36–46. [DOI] [PubMed] [Google Scholar]
- Camacho, P. , Fan H., Liu Z., and He J. Q.. 2016. Small mammalian animal models of heart disease. Am. J. Cardiovasc. Dis. 6:70–80. [PMC free article] [PubMed] [Google Scholar]
- Cannell, M. B. , Berlin J. R., and Lederer W. J.. 1987. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science 238:1419–1423. [DOI] [PubMed] [Google Scholar]
- Cooper, P. J. , Soeller C., and Cannell M. B.. 2010. Excitation‐contraction coupling in human heart failure examined by action potential clamp in rat cardiac myocytes. J. Mol. Cell. Cardiol. 49:911–917. [DOI] [PubMed] [Google Scholar]
- Cordeiro, J. M. , Calloe K., Moise N. S., Kornreich B., Giannandrea D., Di Diego J. M., et al. 2012. Physiological consequences of transient outward K+ current activation during heart failure in the canine left ventricle. J. Mol. Cell. Cardiol. 52:1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo, L. M. , Grantham C. J., and Cannell M. B.. 1990. Kinetics stoichiometry and role of the Na‐Ca exchange mechanism in isolated cardiac myocytes. Nature 345:618–621. [DOI] [PubMed] [Google Scholar]
- Fermini, B. , Wang Z., Duan D., and Nattel S.. 1992. Differences in rate dependence of transient outward current in rabbit and human atrium. Am. J. Physiol. 263:H1747–H1754. [DOI] [PubMed] [Google Scholar]
- Gaborit, N. , Le B. S., Szuts V., Varro A., Escande D., Nattel S., et al. 2007. Regional and tissue specific transcript signatures of ion channel genes in the non‐diseased human heart. J. Physiol. 582:675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles, W. R. , and Imaizumi Y.. 1988. Comparison of potassium currents in rabbit atrial and ventricular cells. J. Physiol. 405:123–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, W. G. , Pham K., and Miksovska J.. 2014. Modulation of the voltage‐gated potassium channel (Kv4.3) and the auxiliary protein (KChIP3) interactions by the current activator NS5806. J. Biol. Chem. 289:32201–32213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox, J. C. , Levi A. J., Lee C. O., and Heap P.. 1993. A method for isolating rabbit atrioventricular node myocytes which retain normal morphology and function. Am. J. Physiol. 265:H755–H766. [DOI] [PubMed] [Google Scholar]
- Herrera, D. , Mamarbachi A., Simoes M., Parent L., Sauve R., Wang Z., et al. 2005. A single residue in the S6 transmembrane domain governs the differential flecainide sensitivity of voltage‐gated potassium channels. Mol. Pharmacol. 68:305–316. [DOI] [PubMed] [Google Scholar]
- Howarth, F. C. , Levi A. J., and Hancox J. C.. 1996. Characteristics of the delayed rectifier potassium current (IK) compared in myocytes isolated from the atrioventricular node and ventricule of the rabbit heart. Pflugers Arch. 431:713–722. [DOI] [PubMed] [Google Scholar]
- Isenberg, G. , and Klockner U.. 1982. Calcium tolerant ventricular myocytes prepared by incubation in a “KB medium”. Pflugers Arch. 395:6–18. [DOI] [PubMed] [Google Scholar]
- Jost, N. , Virag L., Comtois P., Ordog B., Szuts V., Seprenyi G., et al. 2013. Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J. Physiol. 591:4189–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaab, S. , Dixon J., Duc J., Ashen D., Nabauer M., Beuckelmann D. J., et al. 1998. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 98:1383–1393. [DOI] [PubMed] [Google Scholar]
- Kelly, A. , Ghouri I. A., Kemi O. J., Bishop M. J., Bernus O., Fenton F. H., et al. 2013. Subepicardial action potential characteristics are a function of depth and activation sequence in isolated rabbit hearts. Circ. Arrhythm. Electrophysiol. 6:809–817. [DOI] [PubMed] [Google Scholar]
- Levi, A. J. , Hancox J. C., Howarth F. C., Croker J., and Vinnicombe J.. 1996. A method for making rapid changes of superfusate whilst maintaining temperature at 37°C. Pflugers Arch. 432:930–937. [DOI] [PubMed] [Google Scholar]
- Lundby, A. , Jespersen T., Schmitt N., Grunnet M., Olesen S. P., Cordeiro J. M., et al. 2010. Effect of the Ito activator NS5806 on cloned KV4 channels depends on the accessory protein KChIP2. Br. J. Pharmacol. 160:2028–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meedech, P. , Saengklub N., Limprasutr V., Kalandakanond‐Thongsong S., Kijtawornrat A., and Hamlin R. L.. 2015. Transmural dispersion of repolarization and cardiac remodelling in ventricles of rabbit with right ventricular hypertrophy. J. Pharmacol. Toxicol. Methods 71:129–136. [DOI] [PubMed] [Google Scholar]
- Milani‐Nejad, N. , and Janssen P. M.. 2014. Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacol. Ther. 141:235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson, J. S. , and Hancox J. C.. 1999. Characteristics of a transient outward current (sensitive to 4‐aminopyridine) in Ca‐tolerant myocytes isolated from the rabbit atrioventricular node. Pflugers Arch. 438:68–78. [DOI] [PubMed] [Google Scholar]
- Nerbonne, J. M. 2000. Molecular basis of functional voltage‐gated K+ current diversity in the mammalian myocardium. J. Physiol. 525:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne, J. M. , and Kass R. S.. 2005. Molecular physiology of cardiac repolarization. Physiol. Rev. 85:1205–1253. [DOI] [PubMed] [Google Scholar]
- Nguyen, A. , Kath J. C., Hanson D. C., Biggers M. S., Canniff P. C., Donovan C. B., et al. 1996. Novel nonpeptide agents potently block the C‐type inactivated conformation of Kv1.3 and suppress T cell activation. Mol. Pharmacol. 50:1672–1679. [PubMed] [Google Scholar]
- Niwa, N. , and Nerbonne J. M.. 2010. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J. Mol. Cell. Cardiol. 48:12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara, T. , Virag L., Varro A., and Rudy Y.. 2011. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS.Comput. Biol. 7:e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patocskai, B. , Barajas‐Martinez H., Hu D., Gurabi Z., Koncz I., and Antzelevitch C.. 2016. Cellular and ionic mechanisms underlying the effects of cilostazol, milrinone, and isoproterenol to suppress arrhythmogenesis in an experimental model of early repolarization syndrome. Heart Rhythm 13:1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Po, S. , Roberds S., Snyders D. J., Tamkun M. M., and Bennett P. B.. 1993. Heteromultimeric assembly of human potassium channels. Molecular basis of a transient outward current? Circ. Res. 72:1326–1336. [DOI] [PubMed] [Google Scholar]
- Radicke, S. , Cotella D., Graf E. M., Banse U., Jost N., Varro A., et al. 2006. Functional modulation of the transient outward current Ito by KCNE beta‐subunits and regional distribution in human non‐failing and failing hearts. Cardiovasc. Res. 71:695–703. [DOI] [PubMed] [Google Scholar]
- Rose, J. , Armoundas A. A., Tian Y., DiSilvestre D., Burysek M., Halperin V., et al. 2005. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am. J. Physiol. Heart Circ. Physiol. 288:H2077–H2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah, R. , Ramirez R. J., Oudit G. Y., Gidrewicz D., Trivieri M. G., Zobel C., et al. 2003. Regulation of cardiac excitation‐contraction coupling by action potential repolarization: role of the transient outward potassium current (Ito). J. Physiol. 546:5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singarayar, S. , Bursill J., Wyse K., Bauskin A., Wu W., Vandenberg J., et al. 2003. Extracellular acidosis modulates drug block of Kv4.3 currents by flecainide and quinidine. J. Cardiovasc. Electrophysiol. 14:641–650. [DOI] [PubMed] [Google Scholar]
- Suzuki, T. , Morishima M., Kato S., Ueda N., Honjo H., and Kamiya K.. 2013. Atrial selectivity in Na+ channel blockade by acute amiodarone. Cardiovasc. Res. 98:136–144. [DOI] [PubMed] [Google Scholar]
- Swartz, K. J. 2004. Towards a structural view of gating in potassium channels. Nat. Rev. Neurosci. 5:905–916. [DOI] [PubMed] [Google Scholar]
- Szel, T. , and Antzelevitch C.. 2014. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J. Am. Coll. Cardiol. 63:2037–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamargo, J. , Caballero R., Gomez R., Valenzuela C., and Delpon E.. 2004. Pharmacology of cardiac potassium channels. Cardiovasc. Res. 62:9–33. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Feng J., Shi H., Pond A., Nerbonne J. M., and Nattel S.. 1999. Potential molecular basis of different physiological properties of the transient outward K+ current in rabbit and human atrial myocytes. Circ. Res. 84:551–561. [DOI] [PubMed] [Google Scholar]
- Witzel, K. , Fischer P., and Bahring R.. 2012. Hippocampal A‐type current and Kv4.2 channel modulation by the sulfonylurea compound NS5806. Neuropharmacology 63:1389–1403. [DOI] [PubMed] [Google Scholar]
- Yeola, S. W. , and Snyders D. J.. 1997. Electrophysiological and pharmacological correspondence between Kv4.2 current and rat cardiac transient outward current. Cardiovasc. Res. 33:540–547. [DOI] [PubMed] [Google Scholar]