

Due to the scarcity of comparative data for cells from different brain regions in vitro, we demonstrated that neurons isolated from distinct brain areas exhibit unique behaviors in vitro. Moreover, in vivo proper brain function is dependent on the connection and communication of several brain regions, underlining the importance of developing multiregional brain in vitro models. We introduced a novel brain-on-a-chip model, implementing essential in vivo features, such as different brain areas and their functional connections.
Keywords: brain-on-a-chip, different brain regions, protein expression, electrophysiology, metabolism
Abstract
Brain in vitro models are critically important to developing our understanding of basic nervous system cellular physiology, potential neurotoxic effects of chemicals, and specific cellular mechanisms of many disease states. In this study, we sought to address key shortcomings of current brain in vitro models: the scarcity of comparative data for cells originating from distinct brain regions and the lack of multiregional brain in vitro models. We demonstrated that rat neurons from different brain regions exhibit unique profiles regarding their cell composition, protein expression, metabolism, and electrical activity in vitro. In vivo, the brain is unique in its structural and functional organization, and the interactions and communication between different brain areas are essential components of proper brain function. This fact and the observation that neurons from different areas of the brain exhibit unique behaviors in vitro underline the importance of establishing multiregional brain in vitro models. Therefore, we here developed a multiregional brain-on-a-chip and observed a reduction of overall firing activity, as well as altered amounts of astrocytes and specific neuronal cell types compared with separately cultured neurons. Furthermore, this multiregional model was used to study the effects of phencyclidine, a drug known to induce schizophrenia-like symptoms in vivo, on individual brain areas separately while monitoring downstream effects on interconnected regions. Overall, this work provides a comparison of cells from different brain regions in vitro and introduces a multiregional brain-on-a-chip that enables the development of unique disease models incorporating essential in vivo features.
NEW & NOTEWORTHY Due to the scarcity of comparative data for cells from different brain regions in vitro, we demonstrated that neurons isolated from distinct brain areas exhibit unique behaviors in vitro. Moreover, in vivo proper brain function is dependent on the connection and communication of several brain regions, underlining the importance of developing multiregional brain in vitro models. We introduced a novel brain-on-a-chip model, implementing essential in vivo features, such as different brain areas and their functional connections.
korbinian brodmann (1868–1918) was one of the first scientists to categorize the brain into 52 distinct areas based on cellular organization, and confirmed the already strongly believed localist view, which supported the opinion that the brain consists of functionally and structurally distinct regions (Finger 2001). Decades later, much scientific effort has been spent confirming these views, e.g., observations of brain region-dependent microenvironments and cell compositions (Dauth et al. 2016; Herculano-Houzel 2009), and gaining more detailed information about brain organization and function by developing in vitro methods to culture nerve cells. While these in vitro models play an essential role in the understanding of basic physiological processes of nervous system cells, as well as help in encoding disease mechanisms (Karetko and Skangiel-Kramska 2009), there is still a lack of information comparing cells from different brain areas in vitro, mitigating our efforts to understand the building blocks of in vitro models.
Another often overlooked feature in the field of brain in vitro models is that the brain is unique in its structural and functional organization, and that the interactions and communication between different brain areas are essential components of proper brain function (Asher et al. 2001; Bradbury et al. 2002; Buzsáki and Draguhn 2004; Clark et al. 2007; Luna et al. 2007; Yip et al. 2006). There are currently very few in vitro models incorporating cells from two different brain regions (Kato-Negishi et al. 2013; Peyrin et al. 2011). Advances in the induced pluripotent stem cell (iPSC) field also give rise to the high potential of brain organoids, currently restricted to only a few brain structures, but supporting the notion that it is important to recapitulate different brain regions in vitro (Qian et al. 2016). These organoids have their own advantages and disadvantages, but represent a promising in vitro model (Lancaster et al. 2013; Qian et al. 2016). Not only is the connectivity between distinct brain regions essential for physiology, but it is often disturbed in brain pathology (Seeley et al. 2009). One example is schizophrenia, where the prefrontal cortex (pfCx), hippocampus (Hip) and amygdala (Amy) are affected, including molecular alterations, as well as an abnormal connectivity and communication between these regions (Benes 2009).
In this work, we assessed the differences between in vitro cultures originating from different rat brain regions (pfCx, Hip, and Amy) by comparing their cell composition, such as neuronal and nonneuronal cells, protein expression levels, metabolism, and electrophysiology and observed a unique profile for each of these parameters in vitro. In this study, rat brain cells are used, because primary human Amy cells are not currently available, and iPSCs cannot yet generate cells with a specific brain region identity (e.g., pfCx, Hip, Amy). Moreover, animal models have proven invaluable to better understand brain diseases (Asher et al. 2001; Lipska and Weinberger 2000), and animal models, as well as rodent in vitro models, are still commonly used for drug development and safety assessments (Dubreuil et al. 2006; Lecanu and Papadopoulos 2013; Ramnauth et al. 2012). To address the importance of the presence and connection of different brain areas in vivo, we developed a multiregional brain-on-a-chip, consisting of three separated areas of neuronal cell bodies derived from the pfCx, Hip, and Amy that are functionally connected through axons. Both, cell composition and electrophysiology were altered in the brain-on-a-chip model compared with separately cultured cells. The primary advantages of this new model are the presence of functionally interconnected brain areas and the ability to treat one brain region specifically with a drug and assess the effects on parameters, such as electrophysiology, on all interconnected brain areas. Here, we used phencyclidine (PCP) to induce a schizophrenia-like state in vitro (Adachi et al. 2013) and analyzed the effect on the electrical behavior of all three brain regions in vitro. The brain-on-a-chip model presented here can be easily adapted to human neuron cultures when these brain region-specific populations become available.
MATERIALS AND METHODS
Primary Neuronal and Glial Cell Harvest and Culture
Neurons from the pfCx, the Hip, and the Amy were isolated from 2-day-old female and male neonatal Sprague-Dawley rats (Charles River Laboratories, Boston, MA). The 2-day-old pups were decapitated without prior anesthesia. All procedures were approved by the Harvard Animal Care and Use Committee. Using 2-day-old neonatal rats for the isolation of neurons is a widespread and commonly used method. Reasons include a higher percentage of neurogenesis (and therefore pluripotent cells) and the ability to better adjust to a new environment and survive the isolation procedure (“regenerative potential”) in the younger brain (Anderson and Anderson 1996; Nunez 2008; Oberdoerster 2001). Brain regions were surgically isolated and minced in Hibernate-A solution supplemented with B-27 (×50, diluted 1:50 to achieve ×1 B27) and glutamax (0.5 mM; HABG) (all GIBCO Life Technologies, Grand Island, NY), followed by digestion with papain (16 mg in 8 ml, ~34 U/ml; Worthington Biochemical, Lakewood, NJ) for 30 min at 37°C. Tissues from the Hip and the pfCx were isolated from the same set of animals and used for neuronal isolation. Tissue from the Amy was isolated from a separate set of animals. The digested tissue was then transferred to fresh HABG and mechanically disrupted using silane-treated glass pipettes. The mechanical disruption was done by pipetting the tissue up and down for ~10 times. After the remaining tissue settled down, the supernatant was collected and transferred to a fresh tube. These steps were repeated two more times. The collected supernatants were then filtered through a nylon filter of 40-µm pore size (BD Bioscience, San Jose, CA) and centrifuged at 200 g for 5 min. The supernatant was removed, and the pellet was resuspended in prewarmed neurobasal A medium supplemented with B-27 (×1), glutamax (0.5 mM) and gentamycin (10 µg/ml; all GIBCO Life Technologies, Grand Island, NY). Neuronal cells were counted using a Moxi Mini Automated Cell Counter (Orflo, Ketchum, ID) and seeded at the desired density (1.6 K cells/mm2) on substrates coated with 100 µg/ml poly-l-lysine (PLL, Sigma, St. Louis, MO). After 1 h, cells were washed once with HABG to remove nonadherent cells. All samples were cultured in neurobasal A medium supplemented with B-27 (×1), glutamax (0.5 mM) and gentamycin (10 µg/ml) and incubated under standard conditions at 37°C and 5% CO2. Media was replaced by removing one-half and adding fresh media every 72 h until experiments were executed.
Immunofluorescent Staining and Microscopy
Cells were washed two times with prewarmed PBS, fixed for 10 min with prewarmed 4% paraformaldehyde, permeabilized for 10 min with 0.05% Triton X-100 in PBS at room temperature, and blocked with 3–5% bovine serum albumin (Jackson ImmunoResearch, West Grove, PA) in PBS for 30 min at room temperature. The blocking solution was aspirated away, and the primary antibody solution was immediately added and incubated for 1.5 h at 37°C or overnight at 4°C. The primary antibodies used were anti-βIII-tubulin (1:200; Sigma, St. Louis, MO), anti-neurofilament (NF) (1:100; Abcam, Cambridge, MA), anti-glial fibrillary acidic protein (GFAP; 1:200; Abcam, Cambridge, MA), anti-glutamate decarboxylase (GAD) 1/GAD67 (1:100; Novus Biologicals, Littleton, CO), and anti-vesicular glutamate transporter (Vglut) 1 (1:100; Abcam, Cambridge, MA). Primary antibodies were diluted in 0.5% BSA in PBS solution. Following primary staining, cells were washed three times with PBS, and the secondary staining solution consisting of either goat anti-mouse/rabbit/chicken conjugated to Alexa-Fluor 488, goat anti-mouse/rabbit/chicken conjugated to Alexa-Fluor 546, or goat anti-mouse/rabbit/chicken conjugated to Alexa-Fluor 633 (Molecular Probes, Grand Island, NY), and 4′,6-diamidino-2-phenylindole (Molecular Probes Life Technologies, Grand Island, NY) was added to the cells for 1 h at 37°C. Samples were then washed three times with PBS. For glass bottom samples, the glass was removed from the dish and placed on a glass slide. ProLong Gold Antifade reagent (Molecular Probes Life Technologies, Grand Island, NY) was added to preserve the samples, and glass coverslips are affixed using transparent nail polish. Prepared slides were either imaged immediately or stored at 4°C. Imaging was performed with an Olympus confocal microscope or an Olympus VS120 Slide Scanner (both Olympus, Center Valley, PA) with appropriate filter cubes. The GAD, Vglut, and GFAP positive cells were counted manually for each image using ImageJ.
Proteomics
Cell preparation.
Cells were harvested as detailed above. After 14 days in culture, medium was removed, cells were washed with PBS, and incubated with RIPA buffer (Sigma, St. Louis, MO) complemented with protease inhibitors (Complete Mini, Roche, Basel, Switzerland) and 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF, Sigma, St. Louis, MO) on ice for 5 min. Afterwards, cells were scraped off the surface with a cell scraper and lysed cells were transferred to a microcentrifuge tube on ice. Samples were centrifuged at 17,000 g for 30 min at 4°C. Supernatants were transferred to a fresh Eppendorf tube and centrifuged again at 17,000 g for 30 min at 4°C. The resulting supernatants were used for analysis.
Tissue preparation.
Tissue samples were harvested from adult Sprague-Dawley rats (Charles River Laboratories, Boston, MA). All procedures were approved by the Harvard Animal Care and Use Committee. Adult rats were killed using CO2 and cervical dislocation. Two-day-old pups were decapitated. Brains were dissected out of the skull, and the pfCx, the Hip, and the Amy were further surgically dissected and snap frozen in liquid nitrogen. Samples were thawed and homogenized using a hand homogenizer (Cole-Parmer, Vernon Hills, IL) in RIPA buffer (Sigma, St. Louis, MO) and complemented with protease inhibitors (Complete Mini, Roche, Basel, Switzerland) and AEBSF (Sigma, St. Louis, MO). All steps were conducted on ice. After homogenization, samples were embedded in ice and placed on a rocking platform shaker for 30 min. Afterwards samples were centrifuged at 17,000 g for 30 min at 4°C. Supernatants were transferred to a fresh Eppendorf tube and centrifuged again at 17,000 g for 30 min at 4°C. The resulting supernatants were used for analysis.
Tandem mass tag labeling and electrostatic repulsion-hydrophilic interaction chromatography off-line fractionation.
Samples were measured to 100 µg of total protein in each sample. Samples were digested using a slightly modified filter-aided sample preparation protocol, as previously published (Wiśniewski et al. 2009). Subsequently, all samples were labeled by tandem mass tags (TMT) 10plex reagent (product no. 90061, Thermo Fisher, San Jose, CA), according to manufacturer protocol. The TMT10plex reagent set enables up to 10 different peptide samples prepared from cells or tissues to be labeled in parallel and then combined for analysis. For each sample, a unique reporter mass in the low-mass region of the high-resolution tandem mass spectrometry (MS/MS) spectrum is used to measure relative protein expression levels during peptide fragmentation and MS/MS. The resulting labeled 10 samples were pooled into one sample for an off-line fractionation. The high-performance liquid chromatography (HPLC) 1200 Agilent system with fraction collector (Agilent Technologies, Santa Clara, CA) was used for an electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) separation using PolyWAX LP columns (200 × 2.1 mm, 5 µm, 300 Å; PolyLC, Columbia, MD). ERLIC utilizes both hydrophilic interaction and electrostatic forces. By adjusting the pH, salt type, salt concentration, and organic solvent compositions in the mobile phase, an isocratic separation of a mixture of charged analytes (e.g., peptides, amino acids, and nucleotides) can be achieved (Alpert 2008). Samples were fractionated to a total of 20 fractions on a 70-min liquid chromatography gradient. Each fraction was submitted for mass spectrometry analysis.
Mass spectrometry and data analysis.
Samples were run on Orbitrap Velos Pro (Thermo Fisher, San Jose, CA) for all analytical runs. Orbitrap Velos Pro is a hybrid ion trap-Orbitrap mass spectrometer which is ideal for identification of low-level proteins in complex matrices, rapid quantitation of isobarically labeled peptides, and structural elucidation of metabolites. Samples were injected from autosampler of HPLC (Waters, NanoAquity, Milford, MA) into trapping column (75 µm column inner diameter, 5 cm packed with C18 media on 5-µm beats on 200 Å poros, from Michrom Bioresources, Auburn, CA). After binding in trapping column, peptides were washed for 15 min with buffer A (0.1% formic acid in water) and then eluted to an analytical column (75 µm column inner diameter, 20 cm packed with 3-µm C18 media beats on 100 Å poros) with a gradient from 2 to 32% of buffer B (0.1% formic acid in acetonitrile) over a 90-min gradient for each fraction. Each sample was run on Orbitrap Velos instrument with a high-collisional energy/collisional-induced dissociation (HCD/CID) top 20 method that isolates and fragments top ions that came at a particular time of the chromatogram from nanoflow HPLC (HCD fragmentation in Orbitrap was followed by CID event in the ion trap part of the instrument). HCD/CID is used to sequence the peptides present in the samples. The Orbitrap Velos Pro instrument was set up to run the TOP 20 method for MS/MS in ion trap with an exclusion function turned on after MS1 scan in Orbitrap with 60 K resolving power at a mass of 400 m/z. For TMT10plex labeling, MS2 data were acquired with 30 K resolving power; for TMT6plex, MS2 data were collected with 15 K resolving power. Obtained runs were analyzed by Proteome Discoverer 1.4.1.14 (Thermo Fisher, San Jose, CA) software with Percolator version 2.05 (University of Washington) as a statistical data package. Searches were done against rat proteome database filtered out of the Uniprot database by species specificity, and common contaminants that were added to this database. Searches were done with trypsin enzyme specificity, allowing two missed cleavages. Possible modification included in the search parameters were: protein NH2-terminus acetylation, methionine oxidation, deamidation of asparagine, and glutamine amino acids were put as a variable modification as well as for static modifications NH2-terminus of peptide TMT6plex labeling and lysine side chains with TMT6plex labeling group. The database search criteria were held at 1% false discovery rate on both protein and on peptide levels for all output reported data. All data outputs were filtered to 40% maximum co-isolation of MS1, allowing TMT quantitation results to be kept under control from co-isolation effect.
Gene expression dynamics inspector.
Visual representations of the global protein expression profiles of the pfCx, Hip, and Amy neuron populations were created using Gene Expression Dynamics Inspector (GEDI) version 2.1 software (Eichler et al. 2003). This program uses a self-organizing map (SOM) algorithm to reduce data dimensionality by classifying proteins with similar expression profiles into discrete groups that are organized into distinct two-dimensional mosaics. Each tile of these mosaics represents a cluster of proteins, and the mapping of proteins to these tiles is conserved across samples to facilitate comparison. Coloration of each mosaic tile indicates the fold change value of the centroid learned during SOM training. For the proteomics data collected in this study, GEDI mosaic grid size was set to 18 × 20. SOM training occurred in two phases with 200 iterations in the first phase, and 800 iterations in the second phase. All other SOM parameter settings were set to default values.
Proteomaps.
Proteomaps illustrating the composition and abundance of the functional categories represented in the pfCx, Hip, and Amy neuron proteomes were constructed using proteomaps version 2.0 software (Liebermeister et al. 2014). These maps are constructed from Voronoi diagrams that are divided into polygons representing top level KEGG orthology biological process terms, which are, in turn, subdivided based on the subcategories represented in the protein expression data set. The area of the individual polygons comprising the map is a function of the abundance of the proteins they represent. The coloration of the polygons facilitates visual distinction of the biological processes represented in the proteomaps and has no quantitative interpretation.
Principal component analysis.
Principal component analysis (PCA) was used to identify proteins in the pfCx, Hip, and Amy mass spectrometry data whose expression levels exhibited the greatest variability between the three neuron populations tested (Ringnér 2008). PCA was performed using the implementation provided in the scikit-learn machine learning Python package, version 0.17-4. Scatterplots of the fold change values projected onto the two dimensions that explain the most variance, color coded according to their associated Gene Ontology biological process term, were created using the Python package Matplotlib, version 1.5.1-5.
Correlation analysis.
Pearson product moment correlation (Brangwynne et al. 2006) was used to assess the degree of linear correlation between pairwise comparisons of the fold change protein expression values collected for pfCx, Hip, and Amy neuron cultures. Pairwise comparisons were performed using the corr() function provided in the PANDAS version 0.18.0-7 Python package, and visualized using the Seaborn statistical data visualization Python package, version 0.7.1.
Ingenuity pathway analysis.
QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, https://www.qiagenbioinformatics.com/) was used to identify pathways for which a substantial number of upstream regulators are present in the protein expression data set, as inferred from z-score analysis based on prior knowledge stored in the Ingenuity Knowledge Base. This analysis examines the number of known targets.
Protein-protein interaction network analysis.
Protein-protein interaction networks were constructed using the Search Tool for Retrieval of Interacting Genes/Proteins (STRING) database, version 10.0 (Szklarczyk et al. 2015). The STRING database stores information about functional relationships between proteins that are derived from a number of sources. For the analyses performed in this study, only known experimental interactions imported from primary database sources, and pathway knowledge parsed from manually curated databases, were considered. Edges in the network graphs were chosen to represent the confidence that the interaction between two proteins represents a true functional interaction, as judged from evidence provided by the knowledge sources considered. The length of the edges between each pair of protein nodes in the protein-protein interaction networks was computed using a spring-like model to cluster higher confidence protein-protein interactions closer together into functionally-related clusters. Only high-confidence interactions (i.e., interaction scores > 0.7) were considered in this study.
Seahorse Mitochondrial Respiration Measurements
Oxygen consumption rate (OCR) was measured using the Seahorse XF96 equipment (Seahorse Bioscience, North Billerica, MA). The Seahorse XF Cell Mito Stress Test was used to measure key parameters of mitochondrial function by directly measuring the OCR of cells. The Seahorse XF Cell Mito Stress Test uses modulators of respiration to target components of the electron transport chain (ETC) in the mitochondria to observe key parameters of metabolic function. The compounds [oligomycin, carbonyl cyanide-4 (trifluoromethoxy)phenylhydrazone (FCCP), and a mix of rotenone and antimycin A] (Seahorse Bioscience, North Billerica, MA) are serially injected to measure ATP production, maximal respiration, and nonmitochondrial respiration, respectively. The proton leak is then calculated using these parameters and basal respiration. Each of the modulators used targets a specific component of the ETC. Oligomycin inhibits complex V of ATP synthase. The decrease in OCR that follows the injection of oligomycin correlates to the mitochondrial respiration associated with cellular ATP production. FCCP is an uncoupling agent that collapses the proton gradient and disrupts the membrane potential of mitochondria. In turn, electron flow through the ETC is uninhibited, and oxygen is maximally consumed by complex IV. The last injection is a mix of rotenone, inhibiting complex I, and antimycin A, inhibiting complex III. This combination shuts down mitochondrial respiration and enables the calculation of nonmitochondrial respiration driven by processes taking place outside the mitochondria (http://www.agilent.com/cs/library/usermanuals/public/XF_Cell_Mito_Stress_Test_Kit_User_Guide.pdf). The procedure was performed according to the manufacturers’ manual (see above). Briefly, neurons from the pfCx, the Hip, and the Amy were seeded at the same density (100 K/well) and cultured for 2 wk. Just before the assay was carried out, the cell culture medium was replaced by the assay medium, which contains 100 ml aCSF (440 mg BSA, 100 mg glucose, 0.5 mM HEPES). The solution pH was adjusted to 7.4, and for 10 ml assay medium 110 mg sodium pyruvate was added. For the next step, cells were placed into a 37°C non-CO2 incubator for 45 min to 1 h. Afterwards, the plate was placed into the Seahorse apparatus after a calibration run was completed, and the assay was run.
Soft Photolithography and Polydimethylsiloxane Mask Fabrication
A photomask with the desired patterns (line pattern: 15 µm × 15 µm or 10 µm × 15 µm) was created using computer-assisted design software and standard photolithography methods, and then the mask was used to shadow a wafer glass covered with a photoresist when exposed to ultraviolet light. The thickness of the photoresist layer determines the depth of the grooves, and the width is determined by the photomask line patterns when the light-exposed areas of the photoresist are dissolved away. Specifically, silicon wafers (University Wafer) were treated for 10 s with plasma, and SU-8-2002 was spin coated on top of them. Afterwards, wafers were prebaked for 30 s at 65°C, 1 min at 95°C, and 30 s at 65°C on a hot plate. A transparency photomask was used to produce the master by exposing the wafer to UV light for ~10 s. Wafers were then postbaked at 65°C for 30 s, 2 min at 95°C, and 30 s at 65°C on a hot plate. Afterwards, wafers were developed in propylene glycol monomethyl ether acetate for ~90 s. Then wafers were incubated in isopropanol for another 1 min, before air drying. Lastly the wafers were incubated with silane in vacuum overnight. This etched surface was then used as a “master” form onto which liquid polydimethylsiloxane (PDMS) silicon rubber is cast and allowed to cross-link. Specifically, a disposable plastic cup was filled with Sylgard 184 silicone elastomer base, and the curing agent had a 10:1 weight ratio. The solution was poured onto the fabricated wafer and degassed to remove air bubbles. The solution was cured at 65°C for 4 h. The cured PDMS was removed from the wafer, and the desired patterns were laser cut from the PDMS. The resulting stamp masks were washed in isopropanol and sonicated in ethanol before use.
Multielectrode Array Recording of Neuronal Electrical Activity
Preparing multielectrode arrays for neuronal culture.
Multielectrode arrays (MEAs) (Multichannel Systems, Germany) were washed for 24 h in 1% Tergezyme (Alconox, White Plains, NY) and another 24 h in deionized H2O. Afterwards, MEAs were dried and UVOzone treated (UVO Cleaner model NO 342, Jelight Company, Irvine, CA) for 7 min. Then the MEAs were incubated with 100 µg/ml PLL (Sigma, St. Louis, MO) for 1 h and washed three times with sterile PBS (GIBCO Life Technologies, Grand Island, NY). Cells were seeded at the desired density and cultured under standard conditions at 37°C and 5% CO2. Media was replaced by removing one-half and adding fresh media every 72 h until experiments were performed.
Recording.
Neuronal cultures were grown on 60MEA200/30iR-ITO for up to 20 days. Once neuronal cultures exhibited robust spontaneous firing activity, they were considered “mature” (Cullen et al. 2010; Hinard et al. 2012). Our neurons started firing around day 10 in culture and established a robust spontaneous firing activity after 14 days in vitro (DIV) and maintained this robust firing activity for several weeks. The MEAs were recorded with the MEA2100 (Multichannel Systems, Reutlingen, Germany) using the MC_Rack software. Spontaneous action potential discharge activity was recorded with a sampling rate of 25 kHz for 5 min.
Analysis.
The recorded traces were analyzed using MC_Rack software (Multichannel Systems). All data streams were 125-Hz Bessel fourth-order high-pass filtered and 50-Hz Bessel fourth-order band-pass filtered. Spike detection was performed with a threshold of 5 SD difference from noise. Quantification of recordings was done with NeuroExplorer to extract interspike intervals, frequency, bursts, etc. Matlab (MATLAB 8.0 and Statistics Toolbox 8.1, The MathWorks, Natick, MA) was used to generate raster plots.
Microcontact Printing
Washed and sterilized stamp masks were air-dried under sterile conditions and incubated with a PLL and laminin (both Sigma, St. Louis, MO) solution (100 µg/ml PLL and 25 µg/ml laminin) for 1 h. Afterwards, the stamp masks were lowered onto sterile coverslips or MEAs and gently attached to the surface. The holes of the stamp masks were filled with PLL and incubated for 1 h. Afterwards PLL was removed, and holes were washed with PBS three times before cells were seeded at the desired density (also see Fig. 7). One hour after seeding, masks were removed, and coverslips or MEAs were gently washed with HABG (as described above).
Fig. 7.

Novel brain-on-a-chip model comprised of different brain regions. A–D: PDMS mask preparation and seeding procedure for brain-on-a-chip model. A: first an ~1-mm-thick PDMS sheet is laser cut to receive the masks, with 3 wells being 1 mm apart. Line features are engraved on the bottom side of the PDMS mask. B: the line features are then coated with PLL/laminin and air-dried, before being stamped on a coverslip or a MEA. C: wells are filled with PLL to coat the coverslip or MEA surface. D: PLL is washed away before cells from the different brain regions are seeded in the separated wells of the mask. One hour after seeding, masks are removed. Cells will now start growing axons using the microcontact printed PLL/laminin lines as physical cues to connect to the cells from the other brain regions. E: example image of the in vitro model 14 DIV immunostained for βIII tubulin (green) and GFAP (red). Close-up images of axons that have been grown over the 1-mm gap on the microcontact printed PLL/laminin lines connecting the different brain regions (1, 2). The multiregional brain-on-a-chip was stained for βIII-tubulin (green), GFAP (red), and DAPI as a nuclear counterstain (F–H); for βIII tubulin (green), glutamate decarboxylase 1 (GAD1, red), and DAPI (I–K); and for GFAP (green), vesicular glutamate transporter 1 (Vglut, red), and DAPI (L–N). O–Q: quantification of the number (percentage of total cells) of GFAP-, GAD1-, and Vglut-positive cells in pfCx (O), Hip (P), and Amy (Q) areas of the multiregional brain model in comparison to the number of GFAP-, GAD1-, and Vglut-positive cells in pfCx, Hip, and Amy cultures seeded and cultured separately (from Fig. 2). Values are means ± SE; GFAP: n = 31–46 images/region, 4 coverslips; GAD: n = 46–54 images, 4 coverslips; Vglut: n = 20–23 images, 4 coverslips. Scale bars = 1 mm (E), 500 μm (1, 2), 100 μm (F–N). Significance levels are presented in Table 1.
PCP Experiments
Phencyclidine hydrochloride (PCP; P3029, Sigma, St. Louis, MO) was reconstituted in H2O as a stock solution of 5 mg/ml. Neuronal cultures were dosed either with 5 µM 1 h after seeding for 24 h or with 10 µM or 25 µM at 12 DIV. If cells were dosed with PCP at 1 h after seeding, spontaneous electrical activity was measured at DIV 11 until DIV 20 daily. If PCP was added to DIV 12 cultures, electrical activity was measured before the addition of PCP, as well as at several time points after PCP addition (3 h, 6 h, 24 h, 48 h, 72 h, 96 h, 120 h). Electrical activity was measured 5 min for each time point or day (see also Multielectrode Array Recording of Neuronal Electrical Activity above). Dosing regimens for DIV 12 cells were chosen in accordance with procedures described previously (Adachi et al. 2013). A lower PCP concentration was used for the 1-h time point due to the sensitivity of freshly isolated neurons to external factors.
Numbers of Samples
The number of samples is as follows: cell composition separately seeded cultures (n = 2–4 coverslips, 8–20 images); protein expression (n = 1); metabolic activity (n = 3–7 plates, 60–140 wells); electrophysiology (n = 3 MEAs/brain region, 60 electrodes each, 5-min recording each); brain-on-a-chip cell composition (n = 4 coverslips, 20–54 images); brain-on-a-chip electrophysiology (n = 3 MEAs, 6–20 electrodes, 5-min recordings); and PCP experiments (n = 4–5 MEAs/per condition, 8–20 electrodes/MEA, 5-min recordings).
Statistics
Multifactorial one-way ANOVA with a Bonferroni post hoc test was used to statistically analyze results. One-way ANOVA tests together with Bonferroni post hoc tests were conducted for statistical comparisons as our data consist of a single dependent variable (e.g., number of cells) and multiple independent variables (e.g., multiple brain regions, multiple cell type-specific markers). The ANOVA test was chosen over the two-tailed Student’s t-test because multiple t-tests cannot be performed on different conditions without inflating type 1 errors. All values depicted in Table 1 are P values resulting from the second step Bonferroni post hoc test.
Table 1.
|
Fig. 2 Individual Cultures |
Fig. 7 Model |
|||||
|---|---|---|---|---|---|---|
| pfCx | Hip | Amy | pfCx | Hip | Amy | |
| GFAP vs. GAD | 5.1E-21* | 4.4E-9* | 2.6E-11* | 2.8E-12* | 9.5E-15* | 0.3 |
| GFAP vs. Vglut | 1.5E-23* | 0.2 | 4.2E-7* | 2.9E-4* | 2.3E-6* | 0.003* |
| GAD vs. Vglut | 0.004* | 0.006* | 1 | 0.007* | 0.02* | 0.2 |
RESULTS
To comparatively evaluate cells from different brain regions in vitro, we analyzed the cell composition, protein expression levels, metabolism, and electrical activity of neuronal cultures from the pfCx, Hip, and Amy (Fig. 1). As a next step, we introduced a novel multiregional brain-on-a-chip model, which consists of three locally separated areas with cells from the pfCx, Hip, and Amy. The areas containing the cell bodies are physically separated, but the areas are functionally connected through axons. The cell composition and electrical activity of neuronal cultures, which were cultured separately, were compared with the multiregional brain-on-a-chip (Fig. 1). Furthermore, we introduced the possible application of the brain-on-a-chip model to treat brain regions separately with a drug and subsequently analyze the effect on all interconnected regions in vitro (not depicted in Fig. 1).
Fig. 1.

Project overview. The prefrontal cortex, the hippocampus, and the amygdala were dissected from 2-day-old Sprague-Dawley rats. Neurons (and astrocytes) were isolated from each brain region and either cultured individually (middle left) or together (middle right) as a multiregional brain-on-a-chip model. Individual cultures were analyzed regarding their protein expression (Fig. 2), cell composition (Figs. 3 and 4), metabolic activity (Fig. 5), and electrical activity (Fig. 6). The multiregional brain-on-a-chip consisting of all three brain regions was analyzed concerning its cell composition (Fig. 7) and electrical activity (Fig. 8). Results were compared between the multiregional brain-on-a-chip and the individual cultures.
Neuronal and Glial Markers In Vitro Are Expressed in a Brain Region Dependent Manner
Astrocytes and neurons are the main cell types in the brain, and their amounts differ in a brain region-dependent manner in vivo. Furthermore, neurons come in different types, including different morphology, function, and neurotransmitter preference (Herculano-Houzel 2009; Kandel et al. 2000). To elucidate whether cell composition in vitro differs in a brain region-dependent manner, we cultured brain cells isolated from the neonate rat pfCx, Hip, and Amy separately for 14 days. After 14 DIV, cells were fixed and immunostained for various brain relevant markers: GFAP (astrocytes), GAD (GABAergic neurons), and Vglut (glutamatergic neurons). The abundance of these markers was assessed and quantified for each culture (Fig. 2). Figure 2, A–J, depicts example images from each brain region in vitro and for each marker used. Moreover, the number of cells that were positive for GFAP, GAD, and Vglut was quantified and normalized to the total number of cells (Fig. 2J). Cells from the pfCx and the Hip were similar in their amount of the analyzed markers, with around 30% percent of the culture being GFAP-positive cells, 18–22% being Vglut-positive, and the lowest number (13–15%) being GAD-positive. Cells from the Amy, however, showed a very different marker distribution compared with pfCx and Hip cells. A small proportion of the cultured cells (around 23%) were GFAP positive. GAD-positive cells accounted for ~28% of the cell population, and Vglut-positive cells around 33% (Fig. 2J). To investigate whether the differences between brain regions for the markers assessed were significant, one-way ANOVA (post hoc test: Bonferroni) was performed, and significance levels are shown in Table 1. While each culture exhibited a unique cell composition profile, the largest differences were observed between the pfCx and the Amy cultures, as well as between Hip and Amy cultures (Table 1, asterisk = signifcant, no asterisk = not significant). Overall, these results suggest that the expression and distribution of neuronal and glial markers in vitro is highly dependent on the brain region from which the cells originated.
Fig. 2.

Neuronal and glial markers in independent neuronal cultures derived from different brain regions. A–I: independent neuronal cultures isolated from the pfCx, the Hip, and the Amy were fixed 14 DIV and stained for βIII tubulin (neurons, green), glial fibrillary acidic protein (GFAP; astrocytes, red or green), and 4′,6-diamidino-2-phenylindole (DAPI; nucleus, blue) (A–C); βIII tubulin (green), glutamate decarboxylase (GAD; GABAergic neurons, red), and DAPI (blue) (D–F); and GFAP (green), vesicular glutamate transporter (Vglut, glutamatergic neurons, red), and DAPI (blue) (G–I). Scale bars = 100 μm. J: quantification of the amount of cells positive for GFAP, GAD, and Vglut for cells isolated from the pfCx, the Hip, and the Amy are shown as percentage of total cell number, ranging from around 10% to 35%, depending on the cell type. Values are means ± SE. Significance levels are presented in Table 1. GFAP and GAD: n = each region 17–20 images, 4 coverslips; Vglut: n = 8–10 images, 2 coverslips.
Protein Expression In Vitro Is Brain Region Dependent
Protein expression levels of cells isolated from the pfCx, Hip, and Amy were measured using mass spectrometry, and all samples were normalized to neonate pfCx tissue. GEDI analysis was performed (Eichler et al. 2003) on all samples to create visual representations of the global expression profiles and compare the differences between the pfCx, Hip, and Amy neuron populations (Fig. 3, A–D). The protein allocation map for the GEDI mosaics shows how the software assigns proteins to each square based on a SOM algorithm trained using the fold change values from the mass spectrometry measurements (Fig. 3D). Neuronal cultures from the Hip and the Amy exhibited overall similar protein expression profiles, whereas clear differences were observed between these regions to the pfCx, which seems to exhibit a similar protein expression profile as the neonate pfCx (Fig. 3, A–C).
Fig. 3.

Protein expression analyses of different brain regions in vitro. Protein expression was measured via mass spectrometry in 14 DIV old neuronal cultures derived from the prefrontal cortex (pfCx; A), hippocampus (Hip; B), and amygdala (Amy; C), and global protein expression profiles were visualized using gene expression dynamics inspector (GEDI) software. A self-organizing map algorithm was trained on the data set and used to generate mosaics visualizing the expression profiles for each brain region, wherein each tile of the mosaic represents a group of proteins with similar expression profiles. Red colored tiles indicate a high-fold change in protein expression relative to the control (neonate pfCx), whereas blue represents low-fold change. D: proteins were allocated to each tile of the GEDI mosaic according to their expression profile. Red tiles indicate a large number of assigned proteins, whereas blue colored tiles indicate fewer assigned proteins. Pearson product-moment correlation was used to assess the linear correlation between the global expression profiles measured for Hip vs. Amy (r = 0.87) (E), pfCx vs. Amy (r = 0.80) (F), and pfCx vs. Hip (r = 0.86) (G). Proteomaps were used to visualize the KEGG orthology biological process terms associated with proteins exhibiting twofold or greater expression levels in the pfCx (H), Hip (I), and Amy (J) brain regions using Voronoi diagrams, wherein each polygon represents an individual protein, and polygon area is a function of expression level. K: the polygons in the proteomaps are subdivided and color coded according to the top level KEGG orthology biological process terms represented in the protein expression profiles for each brain region.
Pearson correlation analysis allowed us to quantify the degree of difference between the protein expression profiles of the pfCx, Hip, and Amy populations. These results revealed a linear correlation (r > 0.80) in the expression values between each pairwise comparison of the cell types (Fig. 3, E–G). The strongest correlation was observed between the Amy and Hip expression profiles (Fig. 3E), whereas the Amy and Hip expression profiles both exhibited less correlation to the pfCx expression profile. Taken together with the GEDI mosaics, these results suggest that the Amy and Hip neuron populations exhibit proteomes with a distinct subpopulation of proteins corresponding to a biological component or process that is highly expressed in these cells, but not in the pfCx population.
To identify the biological processes associated with differentially expressed proteins between the pfCx, Hip, and Amy neuron populations, we subjected the proteins exhibiting a twofold change or greater expression level to proteomaps analysis (Liebermeister et al. 2014). These maps illustrate the top level gene ontology terms associated with the proteins identified in the expression data and organize them according to a Voronoi diagram to facilitate easy distinction of major biological process groups and their related subgroups (Fig. 3, H–K). The proteomaps for the pfCx, Hip, and Amy cultures show that proteins associated with transcription/translation (i.e., blue areas of the map) and metabolism (i.e., yellow-brown areas of the map) account for the majority of the proteome for these cells, with subtle differences in distinct brain regions in the expression levels of proteins associated with these biological process groups. The animated proteomaps (Supplemental Movies S1–S3; supplemental material for this article is available online at the journal website) further show the hierarchy of the biological processes in increasing amount of detail (see also Supplemental Tables S1–S3). Taken together, these results show that the in vitro protein expression profiles for the pfCx, Hip, and Amy exhibit a large degree of similarity, but that subtle differences observed in the expression patterns may reveal unique underlying biological processes that distinguish these brain regions in vitro.
While a substantial proportion of the proteins in the mass spectrometry data set exhibited similar expression values across the pfCx, Hip, and Amy cultures, PCA enabled us to identify proteins associated with the greatest variance in expression levels between the different brain regions (Fig. 4A). The results of the PCA analysis were projected into the two dimensions associated with the greatest variability in the expression data set and plotted on a scatterplot, with each point color coded to represent the top level gene ontology biological process associated with the corresponding protein (listed in the legend of Fig. 4A). By sorting the proteins in the mass spectrometry data set according to the magnitudes of the first and second principal components, we were able to identify the set of proteins that exhibited the greatest differences in expression between the pfCx, Hip, and Amy neuronal cultures, examples of which are listed in Table 2. We sought to determine whether any of these differentially expressed proteins were associated with cellular components or signaling pathways that may shed light on the functional differences observed in the pfCx, Hip, and Amy neuron populations. Thus the mass spectrometry data from these three neuronal populations were subjected to IPA to identify protein interaction networks wherein proteins from our data set were highly represented. The IPA software used z-score analysis to identify and rank the pathways represented in the expression data set, and the top 13 most representative networks are illustrated in Fig. 4B. These networks were largely associated with cytoskeletal signaling and axonal growth, and the z-scores indicate that the pfCx population demonstrated a distinct profile vs. the Hip and Amy populations, which exhibited similar profiles for all of the highly represented pathways shown.
Fig. 4.

Protein expression variation and protein interaction network analysis of cells derived from different brain regions. A: principal component analysis (PCA) was used to assess the expression variation and abundance of proteins associated with the primary gene ontology biological process terms represented in the mass spectrometry data for each brain region sample. Examples of proteins exhibiting the highest levels of variance between the prefrontal cortex (pfCx), hippocampus (Hip), and amygdala (Amy) samples are shown in Table 2. B: ingenuity pathway analysis (IPA) was used to identify biological pathways associated with proteins identified in the mass spectrometry data for the pfCx, Hip, and Amy brain regions, and a heatmap visualizing the top 13 most represented pathways is shown. The heatmap is color-coded according to z-scores, indicating the level of enrichment for each pathway based on the expression profiles for each brain region. C: protein-protein interaction networks were constructed using the STRING database to identify protein complexes represented in the mass spectrometry data associated with proteins exhibiting both the most variance in expression between brain regions (neurofilament assembly and glutathione metabolism) and the most similar expression levels between brain regions (SNARE complex and synaptic vesicle cycling). The length of the edges between each pair of protein nodes in the protein-protein interaction networks was computed using a spring-like model to cluster higher confidence protein-protein interactions closer together into functionally-related clusters.
Table 2.
Differences in protein expression in cells from different brain regions
| Accession No. | Description | pfCx | Hip | Amy |
|---|---|---|---|---|
| Q8MHH3 | MHC class I RT1-Au heavy chain (fragment) | 3.95 | 10.9 | 27.24 |
| Q31274 | Rat MHC class I RT1 (RT16) mRNA (u haplotype), 3′ end (fragment) | 3.67 | 26.39 | 41.52 |
| M0R4H5 | PDZ and LIM domain protein 4 | 4.04 | 15.49 | 29.81 |
| Q2IBC5 | Caveolin-2 | 5.92 | 25.17 | 35.15 |
| A1A5Q1 | Parp9 protein | 5.3 | 16.24 | 24.96 |
| G3V8L7 | Integrin αM | 3.06 | 9.49 | 37.22 |
| Q6R5Q0 | Activator protein-2γ | 10.96 | 70.58 | 38.29 |
| Q5FVQ0 | Zinc transporter ZIP8 | 10.35 | 36.05 | 28.11 |
| B1WCA2 | Tetratricopeptide repeat domain 25 | 8.4 | 67.38 | 54.98 |
| D3ZAB1 | Lactotransferrin (predicted) | 8.33 | 86.84 | 25.71 |
| F1LQ54 | Protein Pcaf (fragment) | 5.61 | 23.63 | 14.95 |
Principal component analysis (unitless) was used to assess the expression variation and abundance of proteins associated with the primary gene ontology biological process terms represented in the mass spectrometry data for each brain region sample. Examples of proteins exhibiting the highest levels of variance between the pfCx, Hip, and Amy samples are shown.
To obtain a more detailed idea about the changes and similarities in protein expression between the different brain regions, we used the protein-protein interaction network analysis software STRING (Szklarczyk et al. 2015) to create protein-protein interaction networks from the set of proteins that demonstrated the greatest and least variance between the pfCx, Hip, and Amy neuron cultures as determined from the PCA (Fig. 4C). The length of the edges between each pair of protein nodes in the protein-protein interaction networks was computed using a spring-like model to cluster higher confidence protein-protein interactions closer together into functionally-related clusters. We found that interaction networks corresponding to NF assembly and glutathione metabolism (Fig. 4C, top) were present in the set of proteins showing the greatest variance between the three brain regions examined. In contrast, interaction networks associated with the SNARE (soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors) complex and synaptic vesicle cycling (Fig. 4C, bottom) were among the proteins exhibiting similar expression levels across the pfCx, Hip, and Amy neuron cultures. Interestingly, the proteins that are involved in NF assembly and glutathione metabolism appear to be differentially expressed in a brain region-dependent manner. This suggests a difference in brain region neuronal activity pattern, which in turn may result in less reactive oxygen species (ROS) and less need for glutathione. Conversely, proteins involved in the SNARE complex and synaptic vesicle cycling appear to show similar expression levels between the pfCx, Hip, and Amy cultures, as expected, considering that synaptic vesicle transmission of neurotransmitters is a fundamental aspect of neuron function.
Although not the focus of this study, we depicted a brief example list of protein expression levels comparing in vitro vs. in vivo, i.e., comparing pfCx, Hip, and Amy cells in vitro vs. pfCx, Hip, and Amy neonate and adult rat tissue (Table 3). As expected, differences between cells and tissue become obvious either in a difference of expression levels, e.g., for protein S100a13 or apolipoprotein B-100, or in the lack of proteins in either tissue or in vitro system, e.g., glutathione S-transferase α-3, aquaporin-1, or parvalbumin-α (Table 3). Sometimes, proteins were expressed as different isoforms or subunits in either in vitro system or tissue, e.g., Parp9 was expressed in cells, but not in tissue samples, whereas Parp14 was expressed in tissue, but not in vitro (Table 3). Depending on the study to be conducted, the expression (or the lack thereof) of the protein(s) of interest should be evaluated carefully. A similar study was conducted previously, comparing extracellular matrix proteins in rat tissue from different brain regions and cells in vitro of the same brain areas, finding differences in the expression of these proteins in a brain region- and system-dependent manner (Dauth et al. 2016).
Table 3.
Comparison of protein expression levels of pfCx, Hip, and Amy cells in vitro vs. pfCx, Hip, and Amy neonate and adult rat tissue
| Accession No. | Description | pfCx Neurons | pfCx Adult | Hip Neurons | Hip Neonate | Hip Adult | Amy Neurons | Amy Neonate | Amy Adult |
|---|---|---|---|---|---|---|---|---|---|
| P04904 | Glutathione S-transferase α-3 | 18.280 | 50.413 | 23.045 | |||||
| D3ZTB5 | Protein S100a13 | 10.633 | 3.906 | 12.423 | 10.653 | 2.013 | 17.030 | 12.373 | 1.342 |
| F1M6Z1 | Apolipoprotein B-100 | 7.219 | 7.286 | 53.583 | 1.346 | 3.968 | 31.601 | 2.267 | 1.896 |
| Q5U362 | Annexin | 5.764 | 4.325 | 24.645 | 6.571 | 2.332 | 21.381 | 14.900 | 1.055 |
| P07171 | Calbindin | 5.615 | 5.208 | 16.588 | 5.028 | 2.126 | 10.001 | 10.705 | 2.362 |
| P53987 | Monocarboxylate transporter 1 | 5.487 | 4.960 | 17.966 | 4.593 | 3.558 | 24.181 | 8.146 | 2.429 |
| A1A5Q1 | Parp9 protein | 5.299 | 16.243 | 24.964 | |||||
| F1LZ05 | Protein Parp14 | 3.225 | 3.503 | 2.197 | 4.710 | 1.811 | |||
| P29975 | Aquaporin-1 | 4.348 | 5.515 | 5.205 | |||||
| P31000 | Vimentin | 4.212 | 4.066 | 15.962 | 0.661 | 3.265 | 20.943 | 0.954 | 1.247 |
| Q8MHH3 | MHC class I RT1-Au heavy chain (fragment) | 3.952 | 10.896 | 27.241 | |||||
| O35397 | Caspase-6 | 3.611 | 16.186 | 14.304 | |||||
| Q9QZK8 | Deoxyribonuclease-2-α | 3.422 | 4.498 | 15.339 | 2.569 | 2.811 | 17.070 | 4.128 | 2.066 |
| P85972 | Vinculin | 3.392 | 4.958 | 10.984 | 3.117 | 2.852 | 18.603 | 5.102 | 2.124 |
| P19527 | Neurofilament light polypeptide | 3.261 | 7.898 | 12.352 | 6.228 | 2.603 | 14.804 | 13.521 | 1.900 |
| P10860 | Glutamate dehydrogenase 1, mitochondrial | 1.559 | 5.391 | 4.701 | 6.994 | 2.565 | 6.191 | 9.749 | 2.143 |
| D3ZKR3 | Glyceraldehyde-3-phosphate dehydrogenase | 1.326 | 4.647 | 6.244 | 6.243 | 2.836 | 7.450 | 10.223 | 2.213 |
| M0RDJ4 | Glia maturation factor beta | 1.155 | 3.515 | 4.319 | 4.344 | 2.451 | 5.548 | 6.356 | 2.582 |
| P97610 | Synaptotagmin-12 | 0.689 | 4.718 | 2.210 | 8.046 | 2.186 | 3.292 | 11.398 | 1.853 |
| P19490 | Glutamate receptor 1 | 0.689 | 3.107 | 2.219 | 15.078 | 3.333 | 2.864 | 10.894 | 2.670 |
| G3V733 | Synapsin-2 | 0.551 | 3.195 | 0.881 | 14.895 | 2.351 | 1.756 | 21.945 | 1.869 |
| Q5M7V8 | Thyroid hormone receptor-associated protein 3 | 0.757 | 1.671 | 2.244 | 1.011 | 2.565 | 2.743 | 1.694 | 1.690 |
| Q2IBC5 | Caveolin-2 | 5.918 | 25.166 | 35.149 | |||||
| D3ZAB1 | Lactotransferrin (predicted) | 8.327 | 86.844 | 25.711 | |||||
| G3V8L7 | Integrin αM | 3.058 | 9.492 | 37.225 | |||||
| P84076 | Neuron-specific calcium-binding protein hippocalcin | 4.512 | 17.821 | 2.199 | 14.490 | 1.818 | |||
| P02625 | Parvalbumin-α | 5.842 | 8.678 | 2.570 | 9.673 | 2.003 |
Values are in fold change. Protein expression levels of example proteins were compared between the pfCx, the Hip, and the Amy from neonate and adult rat brain, as well as from cells in vitro isolated from the respective brain regions. The neonate pfCx served as a control for all mass spectrometry presented here.
Metabolism In Vitro Is Brain Region Dependent
The energy within cells is mainly produced by the mitochondria as part of the respiration process, and it is characterized by different parameters, such as ATP production, basal respiration, proton leak, in addition to nonmitochondrial respiration. Moreover, the cells’ metabolism is directly coupled to other cell functions, including, but not limited to, electrical activity, cell growth, and cell proliferation (Cherry et al. 2016). Metabolism of neuronal cultures from different brain regions was analyzed by measuring the OCR (Fig. 5). Basal respiration, proton leak, ATP production, and nonmitochondrial respiration were significantly different in Amy cultures compared with cultures from the pfCx and the Hip. More specifically, basal respiration was the same in pfCx and Hip cultures, but three times lower in the Amy cells. Furthermore, the proton leak was highest in Hip cultures (20 pmol/min), slightly lower in pfCx cultures (12.5 pmol/min), and significantly lower in the Amy (2.5 pmol/min). ATP production was similar in cultures from the pfCx and the Hip, but more than two times lower in Amy cultures. Lastly, for the nonmitochondrial respiration, a decrease by almost two times was observed in Amy cultures compared with pfCx and Hip cells. Overall, all analyzed parameters were lower in neuronal cells from the Amy, suggesting that these cells are overall less active in vitro and therefore exhibited a lower energy demand (Fig. 5), which is well in line with the observation that especially the expression of glutathione metabolism proteins, which are needed more in active cells (Yermolaieva et al. 2000), showed a high variance between the samples (Fig. 4).
Fig. 5.
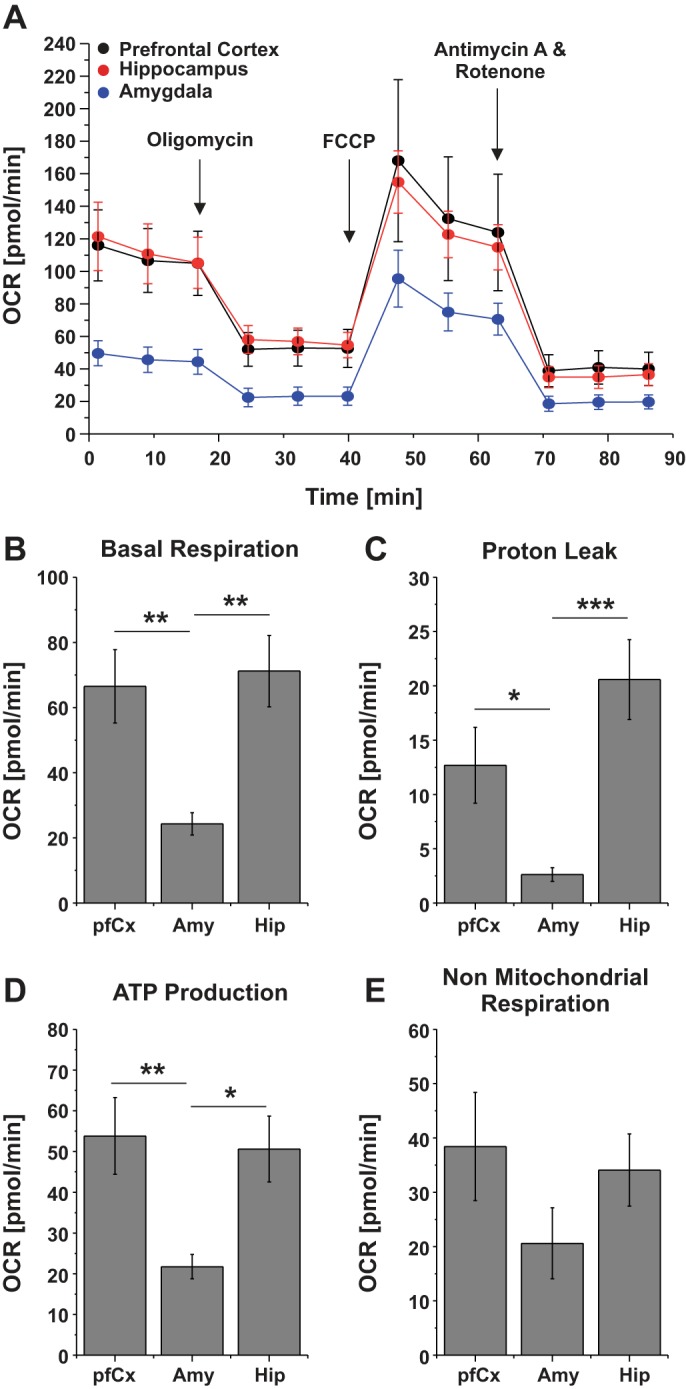
Metabolic activity of cells derived from different brain regions. A: the oxygen consumption rate (OCR) of neuronal cultures from the pfCx, the Hip, and the Amy was measured after 14 DIV. Modulators of respiration were used to target components of the electron transport chain in the mitochondria to observe key parameters of metabolic function. B: basal respiration is assessed before the compounds (oligomycin, FCCP, and a mix of rotenone and antimycin A) and are serially injected to measure and calculate the proton leak (C), ATP production (D), and nonmitochondrial respiration (E). Values are means ± SE; n = 4 plates, 80 wells (pfCx); 3 plates, 60 wells (Hip); and 7 plates, 140 wells (Amy). All samples were seeded at the same cell density (100 K/well). Significances are indicated as follows: *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Electrical Activity In Vitro Is Brain Region Dependent
We have shown thus far that cells from different brain regions cultured in vitro exhibited unique brain region-dependent cell composition, protein expression, and metabolism. We were further interested in whether in vitro cultures of cells from different brain regions were inherently different in their electrical activity. Neuronal cells from different brain regions were cultured on MEAs for 18–21 days, and electrical activity was recorded. Waveform examples are depicted over 50 s for the pfCx (Fig. 6A), Hip (Fig. 6C), and Amy (Fig. 6E). The overall amount of spikes in each region is shown within a raster plot (Fig. 6, B, D, and F). Each spike is represented by a line. Each row represents one electrode. The results indicate that the Amy cultures exhibit overall significantly less spikes compared with pfCx and Hip cells. Electrical activity of each region was analyzed using either a band-pass (50 Hz, Fig. 6G) or high-pass filter (125 Hz, Fig. 6H) to distinguish between high (signal from a couple of cells within the network, local) and low frequencies (field potentials). Results indicated a similar interspike interval for band-pass filtered data for the pfCx and the Amy, whereas the Hip cultures showed more spikes with longer interspike intervals (Fig. 6G), showing that the Hip cultures exhibited more field potentials, which are approximately 1 s apart from each other. High-pass filtered data suggested again a similarity between pfCx and Amy cultures, whereas Hip cultures exhibited more spikes in general (Fig. 6H), correlating well with our observation that overall the frequency is highest in Hip cells (Fig. 6M). Numbers of spikes within a 5-min range were observed to be significantly higher in the Hip compared with the Amy in vitro. A higher number of spikes were also found in the pfCx compared with the Amy (Fig. 6I). In a burst, Hip cultures exhibited a longer interspike interval compared with both the pfCx and the Amy cultures (Fig. 6J), which correlates well with our findings that field potentials in the Hip cultures are exhibiting longer intervals compared with the other two regions (Fig. 6G). Time between bursts was observed to be highest in Amy cultures compared with both Hip and pfCx cultures (Fig. 6K). Burst frequency was observed to be lowest in Hip cultures and highest in pfCx cultures (Fig. 6L). Hip cultures exhibited the highest overall frequency, followed by the pfCx and the Amy (Fig. 6M). The Amy showed the lowest overall frequency, which correlates well with our findings that Amy cells exhibited the lowest basal respiration, ATP production, and proton leak (Fig. 5), suggesting an overall lower activity of Amy cells in vitro compared with pfCx and Hip cultures. Overall, these results suggest that in vitro electrophysiological features, such as frequency, waveform, and interspike intervals, were dependent on the brain region from which cells were derived, suggesting an intrinsically different electrophysiological behavior in vitro. Interestingly, there also seems to be a difference between field potentials (network activity, low frequencies) and overall spikes (high frequencies, signal from a couple of cells within the network) in a brain region-dependent manner, suggesting the presence of several communication patterns in vitro, which is well in line with in vivo results reporting that low frequencies modulate activity over large spatial regions in long temporal windows, and that high frequencies modulate activity over small spatial regions and short temporal windows (Buzsáki and Draguhn 2004; von Stein and Sarnthein 2000).
Fig. 6.

Electrophysiology of prefrontal cortex, hippocampal, and amygdala neurons in vitro. Electrical activity was recorded from 18 DIV neuronal cultures. A representative example of a typical waveform of PfCx (A), Hip (C), and Amy (E) recordings, as well as the corresponding raster plots (B, D, and F) for the respective regions are depicted. Each line in the raster blot represents a neuronal spike (electrical firing activity). G and H: interspike intervals were measured from the pfCx cells (red), Hip cells (green), and Amy cells (blue) using a band-pass filter (50 Hz; G) or a high-pass filter (125 Hz; H). I–M: recordings were further used to analyze number of spikes/5 min (I), interspike interval in a burst (J), interburst interval (K), frequency in a burst (L), and overall frequency (M). Significance levels were analyzed using ANOVA one-way analysis with a Bonferroni post hoc test. Values are means ± SE; n = 3 MEAs/brain region, 60 electrodes each, 5-min recordings. Significances are indicated as follows: *P ≤ 0.05, ***P ≤ 0.001.
A Novel Brain-on-a-Chip Interconnecting Cells from Different Brain Regions and the Influence on Cell Composition
In vivo, proper brain function relies on the communication between different brain regions. Together with our results that in vitro cells from different brain regions exhibit different traits, these observations underline the importance of developing brain in vitro models that recapitulate the complexity of brain region interactions in vivo and suggest that the presence of cells from other brain regions and their connections might have an influence on parameters such as cell composition and electrophysiology. Therefore, we developed a multiregional brain-on-a-chip model (Fig. 7), where cells from different brain regions were seeded in separate compartments, but grew axonal connections and thereby connected to each other (Fig. 7, A–E). Briefly, a PDMS mask consisting of three separate compartments was attached to a coverslip. Cells from each brain region (pfCx, Hip, Amy) were seeded into each compartment and allowed to adhere to the culture substrate before the mask was removed. After removal of the PDMS mask, cell bodies stayed in their area and started extending axons toward the other cell areas (i.e., toward the other brain regions). After a few days, axons had grown over the single gap that existed between the different cell areas and thereby connected the distinct cell regions (Fig. 7). More details can be found in materials and methods.
As previously demonstrated in this study, cells from distinct brain regions exhibited unique characteristics in vitro. The multiregional brain-on-a-chip model now enables us to investigate whether the presence of cells from different brain regions influences characteristics such as cell composition or electrical activity in vitro. As was demonstrated in Fig. 2, separately seeded cells exhibited a different abundance of GFAP, GAD, and Vglut, depending on from which brain region cells were derived. The same analysis was performed for our brain-on-a-chip model for each brain region (Fig. 7, F–Q). Overall, we observed that the presence of cells from other brain areas in vitro influences the abundance of various neuronal and nonneuronal markers as is depicted in Fig. 7, O–Q. Compared with the separately seeded pfCx cells, within this brain area of the brain-on-a-chip model we observed 10% less Vglut-positive cells, about the same amount of GAD-positive cells, and ~5% less GFAP-positive cells. For the Hip area, we determined between 2 and 5% more Vglut- and GAD-positive cells, and ~7% less GFAP-positive cells in the brain-on-a-chip model compared with the separately seeded cells. For the Amy, we saw 10% less Vglut-positive cells, ~4% less GAD-positive cells, and almost 10% more GFAP-positive cells in the brain-on-a-chip model compared with separately seeded cells (Fig. 7, O–Q). Overall, these results suggest that the cell composition of these cultures was influenced by the presence of cells from other brain regions.
The Presence of Cells from Other Brain Regions Influences Electrical Activity In Vitro
Electrical activity of the multiregional brain-on-a-chip model was assessed (Fig. 8) in a manner similar to the individually seeded cultures previously (Fig. 5). An example of a raw MEA signal from one multiregional brain model is shown in Fig. 8B. Electrodes covered by pfCx cells are outlined in red, by Hip cells in blue, and by Amy cells in orange (Fig. 8B). Outline colors correspond to the colors used in Fig. 8A. Within the multiregional brain-on-a-chip, distinct cell populations exhibited distinct waveforms (Fig. 8B). To test if the distinct cell populations were communicating with each other, we analyzed the cross-correlation of their firing activity (Fig. 8C). Red indicates a high cross-correlation; blue none. We observed a clear cross-correlation within each population, but also across distinct populations (Fig. 8C), suggesting that the different cell populations were communicating with each other. Analyzing the recordings using a band-pass filter, we observed a distinct difference in the interspike interval behavior of the Hip cell population, with more spikes having a shorter time period between them (Fig. 8D). Using a high-pass filter revealed in general more spikes for the Hip cell population compared with the other two (Fig. 8E). Raster plots showed similar frequency of the Hip and pfCx cell populations and a lower frequency of the Amy cell population (Fig. 8, G, I, and K). Example waveforms for each cell population are shown in Fig. 8, F, H, and J. Comparing the number of spikes, we did not find a significant difference between the cell populations (Fig. 8L). Interspike intervals appeared to be longer for the Hip compared with the other two regions (Fig. 8M). Overall frequency and interburst intervals appeared to be similar between all three cell populations (Fig. 8, N and O). The frequency in a burst was significantly higher in the pfCx cell population compared with both other regions (Fig. 8P). Overall, these results suggest that, within our multiregional brain-on-a-chip model, different cell populations were communicating with each other, they exhibited distinct electrophysiological behavior (e.g., waveforms, interspike interval), and that, compared with the separately seeded cultures, they altered some of their electrophysiological behavior (e.g., frequency, interspike intervals). This in turn suggests that the presence of cells from other brain regions and their connections influenced electrophysiology in vitro.
Fig. 8.

Electrophysiology of the multiregional brain-on-a-chip model. Electrical activity was recorded from 18 DIV multiregional brain-on-a-chip models. A: multiregional brain-on-a-chip immunostained for βIII-tubulin (green) and glial fibrillary acidic protein (GFAP, red) depicts pfCx (red outline), Hip (blue outline), and Amy cells (orange outline). B: example of an electrical recording (MEA, 60 electrodes) for the multiregional brain model is depicted. Electrodes recording pfCx cell activity are outlined in red, Hip electrodes are outlined in blue, and Amy electrodes are outlined in orange. C: cross-correlation analysis of the different regions of the brain-on-a-chip. Red color indicates a high correlation between network activity, while blue indicates a low correlation. D and E: interspike intervals were measured from pfCx area (red), Hip area (green), and Amy area (blue) using a band-pass filter (50 Hz; D) or a high-pass filter (125 Hz; E). F–K: representative example of a typical waveform. pfCx (F), Hip (H), and Amy (J) recordings are depicted. G, I, and K: raster plots are shown for each brain area, with each line representing a spike. L–P: recordings were further used to analyze number of spikes/5 min (L), interspike interval in a burst (M), interburst interval (N), frequency in a burst (O), and overall frequency (P). n = 3 MEAs, 6–20 electrodes/area, 5-min recordings. Significance levels were analyzed using ANOVA one-way analysis with a Bonferroni post hoc test. Significances are indicated as follows: *P ≤ 0.05, **P ≤ 0.001.
Drug Application
The three brain regions that were chosen for the multiregional brain-on-a-chip, i.e., pfCx, Hip, and Amy, and their connections are implicated in the disease of schizophrenia (Benes 2010). Hence, PCP was chosen as a test compound since it has been shown to induce schizophrenia-like symptoms in healthy individuals and animal models due to its inhibitory action on N-methyl-d-aspartate receptors (Adachi et al. 2013). One area each of the multiregional brain model was treated with 5 µM PCP at seeding (Fig. 9A) and electrical activity was measured daily from DIV 11 until DIV 20. A control sample with no PCP addition was measured in parallel on the same days (Fig. 9A). Figure 9 shows example traces of spontaneous electrical activity for the pfCx (Fig. 9B), the Hip (Fig. 9C), and the Amy (Fig. 9D) for DIV 12, DIV 15, and DIV 18. For all samples that were treated with PCP, traces for the Hip show differences in their amplitude and seem less regular compared with the control (Fig. 9C). For Amy, the control shows a higher frequency compared with the dosed conditions (Fig. 9D). For the pfCx, it seems that, especially when the pfCx itself was dosed, we observed a higher frequency compared with the control (Fig. 9A). Differences are also apparent when comparing the electrical activity of the same regions for different dosing conditions. Cortical traces differ in their amplitude and frequency, depending on which area was dosed with PCP (Fig. 9A). For the Hip, all conditions dosed with PCP show lower amplitude of the signal compared with the control; moreover, the frequency appeared to be different between the varying dosing conditions (Fig. 9C). We further analyzed the overall frequency and active electrodes of all conditions and brain regions for the different time points (Fig. 10). The main differences (a higher frequency) were seen in the Hip when either the Hip itself or the Amy was dosed. Active electrodes showed an overall increase in numbers the first few days and then a plateau phase. Numbers of active electrodes differed the most between regions when PCP was added to the Hip, but these differences correlated well with what we saw in the control condition (Fig. 10). Overall, it became apparent that it matters which brain region was dosed, since the wave patterns differed depending on which brain region was treated, underlining the advantage of this model to apply drugs to individual brain areas separately while analyzing downstream effects on interconnected regions.
Fig. 9.

Assessment of electrical activity of the different brain areas of the multiregional brain-on-a-chip model after phencyclidine dosage. A: the multiregional brain-on-a-chip model was dosed with 5 µM phencyclidine (PCP) at seeding, and electrical activity was measured at DIV 12 until DIV 20. A control with no PCP addition was measured in parallel on the same days. Example traces for the pfCx (B), the Hip (C), and the Amy (D) of DIV 12, DIV 15, and DIV 18 are shown for each dosage condition (A). Traces represent one electrode for the different time points and depict 60 s of a 5-min recording. N = 4–5 MEAs/condition, 8–20 electrodes/area, 5-min recordings/time point.
Fig. 10.

Assessment of electrical activity of the multiregional brain-on-a-chip model dosed with phencyclidine (PCP) at seeding. The brain-on-a-chip model was dosed with 5 µM PCP at seeding, and electrical activity was measured at DIV 11 until DIV 20. PCP was added to one brain area only (either pfCx, or Hip, or Amy, see Fig. 9). A control with no PCP addition was measured in parallel on the same days. A–D: mean frequency was analyzed for each condition and day and normalized to DIV 15 frequency. Frequency measured within the pfCx area is represented by black dots/lines, Hip area by red, and Amy area by blue. E–H: active electrodes were plotted over time for all the conditions. Active electrodes represent the percentage of electrodes that show an electrical signal compared with the electrodes that are covered by cells for each brain area. Values are means ± SE; n = 4–5 MEAs, 8–20 electrodes/area, 5-min recordings/time point.
We investigated a separated condition, where cultures of the multiregional brain-on-a-chip were dosed at DIV 12 with either 10 µM or 25 µM PCP (or no PCP as a control), and electrical activity was measured before PCP addition and at several time points thereafter. Frequency generally decreased right after PCP addition and increased until around 48 h after PCP addition and decreased again afterwards (Fig. 11). Frequency was overall higher in the Amy, assuming a different effect of PCP on Amy connections (inhibitory connections vs. excitatory connections). Comparing the example traces, it becomes apparent that, in the control, the electrical signal became more mature over time in terms of frequency and amplitude (Fig. 11), whereas this did not happen for the PCP dosed cultures, confirming an effect of PCP on electrical activity regarding amplitude and frequency, which might not become so apparent when looking at overall frequency only.
Fig. 11.

Assessment of electrical activity of the multiregional brain-on-a-chip model dosed at DIV 12. A: the multiregional brain-on-a-chip model was dosed with 10 µM, 25 µM, or no phencyclidine (PCP) at DIV 12, and electrical activity was measured before PCP addition, as well as at several time points after drug dosage (3 h, 6 h, 24 h, 48 h, 72 h, 96 h, and 120 h). B–D: mean frequency of all time points was normalized to the mean frequency measured before drug addition. PfCx area is represented by black dots/lines, hippocampal area by red, and amygdala area by blue. E–G: active electrodes were plotted over time for all the conditions. Active electrodes represent the percentage of electrodes that show an electrical signal compared with the electrodes that are covered by cells for each brain area. H–J: example traces for the pfCx, the Hip, and the Amy are depicted for each condition for the three different time points (before PCP addition, 24 h and 48 h after PCP addition). Traces represent one electrode for the different time points and depict 60 s of a 5-min recording. Values are means ± SE; n = 4–5 MEAs, 8–20 electrodes/area, 5-min recordings/time point.
DISCUSSION
In vivo, brain regions differ in their structure, function, cell composition, and other features (Dauth et al. 2016; Herculano-Houzel 2009; Kandel et al. 2000; Masland 2004; Stevens 1998). In vitro, neurons from different brain regions are commonly isolated, but so far studies usually looked at one region per study only (Lesuisse and Martin 2002; Lorenzo et al. 1992; Whitson et al. 1989). In turn, differences and similarities of different brain regions in vitro are still poorly understood.
Cell Composition of Neuronal Cultures Derived from Different Brain Regions
In vitro, not much is known about whether cells from distinct brain regions differ with respect to their cell composition. As our results demonstrated (Fig. 2), cells derived from distinct brain regions exhibited a different cell composition in vitro, e.g., altering amounts of astrocytes as well as of glutamatergic and GABAergic neurons. Interestingly, when trying to compare these results to in vivo data, it becomes apparent that there is a substantial lack of information concerning quantitative data about neuronal and nonneuronal cell numbers. There are some studies available, mainly looking into neuronal and nonneuronal cell numbers in rodents, primates, and in humans, focusing on areas like the cortex, the Hip, the cerebellum, and the olfactory bulb (Bandeira et al. 2009; Herculano-Houzel 2009; Herculano-Houzel and Lent 2005). For some brain regions, different types of neurons have been described (Masland 2004; Stevens 1998), but there are no quantitative reports on numbers of specific neuronal types (e.g., glutamatergic or GABAergic neurons). Astrocytes have been reported to be more abundant in the adult rat cortex and Hip (no data is available for the Amy), but account for ~1/3 of the neuronal population in 2-day old rats (Bandeira et al. 2009). The latter correlates well with our findings for cells that were cultured individually in vitro for these specific brain regions. We further see that the amount of specific neuronal types differed especially in the Amy compared with the other two brain regions, showing less astrocytes and more GABAergic and glutamatergic neurons. The main two neuronal types that have been described in vivo in the Amy are glutamatergic and GABAergic (Sah et al. 2003), which we also observed in vitro. Overall, the amount and distribution of neurons and astrocytes is brain region dependent in vivo, an important feature that should be kept in mind when evaluating in vitro models of specific brain regions.
Protein Expression, Metabolism and Electrophysiology of Neuronal Cultures from Different Brain Regions
While researchers have started mapping the protein expression of different brain regions of the mouse and rat brain and are eager to decipher the human brain proteome (Kasukawa et al. 2011; Katagiri et al. 2010; Martins-de-Souza et al. 2014; Sharma et al. 2015), in vitro these data are not available. Deciphering the protein expression in vitro is crucial for better understanding the cells with which we are working and understanding differences and similarities between multiple brain regions in vitro. It will also possibly increase our knowledge about how well correlated our in vitro models are to their in vivo counterpart.
Here, we show that there are differences as well as similarities between neuronal cultures in vitro from different brain regions. There have been studies reporting the presence of several proteins in different brain regions in vivo (Katagiri et al. 2010; Sharma et al. 2015; Taraslia et al. 2013), as well as one comprehensive quantitative analysis of gene expression in different brain regions of the mouse brain (Kasukawa et al. 2011). Our analysis revealed several proteins that show a similar expression between the different cultures, but also some proteins that show a high variance in protein expression in a brain region-dependent manner. Examples include proteins from the MHC class I family of proteins, which show a high expression in Amy cells and comparably less in the other two cultures. Other proteins, such as lactotransferrin, is highly expressed in the Hip in vitro and comparably less in cultures from the Amy or pfCx. One study has reported differences in the mRNA levels of proteins involved in the immune function in microglial cells isolated from different brain regions (Ren et al. 1999), supporting our findings that cells from different brain regions show differences in vitro in a brain region-dependent manner.
As shown in the network analysis (Fig. 4), proteins involved in either the NF assembly or the glutathione metabolism seem to be different in their expression levels between the different samples. The glutathione metabolism is important for several things, including, but not limited to, antioxidant defense, nutrient metabolism, protein synthesis, and signal transduction (Wu et al. 2004). It has been shown that functionally active neurons show increased metabolic activity and oxygen consumption, resulting in higher levels of ROS (Yermolaieva et al. 2000). One possible explanation of why high variances particularly occur in this protein network might be that Amy cells in vitro are less metabolically and electrically active compared with cells from the pfCx or the Hip (Figs. 5 and 6) and, therefore, produce less ROS and exhibit lower expression levels of proteins involved in glutathione metabolism.
NFs have been reported to be prominent in large axons, whereas small unmyelinated axons contain only few NFs, and some small neurons even lack morphologically identifiable NFs, suggesting a differential expression of NFs depending on neuronal morphology (Elder et al. 1998). We here have only shown that neuronal types are differentially distributed, depending on the brain regions from which the cells were derived, but have not closely looked at morphological differences of the neurons. Therefore, it might be possible that differences in neuronal morphology in the different cultures leads to a different amount of NFs and in turn to high variances in proteins involved in NF assembly.
On the other hand, proteins that are involved in the SNARE complex and the synaptic vesicle cycling exhibit similar expression levels in all three samples analyzed, suggesting that these protein networks are functioning in a non-brain region-dependent manner in vitro. In exocytosis and neurosecretion, SNARE and associated proteins (including proteins involved in synaptic vesicle cycling) play a critical role in vesicle docking, priming, fusion, and synchronization of neurotransmitter release, which is triggered by action potentials, and inherent to most neurons (Hu et al. 2002; Ramakrishnan et al. 2012; Sudhof 2004). Therefore, it is reasonable that cells derived from different brain regions express proteins involved in these events in similar levels.
Another interesting finding is that most of the proteins that show a twofold or higher expression change compared with the control belong to the metabolic process group. Consequently, we were also interested in whether the metabolic activity of cells from different brain regions differs in vitro. Therefore, we analyzed the OCR of our cultures and found that cells from the Amy had the lowest OCR compared with both other regions, suggesting a lower metabolic activity of these cells in vitro. Generally, it has been reported that increased synaptic signaling between neurons leads to higher energy consumption (Karbowski 2007). In vivo, the performance of different cognitive tasks (e.g., learning, fear conditioning) dictates the metabolic activity of involved brain regions (Knight et al. 2004; Linnman et al. 2012). Furthermore, alterations in metabolic activity in the Amy have been associated with psychiatric disorders, such as posttraumatic stress disorder (Shin et al. 2009). It has also been observed that the lateral Amy exhibits low levels of spontaneous activity in vivo, suggesting a lower energy demand (Sah et al. 2003). These results correlate well with our observations that cells from the Amy exhibit a lower metabolic activity as well as a lower overall frequency of spontaneous activity in vitro. Common in vivo oscillations, such as slow oscillations (<1 Hz), δ- (1–4 Hz), θ- (4–8 Hz), α- (8–12 Hz), β- (12–30 Hz), and γ-bands (>30 Hz) have been reported in vitro too, mainly analyzed in cortical slices (Canolty and Knight 2010). Neighboring frequency bands within the same neuronal network are typically associated with different brain states, but several rhythms can temporally coexist in the same or different structures and interact with each other (Buzsáki and Draguhn 2004; Penn et al. 2016). Interestingly, different frequencies provide distinct temporal windows for processing, whereas different rhythms are associated with different spatial scales and therefore different cell population sizes (Canolty and Knight 2010). It has been suggested that low frequencies modulate activity over large spatial regions in long temporal windows, and that high frequencies modulate activity over small spatial regions and short temporal windows (von Stein and Sarnthein 2000). Our results indicate overall slow oscillations around 1 Hz and lower in neuronal cultures from all brain regions. Intracellular recordings from cortical neurons in cats identified slow oscillations (0.16–0.3 Hz), in addition to δ-oscillations (1–4 Hz), a finding replicated in humans at the electroencephalogram level (Achermann and Borbély 1997; Hinard et al. 2012), and well in line with our in vitro observations. Besides the slow oscillations, we also observed high-frequency burst activity in our neuronal cultures in a brain region-dependent manner (pfCx ~55 Hz, Hip ~25 Hz, Amy ~45 Hz). In vitro studies on brain slices have also reported low-γ (30–80 Hz) and high-γ activity (80–200 Hz), where low-γ activity arises mainly from interactions within the highly interconnected GABAergic (inhibitory) interneuron network, as well as from network interactions between populations of (excitatory) pyramidal cells and local interneuron network (Canolty and Knight 2010; Freeman 1950), suggesting the presence of these networks in our in vitro cultures.
Going a step further, differences between cells and tissue become obvious either in a difference of expression levels, e.g., for protein S100a13 or apolipoprotein B-100, or in the lack of proteins in either tissue or in vitro system, e.g., glutathione S-transferase α-3, aquaporin-1, or parvalbumin-α (Table 3). Sometimes, proteins were expressed as different isoforms or subunits in either in vitro system or tissue, e.g., Parp9 was expressed in cells, but not in tissue samples, whereas Parp14 was expressed in tissue, but not in vitro (Table 3). Depending on the study to be conducted, the expression (or the lack thereof) of the protein(s) of interest should be evaluated carefully. A similar study has been conducted previously, comparing extracellular matrix proteins in rat tissue from different brain regions and cells in vitro of the same brain areas, finding differences in the expression of these proteins in a brain region- and system-dependent manner (Dauth et al. 2016).
Multiregional Brain-on-a-Chip Model
In vivo the brain consists of different brain regions, and the connection and communication thereof is essential for proper brain function (Kandel et al. 2000). Current cell in vitro models in neuroscience allow fine observation of cell phenotypes but lack the highly ordered connectivity of neurons in the brain. While in vivo studies are evidently realistic regarding neuronal architectures, the study of molecular and cellular mechanisms remains a challenge. In contrast, in vitro studies allow molecular and cellular investigation, but so far lacked the physiologically realistic connectivity. The in vitro model introduced in this study reconciles these two concerns by implementing cells from different brain regions and highly ordered axonal connections. In light of obvious differences of neuronal cells from distinct brain regions in vitro, as shown in this study, the importance of developing multiregional brain in vitro models becomes even more apparent.
Organotypic cultures of single brain areas, such as the Hip, are used frequently and offer the possibility for easy gene manipulation and precise pharmacological intervention but maintain synaptic organization that is critical to understand synapse function in a more naturalistic context than routine in vitro systems (Opitz-Araya and Barria 2011). Brain organoids have also been introduced in recent years, serving as another potential in vitro model with the advantage of having patient-specific genetic backgrounds by using iPSCs (Lancaster et al. 2013). Currently, brain organoids are restricted to recapitulating some of the brain features (some structures of the forebrain, hypothalamic structures, midbrain), but organoid studies also support the notion of the importance and necessity to develop in vitro models that consist of different brain regions (Qian et al. 2016). These organoids have their own disadvantages, including but not limited to, the following: 1) size limitations due to a lack of a circulatory system and therefore limitations in oxygen and nutrient exchange (Lancaster et al. 2013); 2) due to the dramatic cortical plate-like structure expansion, progenitor zones in forebrain organoids become gradually depleted after day 100, resulting in problems recapitulating brain development beyond the second trimester; 3) more detailed but important structures of the cortical regions, such as the intermediate zone, the subplate, or the six cortical layers, cannot yet be recapitulated (Qian et al. 2016; Shuler and Hickman 2014); and 4) functional assessments are restricted at the moment to patch-clamp experiments, measuring one cell at a time, thus making it hard to analyze overall network functionality. These limitations will probably be addressed for future studies and do not minimize the big advantages of these in vitro models.
Apart from the just discussed organoids, only very few in vitro models have been introduced so far which implement cells from distinct brain regions. A study by Kato-Negishi (2013) introduced millimeter-sized neural building blocks formed by cells isolated from the rat Hip and cortex, thereby reporting a three-dimensional in vitro model implementing cells from two different brain regions. In another study, axon diodes have been introduced by another group for the reconstruction of oriented neuronal networks in microfluidic chambers. The two-chambered microfluidic device was used to culture neurons from the cortex and the striatum and let them connect through controlled and highly ordered axons (Peyrin et al. 2011). Our in vitro model works along those lines, also producing highly ordered and controlled axonal connections, but through a different method. Instead of building a whole microfluidic device, which can be tedious to manufacture, we use micro-contact printing to introduce physical features to control axonal growth. To our knowledge, this presents the first multiregional brain in vitro model implementing three brain areas. We here show that we are able to produce not only highly ordered axonal connection, but also connections that are functionally active. The electrical activity of the different brain areas is highly cross-correlated, suggesting a functional communication between the distinct regions. Interestingly, the cell composition of the different brain areas of the multiregional-on-a-chip model is different compared with when the regions were seeded separately, suggesting that the presence of cells from different brain areas (and their connection) has an influence on cell composition in vitro. This might be due to specific secreted factors by cells from different brain regions that are used for communication between the different brain areas. It would be an interesting follow-up study to assess if cells from different brain regions secrete a different set of factors and whether these secreted factors can alter cell composition and other parameters when added to a different set of cells (from a different brain area). It would further be interesting to see if a changed cell composition can also be achieved by seeding the same brain area cells (e.g., only Hip cells) in all three compartments of the mask. If a change would occur, the spatial separation and the necessity of longer distance axonal growth compared with separately seeded cells might play a role. In addition to an altered cell composition, we also observed a slightly different electrophysiological behavior compared with when the regions were seeded separately. The overall frequency is slightly lower in the multiregional brain in vitro model and so is the frequency in a burst. Differences were also observed regarding the interspike interval, which is much higher in the multiregional-on-a-chip model for cells isolated from the pfCx and the Amy compared with when the regions were seeded separately. These results correlate well with a recent study showing that the overall frequency of networks is an average of all the frequency within this network and can be changed (Penn et al. 2016). Therefore, by connecting cells with different electrical properties (pfCx, Hip, Amy), the network dynamic can be changed. Interestingly, the wave pattern is highly brain region dependent in both conditions.
Applications for this multiregional brain-on-a-chip model are manifold. In this work, we presented just one example demonstrating one of the strengths and the uniqueness of this in vitro model. Brain regions can be dosed with a particular drug or agent separately from each other. We here dose either of the three brain regions with 5 µM PCP at seeding. The three brain regions that were chosen for this in vitro model, the pfCx, the Hip, and the Amy, and their connections are implicated in the disease of schizophrenia (Benes 2010). Hence, PCP was chosen since it has been shown to induce schizophrenia-like symptoms in healthy individual and animal models due to its inhibitory action on N-methyl-d-aspartate receptors (Adachi et al. 2013). Analyzing example traces of mature networks that were dosed with PCP (as well as the quantification of overall frequency thereof), it becomes apparent that it matters which brain region was dosed, since the wave pattern differs, depending on which brain region was dosed. Alterations in electrical activity have been reported in vivo due to psychological disorders, such as schizophrenia or anxiety disorders (Bitanihirwe et al. 2016). We here do not intend to replicate schizophrenia in vitro: we just want to exemplify a possible application of our in vitro model for possible disease and drug assessments. Another possibility of disease modeling and assessment is to dose mature cultures with different concentrations and analyze the effect on the three interconnected brain areas. Here, we see that PCP in different concentrations has a slightly different effect on the Amy compared with the other two areas. All brain regions, when dosed with PCP at a more mature state, show alterations in their electrical activity wave pattern compared with the control, which was not dosed with PCP, suggesting an effect of PCP on the electrophysiological behavior of all three brain regions, which might not be apparent when looking at overall frequency.
Conclusion
Overall, we present a comprehensive comparison between cells in vitro derived from different brain regions. Our results revealed that, while a lot of similarities exist between these cells, they are inherently different in a brain region-dependent manner. Moreover, we introduced a multiregional brain-on-a-chip model implementing essential in vivo features, which will enable us to develop unique brain disease models. The application example given in this study only represents a very small fraction of the potential applications of the multiregional brain-on-a-chip model. For instance, more complex networks, involving different brain areas or more than three will be possible in the future. Furthermore, this system is also a valuable tool to address developmental issues, such as the dynamics of neural networks and synapse formation and the effect of cells from different brain areas on that matter, and to test biological hypotheses regarding, e.g., neuron maturation and communication. In the future, when iPSCs are reproducibly able to generate brain cells with a specific brain region identity (e.g., pfCx, Hip, Amy), this model can be used as a human in vitro model, making it even more valuable for the study of brain diseases.
GRANTS
This research was sponsored by the Wyss Institute for Biologically Inspired Engineering at Harvard University (John A. Paulson School of Engineering and Applied Sciences) and the Defense Advanced Research Projects Agency under Cooperative Agreement no. W911NF-12-2-0036. This work was also supported by a Congressionally Directed Medical Research Program through the United States Army (no. W81XWH-11-2-0057) and an Emerald Grant entitled “Molecular Mechanisms of Diffuse Axonal Injury.” This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network, which is supported by the National Science Foundation under NSF award no. ECS-0335765. CNS is part of Harvard University. The Neurobiology Imaging Facility is supported in part by the Neural Imaging Center as part of National Institute of Neurological Disorders and Stroke P30 Core Center Grant NS-072030.
DISCLAIMERS
The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Defense Advanced Research Projects Agency, or the US Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.D., B.M.M., T.M., M.K.M., A.M.G., and B.B. performed experiments; S.D., B.M.M., S.P.S., T.M., and M.K.M. analyzed data; S.D., B.M.M., and S.P.S. interpreted results of experiments; S.D. and B.M.M. prepared figures; S.D., B.M.M., and S.P.S. drafted manuscript; S.D., B.M.M., S.P.S., and K.K.P. edited and revised manuscript; S.D., B.M.M., S.P.S., M.A.H., T.M., M.K.M., A.M.G., B.B., and K.K.P. approved final version of manuscript; M.A.H. conceived and designed research.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Harvard Medical School Department of Neurobiology and the Neurobiology Imaging Facility for consultation and instrument availability that supported this work. We acknowledge assistance in the preparation of Fig. 7 by Michael Rosnach.
REFERENCES
- Achermann P, Borbély AA. Low-frequency (<1 Hz) oscillations in the human sleep electroencephalogram. Neuroscience 81: 213–222, 1997. doi: 10.1016/S0306-4522(97)00186-3. [DOI] [PubMed] [Google Scholar]
- Adachi N, Numakawa T, Kumamaru E, Itami C, Chiba S, Iijima Y, Richards M, Katoh-Semba R, Kunugi H. Phencyclidine-induced decrease of synaptic connectivity via inhibition of BDNF secretion in cultured cortical neurons. Cereb Cortex 23: 847–858, 2013. doi: 10.1093/cercor/bhs074. [DOI] [PubMed] [Google Scholar]
- Alpert AJ. Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal Chem 80: 62–76, 2008. doi: 10.1021/ac070997p. [DOI] [PubMed] [Google Scholar]
- Anderson NG, Anderson NL. Twenty years of two-dimensional electrophoresis: past, present and future. Electrophoresis 17: 443–453, 1996. doi: 10.1002/elps.1150170303. [DOI] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Moon LD, Fawcett JW. Chondroitin sulphate proteoglycans: inhibitory components of the glial scar. Prog Brain Res 132: 611–619, 2001. doi: 10.1016/S0079-6123(01)32106-4. [DOI] [PubMed] [Google Scholar]
- Bandeira F, Lent R, Herculano-Houzel S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci U S A 106: 14108–14113, 2009. doi: 10.1073/pnas.0804650106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM. Neural circuitry models of schizophrenia: is it dopamine, GABA, glutamate, or something else? Biol Psychiatry 65: 1003–1005, 2009. doi: 10.1016/j.biopsych.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Benes FM. Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacology 35: 239–257, 2010. doi: 10.1038/npp.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitanihirwe BK, Mauney SA, Woo TU. Weaving a net of neurobiological mechanisms in schizophrenia and unraveling the underlying pathophysiology. Biol Psychiatry 80: 589–598, 2016. doi: 10.1016/j.biopsych.2016.03.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416: 636–640, 2002. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, MacKintosh FC, Kumar S, Geisse NA, Talbot J, Mahadevan L, Parker KK, Ingber DE, Weitz DA. Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J Cell Biol 173: 733–741, 2006. doi: 10.1083/jcb.200601060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science 304: 1926–1929, 2004. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Canolty RT, Knight RT. The functional role of cross-frequency coupling. Trends Cogn Sci 14: 506–515, 2010. doi: 10.1016/j.tics.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry C, Thompson B, Saptarshi N, Wu J, Hoh J. A “mitochondria” odyssey. Trends Mol Med 22: 391–403, 2016. doi: 10.1016/j.molmed.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain 11: 223–230, 2007. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Cullen DK, Gilroy ME, Irons HR, Laplaca MC. Synapse-to-neuron ratio is inversely related to neuronal density in mature neuronal cultures. Brain Res 1359: 44–55, 2010. doi: 10.1016/j.brainres.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauth S, Grevesse T, Pantazopoulos H, Campbell PH, Maoz BM, Berretta S, Parker KK. Extracellular matrix protein expression is brain region dependent. J Comp Neurol 524: 1309–1336, 2016. doi: 10.1002/cne.23965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Marklund N, Deschamps K, McIntosh TK, McKerracher L. Activation of Rho after traumatic brain injury and seizure in rats. Exp Neurol 198: 361–369, 2006. doi: 10.1016/j.expneurol.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Eichler GS, Huang S, Ingber DE. Gene Expression Dynamics Inspector (GEDI): for integrative analysis of expression profiles. Bioinformatics 19: 2321–2322, 2003. doi: 10.1093/bioinformatics/btg307. [DOI] [PubMed] [Google Scholar]
- Elder GA, Friedrich VL Jr, Bosco P, Kang C, Gourov A, Tu PH, Lee VM, Lazzarini RA. Absence of the mid-sized neurofilament subunit decreases axonal calibers, levels of light neurofilament (NF-L), and neurofilament content. J Cell Biol 141: 727–739, 1998. doi: 10.1083/jcb.141.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger S. Origins of Neuroscience: A History of Explorations Into Brain Function. Oxford, UK: Oxford University Press, 2001. [Google Scholar]
- Freeman W. Mass action verus mosaic function of the frontal lobe. Bull Los Angel Neuro Soc 15: 220–224, 1950. [PubMed] [Google Scholar]
- Herculano-Houzel S. The human brain in numbers: a linearly scaled-up primate brain. Front Hum Neurosci 3: 31, 2009. doi: 10.3389/neuro.09.031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano-Houzel S, Lent R. Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. J Neurosci 25: 2518–2521, 2005. doi: 10.1523/JNEUROSCI.4526-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinard V, Mikhail C, Pradervand S, Curie T, Houtkooper RH, Auwerx J, Franken P, Tafti M. Key electrophysiological, molecular, and metabolic signatures of sleep and wakefulness revealed in primary cortical cultures. J Neurosci 32: 12506–12517, 2012. doi: 10.1523/JNEUROSCI.2306-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Carroll J, Rickman C, Davletov B. Action of complexin on SNARE complex. J Biol Chem 277: 41652–41656, 2002. doi: 10.1074/jbc.M205044200. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science. New York: McGraw-Hill, Health Professions Division, 2000, p. xli. [Google Scholar]
- Karbowski J. Global and regional brain metabolic scaling and its functional consequences. BMC Biol 5: 18, 2007. doi: 10.1186/1741-7007-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karetko M, Skangiel-Kramska J. Diverse functions of perineuronal nets. Acta Neurobiol Exp (Wars) 69: 564–577, 2009. [DOI] [PubMed] [Google Scholar]
- Kasukawa T, Masumoto KH, Nikaido I, Nagano M, Uno KD, Tsujino K, Hanashima C, Shigeyoshi Y, Ueda HR. Quantitative expression profile of distinct functional regions in the adult mouse brain. PLoS One 6: e23228, 2011. doi: 10.1371/journal.pone.0023228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri T, Hatano N, Aihara M, Kawano H, Okamoto M, Liu Y, Izumi T, Maekawa T, Nakamura S, Ishihara T, Shirai M, Mizukami Y. Proteomic analysis of proteins expressing in regions of rat brain by a combination of SDS-PAGE with nano-liquid chromatography-quadrupole-time of flight tandem mass spectrometry. Proteome Sci 8: 41, 2010. doi: 10.1186/1477-5956-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato-Negishi M, Morimoto Y, Onoe H, Takeuchi S. Millimeter-sized neural building blocks for 3D heterogeneous neural network assembly. Adv Healthc Mater 2: 1564–1570, 2013. doi: 10.1002/adhm.201300052. [DOI] [PubMed] [Google Scholar]
- Knight DC, Smith CN, Cheng DT, Stein EA, Helmstetter FJ. Amygdala and hippocampal activity during acquisition and extinction of human fear conditioning. Cogn Affect Behav Neurosci 4: 317–325, 2004. doi: 10.3758/CABN.4.3.317. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature 501: 373–379, 2013. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecanu L, Papadopoulos V. Modeling Alzheimer’s disease with non-transgenic rat models. Alzheimers Res Ther 5: 17, 2013. doi: 10.1186/alzrt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol 51: 9–23, 2002. doi: 10.1002/neu.10037. [DOI] [PubMed] [Google Scholar]
- Liebermeister W, Noor E, Flamholz A, Davidi D, Bernhardt J, Milo R. Visual account of protein investment in cellular functions. Proc Natl Acad Sci U S A 111: 8488–8493, 2014. doi: 10.1073/pnas.1314810111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnman C, Zeidan MA, Furtak SC, Pitman RK, Quirk GJ, Milad MR. Resting amygdala and medial prefrontal metabolism predicts functional activation of the fear extinction circuit. Am J Psychiatry 169: 415–423, 2012. doi: 10.1176/appi.ajp.2011.10121780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology 23: 223–239, 2000. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Díaz H, Carrer H, Cáceres A. Amygdala neurons in vitro: neurite growth and effects of estradiol. J Neurosci Res 33: 418–435, 1992. doi: 10.1002/jnr.490330308. [DOI] [PubMed] [Google Scholar]
- Luna M, Duke M, Copperman A, Grunfeld L, Sandler B, Barritt J. Blastocyst embryo transfer is associated with a sex-ratio imbalance in favor of male offspring. Fertil Steril 87: 519–523, 2007. doi: 10.1016/j.fertnstert.2006.06.058. [DOI] [PubMed] [Google Scholar]
- Martins-de-Souza D, Carvalho PC, Schmitt A, Junqueira M, Nogueira FC, Turck CW, Domont GB. Deciphering the human brain proteome: characterization of the anterior temporal lobe and corpus callosum as part of the Chromosome 15-centric Human Proteome Project. J Proteome Res 13: 147–157, 2014. doi: 10.1021/pr4009157. [DOI] [PubMed] [Google Scholar]
- Masland RH. Neuronal cell types. Curr Biol 14: R497–R500, 2004. doi: 10.1016/j.cub.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Nunez J. Primary culture of hippocampal neurons from P0 newborn rats. J Vis Exp 19: 895, 2008. doi: 10.3791/895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerster J. Isolation of cerebellar granule cells from neonatal rats. Curr Protoc Toxicol 12: 12.7.1–12.7.10, 2001. doi: 10.1002/0471140856.tx1207s09. [DOI] [PubMed] [Google Scholar]
- Opitz-Araya X, Barria A. Organotypic hippocampal slice cultures. J Vis Exp 48: 2462, 2011. doi: 10.3791/2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn Y, Segal M, Moses E. Network synchronization in hippocampal neurons. Proc Natl Acad Sci U S A 113: 3341–3346, 2016. doi: 10.1073/pnas.1515105113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrin JM, Deleglise B, Saias L, Vignes M, Gougis P, Magnifico S, Betuing S, Pietri M, Caboche J, Vanhoutte P, Viovy JL, Brugg B. Axon diodes for the reconstruction of oriented neuronal networks in microfluidic chambers. Lab Chip 11: 3663–3673, 2011. doi: 10.1039/c1lc20014c. [DOI] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, Yoon KJ, Jeang W, Lin L, Li Y, Thakor J, Berg DA, Zhang C, Kang E, Chickering M, Nauen D, Ho CY, Wen Z, Christian KM, Shi PY, Maher BJ, Wu H, Jin P, Tang H, Song H, Ming GL. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165: 1238–1254, 2016. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan NA, Drescher MJ, Drescher DG. The SNARE complex in neuronal and sensory cells. Mol Cell Neurosci 50: 58–69, 2012. doi: 10.1016/j.mcn.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramnauth J, Renton P, Dove P, Annedi SC, Speed J, Silverman S, Mladenova G, Maddaford SP, Zinghini S, Rakhit S, Andrews J, Lee DK, Zhang D, Porreca F. 1,2,3,4-tetrahydroquinoline-based selective human neuronal nitric oxide synthase (nNOS) inhibitors: lead optimization studies resulting in the identification of N-(1-(2-(methylamino)ethyl)-1,2,3,4-tetrahydroquinolin-6-yl)thiophene-2-carboximidamide as a preclinical development candidate. J Med Chem 55: 2882–2893, 2012. doi: 10.1021/jm3000449. [DOI] [PubMed] [Google Scholar]
- Ren L, Lubrich B, Biber K, Gebicke-Haerter PJ. Differential expression of inflammatory mediators in rat microglia cultured from different brain regions. Brain Res Mol Brain Res 65: 198–205, 1999. doi: 10.1016/S0169-328X(99)00016-9. [DOI] [PubMed] [Google Scholar]
- Ringnér M. What is principal component analysis? Nat Biotechnol 26: 303–304, 2008. doi: 10.1038/nbt0308-303. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev 83: 803–834, 2003. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron 62: 42–52, 2009. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, Rossner MJ, Mann M, Simons M. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci 18: 1819–1831, 2015. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Lasko NB, Macklin ML, Karpf RD, Milad MR, Orr SP, Goetz JM, Fischman AJ, Rauch SL, Pitman RK. Resting metabolic activity in the cingulate cortex and vulnerability to posttraumatic stress disorder. Arch Gen Psychiatry 66: 1099–1107, 2009. doi: 10.1001/archgenpsychiatry.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuler ML, Hickman JJ. Toward in vitro models of brain structure and function. Proc Natl Acad Sci U S A 111: 13682–13683, 2014. doi: 10.1073/pnas.1414484111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF. Neuronal diversity: too many cell types for comfort? Curr Biol 8: R708–R710, 1998. doi: 10.1016/S0960-9822(98)70454-3. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci 27: 509–547, 2004. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452, 2015. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraslia VK, Kouskoukis A, Anagnostopoulos AK, Stravopodis DJ, Margaritis LH, Tsangaris GT. Proteomic analysis of normal murine brain parts. Cancer Genomics Proteomics 10: 125–154, 2013. [PubMed] [Google Scholar]
- von Stein A, Sarnthein J. Different frequencies for different scales of cortical integration: from local gamma to long range alpha/theta synchronization. Int J Psychophysiol 38: 301–313, 2000. doi: 10.1016/S0167-8760(00)00172-0. [DOI] [PubMed] [Google Scholar]
- Whitson JS, Selkoe DJ, Cotman CW. Amyloid beta protein enhances the survival of hippocampal neurons in vitro. Science 243: 1488–1490, 1989. doi: 10.1126/science.2928783. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362, 2009. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr 134: 489–492, 2004. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Brot N, Weissbach H, Heinemann SH, Hoshi T. Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. Proc Natl Acad Sci U S A 97: 448–453, 2000. doi: 10.1073/pnas.97.1.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip PK, Wong LF, Pattinson D, Battaglia A, Grist J, Bradbury EJ, Maden M, McMahon SB, Mazarakis ND. Lentiviral vector expressing retinoic acid receptor beta2 promotes recovery of function after corticospinal tract injury in the adult rat spinal cord. Hum Mol Genet 15: 3107–3118, 2006. doi: 10.1093/hmg/ddl251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.