

ABSTRACT
Mycobacterium tuberculosis uses a proteasome to degrade proteins by both ATP-dependent and -independent pathways. While much has been learned about ATP-dependent degradation, relatively little is understood about the ATP-independent pathway, which is controlled by Mycobacterium tuberculosis proteasome accessory factor E (PafE). Recently, we found that a Mycobacterium tuberculosis pafE mutant has slowed growth in vitro and is sensitive to killing by heat stress. However, we did not know if these phenotypes were caused by an inability to degrade the PafE-proteasome substrate HspR (heat shock protein repressor), an inability to degrade any damaged or misfolded proteins, or a defect in another protein quality control pathway. To address this question, we characterized pafE suppressor mutants that grew similarly to pafE+ bacteria under normal culture conditions. All but one suppressor mutant analyzed contained mutations that inactivated HspR function, demonstrating that the slowed growth and heat shock sensitivity of a pafE mutant were caused primarily by the inability of the proteasome to degrade HspR.
IMPORTANCE Mycobacterium tuberculosis encodes a proteasome that is highly similar to eukaryotic proteasomes and is required for virulence. We recently discovered a proteasome cofactor, PafE, which is required for the normal growth, heat shock resistance, and full virulence of M. tuberculosis. In this study, we demonstrate that PafE influences this phenotype primarily by promoting the expression of protein chaperone genes that are necessary for surviving proteotoxic stress.
KEYWORDS: Mycobacterium, heat shock, proteasome, proteostasis, tuberculosis
INTRODUCTION
Proteasomes are compartmentalized proteases present in all eukaryotes and archaea and in several classes of bacteria. Proteasomal protease activity is carried by the 20S core particle (CP), a barrel-shaped complex formed by the stacking of four heptameric rings of α- and β-subunits (1, 2). The active site of the 20S CP is located at the center of its inner pore, rendering it unavailable to folded proteins (3, 4). Additionally, a gate region at the distal end of the barrel normally obstructs the path to the active site. Thus, to degrade proteins, the 20S CP must form a complex with one of several potential proteasome activators that simultaneously reposition the gate into an open conformation and recruit specific protein substrates to be degraded (5–8).
Some bacteria, including the pathogen Mycobacterium tuberculosis (M. tuberculosis), encode proteasomes that are highly similar to eukaryotic 20S CPs (9). The first proteasomal degradation pathway discovered in bacteria was the Pup-proteasome system, which guides the ATP-dependent degradation of specific proteins in a manner functionally analogous to the eukaryotic ubiquitin-proteasome system (10). Recently, we discovered a second proteasomal degradation pathway that does not require ATP and depends on a proteasome activator that we named PafE (proteasome accessory factor E) (9). PafE forms rings, caps 20S CPs, and enhances the degradation of peptides and a model unfolded protein in vitro. Another group reported similar in vitro degradation phenotypes and refer to PafE as Bpa (bacterial proteasome activator) (11). We also found that PafE forms a dodecameric structure, which is unique in proteasome biology (9, 12). Moreover, PafE is required both in vitro and in vivo for the robust proteasomal degradation of at least one endogenous substrate, the heat shock protein repressor (HspR). HspR represses the expression of genes encoding several protein chaperones that are essential for the maintenance of proteostasis in M. tuberculosis, including dnaK and clpB. Consistent with a role in regulating HspR activity, an M. tuberculosis pafE mutant exhibits reduced expression of heat shock response genes that are repressed by HspR, slow growth in vitro and in mice, and hypersensitivity to heat shock (9). While these findings suggest that a mechanism for PafE to promote proteostasis during heat stress is through the degradation of HspR, we could not rule out that the PafE-proteasome system might also be important for regulating another proteostasis pathway or for directly degrading misfolded proteins that could be toxic to the cell.
In this work, we sought to determine the physiologic contributions of PafE-mediated HspR degradation versus general protein degradation. To do this, we characterized pafE mutants that had acquired suppressor mutations that restored wild-type growth under standard in vitro conditions. These mutations also restored heat shock resistance to a pafE mutant. We found that the majority of suppressor mutations inactivated HspR. Thus, it appears that the accumulation of HspR in a pafE deletion mutant is the primary cause of the slowed growth phenotype on solid medium and failed recovery after heat shock.
RESULTS
Mutations in hspR rescue the in vitro growth defects of an M. tuberculosis pafE mutant.
We previously demonstrated that a PafE-deficient strain of M. tuberculosis grows slowly on agar and in broth and is sensitive to killing by heat shock (9). However, it was unclear if these phenotypes were due to an inability to degrade misfolded proteins in general, the inability to induce expression of chaperone genes due to the accumulation of HspR, or both. To distinguish between these possibilities, we took advantage of the observation that upon electroporation of a pafE mutant with either the integrative plasmid pAJF381 or variants of the episomal plasmid pSYMP-Strep encoding putative PafE-proteasome substrates (9), we identified a small fraction of colonies that grew conspicuously faster on agar. We isolated 16 of these colonies and further characterized one, MHD1160, as a representative example. MHD1160 exhibited growth similar to the parental and complemented strains on solid medium (Fig. 1A). The complemented pafE strain is a pafE-null mutant with a hyperactive allele of pafE (pafEΔ155–164) that was described previously (12). We used this allele because complementation of a pafE mutation with a single copy of wild-type (WT) pafE is incomplete, most likely due to only partial restoration of the PafE protein to parental levels (9).
FIG 1.

Mutations in hspR rescue the growth defect of an M. tuberculosis pafE mutant. (A) The indicated M. tuberculosis strains were grown to logarithmic phase, diluted, and inoculated onto Middlebrook 7H11 agar for 2 weeks. Tenfold serial dilutions were made and spotted on agar from left (undiluted) to right (100,000-fold dilution). (B) The indicated M. tuberculosis strains were grown in 7H9-ADN, with OD580 recorded at the indicated time points. Error bars represent standard deviations for three replicates. (C) HspR targets accumulate in a pafE suppressor strain. Total cell lysates were prepared from the indicated M. tuberculosis strains. For PafE, HspR, and DlaT, samples were separated by 15% SDS-PAGE and transferred to a nitrocellulose membrane that was cut into three pieces, which were analyzed by immunoblotting with the indicated antibodies. For DnaK and ClpB, the same samples were diluted 1:20, separated by 10% SDS-PAGE, and analyzed by immunoblotting with the indicated antibodies. (D) HspR target genes are derepressed in a pafE suppressor strain. The indicated M. tuberculosis strains were grown to an OD580 of ∼0.7, and RNA was purified and reverse-transcribed into cDNA for quantitative real-time PCR analysis. Gene expression was normalized to dlaT. Statistical analysis was done by nonparametric Student's t test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Error bars represent standard deviations for three replicates. (E) pafE suppressor mutants have mutations in hspR. pafE suppressor mutants were isolated, and the hspR gene from each was PCR amplified and sequenced. Locations of mutations are indicated by arrows on a map showing the major functional domains of HspR. HTH, helix-turn-helix domain; W, wing domain; CTD, C-terminal domain.
A pafE-null mutant can sometimes show a subtle slowed growth defect in broth (9), although in this experiment, it grew as well as the parental strain (Fig. 1B). MHD1160 also grew as well as these two strains (Fig. 1B). We checked by immunoblotting that PafE was absent from MHD1160, indicating that it was not contaminated with WT bacteria (Fig. 1C).
We hypothesized that the slow growth of the pafE mutant is caused by the accumulation of the PafE-proteasome substrate HspR, because elevated levels of HspR are toxic to M. tuberculosis (13). HspR represses transcription by binding directly to HspR-associated inverted repeats (HAIRs) in the promoters of at least three known heat shock-responsive loci: the dnaK operon, clpB, and hsp-acr2 (13, 14). During heat shock, HspR is destabilized and expression of these targets is induced. DnaK and ClpB are well-characterized protein-disaggregating chaperones (15, 16) that are essential in M. tuberculosis under normal growth conditions (17), and Hsp/Acr2 is an alpha-crystallin family small heat shock protein (18–20) that may enhance the disaggregation efficiency of DnaK and ClpB (21). Thus, we hypothesized that loss of HspR function resulted in increased expression of these protein quality control genes to rescue the slow growth phenotype in our suppressor mutants. Immunoblotting of total cell lysates indicated that HspR was still present at elevated levels in MHD1160. However, the levels of the HspR targets DnaK and ClpB were dramatically higher in the suppressor strain, suggesting that HspR was inactive. We performed quantitative real-time PCR to assess expression levels of these HspR target genes and found that dnaK and clpB expression levels were ∼8-fold and ∼4-fold higher, respectively, in MHD1160 than in the pafE mutant (Fig. 1D).
We hypothesized that hspR had spontaneously acquired inactivating missense mutations in the suppressor isolates. We sequenced the hspR gene from each of the 16 suppressor mutants: 15 had mutations in the coding sequence of hspR, suggesting that derepression of HspR targets is the primary means by which the pafE mutant slow growth phenotype was rescued. We identified six specific mutations in the suppressors as follows: methionine 23 to threonine (M23T), glutamine 27 to histidine (Q27H), valine 38 to alanine (V38A, eight clones), arginine 49 to histidine (R49H, three clones), and in MHD1160, proline 40 to threonine (P40T) (Fig. 1E). Initially, no mutation in hspR was detected in two of the suppressor mutants; however, upon whole-genome sequencing of MHD1265, we found there was indeed a mutation in hspR (serine 90 to proline [S90P]). The last mutant had two point mutations in different genes: Rv0260c (serine 141 changed to phenylalanine [S141F]) and Rv0516c (arginine 390 changed to tryptophan [R390W]). Rv260c encodes a putative transcriptional regulator, and Rv0516c encodes Tuf, an iron-regulated translation elongation factor. This mutant notably had slowed growth in broth culture despite its improved growth on agar (Fig. 1B, red line).
HspRP40T is inactive.
We hypothesized that the hspR substitutions resulted in the inactivation of HspR. To test this, we performed both in vitro and in vivo experiments to assess the activity of a representative missense mutant protein, HspRP40T, which was produced in MHD1160. To further test the loss of function of this protein in M. tuberculosis, we took advantage of the previous observation that WT HspR is toxic when overproduced in M. tuberculosis (13). We transformed WT M. tuberculosis with overexpression plasmids encoding either HspR or HspRP40T and inoculated transformants onto Middlebrook 7H11 agar to examine growth. As predicted, bacteria transformed with a plasmid encoding HspR grew extremely slowly; in contrast, those transformed with the plasmid encoding HspRP40T grew robustly (Fig. 2A). Thus, HspRP40T was less toxic than HspR in M. tuberculosis, most likely due to a loss of function.
FIG 2.

HspRP40T is inactive. (A) HspRP40T overexpression is not toxic to M. tuberculosis. WT M. tuberculosis H37Rv was transformed by electroporation with the overexpression plasmid pSYMP carrying either hspR or hspRP40T under the control of the constitutive hsp60 promoter, and growth of transformants was compared on solid Middlebrook 7H11 agar. (B) HspRP40T does not bind to a known DNA target sequence. A DNA probe consisting of the infrared dye IR700 conjugated to 126 bp of the promoter region upstream of dnaK was amplified and mixed with either HspR or HspRP40T. Mixtures were separated by 10% nondenaturing PAGE to determine differences in DNA binding. The retention of probe in the well in lanes containing 8 μM protein likely represents the formation of protein-DNA aggregates. (C) HspRP40T does not dimerize. Left, recombinant purified His6-tagged HspR or HspRP40T was separated on a size exclusion column. Absorbance at 280 nm was recorded and spectra were overlaid for comparison of different species present. Arrows indicate the retention volumes of protein standards carbonic anhydrase (29 kDa) and aprotinin (6.5 kDa). Right, SEC-purified HspR (from the “dimer” SEC peak) and HspRP40T were separated by 15% SDS-PAGE and visualized by Coomassie brilliant blue staining. M, molecular mass markers.
Because all the suppressor mutations we found were located in or near the DNA-binding helix-turn-helix, wing-1, and wing-2 motifs of HspR (Fig. 1E), we hypothesized that these mutations disrupted the ability of HspR to interact with DNA. To test this hypothesis, we performed an electrophoretic mobility shift assay (EMSA) to compare the ability of HspR and HspRP40T to bind to an infrared dye-labeled dnaK promoter, which contains two HAIR motifs with which HspR interacts (14). The addition of HspR resulted in a dose-dependent increase in the amount of shifted probe. However, the position of probe remained unchanged in the presence of HspRP40T, indicating that the P40T substitution ablated its binding ability (Fig. 2B).
HspR belongs to the MerR family of transcription factors, which bind to DNA as dimers (22). We therefore reasoned that the loss of HspRP40T activity could be due to either disruption of direct protein-DNA contacts or loss of dimerization. To test for differences in oligomer state, we performed size exclusion chromatography (SEC) on recombinant His6-tagged HspR or HspRP40T purified from Escherichia coli. HspR migrated as two major species. The first species eluted at the expected retention time for an HspR dimer, with a molecular mass of ∼28 kDa, as has been reported elsewhere (23). The second species eluted later, with a retention time of 21.1 ml. In contrast, HspRP40T migrated as a single species eluting at 21.1 ml (Fig. 2C, left). The retention time of this late species was considerably longer than that of the 6.5-kDa protein aprotinin, which eluted at 19.87 ml during generation of a standard curve. Despite the slow migration of HspRP40T during SEC, purified recombinant His6-tagged HspR and HspRP40T both migrated at the expected size for monomeric HspR (∼14 kDa) in SDS-PAGE gels (Fig. 2C, right). We therefore presume that the late-eluting species represents HspR monomers that fold into a compact shape with a small Stokes radius. These data suggest that the inability of HspRP40T to form dimers was the reason for its inactivity in vitro and in M. tuberculosis.
Suppressor mutations restore heat shock resistance to the pafE mutant.
We previously demonstrated that an M. tuberculosis pafE mutant is sensitive to killing by heat stress (9). Because HspR represses the expression of protein chaperone genes that protect M. tuberculosis from heat shock (13, 14), we proposed that the accumulation of HspR was responsible for the heat shock sensitivity of a pafE mutant. The discovery of pafE suppressors with an inactive hspR allele provided a means to test this hypothesis. To this end, we subjected several of the suppressor strains to a heat shock survival assay. We found that all of the suppressor mutants had resistance to heat shock comparable to that of the parental strain (Fig. 3, top). This result was consistent with previous studies that showed an M. tuberculosis hspR mutant is protected from killing by heat stress, presumably due to increased levels of the DnaK and ClpB proteins (13) (Fig. 3, bottom). The inactivation of HspR function appeared to occur by at least two different mechanisms. We showed that HspRP40T failed to dimerize in vitro (Fig. 2C) but appears otherwise to form stable protein that accumulates in a pafE mutant like WT HspR (Fig. 3). We predict that the HspRV38A mutant may have a similar defect. In contrast, neither HspRQ27H nor HspRS90P accumulated in the pafE strain, suggesting these proteins are inherently less stable than WT HspR. Finally, MHD1268, which does not have a mutation in hspR, had apparently normal levels of all of the chaperones despite having increased levels of WT HspR (Fig. 3, right-most lane).
FIG 3.
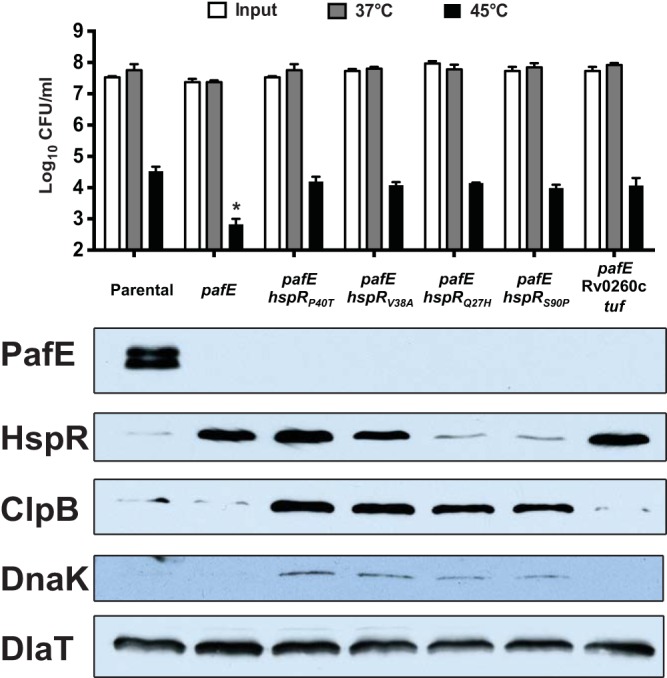
The majority of suppressor mutations are in hspR and restore heat shock resistance. (Top) Heat shock survival assay of M. tuberculosis strains incubated for 24 h at the indicated temperature. CFU were enumerated 2 to 3 weeks later. Statistical analysis was done by nonparametric Student's t test. *, P < 0.05. Error bars represent standard deviations for three replicates. (Bottom) Immunoblots of total cell lysates of the same strains grown at 37°C using antibodies to the proteins indicated on the left. The following strains were used (from left to right, according to the strain collection number): MHD920, MHD932, MHD1160, MHD1202, MHD1203, MHD1265, and MHD1268. Genotypes are listed in Table 1.
Taken together, we conclude that the primary mechanism of slowed growth on solid medium and defective recovery after heat shock of a pafE mutant is due to the accumulation of HspR.
DISCUSSION
In our previous work, we described two in vitro phenotypes that arise when pafE is disrupted in M. tuberculosis: a growth defect and heat shock hypersensitivity (9). Although the accumulation of HspR, a confirmed PafE-proteasome substrate, was a reasonable mechanistic explanation for these phenotypes, this hypothesis remained to be tested. Through suppressor analysis, we identified numerous mutations in hspR that alleviated the repression of three promoters controlling key heat shock chaperones. Loss-of-function hspR mutations restored both normal growth and recovery after heat shock stress, supporting the hypothesis that the hyperrepression of this regulon was responsible for the growth defects of a pafE-null mutant.
To maintain proteostasis, organisms employ three main strategies: preventing protein unfolding, untangling protein aggregates for refolding, and destroying aggregated and misfolded proteins. The best-characterized protein-disaggregating factors are DnaK (Hsp70) and ClpB (Hsp100), which act sequentially to untangle protein aggregates: DnaK first binds to the unfolded protein and then recruits ClpB, which actively pulls individual polypeptides from the aggregate (16). Small heat shock proteins bind to aggregates to increase the efficiency of disaggregation (24). The gene dnaK is essential in M. tuberculosis (17, 25, 26), and deletion of clpB results in bacteria that grow slowly on solid medium (27), implying that the accumulation of protein aggregates cannot be tolerated even under standard laboratory growth conditions. Because these genes are the major targets of HspR and are repressed in a pafE mutant, the HspR-dependent growth defect of this mutant may be caused by an inability to efficiently disaggregate proteins.
Under stressed conditions that result in an increase in unfolded proteins, disaggregation may not be sufficient to maintain proteostasis, necessitating additional mechanisms to survive. One such mechanism is the degradation of damaged and misfolded proteins, which is typically accomplished in bacteria by ATP-dependent proteases, such as ClpP, HslUV, and Lon (28–30). Lon, in particular, specifically recognizes and degrades misfolded proteins (31) and is required to clear abnormal proteins in E. coli (32). Consistent with this role, Lon is required for E. coli to resist killing by heat shock. In contrast to many bacteria and most mycobacteria, the protease Lon is conspicuously absent from M. tuberculosis. Because Lon specifically recognizes and degrades misfolded proteins (31), we speculated that the PafE-proteasome system might play this role in M. tuberculosis. We previously reported that PafE enhanced proteasomal degradation of the model unfolded protein β-casein in vitro, demonstrating that the PafE-proteasome system can degrade unfolded proteins (9). Moreover, our recent finding that the inner channel of PafE is lined with hydrophobic residues (12) further suggests a role for PafE in the accommodation of misfolded substrates, which typically have exposed hydrophobic domains that cannot be well solubilized in the aqueous cytosol. However, our work here suggests proteolysis by the PafE-proteasome system of “random” misfolded proteins is not required for dealing with proteotoxic stress under the conditions tested. Nevertheless, it is still possible that PafE is required for such a role in other situations.
Among the 16 suppressor mutants, one (MHD1268) had missense mutations in Rv0260c and tuf. Interestingly, although MHD1268 had restored growth on solid medium and resistance to heat shock, it had slightly slower growth than the other strains in broth culture (Fig. 1B). Rv0260c is a possible transcription factor of unknown function, while Tuf is a predicted iron-regulated translation elongation factor (33). It remains to be determined which mutation (or both) is responsible for suppressing the heat shock sensitivity and growth defective phenotypes of a strain lacking pafE. Importantly, it seems unlikely that either of these mutations affects the HspR regulon; unlike the other suppressor mutants, MHD1268 did not have a defect in the expression of the HspR regulon, albeit under steady-state conditions (Fig. 3).
The finding that the M. tuberculosis proteasome regulates the expression of heat shock response genes implies that M. tuberculosis has linked two of its important protein quality control systems. It is possible that this provides an efficient means of responding to an acute proteotoxic stress by allowing the rapid removal of the hemistable repressor HspR upon its misfolding. Future studies should provide further insight into how a rapid cooperative effort between the proteasome and chaperone systems is involved in responding to physiologic stresses.
ATP-independent degradation is likely to be less efficient for most substrates, so why have this pathway? Perhaps M. tuberculosis and other bacteria need such a system because they cannot always rely on having sufficient ATP levels under oxygen- or nutrient-deficient conditions. For example, the PafE-proteasome system may provide M. tuberculosis a less energetically costly alternative for handling proteotoxic stress in a hypoxic granuloma in human lungs or allow for the selective production of ATP-consuming chaperones only when they are absolutely necessary to combat increasing proteotoxic stress. Future studies will further illuminate the role of the M. tuberculosis proteasome in what is likely a complex network of protein quality control pathways that are crucial for M. tuberculosis pathogenesis.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, and culture conditions.
Strains, primers, and plasmids used are listed in Table 1. M. tuberculosis strains were grown in Middlebrook 7H9 medium (Difco) with 0.2% glycerol, 0.05% Tween 80, and supplemented with 0.5% bovine serum albumin (BSA), 0.2% dextrose, and 0.085% sodium chloride (ADN). Cultures were grown without shaking in 25- or 75-cm2 vented flasks (Corning Life Sciences) at 37°C. For growth on solid medium, Middlebrook 7H11 agar (Difco) was supplemented with oleic acid-albumin-dextrose-catalase (OADC; Middlebrook Enrichment, BBL). For selection using antibiotics, media were supplemented with 50 μg · ml−1 kanamycin, 50 μg · ml−1 hygromycin, and/or 25 μg · ml−1 streptomycin. Escherichia coli cultures were grown in Luria-Bertani (LB) broth (Difco) at 37°C, and LB agar was used as solid medium. For selection, media were supplemented with 200 μg · ml−1 ampicillin, 100 μg · ml−1 kanamycin, 150 μg · ml−1 hygromycin, and/or 50 μg · ml−1 streptomycin.
TABLE 1.
Bacterial strains, plasmids, and primers
| Strain, plasmid, or primer | Genotype, description, or sequencea | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | F− Φ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) | Gibco, BRL |
| ER2566 | F− λ− fhuA2 [lon] ompT lacZ::T7 gene1 gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::miniTn10)2 R(zgb-210::Tn10) (Tets) endA1 [dcm] | 44 |
| BW25113 | Ampr(Ts); F− DE(araD-araB)567 lacZ4787(Δ)::rrnB-3 LAM− rph-1 DE(rhaD-rhaB)568 hsdR514 | 35 |
| M. tuberculosis | ||
| H37Rv | WT; ATCC 25618 | American Type Culture Collection |
| MHD5 | H37Rv mpa::MycoMarT7 Kanr | 45 |
| MHD802 | H37Rv ΔprcBA::hyg Hygr | 46 |
| MHD920 | H37Rv L5 attB::(Rv3781-Rv3783) Kanr | 9 |
| MHD932 | MHD920 ΔpafE::hyg Kanr Hygr | 9 |
| MHD980 | MHD932/pAJF381 Kanr Hygr Strepr | 9 |
| MHD1091 | MHD932/pAJF381-pafEΔ155–164 Kanr Hygr Strepr | 12 |
| MHD1160 | MHD980 hspRP40T Kanr Hygr Strepr | This study |
| MHD1202 | MHD980 hspRV38A Kanr Hygr Strepr | This study |
| MHD1203 | MHD980 hspRQ27H Kanr Hygr Strepr | This study |
| MHD1256 | MHD932/pSYMP-Strep-PPE12 hspRV38A Kanr Hygr Strepr | This study |
| MHD1257 | MHD932/pSYMP-Strep-PPE12 hspRV38A Kanr Hygr Strepr | This study |
| MHD1258 | MHD932/pSYMP-Strep-PE23 hspRV38A Kanr Hygr Strepr | This study |
| MHD1259 | MHD932/pSYMP-Strep-cyp136 hspRV38A Kanr Hygr Strepr | This study |
| MHD1260 | MHD932/pSYMP-Strep-cyp136 hspRR49H Kanr Hygr Strepr | This study |
| MHD1261 | MHD932/pSYMP-Strep-Rv3480c hspRR49H Kanr Hygr Strepr | This study |
| MHD1262 | MHD932/pSYMP-Strep-Rv3480c hspRR49H Kanr Hygr Strepr | This study |
| MHD1263 | MHD932/pSYMP-Strep-vapC47 hspRV38A Kanr Hygr Strepr | This study |
| MHD1264 | MHD932/pSYMP-Strep-vapC47 hspRM23T Kanr Hygr Strepr | This study |
| MHD1265 | MHD932/pSYMP-Strep-plcB hspRS90P Kanr Hygr Strepr | This study |
| MHD1266 | MHD932/pSYMP-Strep-plcB hspRV38A Kanr Hygr Strepr | This study |
| MHD1267 | MHD932/pSYMP-Strep-Rv2927c hspRV38A Kanr Hygr Strepr | This study |
| MHD1268 | MHD932/pSYMP-Strep-Rv2927c Rv0260cC422T tufC1168T Kanr Hygr Strepr | This study |
| Plasmids | ||
| pET-32a(+) | Ampr; for inducible production of recombinant protein in E. coli | Novagen |
| pET-32a(+)-HspR-G4-His | Ampr; for purification of recombinant His6-HspR | 9 |
| pET-32a(+)-HspRP40T-G4-His | Ampr; for purification of recombinant His6-HspRP40T | This study |
| pKD46 | Ampr(Ts); for arabinose-inducible production of Red recombinase in E. coli | 35 |
| pAJF381 | Strepr; for integration into the M. tuberculosis Tweety attB site | Gift from M. Glickman |
| pSYMP-Strep-tatA | Strepr; episomal plasmid for overexpression of hspR in M. tuberculosis using the constitutive hsp60 promoter | This study |
| pSYMP-Strep-hspR | Strepr; for overproduction of HspR in M. tuberculosis | This study |
| pSYMP-Strep-hspRP40T | Strepr; for overproduction of HspRP40T in M. tuberculosis | This study |
| pSYMP-Strep-PPE12 | Strepr; for overexpression of PPE12-FLAG in M. tuberculosis | This study |
| pSYMP-Strep-PE23 | Strepr; for overexpression of PE23-FLAG in M. tuberculosis | This study |
| pSYMP-Strep-cyp136 | Strepr; for overexpression of cyp136-FLAG in M. tuberculosis | This study |
| pSYMP-Strep-Rv3480c | Strepr; for overexpression of Rv3480c-FLAG in M. tuberculosis | This study |
| pSYMP-Strep-vapC47 | Strepr; for overexpression of vapC47-FLAG in M. tuberculosis | This study |
| pSYMP-Strep-plcB | Strepr; for overexpression of plcB-FLAG in M. tuberculosis | This study |
| pSYMP-Strep-Rv2927c | Strepr; for overexpression of Rv2927c-FLAG in M. tuberculosis | This study |
| Primers | ||
| hspR F NdeI | TTACATATGGCGAAGAACCCAAAGGACGGGGAATCC | |
| hspR R BamHI | TATGGATCCTCACCGGCGCGGTTTCCAGACG | |
| hspR 4G-His R BamHI | TATGGATCCTCAATGATGATGATGATGATGTCCTCCTCCTCCCCGGCGCGGTTTCCAGACG | |
| hspRP40T SOE R | GAGGTGCGCCGCGTGCTGACCAACCCAAGAC | |
| hspRP40T SOE F | GTCTTGGGTTGGTCAGCACGCGGCGCACCTC | |
| dnaK qPCR F | CAACTCCGTCGTCTCGGTTC | |
| dnaK qPCR R | GCGGGCGAACGCGACAATTG | |
| clpB qPCR F | GCTAACCGCGGCGTTACAG | |
| clpB qPCR R | AGTAGCGGTGCGGCGATAC | |
| dlaT qPCR F | CGGGGCACCACCCAGAAG | |
| dlaT qPCR R | CGCCCGCGCCAGACCGGC | |
| dnaK EMSA Fwd IR700 | /5IRD700/GCACGACCAGCGTTAGCATGCTCAG | |
| dnaK EMSA Rev | GTAGAGCTTCTTTCCTGCACTATGATGAG | |
| pSYMP-Strep Fwd | ACCGGCCGTGCGGAATTAAGCCGGCCCGTACCCTGTGAATAGAGGTCCGCTATGCGCTCACGCAACTGGTCCAG | |
| pSYMP-Strep Rev | CAGAGGAACTGCGCCAGTTCCTCCGGATCGGTGAAGCCGGAGAGATCCAGTTATTTGCCGACTACCTTGGTGATC | |
| PPE12_pSYMP_F_NdeI | GAAGGAGATATACATATGGTCGGTTTCGCGTGGTTGC | |
| PPE12-4G-FLAG R HindIII | ATCAAGCTTCTACTTGTCATCGTCATCCTTGTAATCTCCTCCTCCTCCGCGGTGGAAAAAGCCCGACACG | |
| PE23_pSYMP_F_NdeI | GAAGGAGATATACATATGCAGTTCCTGAGCGTGATTCC | |
| PE23-4G-FLAG R HindIII | ATCAAGCTTCTACTTGTCATCGTCATCCTTGTAATCTCCTCCTCCTCCGGAGATCAGCACCGTGACAGC | |
| cyp136_pSYMP_F_NdeI | GAAGGAGATATACATATGGCGACGATCCACCCCCCGGCATACC | |
| cyp136-4G-FLAG R HindIII | ATCAAGCTTCTACTTGTCATCGTCATCCTTGTAATCTCCTCCTCCTCCCCTGGGACGCAGCACGATCG | |
| Rv3480c_pSYMP_F_NdeI | GAAGGAGATATACATATGAGCCAGACGGCCCGGCGGTTGG | |
| Rv3480c-4G-FLAG Rev HindIII | ATCAAGCTTTCACTTGTCATCGTCATCCTTGTAATCTCCTCCTCCTCCGGAACCGAGGCCCGCGGCGCCGTCAG | |
| vapC47_pSYMP_F_NdeI | GAAGGAGATATACATATGATCTATATGGACACCTCGGCCCTGAC | |
| vapC47-4G-FLAG R HindIII | ATCAAGCTTTCACTTGTCATCGTCATCCTTGTAATCTCCTCCTCCTCCCCGGACTGCGCCGGGTGAG | |
| plcB F NdeI | TTACATATGACCCGCCGACAATTTTTTGC | |
| plcB-4G-FLAG R HindIII | ATCAAGCTTTCACTTGTCATCGTCATCCTTGTAATCTCCTCCTCCTCCACAGAGACCGCTGGGAATCC | |
| Rv2927c F NdeI | TTACATATGTACCGAGTCTTTGAAGCGCTGGAC | |
| Rv2927c-4G-FLAG R HindIII | ATCAAGCTTTTACTTGTCATCGTCATCCTTGTAATCTCCTCCTCCTCCGCGCACCGCGTAGTCGTGA |
Tets, tetracycline susceptible; Ts, temperature sensitive; Ampr, ampicillin resistance; Kanr, kanamycin resistance; Hygr, hygromycin resistance; Strepr, streptomycin resistance. /5IRD700/ indicates a 5′ infrared dye on the primer that emits at 700 nm.
We replaced the hygromycin resistance gene in plasmid pSYMP (34) with a streptomycin resistance cassette using the Red recombinase system, as described elsewhere (35). E. coli strain BW25113, with an arabinose-inducible Red recombinase gene carried on the temperature-sensitive plasmid pKD46, was transformed with pSYMP-tatA, and isolates were selected on LB agar containing hygromycin and ampicillin. A 100-ml culture of a single transformant was inoculated from 2 ml of an overnight culture and grown at 30°C in the presence of 1 mM arabinose until the optical density at 580 nm (OD580) reached ∼0.5. Two hundred microliters of electrocompetent cells was mixed with 100 ng of DNA containing the streptomycin resistance gene (aadA), which was amplified by PCR from the plasmid pMV306-Strep with both 5′- and 3′-flanking sequences homologous to the regions flanking the hygromycin resistance gene in pSYMP using primers pSYMP-Strep-Fwd and pSYMP-Strep-Rev (Table 1). The cells were pulsed (2,500 V, 200 Ω, 25 μF) using a BTX ElectroCell manipulator (Harvard Apparatus). One milliliter of LB was added, and bacteria were recovered at 37°C for 1 h and then were left at room temperature for ∼24 h. Bacteria were then inoculated onto LB-streptomycin plates. Transformants were colony purified on LB-streptomycin and then again on both LB-streptomycin and LB-hygromycin plates to check for the loss of hygromycin resistance.
Unless otherwise described, all primers were purchased from Invitrogen, and plasmid sequencing was performed by Genewiz, Inc. to confirm the veracity of all PCR-cloned sequences.
Isolation of M. tuberculosis pafE suppressor mutants.
pafE suppressor mutants that grew similarly to the parental (pafE+) strain were identified in the course of transforming an M. tuberculosis pafE mutant by electroporation with variants of the replicative pSYMP-Strep plasmid or the pAJF381 vector, which integrates into the chromosomal Tweety attB site (Table 1). Bacteria were inoculated onto solid medium with streptomycin. After about 2 weeks at 37°C, colonies were picked, taking care to avoid smaller colonies on the plate, and transferred to larger volumes of 7H9-ADN broth to be cultured and analyzed further.
Purification of recombinant proteins.
M. tuberculosis HspR was purified from E. coli, as previously described (9, 36, 37). Briefly, hspR or hspRP40T was cloned into pET-32a(+) with a C-terminal tetraglycine linker and His6 tag, expression was induced with 1 mM isopropyl-β-d-galactopyranoside (IPTG) for 5 h, and the protein was purified by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography under denaturing conditions using 8 M urea, as described in the QIAexpressionist (Qiagen). Fractions containing HspR were pooled, concentrated, and renatured by separation using an Akta Purifier connected to a 10/300 Superose 6 GL gel filtration column (GE Healthcare) equilibrated with buffer containing 50 mM Tris (pH 7.5), 200 mM KCl, 5 mM EDTA, 5 mM MgCl2, and 10% glycerol. Desired fractions were pooled, concentrated using an Amicon Ultra-4 centrifugal filter unit (Millipore), and frozen at −80°C until use.
Immunoblotting.
Proteins were separated by 10%, 12%, or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as indicated in the figure legends and transferred to 0.2-μm nitrocellulose membranes (GE Healthcare). Antibodies for the detection of PafE and HspR were described previously (9). Antibodies to dihydrolipoamide acyltransferase (DlaT) (38) and ClpB (27) are described elsewhere. Mouse antibodies to M. tuberculosis DnaK were acquired from BEI.
RNA extraction and quantitative real-time PCR.
Three biological replicate cultures of the indicated M. tuberculosis strains were grown in 7H9-ADN to an OD580 of ∼0.7, and RNA was purified and analyzed as described previously (9, 34). Briefly, M. tuberculosis cultures were mixed at a 1:1 (vol/vol) ratio with GTC buffer (5 M guanidine thiocyanate, 25 mM trisodium citrate, 0.5% Sarkosyl, 0.8% β-mercaptoethanol) and bacteria were collected by centrifugation (2,880 × g, 8 min). Pellets were resuspended in 1 ml of TRIzol reagent (Thermo Fisher) per 5 OD equivalents of bacteria, and bacteria were lysed by bead-beating 3 times for 30 s. The TRIzol mixture was added to 300 μl of chloroform and mixed for 30 min to extract RNA. Mixtures were centrifuged (14,000 × g, 15 min, 4°C), and 400 μl of the upper layer was removed and added to 500 μl of ethanol. RNA purification was then performed using the RNeasy column system, as described by the manufacturer (Qiagen). cDNA was prepared from purified RNA using the Reverse transcriptase system as described by the manufacturer (Promega), except for the use of random hexamers from Invitrogen for priming. cDNA was used for quantitative real-time PCR, which was performed with Platinum SYBR green qPCR Supermix-UDG (Thermo Fisher), using a MyiQ iCycler (Bio-Rad). As a control, we quantified dlaT cDNA, as described previously (34). The qPCR primers used are listed in Table 1.
EMSA.
We used a modified version of an infrared detection system (39) to assess direct protein/DNA binding. Briefly, we amplified 126 bp of the promoter region upstream of the dnaK coding sequence from M. tuberculosis chromosomal DNA by the PCR using a primer pair (Table 1), including a forward primer linked to IR700 infrared dye (IDT Technologies). The probe was separated from the chromosomal DNA template using a Qiagen PCR purification kit. Twenty-six nanograms of probe DNA was mixed with increasing amounts of recombinant purified C-terminally His6-tagged HspR or HspRP40T in binding buffer (20 mM HEPES [pH 8], 50 mM KCl, 1 mM MgCl2, 500 μM EDTA, 500 μM dithiothreitol [DTT], 5 μg · ml−1 BSA) with 1 μg of calf thymus DNA (Thermo Fisher) to reduce nonspecific binding. Mixtures were incubated at room temperature (∼25°C) for 30 min and then separated at 4°C on a 10% Mini-Protean TBE precast gel (Bio-Rad) that had been prerun at 100 V for 1 h in 0.5× TBE buffer (50 mM Tris base, 50 mM boric acid, 1 mM EDTA). The gel was then rinsed in phosphate-buffered saline (PBS), and DNA was visualized on an Odyssey Classic infrared imaging system (LI-COR).
Heat shock survival assay.
M. tuberculosis heat shock survival assays were performed as previously described (9). Briefly, the indicated M. tuberculosis strains were grown in 7H9-ADN broth to an optical density at 580 nm (OD580) of ∼0.8, and cells were harvested by centrifugation (2,880 × g, 8 min), resuspended in fresh medium, and then centrifuged at slow speed (115 × g, 8 min) to remove clumped cells. The supernatant containing suspended cells was diluted to OD580 of 0.08 and divided into six 1-ml aliquots into screw-cap microcentrifuge tubes (BioSpec). Three tubes were incubated in a 45°C heat block water bath for 24 h, and three tubes were incubated at 37°C. Each was then serially diluted and inoculated onto 7H11-OADC agar to enumerate CFU.
Whole-genome sequencing.
Genomic DNA was extracted as previously described (40) and sequenced by the NYU School of Medicine Genome Technology Center with an Illumina genome analyzer. Illumina sequencing was performed with paired-end reads of 251 bases and a target coverage of at least three million high-quality bases. We used Burrows-Wheeler Aligner (BWA) (41) to map the reads from the genome sequences against the M. tuberculosis H37Rv accession no. NC_018143.2 reference sequence. BWA outputs were analyzed and annotated using SAMtools (42), GATK (41), and ANNOVAR (43). Single nucleotide polymorphisms (SNPs) in genes annotated as encoding proline-glutamate/proline-proline-glutamate (PE/PPE) genes, integrases, transposases, resolvases, maturases, or phages were removed from the analysis. We noted that our wild-type parental strain and the suppressor mutant strains had shared single nucleotide polymorphisms in Rv0516c (A205C) and fadD26 (G1251C) that were not found in the reference genome-sequenced strain.
ACKNOWLEDGMENTS
We thank Andrew Darwin for helpful comments on a draft version of the manuscript. We thank Volker Briken, Andrew Darwin, Stevan Hubbard, Ian Mohr, and Victor Torres for technical advice and suggestions.
This work was supported by NIH grants AI088075 and HL92774 awarded to K.H.D. and grant AI103268 awarded to B.S. NIH grants F30 AI110067 and T32 AT007180 supported J.B.J. B.S. is a consultant for Regeneron Pharmaceuticals and has received speaking honoraria from Pfizer. K.H.D. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. The Genome Technology Center is a shared resource that is partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center.
REFERENCES
- 1.Kish-Trier E, Hill CP. 2013. Structural biology of the proteasome. Annu Rev Biophys 42:29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomko RJ Jr, Hochstrasser M. 2013. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem 82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. 1997. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature 386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 4.Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. 1995. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science 268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 5.Stadtmueller BM, Hill CP. 2011. Proteasome activators. Mol Cell 41:8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. 2008. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell 30:360–368. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL. 2005. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell 20:687–698. doi: 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 8.Yu Y, Smith DM, Kim HM, Rodriguez V, Goldberg AL, Cheng Y. 2010. Interactions of PAN′s C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO J 29:692–702. doi: 10.1038/emboj.2009.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jastrab JB, Wang T, Murphy JP, Bai L, Hu K, Merkx R, Huang J, Chatterjee C, Ovaa H, Gygi SP, Li H, Darwin KH. 2015. An adenosine triphosphate-independent proteasome activator contributes to the virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 112:E1763–E1772. doi: 10.1073/pnas.1423319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. 2008. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delley CL, Laederach J, Ziemski M, Bolten M, Boehringer D, Weber-Ban E. 2014. Bacterial proteasome activator bpa (rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS One 9:e114348. doi: 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai L, Hu K, Wang T, Jastrab JB, Darwin KH, Li H. 2016. Structural analysis of the dodecameric proteasome activator PafE in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 113:E1983–E1992. doi: 10.1073/pnas.1512094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart GR, Snewin VA, Walzl G, Hussell T, Tormay P, O'Gaora P, Goyal M, Betts J, Brown IN, Young DB. 2001. Overexpression of heat-shock proteins reduces survival of Mycobacterium tuberculosis in the chronic phase of infection. Nat Med 7:732–737. doi: 10.1038/89113. [DOI] [PubMed] [Google Scholar]
- 14.Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, Young DB, Butcher PD. 2002. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 148:3129–3138. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- 15.Finka A, Sharma SK, Goloubinoff P. 2015. Multi-layered molecular mechanisms of polypeptide holding, unfolding and disaggregation by HSP70/HSP110 chaperones. Front Mol Biosci 2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mogk A, Kummer E, Bukau B. 2015. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front Mol Biosci 2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 18.Kennaway CK, Benesch JL, Gohlke U, Wang L, Robinson CV, Orlova EV, Saibil HR, Keep NH. 2005. Dodecameric structure of the small heat shock protein Acr1 from Mycobacterium tuberculosis. J Biol Chem 280:33419–33425. doi: 10.1074/jbc.M504263200. [DOI] [PubMed] [Google Scholar]
- 19.Stewart GR, Newton SM, Wilkinson KA, Humphreys IR, Murphy HN, Robertson BD, Wilkinson RJ, Young DB. 2005. The stress-responsive chaperone alpha-crystallin 2 is required for pathogenesis of Mycobacterium tuberculosis. Mol Microbiol 55:1127–1137. [DOI] [PubMed] [Google Scholar]
- 20.Wilkinson KA, Stewart GR, Newton SM, Vordermeier HM, Wain JR, Murphy HN, Horner K, Young DB, Wilkinson RJ. 2005. Infection biology of a novel alpha-crystallin of Mycobacterium tuberculosis: Acr2. J Immunol 174:4237–4243. doi: 10.4049/jimmunol.174.7.4237. [DOI] [PubMed] [Google Scholar]
- 21.Cashikar AG, Duennwald M, Lindquist SL. 2005. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J Biol Chem 280:23869–23875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heldwein EE, Brennan RG. 2001. Crystal structure of the transcription activator BmrR bound to DNA and a drug. Nature 409:378–382. doi: 10.1038/35053138. [DOI] [PubMed] [Google Scholar]
- 23.Parijat P, Batra JK. 2015. Role of DnaK in HspR-HAIR interaction of Mycobacterium tuberculosis. IUBMB Life 67:816–827. doi: 10.1002/iub.1438. [DOI] [PubMed] [Google Scholar]
- 24.Mogk A, Schlieker C, Friedrich KL, Schonfeld HJ, Vierling E, Bukau B. 2003. Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem 278:31033–31042. doi: 10.1074/jbc.M303587200. [DOI] [PubMed] [Google Scholar]
- 25.Fay A, Glickman MS. 2014. An essential nonredundant role for mycobacterial DnaK in native protein folding. PLoS Genet 10:e1004516. doi: 10.1371/journal.pgen.1004516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lupoli TJ, Fay A, Adura C, Glickman MS, Nathan CF. 2016. Reconstitution of a Mycobacterium tuberculosis proteostasis network highlights essential cofactor interactions with chaperone DnaK. Proc Natl Acad Sci U S A 113:E7947–E7956. doi: 10.1073/pnas.1617644113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaubourgeix J, Lin G, Dhar N, Chenouard N, Jiang X, Botella H, Lupoli T, Mariani O, Yang G, Ouerfelli O, Unser M, Schnappinger D, McKinney J, Nathan C. 2015. Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe 17:178–190. doi: 10.1016/j.chom.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gur E, Ottofueling R, Dougan DA. 2013. Machines of destruction–AAA+ proteases and the adaptors that control them. Subcell Biochem 66:3–33. doi: 10.1007/978-94-007-5940-4_1. [DOI] [PubMed] [Google Scholar]
- 29.Gur E. 2013. The Lon AAA+ protease. Subcell Biochem 66:35–51. doi: 10.1007/978-94-007-5940-4_2. [DOI] [PubMed] [Google Scholar]
- 30.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 31.Gur E, Sauer RT. 2008. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev 22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottesman S, Zipser D. 1978. Deg phenotype of Escherichia coli lon mutants. J Bacteriol 133:844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lechat P, Hummel L, Rousseau S, Moszer I. 2008. GenoList: an integrated environment for comparative analysis of microbial genomes. Nucleic Acids Res 36:D469–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Festa RA, McAllister F, Pearce MJ, Mintseris J, Burns KE, Gygi SP, Darwin KH. 2010. Prokayrotic [sic] ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis. PLoS One 5:e8589. doi: 10.1371/journal.pone.0008589 (Author Correction, 5(7): doi:.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bandyopadhyay B, Das Gupta T, Roy D, Das Gupta SK. 2012. DnaK dependence of the mycobacterial stress-responsive regulator HspR is mediated through its hydrophobic C-terminal tail. J Bacteriol 194:4688–4697. doi: 10.1128/JB.00415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das Gupta T, Bandyopadhyay B, Das Gupta SK. 2008. Modulation of DNA-binding activity of Mycobacterium tuberculosis HspR by chaperones. Microbiology 154:484–490. doi: 10.1099/mic.0.2007/012294-0. [DOI] [PubMed] [Google Scholar]
- 38.Tian J, Bryk R, Itoh M, Suematsu M, Nathan C. 2005. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of alpha-ketoglutarate decarboxylase. Proc Natl Acad Sci U S A 102:10670–10675. doi: 10.1073/pnas.0501605102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jullien N, Herman JP. 2011. LUEGO: a cost and time saving gel shift procedure. Biotechniques 51:267–269. [DOI] [PubMed] [Google Scholar]
- 40.van Soolingen D, Hermans PW, de Haas PE, Soll DR, van Embden JD. 1991. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J Clin Microbiol 29:2578–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chong YH, Jung JM, Choi W, Park CW, Choi KS, Suh YH. 1994. Bacterial expression, purification of full length and carboxyl terminal fragment of Alzheimer amyloid precursor protein and their proteolytic processing by thrombin. Life Sci 54:1259–1268. doi: 10.1016/0024-3205(94)00853-1. [DOI] [PubMed] [Google Scholar]
- 45.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 46.Gandotra S, Lebron MB, Ehrt S. 2010. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog 6:e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]