

Abstract
The hippocampus is a key brain region in the pathophysiology of mesial temporal lobe epilepsy. Long‐term changes of its architecture and function on the network and cellular level are well documented in epilepsy. Astrocytes can control many aspects of neuronal function and their long‐term alterations over weeks, months and years play an important role in epilepsy. However, a pathophysiological transformation of astrocytes does not seem to be required for astrocytes to contribute to epileptic activity. Some of the properties of physiological astrocyte–neuron communication could allow these cells to exacerbate or synchronize neuronal firing on shorter time scales of milliseconds to minutes. Therefore, these astrocyte–neuron interactions are increasingly recognized as potential contributors to epileptic activity. Fast and reciprocal communication between astrocytes and neurons is enabled by a diverse set of mechanisms that could both amplify and counteract epileptic activity. They may thus promote or cause development of epileptic activity or inhibit it. Mechanisms of astrocyte–neuron interactions that can quickly increase network excitability involve, for example, astrocyte Ca2+ and Na+ signalling, K+ buffering, gap junction coupling and metabolism. However, rapid changes of astrocyte neurotransmitter uptake and morphology may also underlie or support development of network hyperexcitability. The temporal characteristics of these interactions, their ability to synchronize neuronal activity and their net effect on network activity will determine their contribution to the emergence or maintenance of epileptic activity.
Abbreviations
- AQP
aquaporin
- Cx
connexin
- GABA
γ‐aminobutyric acid
- NMDA
N‐methyl‐d‐aspartate
- ROCK
Rho‐associated protein kinase
Introduction
How information is transferred and processed in the healthy hippocampus is not determined only by neuronal connectivity or synaptic and neuronal properties. Information processing is also profoundly shaped by rapid and reciprocal astrocyte–neuron communication operating on short time scales of milliseconds to minutes (for a review, Rusakov et al. 2014). Therefore, it is intuitive that long‐term changes of astrocyte structure or function over weeks and months can have an impact on neuronal signalling in the hippocampus. Such astrocytic long‐term changes are a hallmark of mesial temporal lobe epilepsy, a common and disabling disease. A prominent finding in later stages of the disease in patients and animal models is hippocampal astrogliosis. The term describes a number of changes of astrocyte density, morphology, biochemistry and physiology associated with epilepsy but also other diseases (Sofroniew & Vinters, 2009; Pekny & Pekna, 2014). Some of these late and long‐lasting functional modifications like altered K+ and glutamate clearance by astrocytes can contribute to the generation of epileptic activity and seizures. Comprehensive recent reviews and books cover this topic in depth (Carmignoto & Haydon, 2012; Noebels et al. 2012; Crunelli & Carmignoto, 2013; Coulter & Steinhäuser, 2015). Several recent studies suggest a causal role of astrogliosis in epileptogenesis. For example, experimental induction of reactive astrogliosis by viral overexpression of enhanced green fluorescent protein (EGFP) in hippocampal astrocytes led to reduced expression of glutamine synthetase, reduced synaptic inhibition, and as a consequence, network hyperexcitability (Ortinski et al. 2010). Conditional deletion of β1‐integrin also results in astrogliosis, reduced glutamate uptake, altered chloride transporter expression and development of epilepsy (Robel et al. 2015). These studies demonstrate that induction of persistent astrogliosis and the ensuing changes of astrocyte function are sufficient to render neuronal networks susceptible to the development of spontaneous seizures after a ‘latent period’ of days and weeks. They also suggest that any stimulus that initiates the pathophysiological transformation of astrocytes into reactive astrocytes has the potential to result in epilepsy with some delay, which would depend on many factors including the speed of progression of astrogliosis. In addition to long‐term changes of astrocyte physiology, astrocyte–neuron interactions that are physiologically active in the hippocampus may also contribute to the generation or propagation of epileptic activity.
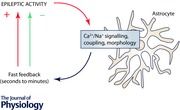
A general view is that epileptic activity and seizures are characterized by an overall increase of neuronal action potential firing, although it is not fully understood how ictal activity emerges and terminates (Bower & Buckmaster, 2008; Truccolo et al. 2011; Walker, 2011). Therefore, any property that allows astrocyte–neuron communication to amplify, prolong or disinhibit neuronal activity could be considered epileptogenic. Indeed, a multitude of astrocyte–neuron interactions that could create excitatory feedback loops between neurons and astrocytes have been documented in the healthy hippocampus. Thus, astrocytes may amplify even mild increases of network activity or contribute to epileptic activity. Experimental data also demonstrate that astrocyte–neuron signalling can occur on a slower time scale than fast neuronal synaptic transmission thereby possibly prolonging periods of increased network activity. In addition to positive feedback, astrocyte–neuron signalling can provide short‐term negative feedback by, for instance, inhibiting excitatory synaptic transmission. The balance of positive and negative astrocyte feedback on epileptic activity could determine the overall effect of astrocyte–neuron interactions on epileptiform activity. The aim of this review is to explore specific astrocyte–neuron interactions that could rapidly promote or counteract generation and propagation of epileptic activity without requiring the long‐lasting astrocyte modifications associated with astrogliosis. The focus will be on mechanisms of astrocyte–neuron signalling documented in the healthy hippocampus that operate on time scales of seconds and minutes and thus establish fast feedback in response to short periods of increased neuronal/epileptic activity.
Astrocyte Ca2+ signalling
Astrocytes are electrically passive cells but respond to a variety of neurotransmitters with cytosolic and often store‐dependent Ca2+ increases. This was first observed for the neurotransmitter glutamate in cultured astrocytes using Ca2+‐sensitive fluorescent indicators (Cornell‐Bell et al. 1990; for a review, Rusakov et al. 2014). In the hippocampus, astrocyte Ca2+ transients can be induced in situ by high frequency stimulation protocols of axonal connections (Porter & McCarthy, 1996; Henneberger et al. 2010) and in vivo using sensory stimulation (Navarrete et al. 2012). They are also associated with epileptiform activity in vitro. For example, induction of paroxysmal depolarizing shifts characteristic for interictal activity (Engel, 2001) in acute hippocampal slices resulted in time‐locked astrocyte Ca2+ transients (Tian et al. 2005). The probability of observing Ca2+ transients was also increased when mixed interictal and ictal‐like activity was acutely induced in hippocampal slices (Fellin et al. 2006). Similarly, ictal‐like network activity was shown to activate somatic Ca2+ signalling in entorhinal and hippocampal astrocytes (Gómez‐Gonzalo et al. 2010). Equivalent observations have been made in vivo. Pharmacological disinhibition of cortical network activity led to an acute increase of neuronal population bursting, to an increased occurrence of astrocyte Ca2+ spikes and to more strongly coordinated Ca2+ signalling in neighbouring astrocytes (Hirase et al. 2004).
Although these studies have focused primarily on somatic Ca2+ transients for technical reasons, they clearly indicate that high frequency network activity and epileptic activity do trigger astrocyte Ca2+ transients. However, improved imaging techniques and the development of genetically encoded Ca2+ indicators have revealed that this might represent only the proverbial ‘tip of the iceberg’. Focal and small‐scale Ca2+ transients occurring in the periphery of astrocytes were more recently shown to occur much more frequently than somatic or large‐scale transients in vitro and in vivo (Di Castro et al. 2011; Kanemaru et al. 2014; Srinivasan et al. 2015). This might be important in the context of epilepsy for at least two reasons. Firstly, short, interictal‐like activity may be a potent trigger of local astrocyte Ca2+ signalling without involvement of the soma because even single synaptic stimuli are sufficient to induce local Ca2+ transients (Panatier et al. 2011). Secondly, the spatial extent of Ca2+ transients triggered by epileptic activity (e.g. local transients extending a couple of micrometres, individual branches or the entire astrocyte) could be an important parameter. It would determine how many of the ∼140,000 synapses an individual astrocyte can cover in the CA1 stratum radiatum (Bushong et al. 2002) could be synchronized as a result of a single astrocyte Ca2+ transient. Therefore, revealing the subcellular and temporal pattern of astrocyte Ca2+ transients will be important to understand how astrocytes promote or counteract development of network hyperexcitability and epileptic activity.
Another important parameter for Ca2+‐dependent mechanisms is the resting Ca2+ concentration. Firstly, it is an important modulator of Ca2+ signalling itself. For example, it sets the level of saturation of intracellular Ca2+ buffers and the open probability of IP3 receptors (Foskett et al. 2007). Secondly, it could determine to what degree Ca2+‐dependent astrocyte–neuron signalling is constitutively active. It is particularly interesting in this regard that the astrocyte resting [Ca2+] is neither constant nor homogeneous within or between cells (Zheng et al. 2015). Similar to the resting [Ca2+] in Alzheimer's disease (Kuchibhotla et al. 2009), the distribution of resting [Ca2+] in astrocytes could be affected by epileptic activity. This would profoundly alter Ca2+‐dependent astrocyte–neuron communication and thereby maintain an altered network excitability. Astrocyte mitochondria represent another emerging key regulator of astrocyte Ca2+ signalling. Their mobility of up to 0.4 μm s−1 within hippocampal astrocytes is reduced and they are located closer to synapses after enhancing neuronal activity (Jackson & Robinson, 2015; Stephen et al. 2015). Epileptic activity may have a very similar effect. Interestingly, disrupting this mobility pattern of astrocytic mitochondria can prolong spontaneously occurring Ca2+ transients (Jackson & Robinson, 2015; Stephen et al. 2015). Therefore, epileptic activity may recruit mitochondria to synapses and constrain local Ca2+ signalling in astrocytes. However, it remains to be tested if these hypothetical mechanisms modulate epileptic activity.
Targets of astrocyte Ca2+ signalling
Astrocyte–neuron communication can involve a diverse set of signalling mechanisms, many of which depend on astrocyte Ca2+ signalling (for a review, Rusakov et al. 2014). In the context of epilepsy, Ca2+‐dependent glutamate release from astrocytes (Parpura et al. 1994) has attracted particular attention. It may amplify epileptic activity by an excitatory feedback loop or even trigger epileptic activity via neuronal N‐methyl‐d‐aspartate (NMDA) receptors (Tian et al. 2005; Fellin et al. 2006; Gómez‐Gonzalo et al. 2010; Crunelli & Carmignoto, 2013). Whether glutamate release from astrocytes directly triggers epileptiform activity has been a matter of debate (Tian et al. 2005; Fellin et al. 2006). An intriguing scenario is that while Ca2+‐dependent release from astrocytes may not trigger epileptic activity in itself, it can recruit neurons into synchronous activity thus lowering the threshold of ictal activity (Gómez‐Gonzalo et al. 2010; Crunelli & Carmignoto, 2013). The exact signalling pathway remains unidentified, however, and plenty of candidates exist (see Fig. 1 for a potential scenario). Nonetheless, astrocyte Ca2+ signalling appears to be largely pro‐epileptic because infusion of the high affinity Ca2+ chelator 1,2‐bis(o‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid (BAPTA) intro astrocyte networks impairs Ca2+ signalling and ictal discharges (Gómez‐Gonzalo et al. 2010). Similarly, expression of dominant‐negative soluble NSF attachment protein receptor (SNARE) in glia delays the onset of epileptiform activity in the hippocampus in vitro and decreases its frequency (Clasadonte et al. 2013). Thus, these two studies demonstrate an important permissive role of astrocyte–neuron signalling for the development of epileptiform activity.
Figure 1. Example of fast positive neuron–astrocyte feedback loops involving glutamate signalling.
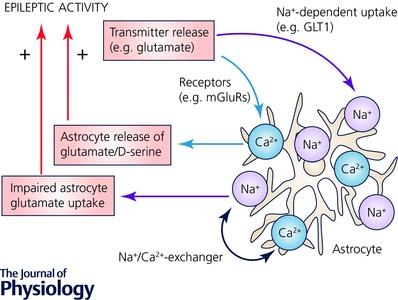
Neuronal epileptic activity can induce robust cytosolic Ca2+ (orange) and Na+ (violet) concentration increases in astrocytes (grey). These can be mediated by glutamate released from neurons acting on, for instance, astrocyte metabotropic glutamate receptors (mGluRs) and its Na+‐dependent uptake through astrocyte glutamate transporters (GLT1). But other signalling molecules may be equally important (see text and Perea et al. 2009; Kirischuk et al. 2012). The spatial extent and duration of Na+ and Ca2+ signals is likely to depend on the pattern and intensity of neuronal activity. Na+ and Ca2+ signalling can mutually influence each other via the Na+/Ca2+ exchanger (NCX). An increase of Na+ after glutamate uptake may lead to NCX operating in reverse mode transporting Ca2+ into the astrocyte. Both astrocyte Ca2+ and Na+ can establish a feedback loop onto neurons using various mechanisms (see text). For example, Ca2+‐dependent glutamate or d‐serine release from astrocytes may strengthen excitatory synaptic transmission and an increase of astrocyte Na+ can reduce the driving force for astrocyte glutamate uptake. Both mechanisms would increase glutamatergic excitation and thereby provide positive feedback on a time scale of seconds and less.
Besides glutamate, other signalling molecules can mediate a positive feedback loop (for a review, Zorec et al. 2012). The NMDA receptor co‐agonist d‐serine is released from astrocytes in a Ca2+‐dependent manner in acute hippocampal slices and in the cortex in vivo (Henneberger et al. 2010; Takata et al. 2011). A brief high‐frequency stimulus lasting only 200 ms, similar to the duration of interictal activity, can increase NMDA receptor co‐agonist site occupancy for at least 20 s (Henneberger et al. 2010). Astrocytic d‐serine supply may thus confer an increased excitability to neurons after an interictal discharge. This could occur even in the, previously reported (Clasadonte et al. 2013), absence of persistent changes of the resting d‐serine concentration. Adenosine receptors could represent additional mediators of a positive feedback loop. Astrocyte Ca2+ signalling can lead to increased basal glutamatergic synaptic transmission involving activation of A2A receptors (Panatier et al. 2011). Although this could be balanced by astrocyte‐mediated presynaptic inhibition through A1 receptors (Pascual et al. 2005; Zorec et al. 2012). In addition, GABAergic synaptic transmission may also be affected because of astrocytic Ca2+ increases. For instance, reducing intracellular Ca2+ in astrocytes by BAPTA reduces γ‐aminobutyric acid (GABA) transporter type 3 (GAT3) levels in astrocytes and increases tonic GABA receptor‐mediated currents. As a consequence, spontaneous inhibitory postsynaptic currents in interneurons but not pyramidal cells are reduced (Shigetomi et al. 2012). The net effect of this mechanism on network activity appears difficult to predict. Nonetheless, it represents a potent modulator of network excitability. Finally, glutamate uptake by astrocytes could be altered after increases of the cytosolic Ca2+ concentration in astrocytes (Fig. 1). The glutamate transporter EAAT2b was recently demonstrated to show strong surface expression in cultured astrocytes and slices but is internalized after increasing [Ca2+] by glutamate exposure (Underhill et al. 2015). This may also increase neuronal excitation because astrocytes take up most of the synaptically released glutamate (Danbolt, 2001). However, the exact time courses of internalisation and required astrocyte Ca2+ signal remain to be established.
As discussed above, direct experimental evidence indicates that astrocyte Ca2+ signalling can promote generation of epileptiform activity (Tian et al. 2005; Gómez‐Gonzalo et al. 2010). Nonetheless, Ca2+ signalling may also counteract the development of epileptic activity. For example, it is required for the transient post‐burst depression of release from excitatory synapses (Andersson & Hanse, 2010) and spontaneous excitatory synaptic transmission can be inhibited by a Ca2+‐dependent boost of astrocyte K+ uptake (Wang et al. 2012). Also, induction of Ca2+ transients can increase GABAergic transmission pre‐ and postsynaptically (Kang et al. 1998). Finally, the extracellular Ca2+ concentration is reduced during seizures (Heinemann et al. 1977) and similar reductions of Ca2+ can trigger astrocyte Ca2+ signalling and activation of interneurons by purinergic signalling (Carmignoto & Haydon, 2012; Torres et al. 2012). These potentially opposing and diverse effects complicate any prediction of how astrocyte Ca2+ signalling affects network activity. As a consequence, further investigation is required to reveal by which specific mechanism Ca2+ dependent astrocyte signalling acutely modulates epileptic activity.
Taken together, astrocytes respond robustly with cytosolic Ca2+ increases to high‐frequency neuronal activity and epileptiform activity. A number of Ca2+‐dependent signalling cascades have been documented in astrocytes of the healthy hippocampus that can establish an excitatory feedback loop operating on the time scale of milliseconds to seconds. Whether this happens primarily at excitatory or inhibitory neurons or both will determine the net effect on network activity. The spatial scale of astrocyte Ca2+ transients could be critical for the ability of astrocytes to rapidly synchronize large synaptic populations. Finally, the kinetics of Ca2+ transients could be important for amplification of epileptic activity. For instance, epileptic neuronal activity can trigger an astrocyte Ca2+ transient that outlasts the neuronal activity. Ca2+ dependent excitatory astrocyte–neuron feedback could then prolong epileptic activity or establish a time window of increased network excitability. However, this would require, for example, that a transmitter released from astrocytes is not quickly depleted. It appears that investigating the kinetics and spatial aspects of Ca2+‐dependent astrocyte–neuron signalling during increased or epileptic network activity will provide new insights into the role of astrocyte signalling in epilepsy.
Astrocyte Na+ signalling
Astrocyte glutamate transporters provide most of the glutamate clearance in the brain and take up glutamate along with Na+ ions (Rose & Ransom, 1996; Danbolt, 2001). Therefore, synaptic release of glutamate is tightly coupled to a cytosolic Na+ increase in astrocytes (Kirischuk et al. 2007, 2012; Langer & Rose, 2009). Similar to astrocyte Ca2+ signalling, a brief burst of neuronal activity of 200 ms is sufficient to elicit robust Na+ transients in hippocampal astrocytes in situ, which can last tens of seconds. Their amplitudes scale with stimulus intensity and reach a few millimolar at the astrocyte soma but can be considerably larger in the astrocyte periphery closer to locally activated axons (Langer & Rose, 2009). Most of the glutamate uptake is likely to take place directly at the synapse where thin, leaf‐like astrocyte processes approach synaptic structures (Medvedev et al. 2014). Therefore, fast and confined Na+ increases in fine astrocyte processes may far exceed the peak Na+ concentrations estimated using diffraction‐limited fluorescence imaging techniques. Another feature of these locally generated Na+ transients is their reliable propagation through individual hippocampal astrocytes and into neighbouring astrocytes (Langer & Rose, 2009). Similar Na+ transients occur time‐locked to epileptiform activity in hippocampal slices (Karus et al. 2015), suggesting that ictal and interictal activity can induce synchronized and widespread Na+ increases in astrocytes that last for tens of seconds (Fig. 1).
An increase of cytosolic Na+ levels can have substantial effects on astrocyte physiology ranging from changing driving forces for glutamate and GABA transport and altered ion homoeostasis to metabolic changes (for a review, Kirischuk et al. 2012). Virtually all of these have the potential to alter network excitability considerably. A recent study estimated that a 12 mm increase of Na+ reduces the driving force for glutamate uptake by about 20% (Karus et al. 2015). At the same time, a Na+ increase of this magnitude could easily reverse GABA uptake in astrocytes and release this inhibitory neurotransmitter (Richerson & Wu, 2003; Kirischuk et al. 2012). A similar argument can be made about the extracellular concentration of the NMDA receptor co‐agonist glycine and its transporter Glyt1b. Because of its stoichiometry Glyt1b could reverse during periods of high intracellular astrocyte Na+ (Roux & Supplisson, 2000) and thereby increase NMDA receptor‐mediated excitation but also glycine receptor‐mediated inhibition at higher concentrations. Glyt1 blockade was recently shown to inhibit epileptic activity (Shen et al. 2015) suggesting that elevated extracellular glycine levels may overall inhibit epileptic activity. Finally, a Na+ increase can trigger the reversal of the Na+/Ca2+ exchanger effectively allowing Ca2+ to enter the astrocyte (Kirischuk et al. 1997) thereby linking Na+ entry to the Ca2+‐dependent mechanisms discussed above.
One experimental approach to study the effects of increased astrocyte Na+ load is to poison astrocyte metabolism selectively by fluorocitrate or related compounds. This approximately doubles the resting [Na+] over an hour (Karus et al. 2015). In vivo, intracerebral injection of fluorocitrate induces epileptiform discharges within 30 min (Willoughby et al. 2003) and epileptiform activity in vitro is also increased by fluoroacetate application in hippocampal slices (Karus et al. 2015). While this is compatible with the idea that elevated Na+ levels could promote development of epileptic activity, metabolic poisoning may affect a host of other astrocyte functions. Thus, astrocyte Na+ signalling in response to neuronal epileptic activity may be involved in establishing a period of increased excitability by positive feedback or synchronization, not unlike astrocyte Ca2+ signalling (Fig. 1). Investigation of specific mechanisms will certainly be challenging because of the relatively limited toolset available for specific manipulation of Na+ signalling and the number of potential mechanisms outlined above that require testing.
Astrocyte gap junction coupling
Extensive gap junction coupling is a distinct property of astrocytes and enables exchange of small, diffusible molecules throughout large astrocyte networks (for a review, Giaume et al. 2010). Diffusion in gap junction coupled astrocyte networks depends on the properties of gap junctions but also on the diffusion within individual cells (Giaume et al. 2010; Anders et al. 2014). The effect of network activity on gap junction coupling appears to be variable and to depend on brain region and experimental protocols (Giaume et al. 2010). A particularly fast modulation of dye coupling was observed by Serrano et al. who demonstrated an increase of gap junction coupling after exposing acute hippocampal slices to NMDA for 2 min (Serrano et al. 2008). This suggests that rapid modulation of diffusion within astrocyte networks is possible. However, induction of epileptiform activity in hippocampal slices obtained from 2‐ to 4‐week‐old mice did not impair dye coupling between astrocytes (Rouach et al. 2008). In contrast, induction of status epilepticus by kainate injection in 8‐ to 9‐week‐old mice reduced gap junction coupling within 4 h (Bedner et al. 2015). These results imply that epileptic activity could rapidly affect gap junction coupling but also that time course and exact conditions remain to be firmly established (for a review, Coulter & Steinhäuser, 2015; Steinhäuser et al. 2015).
Studies on the role of astrocyte gap junction coupling in epilepsy have successfully used genetic models lacking connexins (Cx), which represent the building blocks of astrocyte gap junctions (Cx43 and Cx30). For example, deletion of Cx43 and Cx30 was found to abolish astrocyte coupling, to impair K+ buffering and to lead to spontaneous epileptiform activity in the CA1 region of hippocampal slices (Wallraff et al. 2006). On the other hand, glucose supply through astrocyte networks supports synaptic transmission and also, partially, epileptiform activity (Rouach et al. 2008). Recent in vivo observations also suggest that energy supply via the astrocyte–neuron lactate shuttle maintains epileptic activity (Sada et al. 2015). Therefore, a modulation of gap junction coupling in response to increased or epileptic neuronal activity could result in positive or negative feedback on neuronal activity. A sample scenario assuming a downregulation of gap junction coupling by epileptic activity is illustrated in Fig. 2. Whether a rapid modulation of gap junction coupling can be triggered by short bursts of neuronal or epileptic activity remains to be established.
Figure 2. A possible scenario for astrocyte feedback on epileptic activity through a reduction of astrocyte gap junction coupling.
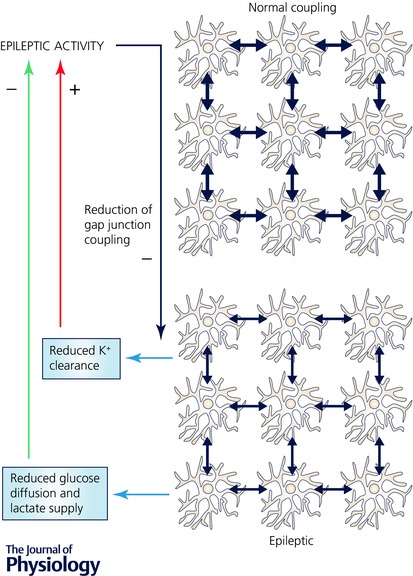
Astrocytes form extensive networks by gap junction coupling (blue arrows). Many metabolites including glucose and lactate, ions and signalling molecules can diffuse through this network. Therefore, a reduction of astrocyte gap junction coupling after development of epilepsy (see text and Bedner et al. 2015) is expected to limit energy supply by astrocytes to neurons and to impair K+ clearance. While limiting energy supply can inhibit ongoing epileptic activity (green arrow), a reduced K+ clearance can increase neuronal excitability and thereby promote epileptic activity (red arrow). The net effect could depend on the temporal properties of gap junction modulation and glucose and lactate depletion (see text).
The time course of the functional consequences of modified coupling also requires consideration. For example, an acute reduction of gap junction coupling may have an immediate effect on astrocyte K+ buffering while a slow energy depletion may take longer to become manifest. A rapid modulation of gap junction coupling is also of interest with respect to astrocyte Na+ and Ca2+ signals, which can propagate through gap junctions. For instance, Na+ or Ca2+ spread in the astrocyte network could be constrained as a result of reduced astrocyte coupling. This could limit the ability of Na+‐ or Ca2+‐dependent astrocyte–neuron communication to synchronize larger populations of synapses and neurons. Complex interactions are also expected because Ca2+ increases could themselves affect gap junction coupling (Enkvist & McCarthy, 1994).
Astrocyte morphology
Many astrocyte–neuron interactions depend on diffusion of signalling molecules or ions and therefore on the spatial relationship between astrocytes, pre‐ and postsynaptic neuronal structures and the extracellular space. Therefore, it is likely that rapid changes of astrocyte morphology on the time scale of seconds and minutes affect astrocyte–neuron interactions. For example, acute swelling of astrocyte processes can constrain extracellular space thereby altering extracellular concentration transients of K+ or other ions and released neurotransmitters (for a review, Syková, 2004). Therefore, rapid astrocyte morphology changes could represent a feedback loop between epileptic neuronal activity and astrocyte‐mediated changes of network excitability.
Indeed, it is well established that both the K+ increase observed during seizures (Heinemann et al. 1977) and the large amount of glutamate released during epileptic activity potently trigger astrocyte swelling and a reduction of extracellular space (for a review, Syková, 2004). Inducing a similar reduction of extracellular space by changing medium osmolality increases the likelihood of the hippocampus to generate epileptiform activity (Dudek et al. 1990). This strongly suggests that astrocyte swelling could represent a positive feedback mechanism by which astrocytes promote epileptic activity (see Fig. 3 for an example). However, an undershoot of extracellular K+ is also often observed after ictal activity (Heinemann et al. 1977). If astrocyte volume purely followed the extracellular K+ concentration, their volume response to epileptic activity would be more complex than simple swelling. A similar point can be made about the spatial distribution of astrocyte volume changes. Astrocytes do not swell homogeneously when challenged with hypotonic solution (Chvátal et al. 2007). Therefore, small perisynaptic processes may respond differently to epileptic activity than larger branches, with different pathophysiological consequences. A spatially heterogeneous astrocyte volume response appears likely, particularly on short time scales of seconds and below, because perisynaptic astrocyte branches are probably exposed to higher neurotransmitter levels.
Figure 3. Rapid astrocyte restructuring in response to epileptic activity could increase neuronal excitation by glutamate signalling and excitability – two examples.
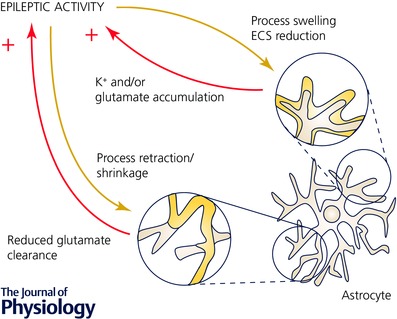
Epileptic and neuronal activity can affect volume and structure of astrocytes. Acute swelling of astrocytes as a result of increased neuronal activity has been described (top right, grey before, yellow swollen, see text). Consequently, the extracellular volume is reduced and the amplitudes of extracellular K+ and glutamate transients during synaptic activity are expected to increase (ECS, extracellular space). This could provide an immediate positive feedback and support development of epileptic activity by increasing neuronal excitability and activation of additional glutamate receptors. High frequency neuronal activity can also acutely, over the time course of minutes, induce astrocyte restructuring (i.e. a fundamental change of the spatial relationship between astrocyte and neuron). For example, a retraction or shrinkage of astrocyte processes (bottom, grey before, yellow after retraction) can increase the distance between neuronal glutamate release and astrocyte glutamate transporters and thus delay or impair glutamate clearance. Released glutamate may then act longer and/or on a larger population of glutamate receptors thereby providing additional excitatory drive to neurons.
Aquaporin 4 (AQP4) is thought to be critical for linking synaptic activity and astrocyte volume by facilitating water entry into astrocytes after uptake of K+ or glutamate (for a review, Nagelhus et al. 2004; Wetherington et al. 2008). The observation of prolonged seizures in AQP4 knockout mice (Binder et al. 2006; Coulter & Steinhäuser, 2015) provides additional evidence for a role of rapid astrocyte volume changes in the generation of epileptic activity. Interestingly, AQP4 was recently shown to be required for astrocyte Ca2+ transients generated in parallel to astrocyte volume responses in cultured astrocytes (Benfenati et al. 2011). This establishes a potential link between neuronal activity and the Ca2+‐dependent mechanisms of astrocyte–neuron communication discussed above.
Astrocyte processes may also extend, retract or move in response to epileptic activity. This alters the spatial relationship between astrocyte processes and neurons and thereby astrocyte–neuron communication. Indeed, fine astrocyte processes can approach or withdraw from neuronal spines on a time scale of minutes (Haber et al. 2006; Bernardinelli et al. 2014). Similar to astrocyte Ca2+ transients, high frequency stimulation paradigms used for induction of synaptic plasticity appear to be particularly effective triggers of astrocyte restructuring in the hippocampus (Wenzel et al. 1991; Henneberger et al. 2008; Bernardinelli et al. 2014; Perez‐Alvarez et al. 2014). As a result, astrocyte–neuron communication can be impaired (Perez‐Alvarez et al. 2014). This is reminiscent of findings in the supraoptic nucleus where physiological changes of astrocyte coverage of neurons during lactation control glutamate uptake and synaptic plasticity (Oliet et al. 2001; Panatier et al. 2006). Fine astrocyte morphology changes on the time scale of minutes have not been explored in the context of epilepsy. Nonetheless, it is tempting to speculate that epileptic activity induces a rapid restructuring of small astrocyte processes affecting, for instance, glutamate uptake. Astrocyte glutamate transporters display a high motility in the astrocyte membrane in general, but not in the vicinity of synapses (Murphy‐Royal et al. 2015). The activity‐dependent process that traps transporters at the synapse is not fully understood. However, it is conceivable that an astrocyte morphological change may release this break on transporter mobility and allow them to diffuse away from sites of synaptic glutamate release. The ensuing reduction of local glutamate uptake could promote epileptic activity. A central signalling pathway controlling astrocyte morphology involves Rho‐associated protein kinase (ROCK). Its pharmacological inhibition promotes a more complex, branched morphology in culture (Abe & Misawa, 2003; Racchetti et al. 2012) and can increase glutamate uptake (Lau et al. 2011). Interestingly, ROCK inhibition also attenuates seizures and hippocampal tissue damage (Jeon et al. 2013). These observations imply that rapid ROCK‐dependent restructuring of astrocytes may play a role in epilepsy (see Fig. 3 for a possible scenario).
For these reasons, rapid astrocyte volume and morphology changes could be potent mediators of a feedback loop that amplifies or prolongs aberrant neuronal activity and thus supports emergence or maintenance of epileptic activity. This may involve various mechanisms including modified K+ or glutamate clearance. In addition, morphology changes can also affect diffusion of Na+ and Ca2+ within astrocytes and between astrocytes coupled via gap junctions. Whether changes of astrocyte morphology and intracellular diffusivity indeed have an impact on astrocyte–neuron communication in the context of epilepsy would be an interesting question to address.
Conclusion
In the healthy hippocampus, astrocytes and neurons reciprocally exchange signals on time scales ranging from milliseconds to minutes using a variety of mechanisms. The aim was to review and explore those that can amplify, prolong or dampen neuronal epileptic activity without requiring the well‐known long‐term changes of astrocyte function seen later in epilepsy. Ca2+‐ and Na+‐dependent mechanisms of astrocyte–neuron communication, modulation of astrocyte gap junction coupling and astrocyte morphology changes all appear to be able to contribute to the emergence and maintenance of epileptic activity. A role of astrocytes in the development of epilepsy may thus entirely rely on mechanisms of astrocyte–neuron communication intrinsic to the healthy brain. Among these mechanisms, especially Ca2+‐dependent astrocyte–neuron signalling is strongly implicated in the generation of epileptiform activity. However, in most cases, arguments can be also presented for an antiepileptic action. Which effect dominates is likely to depend on the selectivity of an astrocyte–neuron interaction for a specific cell type (i.e. excitatory vs. inhibitory neurons and their synapses), its temporal characteristics and spatial extent. Especially the spatial extent could determine how strongly neuronal activity can be synchronized by astrocytes and how the various types of astrocyte–neuron signalling are orchestrated within individual astrocytes and in the astrocyte network.
A significant proportion of the studies discussed above have dissected the mechanisms of astrocyte–neuron communication in reduced preparations like acute hippocampal slices. However, especially in a disease context like epilepsy it will be important to test which types of astrocyte–neuron communication are engaged by brief increases of network activity, interictal or epileptic activity in vivo. Their differential recruitment, for example by interictal activity or towards the end of a seizure, will hint at their pathophysiological significance and could suggest experimental strategies to establish a causal role of astrocyte–neuron interactions in epilepsy. This will add to the challenge of deciphering how a specific type of astrocyte–neuron signalling amplifies or dampens epileptic activity in the face of neuronal network heterogeneity and astrocyte signalling diversity.
Additional information
Competing interests
None.
Funding
This work was supported by the German Research Foundation (DFG, SFB1089 B03 and SPP1757 HE6949/1), the NRW‐Rückkehrerprogramm, the Human Frontiers Science Program (RGY0084/2012) and the German Centre for Neurodegenerative Diseases (DZNE, Bonn, Germany).
Acknowledgements
Helpful comments on an earlier version of the manuscript by Christian Steinhäuser and Elizabeth Matthews (both University of Bonn Medical School, Germany) were highly appreciated.
Biography
Christian Henneberger studied medicine at the Humboldt and Free University Berlin (Germany) where he obtained his degree and defended his thesis in neurophysiology both in 2003. He continued his work on the properties of synaptic transmission in the developing visual system as a postdoctoral fellow at the Institute of Neurophysiology at the Charité (Berlin, AiP research fellowship). After moving to the UCL Institute of Neurology (London, UK), he focused on hippocampal synaptic transmission and plasticity and its dependence on components of the extracellular matrix and astrocyte Ca2+ signalling. Obtaining a UCL Excellence Fellowship allowed him to continue this line of work as a principal investigator at UCL before establishing his own lab in Bonn in 2011. Funded by the NRW‐Rückkehrerprogramm, DFG and HFSP his lab (www.henneberger‐lab.com, Institute of Cellular Neuroscience) investigates how dynamic changes of astrocyte morphology affect astrocyte–neuron interactions at the cellular and synaptic level in healthy brain tissue and in disease.

References
- Abe K & Misawa M (2003). Astrocyte stellation induced by Rho kinase inhibitors in culture. Brain Res Dev Brain Res 143, 99–104. [DOI] [PubMed] [Google Scholar]
- Anders S, Minge D, Griemsmann S, Herde MK, Steinhäuser C & Henneberger C (2014). Spatial properties of astrocyte gap junction coupling in the rat hippocampus. Philos Trans R Soc Lond B Biol Sci 369, 20130600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M & Hanse E (2010). Astrocytes impose postburst depression of release probability at hippocampal glutamate synapses. J Neurosci 30, 5776–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedner P, Dupper A, Hüttmann K, Müller J, Herde MK, Dublin P, Deshpande T, Schramm J, Häussler U, Haas CA, Henneberger C, Theis M & Steinhäuser C (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP & Amiry‐Moghaddam M (2011). An aquaporin‐4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell‐volume control in astrocytes. Proc Natl Acad Sci USA 108, 2563–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardinelli Y, Randall J, Janett E, Nikonenko I, König S, Jones EV, Flores CE, Murai KK, Bochet CG, Holtmaat A & Muller D (2014). Activity‐dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr Biol 24, 1679–1688. [DOI] [PubMed] [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS & Manley GT (2006). Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin‐4 water channels. Glia 53, 631–636. [DOI] [PubMed] [Google Scholar]
- Bower MR & Buckmaster PS (2008). Changes in granule cell firing rates precede locally recorded spontaneous seizures by minutes in an animal model of temporal lobe epilepsy. J Neurophysiol 99, 2431–2442. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ & Ellisman MH (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G & Haydon PG (2012). Astrocyte calcium signaling and epilepsy. Glia 60, 1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvátal A, Anderová M, Hock M, Prajerová I, Neprasová H, Chvátal V, Kirchhoff F & Syková E (2007). Three‐dimensional confocal morphometry reveals structural changes in astrocyte morphology in situ. J Neurosci Res 85, 260–271. [DOI] [PubMed] [Google Scholar]
- Clasadonte J, Dong J, Hines DJ & Haydon PG (2013). Astrocyte control of synaptic NMDA receptors contributes to the progressive development of temporal lobe epilepsy. Proc Natl Acad Sci USA 110, 17540–17545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell‐Bell AH, Finkbeiner SM, Cooper MS & Smith SJ (1990). Glutamate induces calcium waves in cultured astrocytes: long‐range glial signaling. Science 247, 470–473. [DOI] [PubMed] [Google Scholar]
- Coulter DA & Steinhäuser C (2015). Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med 5, a022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V & Carmignoto G (2013). New vistas on astroglia in convulsive and non‐convulsive epilepsy highlight novel astrocytic targets for treatment. J Physiol 591, 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC (2001). Glutamate uptake. Prog Neurobiol 65, 1–105. [DOI] [PubMed] [Google Scholar]
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, Tiret P & Volterra A (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci 14, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A & Tasker JG (1990). Osmolality‐induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: Importance of non‐synaptic mechanisms in hippocampal epileptogenesis. Neurosci Lett 120, 267–270. [DOI] [PubMed] [Google Scholar]
- Engel J (2001). Mesial temporal lobe epilepsy: what have we learned? Neuroscientist 7, 340–352. [DOI] [PubMed] [Google Scholar]
- Enkvist MOK & McCarthy KD (1994). Astroglial gap junction communication is increased by treatment with either glutamate or high K+ concentration. J Neurochem 62, 489–495. [DOI] [PubMed] [Google Scholar]
- Fellin T, Gomez‐Gonzalo M, Gobbo S, Carmignoto G & Haydon PG (2006). Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci 26, 9312–9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung K‐H & Mak D‐OD (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D & Rouach N (2010). Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11, 87–99. [DOI] [PubMed] [Google Scholar]
- Gómez‐Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, de Curtis M, Ratto GM & Carmignoto G (2010). An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol 8, e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber M, Zhou L & Murai KK (2006). Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci 26, 8881–8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Lux HD & Gutnick MJ (1977). Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res 27, 237–243. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Medvedev NI, Stuart MG & Rusakov DA (2008). LTP induction changes the morphology of astrocytes in the CA1 region of the hippocampus in vitro. Program No. 242.7/I2 2008 Neuroscience Meeting Planner. Society for Neuroscience, Washington, DC.
- Henneberger C, Papouin T, Oliet SHR & Rusakov DA (2010). Long‐term potentiation depends on release of d‐serine from astrocytes. Nature 463, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase H, Qian L, Barthó P & Buzsáki G (2004). Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol 2, E96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JG & Robinson MB (2015). Reciprocal regulation of mitochondrial dynamics and calcium signaling in astrocyte processes. J Neurosci 35, 15199–15213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon BT, Jeong EA, Park S‐Y, Son H, Shin HJ, Lee DH, Kim HJ, Kang SS, Cho GJ, Choi WS & Roh GS (2013). The Rho‐kinase (ROCK) inhibitor Y‐27632 protects against excitotoxicity‐induced neuronal death in vivo and in vitro. Neurotox Res 23, 238–248. [DOI] [PubMed] [Google Scholar]
- Kanemaru K, Sekiya H, Xu M, Satoh K, Kitajima N, Yoshida K, Okubo Y, Sasaki T, Moritoh S, Hasuwa H, Mimura M, Horikawa K, Matsui K, Nagai T, Iino M & Tanaka KF (2014). In vivo visualization of subtle, transient, and local activity of astrocytes using an ultrasensitive Ca2+ indicator. Cell Reports 8, 311–318. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA & Nedergaard M (1998). Astrocyte‐mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1, 683–692. [DOI] [PubMed] [Google Scholar]
- Karus C, Mondragão MA, Ziemens D & Rose CR (2015). Astrocytes restrict discharge duration and neuronal sodium loads during recurrent network activity. Glia 63, 936–957. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Kettenmann H & Verkhratsky A (1997). Na+/Ca2+ exchanger modulates kainate‐triggered Ca2+ signaling in Bergmann glial cells in situ. FASEB J 11, 566–572. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Kettenmann H & Verkhratsky A (2007). Membrane currents and cytoplasmic sodium transients generated by glutamate transport in Bergmann glial cells. Pflugers Arch 454, 245–252. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Parpura V & Verkhratsky A (2012). Sodium dynamics: another key to astroglial excitability? Trends Neurosci 35, 497–506. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT & Bacskai BJ (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer J & Rose CR (2009). Synaptically induced sodium signals in hippocampal astrocytes in situ. J Physiol 587, 5859–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, O'Shea R, Broberg B, Bischof L & Beart P (2011). The Rho kinase inhibitor Fasudil up‐regulates astrocytic glutamate transport subsequent to actin remodelling in murine cultured astrocytes. Br J Pharmacol 163, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev N, Popov V, Henneberger C, Kraev I, Rusakov DA & Stewart MG (2014). Glia selectively approach synapses on thin dendritic spines. Philos Trans R Soc Lond B Biol Sci 369, 20140047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy‐Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L & Oliet SHR (2015). Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci 18, 219–226. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Mathiisen TM & Ottersen OP (2004). Aquaporin‐4 in the central nervous system: Cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 129, 905–913. [DOI] [PubMed] [Google Scholar]
- Navarrete M, Perea G, de Sevilla DF, Gómez‐Gonzalo M, Núñez A, Martín ED & Araque A (2012). Astrocytes mediate in vivo cholinergic‐induced synaptic plasticity. PLoS Biol 10, e1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL, Avoli M, Rogawski MA, Olsen RW. & Delgado‐Escueta AV. (eds) (2012). Jasper's Basic Mechanisms of the Epilepsies, 4th edn National Center for Biotechnology Information, Bethesda, MD: http://www.ncbi.nlm.nih.gov/books/NBK50785/ [Accessed 9 September 2015]. [PubMed] [Google Scholar]
- Oliet SH, Piet R & Poulain DA (2001). Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292, 923–926. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG & Coulter DA (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet J‐P, Touquet B, Pollegioni L, Poulain DA & Oliet SHR (2006). Glia‐derived D‐serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784. [DOI] [PubMed] [Google Scholar]
- Panatier A, Vallée J, Haber M, Murai KK, Lacaille J‐C & Robitaille R (2011). Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146, 785–798. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S & Haydon PG (1994). Glutamate‐mediated astrocyte–neuron signalling. Nature 369, 744–747. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla‐Sanchez R, Sul J‐Y, Takano H, Moss SJ, McCarthy K & Haydon PG (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. [DOI] [PubMed] [Google Scholar]
- Pekny M & Pekna M (2014). Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev 94, 1077–1098. [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M & Araque A (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32, 421–431. [DOI] [PubMed] [Google Scholar]
- Perez‐Alvarez A, Navarrete M, Covelo A, Martin ED & Araque A (2014). Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J Neurosci 34, 12738–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT & McCarthy KD (1996). Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci 16, 5073–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racchetti G, D'Alessandro R & Meldolesi J (2012). Astrocyte stellation, a process dependent on Rac1 is sustained by the regulated exocytosis of enlargeosomes. Glia 60, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson GB & Wu Y (2003). Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol 90, 1363–1374. [DOI] [PubMed] [Google Scholar]
- Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B & Sontheimer H (2015). Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci 35, 3330–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR & Ransom BR (1996). Mechanisms of H+ and Na+ changes induced by glutamate, kainate, and D‐aspartate in rat hippocampal astrocytes. J Neurosci 16, 5393–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K & Giaume C (2008). Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322, 1551–1555. [DOI] [PubMed] [Google Scholar]
- Roux MJ & Supplisson S (2000). Neuronal and glial glycine transporters have different stoichiometries. Neuron 25, 373–383. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Bard L, Stewart MG & Henneberger C (2014). Diversity of astroglial functions alludes to subcellular specialisation. Trends Neurosci 37, 228–242. [DOI] [PubMed] [Google Scholar]
- Sada N, Lee S, Katsu T, Otsuki T & Inoue T (2015). Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367. [DOI] [PubMed] [Google Scholar]
- Serrano A, Robitaille R & Lacaille J‐C (2008). Differential NMDA‐dependent activation of glial cells in mouse hippocampus. Glia 56, 1648–1663. [DOI] [PubMed] [Google Scholar]
- Shen H‐Y, van Vliet EA, Bright K‐A, Hanthorn M, Lytle NK, Gorter J, Aronica E & Boison D (2015). Glycine transporter 1 is a target for the treatment of epilepsy. Neuropharmacology 99, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Tong X, Kwan KY, Corey DP & Khakh BS (2012). TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT‐3. Nat Neurosci 15, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV & Vinters HV (2009). Astrocytes: biology and pathology. Acta Neuropathol 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P & Khakh BS (2015). Ca2+ signaling in astrocytes from Ip3r2–/– mice in brain slices and during startle responses in vivo . Nat Neurosci 18, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhäuser C, Grunnet M & Carmignoto G (2015). Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience 323, 157–169. [DOI] [PubMed] [Google Scholar]
- Stephen T‐L, Higgs NF, Sheehan DF, Awabdh SA, López‐Doménech G, Arancibia‐Carcamo IL & Kittler JT (2015). Miro1 regulates activity‐driven positioning of mitochondria within astrocytic processes apposed to synapses to regulate intracellular calcium signaling. J Neurosci 35, 15996–16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syková E (2004). Glia and volume transmission during physiological and pathological states. J Neural Transm 112, 137–147. [DOI] [PubMed] [Google Scholar]
- Takata N, Mishima T, Hisatsune C, Nagai T, Ebisui E, Mikoshiba K & Hirase H (2011). Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J Neurosci 31, 18155–18165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G‐F, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J & Nedergaard M (2005). An astrocytic basis of epilepsy. Nat Med 11, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres A, Wang F, Xu Q, Fujita T, Dobrowolski R, Willecke K, Takano T & Nedergaard M (2012). Extracellular Ca2+ acts as a mediator of communication from neurons to glia. Sci Signal 5, ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truccolo W, Donoghue JA, Hochberg LR, Eskandar EN, Madsen JR, Anderson WS, Brown EN, Halgren E & Cash SS (2011). Single‐neuron dynamics in human focal epilepsy. Nat Neurosci 14, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill SM, Wheeler DS & Amara SG (2015). Differential regulation of two isoforms of the glial glutamate transporter EAAT2 by DLG1 and CaMKII. J Neurosci 35, 5260–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC (2011). The ups and downs of seizure activity. Nat Neurosci 14, 535–536. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K & Steinhäuser C (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 26, 5438–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L & Nedergaard M (2012). Astrocytes modulate neural network activity by Ca2+‐dependent uptake of extracellular K+ . Sci Signal 5, ra26–ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J, Lammert G, Meyer U & Krug M (1991). The influence of long‐term potentiation on the spatial relationship between astrocyte processes and potentiated synapses in the dentate gyrus neuropil of rat brain. Brain Res 560, 122–131. [DOI] [PubMed] [Google Scholar]
- Wetherington J, Serrano G & Dingledine R (2008). Astrocytes in the epileptic brain. Neuron 58, 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby JO, Mackenzie L, Broberg M, Thoren AE, Medvedev A, Sims NR & Nilsson M (2003). Fluorocitrate‐mediated astroglial dysfunction causes seizures. J Neurosci Res 74, 160–166. [DOI] [PubMed] [Google Scholar]
- Zheng K, Bard L, Reynolds JP, King C, Jensen TP, Gourine AV & Rusakov DA (2015). Time‐resolved imaging reveals heterogeneous landscapes of nanomolar Ca2+ in neurons and astroglia. Neuron 88, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorec R, Araque A, Carmignoto G, Haydon PG, Verkhratsky A & Parpura V (2012). Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro 4, AN20110061. [DOI] [PMC free article] [PubMed] [Google Scholar]