

Abstract
Key points
Patients with diabetes show a blunted cardiac inotropic response to β‐adrenergic stimulation despite normal cardiac contractile reserve.
Acute insulin stimulation impairs β‐adrenergically induced contractile function in isolated cardiomyocytes and Langendorff‐perfused hearts.
In this study, we aimed to examine the potential effects of hyperinsulinaemia associated with high‐fat diet (HFD) feeding on the cardiac β2‐adrenergic receptor signalling and the impacts on cardiac contractile function.
We showed that 8 weeks of HFD feeding leads to reductions in cardiac functional reserve in response to β‐adrenergic stimulation without significant alteration of cardiac structure and function, which is associated with significant changes in β2‐adrenergic receptor phosphorylation at protein kinase A and G‐protein receptor kinase sites in the myocardium.
The results suggest that clinical intervention might be applied to subjects in early diabetes without cardiac symptoms to prevent further cardiac complications.
Abstract
Patients with diabetes display reduced exercise capability and impaired cardiac contractile reserve in response to adrenergic stimulation. We have recently uncovered an insulin receptor and adrenergic receptor signal network in the heart. The aim of this study was to understand the impacts of high‐fat diet (HFD) on the insulin–adrenergic receptor signal network in hearts. After 8 weeks of HFD feeding, mice exhibited diabetes, with elevated insulin and glucose concentrations associated with body weight gain. Mice fed an HFD had normal cardiac structure and function. However, the HFD‐fed mice displayed a significant elevation of phosphorylation of the β2‐adrenergic receptor (β2AR) at both the protein kinase A site serine 261/262 and the G‐protein‐coupled receptor kinase site serine 355/356 and impaired adrenergic reserve when compared with mice fed on normal chow. Isolated myocytes from HFD‐fed mice also displayed a reduced contractile response to adrenergic stimulation when compared with those of control mice fed normal chow. Genetic deletion of the β2AR led to a normalized adrenergic response and preserved cardiac contractile reserve in HFD‐fed mice. Together, these data indicate that HFD promotes phosphorylation of the β2AR, contributing to impairment of cardiac contractile reserve before cardiac structural and functional remodelling, suggesting that early intervention in the insulin–adrenergic signalling network might be effective in prevention of cardiac complications in diabetes.
Keywords: ardiac function, beta adrenergic receptor, high fat diet
Key points
Patients with diabetes show a blunted cardiac inotropic response to β‐adrenergic stimulation despite normal cardiac contractile reserve.
Acute insulin stimulation impairs β‐adrenergically induced contractile function in isolated cardiomyocytes and Langendorff‐perfused hearts.
In this study, we aimed to examine the potential effects of hyperinsulinaemia associated with high‐fat diet (HFD) feeding on the cardiac β2‐adrenergic receptor signalling and the impacts on cardiac contractile function.
We showed that 8 weeks of HFD feeding leads to reductions in cardiac functional reserve in response to β‐adrenergic stimulation without significant alteration of cardiac structure and function, which is associated with significant changes in β2‐adrenergic receptor phosphorylation at protein kinase A and G‐protein receptor kinase sites in the myocardium.
The results suggest that clinical intervention might be applied to subjects in early diabetes without cardiac symptoms to prevent further cardiac complications.
Abbreviations
- AR
adrenergic receptor
- AUC
area under the curve
- β2AR
β2‐adrenergic receptor
- EF
ejection fraction
- FS
fractional shortening
- Gi
inhibitory guanine nucleotide‐binding protein
- GRK
G‐protein receptor kinase
- HFD
high‐fat diet
- IR
insulin receptor
- IRS
insulin receptor substrates
- ISO
isoproterenol
- IVRT
isovolumetric relaxation time
- KO
knockout
- maximal dP/dt
maximum values of the first derivative of left ventricular pressure
- minimal dP/dt
minimal values of the first derivative of left ventricular pressure
- NC
normal chow
- PLB
phospholamban
- PKA
protein kinase A
- SERCA
sarco/endoplasmic reticulum Ca2+‐ATPase
- TnI
troponin I
- WT
wild‐type
Introduction
Population‐based studies have shown that patients with type 2 diabetes have two to three times the risk of heart disease when compared with the general population (Bell, 2003; From et al. 2006). The increased incidence of heart failure in diabetic patients persists despite correction for age, hypertension, hypercholesterolaemia and coronary artery disease (Boudina & Abel, 2007). The pathogenesis of diabetic cardiomyopathy is multifactorial. Several hypotheses have been proposed, including metabolic derangements, abnormalities in ion homeostasis, alteration in structural proteins and interstitial fibrosis (Boudina & Abel, 2007). One of the hallmarks in type 2 diabetes is insulin resistance and the associated increase in the concentrations of circulating insulin, free fatty acids and glucose, which are risk factors for diabetic cardiomyopathy (Fang et al. 2004). Many studies have documented the effects of fatty acids and glucose in the myocardium, including dysregulation of metabolism and autophagy and an increase in oxidative stress (Ansley & Wang, 2013; Kubli & Gustafsson, 2014). These alterations not only contribute to cardiac contractile dysfunction, but also promote cardiac remodelling and increase the risk of cardiac ischaemic events (Ansley & Wang, 2013; Mei et al. 2015). In contrast, little is known about the impacts of circulating insulin on diabetic hearts.
The prominent defect in diabetic hearts is diastolic dysfunction, with preservation of the ejection fraction (EF; Desai & Fang, 2008). A large number of diabetic patients also show a decrease in exercise tolerance before the onset of overt cardiac dysfunction (Scognamiglio et al. 1995), which has been attributed to autonomic neuropathy (Vinik & Ziegler, 2007). Others have reported that exercise‐intolerant patients also show a blunted inotropic response to dobutamine stimulation, suggesting that the cardiac β‐adrenergic system is affected (Scognamiglio et al. 1995). In agreement, cardiac preparations from streptozotocin‐induced diabetic rats display reduced inotropic and chronotropic responses to β‐adrenergic stimulation (Vadlamudi & McNeill, 1984; Yu & McNeill, 1991). These studies suggest that exercise intolerance and a blunted inotropic response to adrenergic stimulation can be considered as early signs of diabetic cardiomyopathy, preceeding clinical symptoms of cardiac complications. Mechanistically, in streptozotocin‐induced diabetic rats, the expression of cardiac β1‐adrenergic receptors (β1ARs) is significantly decreased, which may account for the depressed contractile response to adrenergic stimuli (Heyliger et al. 1982). Meanwhile, in leptin receptor‐deficient db/db mice, an impaired cardiac functional reserve capacity during maximal β‐adrenergic stimulation is linked with unfavourable changes in cardiac energy metabolism (Daniels et al. 2010). Therefore, the mechanisms involved in the early stages of myocardial adaptations, such as exercise intolerance and a blunted inotropic response to adrenergic stimulation, prior to an overt decline in mechanical performance, are still incompletely understood.
β‐Adrenergic signalling is essential in the regulation of cardiac function in response to stress. Both β1ARs and β2‐adrenergic receptors (β2ARs) are functionally coupled to the adenylate cyclase system, contributing to the cardiac response to β‐adrenergic stimulation (Dincer et al. 2001). In congestive and ischaemic human heart failure, elevated sympathetic drive can promote compensatory cardiac contractility. But chronic sympathetic stress leads to desensitization of βARs via several molecular mechanisms, including a decrease in the expression of β1ARs and in coupling of β1ARs to adenylate cyclase, and an increase in the expression of inhibitory G‐protein (Gi) and G‐protein receptor kinases (GRKs; Liggett, 2001). The cardiac β2AR signalling is traditionally considered beneficial to the myocardium relative to the cardiac toxicity induced by β1ARs (Bernstein et al. 2005; Grundy, 2015). Phosphorylated β2ARs couple to Gi and promote Akt signalling (Wang et al. 2008), which protects myocytes against apoptosis induced by oxidative stress or chronic sympathetic β1AR signalling (Zhu et al. 2001). However, the same β2AR–Gi coupling can lead to inhibition of adenylate cyclase, which compromises cAMP and protein kinase A (PKA) activity and contributes to cardiac dysfunction in pressure‐overload transaortic constriction models (Du et al. 2000; Zhu et al. 2012). Taken together, these previous findings suggest that changes in β‐adrenergic signalling in the heart may play either a detrimental or a protective role depending on the pathological setting.
We have recently characterized a complex consisting of the insulin receptor (IR) and the β2AR in the heart (Fu et al. 2014). Furthermore, acute insulin stimulation promotes cross‐talk with the β2AR signalling pathway in a Gβγ‐independent, insulin receptor substrate‐dependent G‐protein receptor kinase (GRK)2 phosphorylation of β2ARs. Consequently, insulin impairs β‐adrenergically induced contractile function in isolated cardiomyocytes and in Langendorff‐perfused hearts (Fu et al. 2014). In the present study, we aimed to examine the potential effects of hyperinsulinaemia associated with feeding a high‐fat diet (HFD) on the cardiac IR–β2AR signalling network and the impacts on cardiac adaptation during early stages of diabetes. Mice lacking the β2AR (β2AR‐KO) were used to define whether the β2AR is a contributor to hyperinsulinaemia‐induced βAR signalling dysfunction. We analysed cardiac function at the baseline and after β‐adrenergic stimulation by i.p. injection of isoproterenol after 8 weeks of HFD feeding. Cardiac structural remodelling was analysed using immunohistochemical and biochemical analyses. Our results show that 8 weeks of HFD feeding leads to decreases in cardiac functional reserve in response to β‐adrenergic stimulation without significant alteration of cardiac structure and basal function. This decrease in cardiac functional reserve is associated with significant increases in the levels of phosphorylation of the β2AR at PKA and GRK sites in the heart.
Methods
Ethical approval
The animal care and experimental protocols conform to the principles of UK regulations (Grundy, 2015) and were approved by the institutional animal care and use committee of Tongji Medical College, Huazhong University of Science and Technology and the University of California at Davis (approval numbers 17407 and 19147).
Experimental animals and in vivo treatment
C57BL/6 mice were purchased from Charles River and Beijing HFK Bioscience Co. Ltd, China. In brief, 50 wild‐type (WT) and 50 β2AR global knockout (β2AR‐KO) 5‐ to 6‐week‐old male C57BL/6J mice were randomly assigned to low‐fat or high‐fat diet for 8 weeks (n = 25 each group). The diets used for these studies were from Research Diets (New Brunswick, NJ, USA), with 10% kcal fat (catalogue no. D12450J) used as the control normal chow (NC) diet and 60% kcal fat diet (catalogue no. D12492) used as the HFD. The mice had ad libitum access to food and they were housed in a room with a 12 h light–12 h dark cycle.
Intraperitoneal glucose tolerance test
Mice were fasted for 16 h and then challenged with glucose (1 g kg−1, i.p.). Blood samples were drawn from the tail vein immediately before the glucose challenge and at 30, 60, 90 and 120 min thereafter. Blood glucose concentrations were determined using a glucometer (ACCU‐CHEK, Roche, Mannheim, Germany). The area under the curve (AUC) was calculated to evaluate glucose tolerance.
Insulin tolerance test and blood insulin detection
After a 6 h fast, mice were injected with insulin (0.75 U kg−1, i.p.; Sigma, St Louis, MO, USA). The blood glucose value and AUC were assessed before and at 30, 60, 90 and 120 min after injection of insulin. Blood serum was collected before glucose injection to measure the insulin concentration. Insulin was measured using ALPCO mouse ultrasensitive insulin ELISA kits (Salem, NH, USA) according to the manufacturer's protocol.
Echocardiography
Echocardiography was performed using a Vevo 2100 imaging system from Visual Sonics (Toronto, ON, Canada) with a 22–55 MHz MS550D transducer. Mice were anaesthetized with isofluorane supplemented with 100% O2, and the body temperature, respiratory rate and ECG were constantly monitored. To minimize variation of the data, the heart rate was maintained at 400–450 beats min−1 during the measurements of cardiac function. Cardiac function was recorded at the baseline and after administration of the βAR agonist isoproterenol (ISO, 0.2 mg kg−1, i.p.; Sigma). Systolic function parameters, including the EF and fractional shortening (FS), were measured by two‐dimensional parasternal short‐axis imaging plane of M‐mode traces close to the papillary muscle level. Tissue Doppler imaging mode was applied to measure diastolic function as previously described (Qi et al. 2013; Zhang et al. 2013).
Haemodynamic study
Haemodynamic measurement was carried out with a pressure catheter (SPR‐839; Millar Instruments, Houston, TX, USA) connected to an AD Instruments Power‐Lab 4/30 with Lab Chart Pro 7.0 software (MPVS‐300; AD Instruments, Colorado Springs, CO, USA). In brief, mice were anaesthetized by i.p. injection of ketamine and xylazine (80 and 5 mg kg−1, respectively, Sigma, St Louis, MO, USA) and placed in the supine position on a heat pad. A ventral mid‐line neck incision was made, the carotid artery separated from the vagus nerve and a pressure–volume catheter inserted via the carotid artery tip into the left ventricle. After 5–10 min of stabilization, the baseline pressure was recorded. To measure the β‐adrenergic response, 0.2 mg kg−1 ISO was injected i.p. Measurements and analysis were performed with Lab Chart Pro 7.0.
Adult myocyte‐shortening assay
Adult mouse cardiomyocytes were isolated from WT and β2‐KO mice as indicated and cultured as described previously (Soto et al. 2009). Cells were placed in the middle of a glass‐bottomed dish with beating buffer (mm: NaCl, 120; KCl, 5.4; NaH2PO4, 1.2; MgSO4, 1.2; Hepes, 20; glucose, 5.5; and CaCl2, 1; pH 7.1) and allowed to settle for 10 min. Platinum electrodes were placed near the cells, and the cells were paced at 1 Hz with a voltage of 30 V using an SD9 stimulator (Grass Technology, Warwick, RI, USA) as described by Fu et al. (2015). The contractile shortening events of myocytes were recorded on an inverted microscope (Zeiss AX10; Dublin, CA, USA) at ×20 magnification, which allowed observation of eight to 10 rod‐shaped cardiomyocytes with clear striations per field of view using the MetaMorph program (Molecular Devices, Sunnyvale, CA, USA). The maximal shortening was analysed by MetaMorph and normalized to the baseline.
Western blot analysis
Adult cardiomyocytes isolated from NC and HFD mice were stimulated with ISO (100 nmol l−1) after incubation with or without pertussis toxin (1 mg ml−1 for 3 h). Left ventricular extracts or adult cardiomyocyte lysates were homogenized at 4°C for Western blotting using standard methods previously described (Fu et al. 2014). Membranes were probed with antibodies directed at β1AR (V‐19, 1:500 dilution), β2AR (M‐20, 1:500 dilution), GRK2 (C‐15, 1:1000 dilution) and Gαi (C‐10, 1:1000 dilution) from SCBT (Santa Cruz, CA, USA), phospho‐troponin I (Ser23/24, #4004, 1:2000 dilution) and troponin I (#4002, 1:2000 dilution) from Cell Signaling (Danvers, MA, USA), SERCA (2A7‐A1, 1:1000 dilution; Thermo, Rockford, IL, USA), phospho‐phospholamban (Ser16, 1:5000 dilution; Bradilla, London, UK), phospholamban (MA3‐922, 1:1000 dilution; Affinity Bioreagent, Golden, CO, USA) and γ‐tubulin (T6557, 1:5000 dilution; Sigma‐Aldrich, St Louis, MO, USA). The primary antibodies were revealed with fluorescent‐conjugated secondary antibodies using an Odyssey scanner (Li‐COR Biosciences, Lincoln, NE, USA). The signal was quantified by Image Studio software version 2.1 (Li‐COR Biosciences).
Histology
Mouse hearts were perfused with 4% paraformaldehyde and fixed in paraformaldehyde for 24 h. Fixed hearts were paraffin embedded and serially sectioned at 5 μm on a microtome. Tissue sections were stained with Haematoxylin and Eosin to examine heart morphology. Masson's trichrome staining was used to examine myocardial fibrosis. The blue‐stained areas in Masson's trichrome staining and cardiomyocyte cross‐section in Haematoxylin and Eosin staining were measured using the ImageJ program (National Institutes of Health, Bethesda, MD, USA) by a researcher blinded to the samples. The quantification was performed in five regions of each heart.
Quantitative real‐time PCR analysis
Total RNA was isolated from snap‐frozen hearts using RNAiso Plus (TaKaRa Bio Inc., Kusatsu, Shiga, Japan) according to the manufacturer's protocol. First‐strand cDNA was synthesized from RNA using a SMARTer PCR cDNA Synthesis Kit (TaKaRa Bio Inc.). Real‐time quantitative RT‐PCR was performed with a Step One Plus Real‐Time PCR system thermocycler (Thermo Fisher Scientific, Waltham, MA, USA) using SYBR Premix Ex Taq (Tli RNaseH Plus; TaKaRa Bio Inc.). All the primer sequences are listed in Table 1. The relative expression level of specific mRNA was determined by the comparative cycle threshold (CT) method () normalized to endogenous control GAPDH gene.
Table 1.
Quantitative RT/PCR primer sequences
| Gene | Primer | Sequence |
|---|---|---|
| α‐Myosin heavy chain (α‐MHC) | Forward | 5′‐CAACGCCAAGTGTTCCTC‐3′ |
| Reverse | 5′‐AGCTCTGACTGCGACTCCTC‐3′ | |
| β‐Myosin heavy chain (β‐MHC) | Forward | 5′‐ATGTGCCGGACCTTGGAAG‐3′ |
| Reverse | 5′‐CCTCGGGTTAGCTGAGAGATCA‐3′ | |
| Atrial natriuretic peptide (ANP) | Forward | 5′‐TCGTCTTGGCCTTTTGGCT‐3′ |
| Reverse | 5′‐TCCAGGTGGTCTAGCAGGTTCT‐3′ | |
| Brain natriuretic peptide (BNP) | Forward | 5′‐CTCCTGAAGGTGCTGTCC‐3′ |
| Reverse | 5′‐GCCATTTCCTCCGACTTT‐3′ | |
| α‐Smooth muscle actin (αSMA) | Forward | 5′‐GTCCCAGACATCAGGGAGTAA‐3′ |
| Reverse | 5′‐TCGGATACTTCAGCGTCAGGA‐3′ | |
| Connective tissue growth factor (CTGF) | Forward | 5′‐CACAGAGTGGAGCGCCTGTTC‐3′ |
| Reverse | 5′‐GATGCACTTTTTGCCCTTCTTAATG‐3′ | |
| Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) | Forward | 5′‐CATGGCCTTCCGTGTTCCTA‐3′ |
| Reverse | 5′‐CCTGCTTCACCACCTTCTTGAT‐3′ |
Statistical analysis
All statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA, USA). Statistical analyses were performed using Student's two‐tailed unpaired t test, one‐way ANOVA followed by Tukey's post hoc test or two‐way repeated‐measures ANOVA with Bonferroni multiple comparisons test. A value of P < 0.05 was considered to indicate statistical significance. All data are expressed as means ± SEM. The sample size for each group is shown in the figure legends or tables.
Results
We fed C57BL/6J wild‐type mice with 60% HFD for 8 weeks. The HFD‐fed mice exhibited significant body weight gain compared with the mice fed with NC, which was associated with elevated fasting blood glucose and insulin (Fig. 1 A–C). The HFD mice also exhibited significant glucose intolerance and mild insulin intolerance relative to NC control animals (Fig. 1 D and E). The overall features indicated a stage of prediabetes or an early stage of diabetes associated with obesity. At this stage, echocardiographic analysis revealed that cardiac contractile function, including the systolic indices of EF and FS and the diastolic parameter isovolumetric relaxation time (IVRT), was normal in HFD animals relative to NC control animals (Fig. 1 F–H and Table 2). Consistent with the observed cardiac function, HFD hearts were grossly normal in morphology, with mild hypertrophy (Fig. 2 A and B). We also examined the cardiac contractile reserve in response to β‐adrenergic stimulation, which is usually impaired in patients with diabetes and/or metabolic syndrome (Scognamiglio et al. 1998). Despite the grossly normal cardiac structure and function, the cardiac contractile reserve in response to the β‐adrenergic agonist isoproterenol was impaired in HFD animals relative to NC control mice (Fig. 1 F and G and Table 2). Further examination showed that there was no significant change in expression of regulatory proteins in excitation–contraction coupling, including troponin I (TnI), phospholamban (PLB) and sarco/endoplasmic reticulum calcium ATPase 2 (SERCA2a; Fig. 2 C and D). Moreover, the basal levels of phosphorylation of PLB and TnI by PKA, which is essential for excitation–contraction coupling, were not altered in HFD hearts relative to NC control hearts (Fig. 2 C and D). Together, these observations suggest that 8 weeks of HFD feeding induced mild or early diabetes in mice, which was associated with impaired contractile reserve to adrenergic stress despite overall normal cardiac structure and function.
Figure 1. Feeding high‐fat diet (HFD) for 8 weeks induces impairment of cardiac functional reserve in response to adrenergic stimulation.
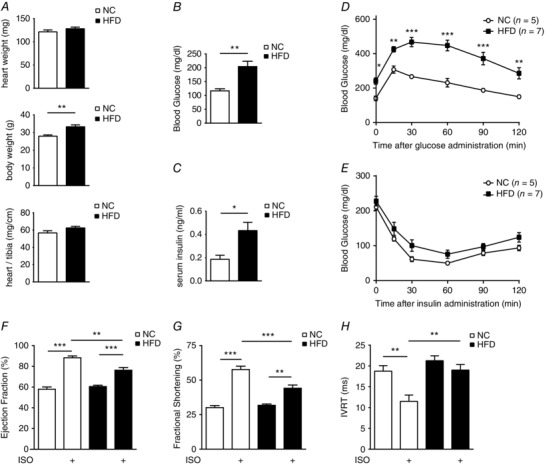
A–C show body weight (A), fasting blood glucose (B) and fasting serum insulin concentrations (C) after 8 weeks of normal chow (NC) or high‐fat diet (HFD) feeding. * P < 0.05 and ** P < 0.01 between HFD and NC groups (n = 10). The HFD induces glucose intolerance (D) and mild insulin intolerance (E) when compared with NC control animals. * P < 0.05 and ** P < 0.01 by two‐way repeated‐measures ANOVA with Bonferroni multiple comparisons test (n = 5–7). F–H, cardiac contractile function of the mice before and after β‐adrenergic stimulation [isoproterenol (ISO), 0.2 mg kg−1, i.p.] was measured by echocardiography after 8 weeks of HFD. Data show ejection fraction (F) and fractional shortening (G) measured by M‐mode, and isovolumetric relaxation time (IVRT; H) measured by tissue Doppler image mode. ** P < 0.01 and *** P < 0.001 (n = 8) by one‐way ANOVA followed by Tukey's post hoc test.
Table 2.
Echocardiographic characteristics of NC and HFD mice treated with β‐adrenergic stimulation (ISO, 0.2 mg kg−1, i.p.)
| Paramter | NC | NC + ISO | HFD | HFD + ISO |
|---|---|---|---|---|
| HR (beats min−1) | 419 ± 14.42 | 603 ± 9.16*** | 411 ± 21.40 | 617 ± 13.10*** |
| IVS;d (mm) | 0.67 ± 0.05 | 0.83 ± 0.09 | 0.69 ± 0.06 | 0.81 ± 0.06 |
| IVS;s (mm) | 0.94 ± 0.10 | 1.39 ± 0.12* | 1.28 ± 0.36 | 1.27 ± 0.12 |
| LVID;d (mm) | 3.67 ± 0.07 | 3.18 ± 0.08 | 3.64 ± 0.19 | 3.23 ± 0.26 |
| LVID;s (mm) | 2.53 ± 0.09 | 1.41 ± 0.10* | 2.32 ± 0.26 | 1.69 ± 0.29 |
| LVPW;d (mm) | 0.73 ± 0.06 | 0.72 ± 0.07 | 0.79 ± 0.03 | 0.82 ± 0.11 |
| LVPW;s (mm) | 1.05 ± 0.06 | 1.17 ± 0.11 | 1.09 ± 0.07 | 1.2 ± 0.15 |
| EF (%) | 57.94 ± 2.22 | 88.25 ± 1.79*** | 60.54 ± 1.27 | 76.25 ± 2.58***## |
| FS (%) | 30.07 ± 1.53 | 57.66 ± 2.45*** | 31.81 ± 0.92 | 44.14 ± 6.15***## |
| LV mass (mg) | 88.04 ± 6.70 | 73.8 ± 7.06 | 94.91 ± 4.59 | 80.76 ± 6.67 |
| LV mass (corrected) | 68.84 ± 5.36 | 59.04 ± 5.65 | 75.93 ± 3.67 | 64.61 ± 5.33 |
| LV Vol;d (μI) | 58.43 ± 2.12 | 38.4 ± 3.82*** | 62.2 ± 3.46 | 40.91 ± 3.64*** |
| LV Vol;s (μI) | 24.66 ± 1.80 | 4.38 ± 0.75*** | 24.44 ± 1.22 | 9.9 ± 1.73*** |
Results are expressed as means ± SEM (n = 8 per group). * P < 0.05, ** P < 0.01 and *** P < 0.001 compared with the control group. ## P < 0.01 compared with the NC + ISO group. Abbreviations: EF, ejection fraction; FS, fractional shortening; HFD, high‐fat diet; HR, heart rate; ISO, isoproterenol; IVS;s and IVS;d, interventricular septal wall thickness at systole and diastole; LVID;s and LVID;d, left ventricular dimension at systole and diastole; LV mass, left ventricular mass; LV mass (corrected), LV mass corrected for body surface area LVPW;s and LVPW;d, posterior wall thickness at systole and diastole; LV Vol;s and LV Vol;d, left ventricle volume at systole and diastole; and NC, normal chow.
Figure 2. Mice after 8 weeks of HFD feeding display normal cardiac gene expression and morphology.
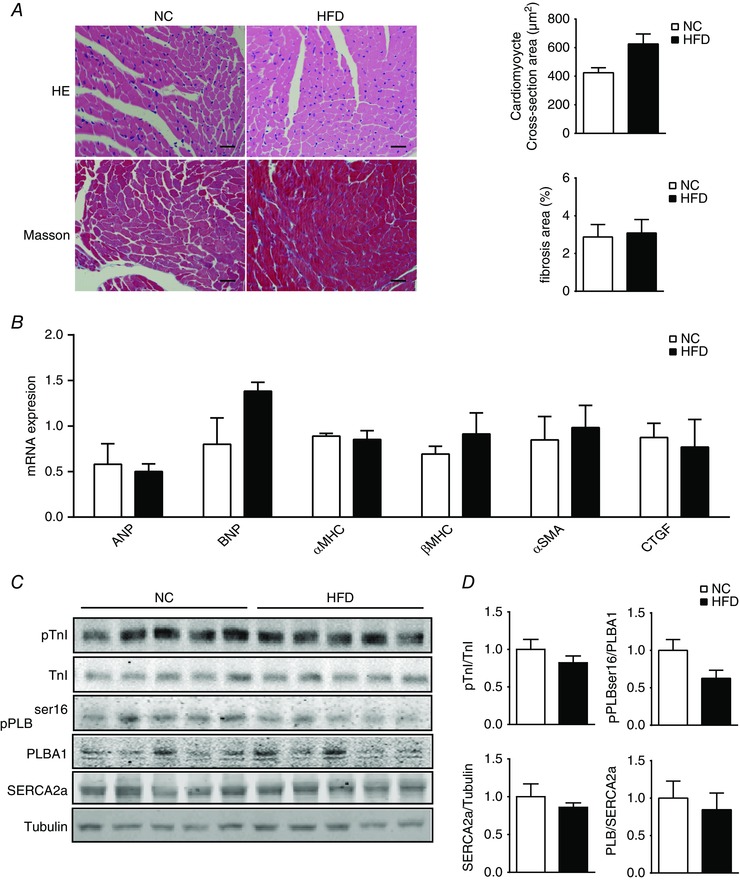
A, heart sections from NC and HFD mice were stained with Haematoxylin and Eosin (HE; scale bar = 100 μm) to examine heart morphology and stained with Masson's trichrome (scale bar = 100 μm) to examine fibrosis. The cardiomyocyte cross‐sectional areas and fibrosis‐positive areas were quantified and plotted (n = 3). B, RT‐PCR showing left ventricular expression of genes involved in cardiac hypertrophy and fibrosis in mice after 8 weeks of HFD. C and D, Western blots showing protein kinase A (PKA) phosphorylation of phospholamban (PLB) and troponin I (TnI), and SERCA2a expression in NC and HFD mouse hearts. [Color figure can be viewed at wileyonlinelibrary.com]
We then set out to examine potential alterations in cardiac adrenergic signalling pathways in the HFD mice. The HFD hearts displayed normal expression of β1AR, β2AR and G‐protein. G‐Protein receptor kinase 2, a kinase that regulates cardiac adrenergic signalling and is usually upregulated in heart failure, was not altered either (Fig. 3 A and B). However, the β2ARs in HFD hearts displayed increased levels of phosphorylation at both the PKA site serine 261/262 and the GRK site serine 355/356 relative to NC control hearts (Fig. 3 C and D). To assess adrenergic signalling directly in cardiomyocytes, we isolated adult myocytes from NC and HFD mice to perform a contractile shortening assay. Myocytes from both NC and HFD mice exhibited similar contractile shortening at baseline (Fig. 3 E). However, myocytes from HFD mice exhibited a reduced contractile response to isoproterenol stimulation relative to NC control myocytes (Fig. 3 E). Consistently, myocytes from HFD mice exhibited a reduced PKA phosphorylation of PLB at serine 16 in response to isoproterenol stimulation relative to NC control myocytes (Fig. 3 F). We have recently shown that acute insulin stimulation promotes β2AR coupling to Gi in cardiomyocytes (Fu et al. 2014). Inhibition of Gi with pertussis toxin increased the isoproterenol‐induced PKA phosphorylation of PLB in cardiomyocytes from HFD mice (Fig. 3 G), indicating that isoproterenol triggered βAR activation of Gi in HFD cardiomyocytes. As a control, pertussis toxin did not affect isoproterenol‐induced phosphorylation of PLB in NC cardiomyocytes. Together, these data suggested that HFD feeding promoted desensitization of cardiac βAR via phosphorylation at both PKA and GRK sites, which resulted in a reduced contractile shortening response to adrenergic stress in the cardiomyocytes. Consequently, the reduced myocyte shortening is likely to contribute to the impaired cardiac contractile reserve in response to β‐adrenergic stimulation in vivo (Fig. 1).
Figure 3. Feeding the HFD for 8 weeks induces an impaired contractility response to adrenergic stimulation in cardiomyocytes.
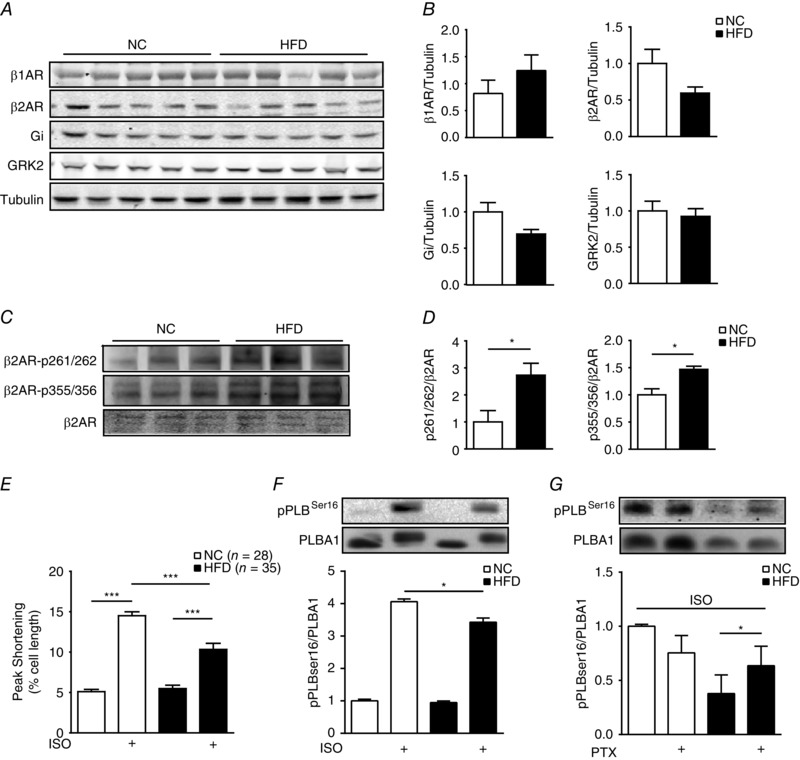
A and B, Western blots show the expression of cardiac β1‐adrenergic receptor (β1AR), β2‐adrenergic receptor (β2AR), G‐protein and G‐protein receptor kinase (GRK)2 in NC and HFD hearts. C and D, Western blots show the phosphorylation of β2AR at both PKA and GRK sites in NC and HFD hearts. * P < 0.05 by Student's t test between paired groups. E, data show contractile function in response to β‐adrenergic stimulation (ISO, 100 nm) in adult cardiac myocytes isolated from NC and HFD mice. *** P < 0.001 by one‐way ANOVA followed by post hoc Tukey's test. F and G, data show PKA phosphorylation of PLB in response to β adrenergic stimulation (ISO, 100 nm) in the absence or presence of pertussis toxin (PTX, 1 μg ml−1, 3 h) in adult cardiomyocytes isolated from NC and HFD mice (n = 3). * P < 0.05 by Student's t test between paired groups.
We then used mice lacking the β2AR gene to assess the role of the β2AR in HFD‐induced impairment of contractile reserve in hearts. After HFD feeding, β2AR‐KO mice exhibited significant body weight gain compared with the β2AR‐KO NC control mice, which was associated with elevated resting blood glucose and insulin, similar to those in WT mice (Fig. 4 A–C). The HFD‐fed β2AR‐KO mice also exhibited significant glucose intolerance and mild insulin intolerance relative to NC control animals (Fig. 4 D and E). Overall, echocardiographic analysis revealed that cardiac function was normal in HFD β2AR‐KO mice, including the systolic indices of EF and FS and the diastolic parameter IVRT (Fig. 4 F–H). In contrast to WT mice, mice lacking the β2AR exhibited normal cardiac contractile reserve in response to β‐adrenergic stimulation after HFD feeding (Fig. 4 F and G). There was no significant change in expression of TnI, PLB and SERCA2a pump in HFD β2AR‐KO hearts compared with NC β2AR‐KO mice (Fig. 5 A and B). The phosphorylation of TnI and PLB was also preserved in HFD‐fed β2AR‐KO hearts (Fig. 5 A and B). Moreover, HFD β2AR‐KO hearts exhibited normal expression of β1AR, G‐protein and GRK2 compared with NC β2AR‐KO mice (Fig. 5 C and D). Consistently, myocytes isolated from HFD β2AR‐KO mice exhibited normal contractile shortening in the resting state and after stimulation with isoproterenol (Fig. 5 E). Together, these data indicated that deletion of β2AR gene ameliorated HFD‐induced impairment of contractile reserve in hearts.
Figure 4. Deletion of the β2AR gene normalizes cardiac contractile response to adrenergic stimulation in HFD mice.
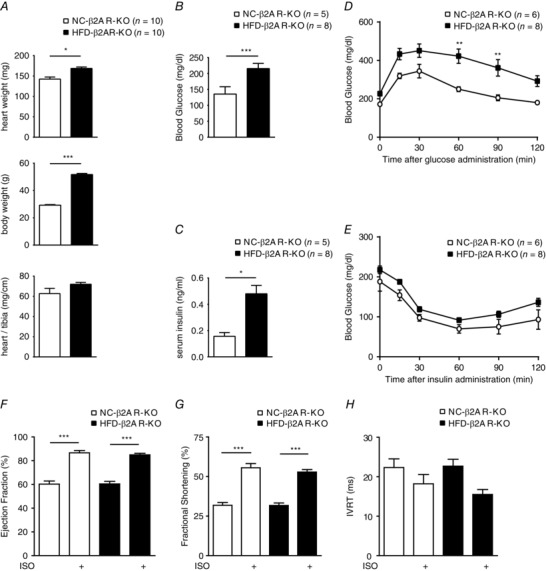
A–C show body weight (A), fasting glucose (B) and insulin concentrations (C) in mice lacking the β2AR (β2AR‐KO) after 8 weeks of NC or HFD feeding. * P < 0.05 and *** P < 0.001 for HFD vs NC. Eight weeks of HFD feeding induces glucose intolerance (D) and mild insulin intolerance (E) when compared with NC control animals. *P < 0.05 and **P < 0.01 between groups by two‐way repeated‐measures ANOVA with Bonferroni multiple comparisons test. F–H, after 8 weeks of HFD feeding, cardiac contractile function in response to β‐adrenergic stimulation (ISO, 0.2 mg kg−1, i.p.) was examined by echocardiography in the β2AR‐KO mice. Data show ejection fraction (F) and fractional shortening (G) measured by M‐mode and isovolumetric relaxation time (H) measured by tissue Doppler image mode (n = 8). *** P < 0.001 by one‐way ANOVA followed by Tukey's post hoc test.
Figure 5. Deletion of the β2AR gene normalizes myocyte contractile response to adrenergic stimulation after HFD feeding.
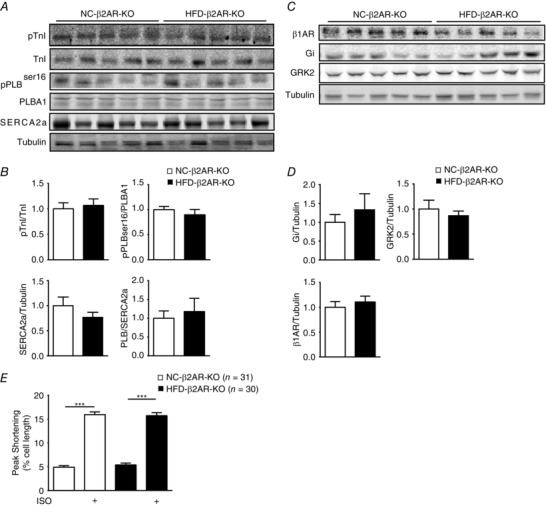
Western blots show PKA phosphorylation of PLB and TnI, and SERCA2a expression (A and B), as well as expression of cardiac adrenergic signalling regulatory proteins in the hearts of β2AR‐KO mice fed NC and HFD (C and D). E, myocyte contractile shortening in response to β‐adrenergic stimulation (ISO, 100 nm) was examined in NC and HFD β2AR‐KO cardiomyocytes. *** P < 0.001 by one‐way ANOVA followed by Tukey's post hoc test.
We also assessed cardiac function directly with haemodynamic studies in vivo. In WT mice, 2 months of HFD feeding did not affect contractile properties at the baseline, including maximal dP/dt (maximum values of the first derivative of left ventricular pressure 7789 ± 699.9 vs. 7078 ± 748.5 mmHg s−1), minimal dP/dt (minimum values of the first derivative of left ventricular pressure −5962 ± 640.3 vs. −5502 ± 461.5 mmHg s−1) and cardiac output (6380 ± 758.3 vs. 5078 ± 1164 μl min−1), but significantly impaired cardiac contractility in response to administration of isoproterenol, including maximal dP/dt (16,973 ± 506.9 vs. 14,472 ± 432.8 mmHg s−1), minimal dP/dt (−10,466 ± 673.6 vs. −8710 ± 673.6 mmHg s−1) and cardiac output (16,648 ± 668.2 vs. 12,203 ± 1548 μl min−1; Fig. 6 A–D). In comparison, deletion of β2AR normalized cardiac contractility in HFD mice, including maximal dP/dt (8196 ± 724.3 vs. 10,504 ± 1212 mmHg s−1), minimal dP/dt (−6910 ± 613.4 vs. −8808 ± 978.5 mmHg s−1) and cardiac output (7274 ± 1502 vs. 9733 ± 1721 μl min−1), and rescued cardiac contractile reserve in response to administration of isoproterenol, including maximal dP/dt (20,046 ± 746.7 vs. 20,576 ± 792.8 mmHg s−1), minimal dP/dt (−11,339 ± 769.9 vs −12,053 ± 1170 mmHg s−1) and cardiac output (19,388 ± 2871 vs. 16,719 ± 2161 μl min−1; Fig. 6 A–D).
Figure 6. Deletion of the β2AR gene normalizes the cardiac functional reserve response to adrenergic stimulation after HFD feeding.
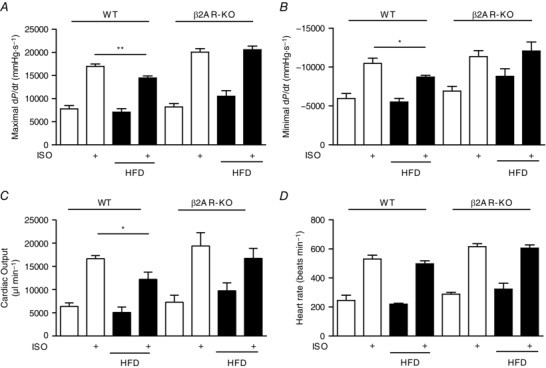
After HFD feeding, cardiac haemodynamics were measured in wild‐type (WT) and β2AR‐KO mice before and after i.p. injection of ISO (0.2 mg kg−1). The maximal dP/dt (A), minimal −dP/dt (B), cardiac output (C) and heart rate (D) were recorded and plotted in bar graphs (n = 6). * P < 0.05 and ** P < 0.01, by Student's unpaired t test between groups.
Discussion
β‐Adrenergic receptors are the key regulators of cardiac function in response to stress. In heart diseases, βARs are often desensitized via phosphorylation as a result of elevated sympathetic drive (Brum et al. 2006). Here, we show that 2 months of HFD feeding promotes phosphorylation of cardiac β2ARs at both PKA and GRK sites, attenuation of myocyte contractile shortening in vitro and impairment of cardiac contractile reserve in vivo in response to adrenergic stimulation. The HFD‐induced attenuation of the adrenergic response occurs without significant alteration of the cardiac structure and basal function. These data provide direct evidence that 2 months of HFD feeding promotes phosphorylation of β2ARs, which is correlated with the compromised cardiac stress response that is present in HFD‐fed mice before structural remodelling occurs in hearts.
In a classic view, during augmented circulatory demands in clinical conditions such as diabetes mellitus, the increased sympathetic activity and subsequent βAR stimulation is an important physiological mechanism to enhance cardiac performance. However, the chronic increase of cardiac adrenergic drive could in turn promote β1AR desensitization, internalization and/or downregulation (Brodde et al. 2006), contributing to the development and progression of diabetic cardiomyopathy (Aneja et al. 2008; Tillquist & Maddox, 2012) and heart failure (Lymperopoulos et al. 2013; Florea & Cohn, 2014). In comparison, β2ARs are known to couple functionally to both stimulatory G protein (Gs) and Gi to modulate cAMP production (Grundy, 2015), and the phosphorylation of β2AR promotes a coupling switch from Gs to Gi (Daaka et al. 1997), which induces Akt signalling to protect myocytes against apoptosis (Zhu et al. 2001). However, the same β2AR–Gi coupling can lead to inhibition of cAMP production by adenylate cyclase and subsequent PKA activity, which has been shown to contribute to cardiac dysfunction in pressure‐overload transaortic constriction models (Du et al. 2000; Zhu et al. 2012). In the present study, HFD feeding promoted phosphorylation of cardiac β2ARs at PKA and GRK sites, decreased PKA phosphorylation of PLB and attenuated myocyte shortening and cardiac contractile reserve in response to β‐adrenergic stimulation. Incubation with the Gi inhibitor pertussis toxin increased isoproterenol‐induced phosphorylation of PLB in isolated HFD cardiomyocytes, confirming that isoproterenol triggered receptor activation of Gi to shunt the β2AR signal away from PKA activation. Deletion of the β2AR restored the contractile response in cardiomyocytes isolated from HFD mice and normalized contractile reserve in response to adrenergic stress. Together, these data indicate that HFD feeding promoted both phosphorylation of β2ARs and the receptor coupling to inhibitory G‐protein to diminish contractile reserve in the mice.
β‐Adrenergic receptors are known to undergo heterologous desensitization mediated by secondary messenger‐dependent kinases, including PKA and protein kinase C (Chuang et al. 1996; Gainetdinov et al. 2004). Recent studies show that a growing list of neurohormonal factors and cytokines can promote desensitization of cardiac βARs and contribute to cardiac dysfunction via divergent cross‐talk mechanisms in vivo (Vasudevan et al. 2013; Tilley et al. 2014). For example, tumour necrosis factor α promotes GRK‐dependent phosphorylation of β2ARs, which attenuates adrenergic signalling and exacerbates cardiac dysfunction in a pressure‐overload model with transaortic constriction (Vasudevan et al. 2013). Block of tumour necrosis factor α signalling ameliorates cardiac dysfunction in mice. Interestingly, tumour necrosis factor α induces GRK2‐mediated desensitization of βARs in a Gβγ‐independent fashion. Coincidentally, insulin also promotes a Gβγ‐independent GRK2 phosphorylation of β2AR in the heart, which is dependent on IR and insulin receptor substrates (Daaka et al. 1997; Fu et al. 2014). These observations suggest that GRK2 functions as a key factor to promote desensitization of cardiac βARs independent of sympathetic drive. As a result, tumour necrosis factor α and insulin are linked to cardiac adrenergic regulation by promoting βAR phosphorylation and/or desensitization, which can either directly induce cardiac dysfunction or exacerbate cardiac pathophysiology in heart failure. Notably, recent clinical trials of diabetic medications (e.g. glyburide) in heart failure patients show adverse effects (Basu et al. 2007; Pantalone et al. 2012), suggesting that aggressive metabolic controls with insulin secretagogues might exacerbate cardiac symptoms. This may be attributable to these medications driving IR‐mediated phosphorylation of β2AR. On the other hand, a hyperinsulinaemia‐driven phosphorylation of β2ARs might offer an explanation for the lack of benefit from β‐blocker therapy in heart failure patients with diabetes mellitus (Haas et al. 2003). Together, these studies suggest the therapeutic potential of targeting hyperinsulinaemia and the Gβγ‐independent GRK2 signalling pathway in heart failure associated with diabetes.
In summary, we show that cardiac β‐adrenergic signalling is impaired in the early stages of diabetes after 8 weeks of HFD feeding, which occurs before structural change in the heart. This study also suggests that clinical intervention might be applied to subjects with early diabetes without cardiac symptoms to prevent further cardiac complications.
Additional information
Competing interests
None declared.
Author contributions
Q.F, Y.K.X. and N.C. conceived and designed the experiments. Q.F., Y.T.H., Q.W., Y.L., B.X., N.L. and S.K. collected, assembled, analysed and interpreted the data. Q.F. and Y.K.X. wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by China National Natural Science Foundation of China grants 81102438 and 81473212 (Q.F.) and 81428022 (Y.K.X.) and by National Insitute of Health grants HL127764 and HL112413 (Y.K.X.) and S10 OD10389 (Y.K.X.). Y.K.X. is an established investigator of the American Heart Association and a Shanghai Eastern Scholar.
Acknowledgements
We thank Toni West for English proofreading.
Q. Fu and Y. K. Xiang contributed equally to this study.
This is an Editor's Choice article from the 15 March 2017 issue.
Contributor Information
Qin Fu, Email: fuqin@mails.tjmu.edu.cn.
Yang K. Xiang, Email: ykxiang@ucdavis.edu
References
- Aneja A, Tang WH, Bansilal S, Garcia MJ & Farkouh ME (2008). Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med 121, 748–757. [DOI] [PubMed] [Google Scholar]
- Ansley DM & Wang B (2013). Oxidative stress and myocardial injury in the diabetic heart. J Pathol 229, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Charkoudian N, Schrage W, Rizza RA, Basu R & Joyner MJ (2007). Beneficial effects of GLP‐1 on endothelial function in humans: dampening by glyburide but not by glimepiride. Am J Physiol Endocrinol Metab 293, E1289–E1295. [DOI] [PubMed] [Google Scholar]
- Bell DS (2003). Heart failure: the frequent, forgotten, and often fatal complication of diabetes. Diabetes Care 26, 2433–2441. [DOI] [PubMed] [Google Scholar]
- Bernstein D, Fajardo G, Zhao M, Urashima T, Powers J, Berry G & Kobilka BK (2005). Differential cardioprotective/cardiotoxic effects mediated by β‐adrenergic receptor subtypes. Am J Physiol Heart Circ Physiol 289, H2441–H2449. [DOI] [PubMed] [Google Scholar]
- Boudina S & Abel ED (2007). Diabetic cardiomyopathy revisited. Circulation 115, 3213–3223. [DOI] [PubMed] [Google Scholar]
- Brodde OE, Bruck H & Leineweber K (2006). Cardiac adrenoceptors: physiological and pathophysiological relevance. J Pharmacol Sci 100, 323–337. [DOI] [PubMed] [Google Scholar]
- Brum PC, Rolim NP, Bacurau AV & Medeiros A (2006). Neurohumoral activation in heart failure: the role of adrenergic receptors. Anais da Academia Brasileira de Ciencias 78, 485–503. [DOI] [PubMed] [Google Scholar]
- Chuang TT, Iacovelli L, Sallese M & De Blasi A (1996). G protein‐coupled receptors: heterologous regulation of homologous desensitization and its implications. Trends Pharmacol Sci 17, 416–421. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM & Lefkowitz RJ (1997). Switching of the coupling of the β2‐adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91. [DOI] [PubMed] [Google Scholar]
- Daniels AM, van Bilsen BJ, Janssen AE, Brouns JP, Cleutjens TH, Roemen G, Schaart J, van der Velden GJ, van der Vusse FA & van Nieuwenhoven FA (2010). Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiol (Oxf) 200, 11–22. [DOI] [PubMed] [Google Scholar]
- Desai A & Fang JC (2008). Heart failure with preserved ejection fraction: hypertension, diabetes, obesity/sleep apnea, and hypertrophic and infiltrative cardiomyopathy. Heart Failure Clinics 4, 87–97. [DOI] [PubMed] [Google Scholar]
- Dincer UD, Bidasee KR, Guner S, Tay A, Ozcelikay AT & Altan VM (2001). The effect of diabetes on expression of β1‐, β2‐, and β3‐adrenoreceptors in rat hearts. Diabetes 50, 455–461. [DOI] [PubMed] [Google Scholar]
- Du XJ, Autelitano DJ, Dilley RJ, Wang B, Dart AM & Woodcock EA (2000). β2‐Adrenergic receptor overexpression exacerbates development of heart failure after aortic stenosis. Circulation 101, 71–77. [DOI] [PubMed] [Google Scholar]
- Fang ZY, Prins JB & Marwick TH (2004). Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev 25, 543–567. [DOI] [PubMed] [Google Scholar]
- Florea VG & Cohn JN (2014). The autonomic nervous system and heart failure. Circ Res 114, 1815–1826. [DOI] [PubMed] [Google Scholar]
- From AM, Leibson CL, Bursi F, Redfield MM, Weston SA, Jacobsen SJ, Rodeheffer RJ & Roger VL (2006). Diabetes in heart failure: prevalence and impact on outcome in the population. Am J Med 119, 591–599. [DOI] [PubMed] [Google Scholar]
- Fu Q, Xu B, Liu Y, Parikh D, Li J, Li Y, Zhang Y, Riehle C, Zhu Y, Rawlings T, Shi Q, Clark RB, Chen X, Abel ED & Xiang YK (2014). Insulin inhibits cardiac contractility by inducing a Gi‐biased β2‐adrenergic signaling in hearts. Diabetes 63, 2676–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Xu B, Parikh D, Cervantes D & Xiang YK (2015). Insulin induces IRS2‐dependent and GRK2‐mediated β2AR internalization to attenuate βAR signaling in cardiomyocytes. Cell Signal 27, 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ & Caron MG (2004). Desensitization of G protein‐coupled receptors and neuronal functions. Annu Rev Neurosci 27, 107–144. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas SJ, Vos T, Gilbert RE & Krum H (2003). Are β‐blockers as efficacious in patients with diabetes mellitus as in patients without diabetes mellitus who have chronic heart failure? A meta‐analysis of large‐scale clinical trials. Am Heart J 146, 848–853. [DOI] [PubMed] [Google Scholar]
- Heyliger CE, Pierce GN, Singal PK, Beamish RE & Dhalla NS (1982). Cardiac alpha‐ and beta‐adrenergic receptor alterations in diabetic cardiomyopathy. Basic Res Cardiol 77, 610–618. [DOI] [PubMed] [Google Scholar]
- Kubli DA & Gustafsson AB (2014). Cardiomyocyte health: adapting to metabolic changes through autophagy. Trends Endocrinol Metab 25, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett SB (2001). β‐Adrenergic receptors in the failing heart: the good, the bad, and the unknown. J Clin Invest 107, 947–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G & Koch WJ (2013). Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 113, 739–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Thompson MD, Cohen RA & Tong X (2015). Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta 1852, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantalone KM, Kattan MW, Yu C, Wells BJ, Arrigain S, Jain A, Atreja A & Zimmerman RS (2012). Increase in overall mortality risk in patients with type 2 diabetes receiving glipizide, glyburide or glimepiride monotherapy versus metformin: a retrospective analysis. Diabetes Obes Metab 14, 803–809. [DOI] [PubMed] [Google Scholar]
- Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, Dostal DE, White MF, Baker KM & Guo S (2013). Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes 62, 3887–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scognamiglio R, Avogaro A, Casara D, Crepaldi C, Marin M, Palisi M, Mingardi R, Erle G, Fasoli G & Dalla Volta S (1998). Myocardial dysfunction and adrenergic cardiac innervation in patients with insulin‐dependent diabetes mellitus. J Am Coll Cardiol 31, 404–412. [DOI] [PubMed] [Google Scholar]
- Scognamiglio R, Fasoli G, Ferri M, Nistri S, Miorelli M, Egloff C, Buja G, Fedele D & Dalla‐Volta S (1995). Myocardial dysfunction and abnormal left ventricular exercise response in autonomic diabetic patients. Clin Cardiol 18, 276–282. [DOI] [PubMed] [Google Scholar]
- Soto D, De Arcangelis V, Zhang J & Xiang Y (2009). Dynamic protein kinase A activities induced by β‐adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res 104, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley DG, Zhu W, Myers VD, Barr LA, Gao E, Li X, Song J, Carter RL, Makarewich CA, Yu D, Troupes CD, Grisanti LA, Coleman RC, Koch WJ, Houser SR, Cheung JY & Feldman AM (2014). β‐Adrenergic receptor‐mediated cardiac contractility is inhibited via vasopressin type 1A‐receptor‐dependent signaling. Circulation 130, 1800–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillquist MN & Maddox TM (2012). Update on diabetic cardiomyopathy: inches forward, miles to go. Curr Diab Rep 12, 305–313. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RV & McNeill JH (1984). Effect of experimental diabetes on isolated rat heart responsiveness to isoproterenol. Can J Physiol Pharmacol 62, 124–131. [DOI] [PubMed] [Google Scholar]
- Vasudevan NT, Mohan ML, Gupta MK, Martelli EE, Hussain AK, Qin Y, Chandrasekharan UM, Young D, Feldman AM, Sen S, Dorn GW, Dicorleto PE 2nd & Naga Prasad SV (2013). Gbetagamma‐independent recruitment of G‐protein coupled receptor kinase 2 drives tumor necrosis factor α‐induced cardiac β‐adrenergic receptor dysfunction. Circulation 128, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinik AI & Ziegler D (2007). Diabetic cardiovascular autonomic neuropathy. Circulation 115, 387–397. [DOI] [PubMed] [Google Scholar]
- Wang Y, De Arcangelis V, Gao X, Ramani B, Jung YS & Xiang Y (2008). Norepinephrine‐ and epinephrine‐induced distinct β2‐adrenoceptor signaling is dictated by GRK2 phosphorylation in cardiomyocytes. J Biol Chem 283, 1799–1807. [DOI] [PubMed] [Google Scholar]
- Yu Z & McNeill JH (1991). Altered inotropic responses in diabetic cardiomyopathy and hypertensive‐diabetic cardiomyopathy. J Pharmacol Exp Ther 257, 64–71. [PubMed] [Google Scholar]
- Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY & Chen X (2013). Cardiotoxic and cardioprotective features of chronic β‐adrenergic signaling. Circ Res 112, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Petrashevskaya N, Ren S, Zhao A, Chakir K, Gao E, Chuprun JK, Wang Y, Talan M, Dorn GW, Lakatta EG 2nd, Koch WJ, Feldman AM & Xiao RP (2012). Gi‐biased β2AR signaling links GRK2 upregulation to heart failure. Circ Res 110, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK & Xiao RP (2001). Dual modulation of cell survival and cell death by β2‐adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci USA 98, 1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]