

Abstract
Astrocytes comprise half of the cells in the brain. Although astrocytes have traditionally been described as playing a supportive role for neurons, they have recently been recognized as active participants in the development and plasticity of dendritic spines and synapses. Astrocytes can eliminate dendritic spines, induce synapse formation, and regulate neurotransmission and plasticity. Dendritic spine and synapse impairments are features of many neurological disorders, including autism spectrum disorder, schizophrenia, and Alzheimer's disease. In this review we will present evidence from multiple neurological disorders demonstrating that changes in astrocyte–synapse interaction contribute to the pathologies. Genomic analysis has connected altered astrocytic gene expression with synaptic deficits in a number of neurological disorders. Alterations in astrocyte‐secreted factors have been implicated in the neuronal morphology and synaptic changes seen in neurodevelopmental disorders, while alteration in astrocytic glutamate uptake is a core feature of multiple neurodegenerative disorders. This evidence clearly demonstrates that maintaining astrocyte–synapse interaction is crucial for normal central nervous system functioning. Obtaining a better understanding of the role of astrocytes at synapses in health and disease will provide a new avenue for future therapeutic targeting.
Keywords: astrocyte, disease, synapse
Abbreviations
- Aβ
amyloid‐β
- AD
Alzheimer's disease
- AMPAR
AMPA receptor
- APP
amyloid precursor protein
- CS
Costello's syndrome
- DISC‐1
disrupted in schizophrenia‐1
- DS
Down's syndrome
- FMRP
fragile X mental retardation protein
- FXS
fragile X syndrome
- GLAST
glutamate aspartate transporter
- GLT‐1
glutamate transporter 1
- Gpc
glypican
- HD
Huntington's disease
- HRAS
Harvey rat sarcoma viral oncogene homolog
- IGF‐1
insulin‐like growth factor 1
- iPSC
Induced pluripotent stem cells
- KO
knock out
- LTD
long‐term depression
- LTP
long‐term potentiation
- MAPK
mitogen‐activated protein kinase
- MeCP2
methyl CpG binding protein 2
- mGluR
metabotropic glutamate receptor
- NAc
nucleus accumbens
- NF1
neurofibromatosis 1
- NMDAR
NMDA receptor
- PAP
perisynaptic astrocyte process
- PD
Parkinson's disease
- PFC
prefrontal cortex
- RTT
Rett's syndrome
- SPARC
secreted protein acidic and rich in cysteine
- SNr
substantia nigra pars reticulate
- TNF‐α
tumour necrosis factor‐α
- TSP
thrombospondin
Introduction
Astrocytes have recently been recognized as a crucial player at the synapse in the central nervous system (CNS). As part of the tripartite synapse, astrocytes can detect and respond to neuronal activity, influencing the formation and plasticity of the synapse during development and into adulthood (Araque & Perea, 2004; Yang et al. 2009). Multiple central nervous system disorders have been connected to alterations in dendritic spines and synaptic impairment (Penzes et al. 2011). In this review we first provide a background on synaptogenesis and the role of astrocytes at synapses in the healthy brain, followed by an in‐depth discussion of how astrocyte dysfunction may contribute to the phenotype of a variety of disorders, from neurodevelopmental to neurodegenerative. In particular, we highlight how changes in astrocytic protein factor secretion may significantly impact the development and stability of the synapse. A better understanding of these changes in astrocytes, and how their secreted factors impact synaptic function, will likely prove crucial to developing new and effective therapies for these disorders.
Synapses and dendritic spines
Synapses are specialized structures between neurons that enable neuronal communication. Neurotransmitters are released from the presynaptic axon into the synaptic cleft, and bind and activate receptors on the postsynaptic dendrite to pass on the signal (Hering & Sheng, 2001). Synapse formation and subsequent modification of synapse structure and strength are controlled by diverse factors, including factors secreted by neighbouring astrocytes (discussed below). Many excitatory glutamatergic synapses form onto postsynaptic spines, which are actin‐rich structures that protrude from the dendritic shaft and contain the post‐synaptic machinery (Kasai et al. 2010; Verpelli et al. 2012). Spine size and morphology regulate synaptic strength and determine the efficiency of synaptic transmission. For example, the larger mushroom‐shaped spines are relatively stable and contain functional synapses, while thin filopodia‐type spines are immature and unstable (Yoshihara et al. 2009). Spines are rapidly formed and eliminated in the developing brain (Hotulainen & Hoogenraad, 2010; Shirao & González‐Billault, 2013), and are more stable in the adult brain (Alvarez & Sabatini, 2007). It has been proposed that loss of synapse and spine stability at later life stages may contribute to neurodegenerative disorders where memory, learning and cognition are compromised (Kasai et al. 2010; Koleske, 2013).
In the scope of this review we will focus on glutamatergic synapses, as glutamate is the major excitatory neurotransmitter in the mammalian CNS. The two major classes of ionotropic glutamate receptors on the postsynaptic neuron are AMPA receptors (AMPARs) and NMDA receptors (NMDARs). AMPARs are activated by glutamate binding (Shepherd & Huganir, 2007). NMDARs require binding of both glutamate and a co‐agonist (d‐serine or glycine) before the channel will open to Na+, K+ and Ca2+ ions. These receptors play critical roles in the development and plasticity of most excitatory synapses (Dongen, 2009), highlighted by the fact that a number of neurological disorders associated with synaptic dysfunction have alterations in NMDAR and AMPAR expression, trafficking, and signalling (discussed below). A change in synaptic strength in response to experience is the cellular phenomenon underlying learning and memory in the brain. Long‐term potentiation (LTP) is the long‐lasting strengthening of a synaptic connection, while long‐term depression (LTD) weakens synapses (for more information on LTP and LTD see Malenka & Bear, 2004). Activation of neurons through NMDAR receptors plays an important role in most forms of LTP, as NMDAR opening allows intracellular Ca2+ concentrations to rise, which activates downstream signalling cascades and increases AMPAR levels at synapses (Lüscher & Malenka, 2012). Several pathological conditions are also linked to defects in metabotropic glutamate receptors (mGluRs), which are G‐protein‐coupled receptors expressed by neurons and glial cells. Astrocytes express mGluR5 (Ca2+ pathway) and mGluR3 (adenylate cyclase pathway) (Mukherjee & Manahan‐Vaughan, 2013).
Astrocytes and synapses
Astrocytes represent a major glial cell type in the brain. They are highly process‐bearing cells, and astrocyte processes (known as perisynaptic astrocyte processes; PAPs) contact many synapses, putting them in a position to interact with neurons. The combination of an astrocyte process with the pre‐ and post‐synaptic compartments is known as the tripartite synapse (Araque et al. 1999). PAPs are motile, and their motility and synaptic coverage regulates the structure of dendritic spines, the efficiency of synaptic transmission, synaptic plasticity and synaptic stability (Bernardinelli et al. 2014). PAPs can promote synaptic pruning through interaction with the synapse in both development and the adult brain (Perez‐Alvarez et al. 2014). Any event that compromises PAPs may impair synaptic transmission and lead to loss of spines, a hallmark of numerous neurological disorders.
In the developing brain astrocytes secrete a number of factors that regulate neuronal synapse formation and function including thrombospondin, hevin, glypicans, SPARC (secreted protein acidic and rich in cysteine) and tumour necrosis factor‐α (TNF‐α) (Allen, 2014). Thrombospondin and hevin induce the formation of structurally mature synapses, but they lack functionality (Christopherson et al. 2005; Kucukdereli et al. 2011). The heparan sulphate proteoglycan glypican 4 (Gpc4) recruits AMPARs to the surface of dendrites, inducing synapse formation and synaptic activity (Allen et al. 2012). Not all astrocyte‐secreted factors have positive synaptogenic functions; for example, SPARC blocks synapse formation and decreases AMPAR levels at synapses (Jones et al. 2011; Kucukdereli et al. 2011).
Unlike neurons, astrocytes are not electrically excitable, and one major type of astrocyte signalling is via alterations in intracellular Ca2+ levels (Perea et al. 2009). Astrocytes release gliotransmitters (such as ATP, adenosine and d‐serine) that act to regulate synaptic transmission and plasticity (Allen, 2014). Ongoing synaptic transmission is regulated by neurotransmitter uptake transporters expressed on astrocytes that clear glutamate from the synaptic cleft (glutamate transporter 1 (GLT‐1) and glutamate aspartate transporter (GLAST)) (Perego et al. 2000). Glutamate is converted to glutamine in astrocytes by glutamine synthetase, and the glutamine is then recycled to neurons to synthesize glutamate in order to maintain synaptic transmission (Danbolt, 2001). Excessive glutamate release at the synapse and/or deficits in glutamate clearance can lead to excitotoxicity, which has been linked to a number of neurological disorders that show synaptic dysfunction (Jia et al. 2015). In addition to glutamate uptake, astrocytes modulate neuronal networks by buffering extracellular K+ through Kir4.1 channels (Chever et al. 2010).
Astrocyte–synapse interactions in diseases
Given the role of synaptic dysfunction in numerous neurological disorders, and the strong role that astrocytes play in regulating synaptic function both in development and the adult, it is likely that alterations in astrocyte function may contribute to some of the pathology of neurological disorders. We will discuss a number of neurological disorders that have synaptic defects, and the known connections to astrocytic function. We aim to highlight novel potential targets for therapeutic consideration by elucidating the role of astrocytes in disease (Table 1).
Table 1.
Diseases and disorders associated with dendritic spine and synaptic defects, and the known roles of astrocytes in their pathology
| Disease | Synaptic/spine defect | Astrocytic involvement | References |
|---|---|---|---|
| Rett's syndrome | Decreased spine density, altered glutamate clearance at the synapse | WT astrocyte‐conditioned media rescues phenotype in RTT neurons; restoration of MeCP2 expression to astrocytes attenuates disease phenotype in vivo; MeCP2‐null astrocytes show downregulation of glutamate transporters | Ballas et al. 2009; Okabe et al. 2012; Williams et al. 2014 |
| Fragile X syndrome | Immature, thin spines; impaired synaptic protein clustering; impairments in GluA1 delivery to the synapse; alterations in LTD | WT astrocytes prevent the FXS phenotype in mutant neurons in vitro, while FXS astrocytes can induce the mutant phenotype in WT neurons; FXS astrocytes show alterations in GLT‐1 and mGluR expression; pharmacological inhibition of mGluR5 rescues dendritic spine defects and behavioural symptoms in mice | Jacobs & Doering, 2010 a, b; Michalon et al. 2012 |
| Down's syndrome | Reduced number of spines; immature spine morphology; reduced synapse number | Astrocyte‐secreted TSP‐1 regulates spine density and structural synapse formation; DS astrocytes show defects in APP metabolism, which may impact normal dendritic growth and development | Garcia et al. 2010; Chen et al. 2014; Hibaoui et al. 2014 |
| The RASopathies (NF1, CS) | Aberrant expression of synapse‐related genes, including NMDAR1, AMPAR4 and mGluR5, in NF1 | CS patient‐derived astroglial iPSCs overproduce extracellular matrix remodelling factors and proteoglycans | Krencik et al. 2015 |
| Stroke | Reduction in synapse density | TSP‐1/2 is upregulated during ischaemic stroke and is involved in synaptic recovery after the insult | Gleichman & Carmichael, 2014; Liauw et al. 2008. |
| Alzheimer's disease | Synaptic loss and reduction of spine density | Intracellular accumulation of Aβ in astrocytes, which impairs glutamate reuptake and triggers excitotoxic pathways | Duan et al. 1999; Matos et al. 2008; Rao et al. 2013; Talantova et al. 2013. |
| Parkinson's disease | Degeneration of dopaminergic and glutamatergic synapses in the striatum | Remodelling of PAP; increase of spontaneous Ca2+ activity of astrocytes | Bosson et al. 2015 |
| Huntington's disease | Synapse loss and spine instability | Decrease of astrocytic Kir4.1 potassium ion channel causes depolarization of medium spiny neurons and excitotoxicity | Shin et al. 2005; Tong et al. 2014. |
| Schizophrenia | Decreased spine density in pyramidal neurons | Mutant astrocytic DISC‐1 downregulates d‐serine, impairing NMDAR activation | Tanahashi et al. 2012; Ma et al. 2013 |
| Addiction | Increased spine density in NAc and PFC; increased synaptic activity between NAc and PFC | GLT‐1 downregulation that impairs glutamate reuptake by astrocytes, leading to excitotoxicity | Scofield et al. 2014; Duseja et al. 2015 |
| Epilepsy | Enhanced excitatory connectivity in the neocortex and hippocampus | Gabapentin, an anti‐epileptic drug, inhibits α2δ1, the receptor for astrocyte‐secreted TSP‐1, inhibiting excitatory synapse formation during development and possibly reducing excessive excitatory connectivity during epileptogenesis | Luo et al. 2001; Eroglu et al. 2009; Crunelli et al. 2015; |
Neurodevelopmental disorders
Based on the strong connection between astrocytes and normal synaptic development, it is likely that astrocyte dysfunction may play a role in neurodevelopmental disorders associated with changes in dendritic spines and synapse structure and function. Below, we highlight several disorders associated with defects in spine and synapse development, and describe the evidence for astrocytic involvement.
Rett's syndrome
Rett's syndrome (RTT) is a genetic disorder caused by mutations to methyl‐CpG binding protein 2 (MeCP2) on the X‐chromosome (Amir et al. 1999). It presents as a progressive, severe neurological disorder, affecting predominantly females (Hagberg et al. 1983). Children develop normally for 6–18 months before experiencing a regression of motor and language skills and the loss of purposeful hand movements. As regression continues, many symptoms of autism arise, including hand stereotypies, intellectual disabilities, and respiratory irregularities (Jeffrey et al. 2010). MeCP2 is an important regulator of synaptogenesis and synaptic pruning (Calfa et al. 2011). RTT pathology is associated with abnormalities in dendritic morphology, including reductions in dendritic complexity and decreased spine density (Xu et al. 2014).
Recent studies have implicated astrocyte dysfunction in these pathologies. Studies in mice or cells derived from patient‐obtained induced pluripotent stem cells (iPSCs) have demonstrated that RTT astrocytes fail to support typical development of wild‐type neurons in vitro, while conditioned media from wild‐type astrocytes rescues dendritic defects in RTT neurons (Ballas et al. 2009; Freitas et al. 2014; Williams et al. 2014). It is particularly interesting that this effect is seen with the application of conditioned media, as it indicates that contact is not required for the rescue. Systemic delivery of MeCP2 protein rescues some of the behavioural phenotype in mice (Garg et al. 2013), and importantly, rescuing MeCP2 expression selectively in astrocytes attenuates disease outcomes in vivo (Lioy et al. 2011), highlighting the importance of normal astrocytic function in this disorder. In a study examining astrocytes differentiated from RTT patient iPSCs, researchers noted that the application of insulin‐like growth factor 1 (IGF‐1) was capable of partially rescuing the neuronal phenotype (Williams et al. 2014), though it is unclear what, if any, astrocytic changes may play a role in this effect. Additional research has demonstrated that MeCP2 plays a role in glutamate clearance by astrocytes through regulation of the glutamate transporters GLT‐1 and GLAST, and glutamine synthetase (Okabe et al. 2012). MeCP2‐null astrocytes demonstrate impaired downregulation of transporter expression and higher levels of glutamine synthetase protein following exposure to glutamate in vitro, and these changes may lead to alterations in glutamate clearance at the synapse, and thus synaptic dysfunction (Okabe et al. 2012). More recent work in mice has found that MeCP2‐deficient medullary astrocytes have impaired CO2 sensitivity, which may contribute to the respiratory irregularities seen in patients with this disorder. Restoration of MeCP2 to these astrocytes leads to normal respiratory patterns (Turovsky et al. 2015). Taken together there is compelling evidence that defects in astrocytes are contributing to the alterations in neuronal function seen in Rett's syndrome, and altering astrocytic function may provide a novel therapeutic target.
Fragile X syndrome
Like RTT, fragile X syndrome (FXS) is an X‐linked genetic disorder. The disease is caused by a failure to express fragile X mental retardation protein (FMRP) due to an expansion in the CGG trinucleotide repeat in the fragile X mental retardation 1 gene (Fmr1) (Yudkin et al. 2014). FXS is associated with an autism‐like phenotype in many individuals with the disorder, including hand stereotypies, intellectual disability and social anxiety (McDuffie et al. 2015). FXS has been linked to defects in synaptic development and plasticity (Berry‐Kravis, 2014). Post‐mortem analysis of neocortical morphology has found that Fragile X patients have long, thin and immature‐appearing dendritic spines (Rudelli et al. 1985; Hinton, 2015), a morphology also seen in Fmr1 knock‐out (KO) mice (Irwin et al. 2000; Nimchinsky et al. 2001). FMRP is highly expressed in the brain (Feng et al. 1997; Pacey & Doering, 2007), and is essential for spine maturation.
Co‐culture studies have demonstrated roles for astrocytes in FXS. Wild‐type hippocampal neurons grown in culture with astrocytes from an FXS mouse show abnormal dendritic morphology and impaired synaptic protein clustering at 7 but not 21 days in vitro (Jacobs & Doering, 2010 a,b), indicating a delay in normal neuronal development. In contrast, FXS neurons that are grown on wild‐type astrocytes develop normally. Alterations in AMPAR presence at the synapse have been noted in FXS (Berry‐Kravis, 2014). Astrocytes can regulate synaptic recruitment of AMPARs via secretion of Gpc4 (Allen et al. 2012), but it is not yet known whether alterations in Gpc4 play a role in FXS. Glutamate clearance dysfunction may also play a role in the pathology of the disease. In Fmr1 KO mice, reduced GLT‐1 expression leads to a reduction in glutamate reuptake by astrocytes (Higashimori et al. 2013). In addition, mGluRs have been shown to play a role in FXS. In the absence of FMRP, excessive mGluR signalling at the synapse has been linked to abnormal dendritic spine morphology and maturation (Berry‐Kravis, 2014) and alterations in LTD. This hypothesis is supported by work demonstrating that chronic pharmacological inhibition of mGluR5 rescues the abnormal dendritic spine phenotype and leads to significant recovery from cognitive deficits associated with FXS in mice (Michalon et al. 2012). On the other hand, Higashimori et al. (2013) have found that Fmr1 KO leads to a reduction in astrocytic mGluR5 expression, which in turn contributes to reduced GLT‐1 expression. It therefore appears that where mGluRs are expressed can have a significant and differential effect in FXS, complicating potential therapies. Future work on the role of astrocytes in the morphological deficits in FXS, particularly during LTD, may provide better insight into potential treatments.
Down's syndrome
Abnormalities in dendritic spine morphology are characteristic of Down's syndrome (DS) (Marin‐Padilla, 1976; Suetsugu, 1980; Takashima et al. 1989). DS is the most common genetic cause of intellectual disability (Graber et al. 2012; Ross & Olsen, 2014) and is due to trisomy of chromosome 21. Neurons in DS brains have reduced numbers of spines, and these spines frequently show a long, tortuous morphology (Benavides‐Piccione et al. 2004), which has been linked to the development of Alzheimer's disease (AD) in adults with DS (Ferrer & Gullotta, 1990). A recent study on iPSCs derived from monozygotic twins, where one twin had DS while the other did not, demonstrated that DS lineage cells show increased astroglial and oligodendrocyte cell populations, along with decreased neurogenesis. Neuronal cells derived from DS iPSCs show deficits in dendritic development and reduced expression of both pre‐ and postsynaptic proteins, including postsynaptic density protein 95 (PSD95) and synapsin (Hibaoui et al. 2014).
Astrocytes have been implicated in this pathology through co‐culture studies, identifying thrombospondin (TSP)‐1 as a critical secreted astrocytic factor – decreased TSP‐1 release by DS astrocytes leads to alterations in spine density and morphology in DS (Garcia et al. 2010) (Fig. 1). In addition, hippocampal neurons grown on top of TSP‐1 KO astrocytes exhibit a dramatic increase in filopodia‐like spines, much like the pathology described in DS, and the addition of TSP‐1 to neuronal cultures grown in DS astrocyte‐conditioned media can reverse the reduced synapse number seen in culture (Garcia et al. 2010). While this research demonstrates a clear role for astrocytes in this disorder in vitro, it remains unknown whether or not application of TSP‐1 or overexpression of astrocytic TSP‐1 might rescue the DS spine pathology in vivo. Further research on DS astroglial cells derived from iPSCs has found that these cells exhibit increased levels of S100B, glial fibrillary acidic protein (GFAP), and reactive oxygen species, indicating that they are in a reactive state (Chen et al. 2014). Medium collected from these cells could not support normal neurite outgrowth or synapse formation, resulting in abnormal dendritic and spine morphology. This phenotype was partially rescued through the application of minocycline (Chen et al. 2014), potentially through anti‐inflammatory actions, indicating promise for therapeutic treatment. DS astrocytes also demonstrate deficits in amyloid precursor protein (APP) metabolism and secretion, associated with mitochondrial dysfunction (Busciglio et al. 2002), which may contribute to the comorbidity of DS and AD (Takashima et al. 1989). Increased deposition of APP during development in DS brains may result in inflammatory responses that hinder normal dendritic growth and development (Benavides‐Piccione et al. 2004). Further research into the connection between APP deposition, inflammation, and dendritic alterations may reveal a role for astrocytes in this pathology, but at this time there are many questions to be answered about the connection between astrocyte dysfunction and DS‐linked AD.
Figure 1. Astrocytes in synaptogenesis.
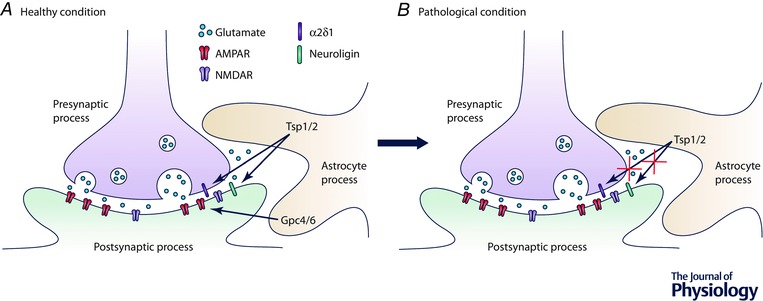
A, astrocytes induce synapse formation by secreting factors such as TSP1/2, which binds to α2δ1 and neuroligin to induce structural synapse formation, and Gpc4/6, which recruits GluA1‐containing AMPA receptors to the synapse. B, there is strong evidence through co‐culture experiments that astrocyte secreted protein factors play a role in many neurodevelopmental disorders. In Down's syndrome, for example, it has been found that astrocytes secrete lower levels of TSP‐1, which may lead to changes in the structure of the synapse, though the exact mechanism of this action is not yet known.
The RASopathies
The RASopathies are neurodevelopmental disorders caused by alterations in RAS pathway signalling. They have diverse phenotypes, but common characteristics include abnormal craniofacial morphology, cognitive impairment and cardiac abnormalities (Rauen, 2013). RAS/mitogen‐activated protein kinase (MAPK) signalling pathways have been implicated in synaptic trafficking of AMPARs in synaptic plasticity (Gu & Stornetta, 2007). Neurofibromatosis 1 (NF1) is a RASopathy characterized by learning disabilities, with a mutation in the NF1 gene affecting regulation of RAS/MAPK and cAMP signalling. DNA microarray analysis of NF1+/– mouse hippocampus has demonstrated aberrant expression of over 200 synapse‐related genes, including NMDAR1, GluA4, and mGluR5 (Park et al. 2009).
In mouse models of RASopathies including NF1, Noonan syndrome, and Costello's syndrome (CS), astrocytes have an accelerated rate of development and/or proliferation (Krencik & Ullian, 2013). CS, which results from a mutation in Harvey rat sarcoma viral oncogene homologue (HRAS), is characterized by hyperactivation of the RAS pathway, and patients show developmental delays and intellectual disability (Tidyman & Rauen, 2009). Astroglial cells differentiated from iPSCs derived from CS patient fibroblasts show accelerated maturation compared to wild‐type counterparts, and demonstrate a dramatic increase in cell area. Transcriptional analysis found that these cells overproduce extracellular matrix remodelling factors and proteoglycans (including chondroitin sulfate proteoglycans, heparan sulphate proteoglycans, SPARC, TSP‐1, and Gpc6) (Krencik et al. 2015), indicating that there may be hyperactivation of astrocyte‐to‐neuron signalling in this disorder. Co‐culturing these astroglia with neurons or the addition of their conditioned media to neurons led to higher synaptic density and increased neurite outgrowth. In addition, mice expressing mutant HRAS selectively in astrocytes showed increased accumulation of perineuronal net proteoglycans in the cerebral cortex (Krencik et al. 2015). This accelerated maturation of astrocytes and increased production of synaptogenic factors is the opposite phenotype to that seen in the other neurodevelopmental disorders we discussed, where in those cases astrocytes decreased production of pro‐synaptogenic factors. This suggests that the correct timing of the production and release of synaptogenic factors from astrocytes is crucial for the formation of fully functioning neuronal circuits, and that being too early or too late is sufficient to contribute to neurodevelopmental disorders.
Summary of neurodevelopmental disorders
Many of the neurodevelopmental disorders discussed above manifest with abnormal dendritic branching, immature and thin dendritic spines, and reduced synapse number. These defects can be rescued when mutant neurons are co‐cultured with wild‐type astrocytes, demonstrating a critical role for astrocytes in these disorders. The fact that these phenotypes can be partially rescued through application of wild‐type astrocyte‐conditioned media indicates that cell‐to‐cell contact is not required for these effects, and much work remains to be done to identify the changes in astrocyte secretion in each of these disorders to enable therapeutic targeting. Further studies using in vivo models of astrocyte‐specific manipulation (KO or rescue) of genes involved in neurodevelopmental disorders will give important insight into the in vivo importance of astrocytes in the pathology of each of these disorders.
Neurodegenerative disorders
Neurodegenerative diseases represent an important health threat, and are characterized by degeneration of the structure and function of neurons. Neurodegeneration can eventually lead to loss of cognitive and/or motor function and currently there are few effective treatments to stop the progression of these diverse disorders. Recent research has been focused on the role of astrocytes in triggering detrimental mechanisms such as excitotoxicity pathways that lead to neuronal death during neurodegeneration.
Stroke
Ischaemic stroke is a condition characterized by an abrupt loss of blood flow to the whole brain (global ischaemia) or a certain area of the brain (focal ischaemia). Haemorrhagic stroke, less common than ischaemic stroke, consists of the rupture of a blood vessel that causes accumulation of blood in a given area. In either case, glucose and oxygen are not delivered to the cells within the affected area (Nolte, 1999).
Astrocytes are important players during stroke and there are numerous studies about the action of astrocytes during and in response to stroke. These include an increased inflammatory response, formation of a glial scar, changes in glutamate uptake, blood–brain barrier repair and blood vessel restoration (Gleichman & Carmichael, 2014). Recently it has been suggested that astrocyte‐secreted factors can play a role in the recovery of neuronal synapses after a stroke. The astrocyte synaptogenic proteins TSP‐1 and TSP‐2 are upregulated after ischaemia (Liauw et al. 2008). During ischaemic stroke there is a significant reduction in synaptic density, with a gradual recovery after the insult in the area surrounding the core of the ischaemic stroke (Li & Murphy, 2008). In TSP‐1/2 KO mice synaptic recovery post‐insult is not as efficient as in wild‐type mice, suggesting that upregulation of TSP‐1/2 in astrocytes normally contributes to post‐stroke recovery of synapse number (Liauw et al. 2008). Moreover, a key feature in neurons during ischaemia‐like conditions is an alteration in surface AMPAR subunit expression (Blanco‐Suarez & Hanley, 2014), which determines neuronal Ca2+ permeability and can activate pathways that lead to neuronal death (Liu & Zukin, 2007). Some astrocytic secreted factors mediate AMPAR recruitment to the postsynaptic density (Allen, 2014), and therefore may represent important targets to prevent excitotoxicity from occurring. Taken together, these studies suggest that astrocyte‐secreted factors contribute to synaptic recovery after ischaemic stroke.
Alzheimer's disease
Alzheimer's disease (AD) is a common cause of dementia and is well known to affect cognition and memory. It is characterized by the accumulation of intracellular neurofibrillary tangles and amyloid‐β (Aβ) plaques in the extracellular space (Sheng et al. 2012). Additionally, dendritic spine loss has been identified as an early sign of the disease, which occurs prior to the formation of plaques and neuronal death, and contributes to the cognitive deficiency (Moolman et al. 2004). Aβ decreases synaptic function of NMDARs and AMPARs (Hsieh et al. 2006) and promotes the internalization of AMPARs from synaptic sites, contributing to synaptic depression and decreasing spine density (Hsieh et al. 2006).
Normally the release of glutamate to the synaptic cleft triggers glutamate uptake in astrocytes (Duan et al. 1999), but in AD this mechanism is impaired by Aβ‐induced inhibition of GLT‐1 and GLAST (Matos et al. 2008). Aβ induces the release of glutamate from neurons and astrocytes in vitro, which activates extrasynaptic NMDARs due to glutamate spillover (Li et al. 2011), activating excitotoxic pathways that contribute to synapse loss (Talantova et al. 2013) (Fig. 2). Additionally, Aβ is known to induce metabolic impairments in astrocytes, leading to an increased intracellular accumulation of TSP‐1 and a reduction in its release (Rao et al. 2013). A deeper understanding of the role of astrocytes during AD could provide a useful new angle to study the mechanisms that lead to the characteristic spine loss and neuronal degeneration of this devastating disease.
Figure 2. The role of astrocytes in glutamate uptake.
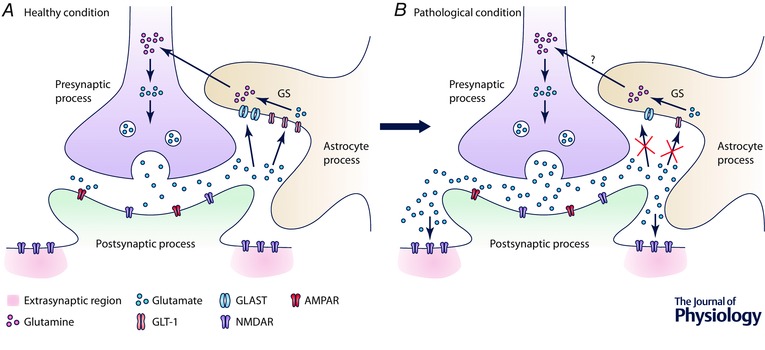
A, glutamate in the extracellular space is taken up by astrocytes through GLT‐1 and GLAST, whose expression in astrocytes can be upregulated by glutamate. The conversion of glutamate into glutamine is catalysed by glutamine synthetase (GS). Glutamine is transported back to the presynaptic neuron to be converted back to glutamate. B, this mechanism is impaired in various CNS disorders, such as RTT, FXS, stroke, AD and addiction. The excess glutamate can activate extrasynaptic NMDARs, leading to activation of excitotoxic pathways that cause synaptic loss and eventual cell death.
Parkinson's disease
Parkinson's disease (PD) is a neurodegenerative disorder characterized by motor deficiencies, including tremor and bradykinesia (Dickson, 2012). The loss of dopaminergic neurons in the substantia nigra pars compacta, which innervates the striatum, is the primary cause of PD. Consequently there is a significant decrease in dendritic spine density on neurons in the striatum (Day et al. 2006; Morales et al. 2015).
In parkinsonian monkeys PAPs undergo structural remodelling leading to a greater association with synapses (Villalba & Smith, 2011); however, the functional purpose of this is not yet understood. Dopamine loss in PD increases spontaneous Ca2+ activity of individual astrocytes and increases the synchronization of the whole astrocytic network (Bosson et al. 2015). The current surgical treatment for severe cases of PD is high frequency stimulation of the subthalamic nucleus, which restores normal activity to neurons in the substantia nigra pars reticulata. In a non‐diseased state acute blockade of dopamine receptors to mimic PD is sufficient to induce astrocyte hyperactivity and synchronicity, which is significantly decreased by high frequency stimulation of the subthalamic nucleus (Bosson et al. 2015). Therefore, dopaminergic transmission impairments affecting substantia nigra pars reticulata neuronal activity have consequences for the astrocytic network, and alterations in astrocytes may be contributing to some of the motor deficits in PD.
Huntington's disease
Huntington's disease (HD) is caused by the accumulation of CAG repeats in the huntingtin gene, which causes motor and occasional cognitive dysfunctions (Ross et al. 2014). There is an underlying neuronal dysfunction that includes spine and synaptic loss and eventual neuronal death (Murmu et al. 2013).
Astrocytes in the striatum show intracellular accumulation of mutant huntingtin at an early stage of the disease before astrogliosis is triggered, which disrupts the function and expression of crucial astrocyte proteins. It has been suggested that astrocytic dysfunction at early stages of HD might represent a good target to protect neurons from insult (Tong et al. 2014), including synapse loss and spine instability. In HD the Kir4.1 potassium ion channel is downregulated in astrocytes (Tong et al. 2014). This causes extracellular K+ levels to increase due to an impairment of K+ buffering in the striatum, which leads to depolarization of medium spiny neurons and subsequent excitotoxicity (Shin et al. 2005; Tong et al. 2014). Loss of Kir4.1 is linked to loss of GLT1, which decreases astrocyte glutamate uptake, promoting the activation of mGluRs and increasing astrocyte Ca2+ signals. Restoring Kir4.1 in vivo rescues the expression of GLT1, and recovers normal Ca2+ and glutamate signalling in astrocytes (Jiang et al. 2016), providing a novel therapeutic target for HD treatment.
Summary of neurodegenerative disorders
Astrocyte dysfunction has in recent years proven to be a common crossroads in neurodegenerative disorders such as stroke, AD, PD and HD. Glutamate reuptake by astrocytes, astrocytic network synchronicity, tripartite synapse structure and astrocyte‐to‐neuron signalling are some of the mechanisms that are impaired in these disorders. Targeting astrocytes may enable the re‐establishment of proper neuronal function following injury (e.g. stroke) or during disease progression (e.g. HD, PD and AD), by regulating the expression of channels and receptors on the neuronal surface. Dissecting these mechanisms in astrocytes will help elucidate the role that astrocytes play in spine and synapse loss observed in neurodegenerative disorders, and may yield insight into new therapies.
Other neurological disorders
The connection between astrocytes and synaptic maintenance and function indicates that astrocytes are ideal candidates for investigating neurological disorders characterized by changes in synapse number and plasticity. As we discuss here, changes in astrocytic function may be connected to the phenotypes of some of these disorders.
Schizophrenia
Schizophrenia is a severe and debilitating disorder characterized by abnormal social behaviour. It manifests with positive symptoms, such as delusions and hallucinations, as well as negative symptoms, including flat affect, lack of motivation and an inability to experience pleasure (Picchioni & Murray, 2007). Post‐mortem studies have found decreased spine density on prefrontal cortical pyramidal neurons in patients with schizophrenia (Glantz & Lewis, 2000).
The gene DISC1 has been strongly linked to the development of schizophrenia, and has been shown to play a role in cell proliferation and dendritic outgrowth (Brandon et al. 2009). There is evidence that its protein, disrupted in schizophrenia‐1 (DISC‐1), is found at the synapse, but its role there is poorly understood. A mouse model expressing a truncated version of DISC‐1 shows a reduction in spine density in the dentate gyrus (Kvajo et al. 2008), and bioinformatics analysis of the DISC‐1 ‘interactome’ indicates that it is likely to play a role in synaptic plasticity (Camargo et al. 2007). DISC‐1 is also present in astrocytes, and mutant astrocytic DISC1 leads to a reduction in d‐serine production by astrocytes in vitro (Ma et al. 2013). d‐Serine is a co‐ligand required for activation of the NMDA receptor (Mothet et al. 2000). Deficient glutamate transmission via NMDARs has been implicated in schizophrenia, suggesting astrocyte release of d‐serine may be contributing to this effect. Whether antipsychotic drugs used to treat schizophrenia have effects on astrocyte function has not been fully examined, although clozapine has been shown to activate astrocytic release of d‐serine, which would enhance NMDA neurotransmission, while haloperidol had no such effect (Tanahashi et al. 2012). Given the importance of astrocytes in maintaining the synapse, and particularly in modulating glutamatergic transmission, it seems likely that they may play a role in the glutamate dysfunction seen in schizophrenia.
Addiction
Addiction causes a behavioral impairment in the control of the consumption of a certain substance, despite the harm that it may cause. Alteration of the dopaminergic circuitry in the prefrontal cortex (PFC) and striatum plays a central role in addiction disorders and contributes to drug‐seeking behaviour and relapse. Additionally, glutamatergic neurotransmission is impaired between PFC and nucleus accumbens (NAc) during addiction (Kalivas, 2009).
During seeking of addictive substances (e.g. heroin, cocaine, nicotine) there is an increase in synaptic activity between the PFC and the NAc in rats, and a consequent increase in glutamate release in the NAc (Scofield & Kalivas, 2014). This occurs along with downregulation of the glutamate transporter GLT‐1 in astrocytes in the NAc, which is normally responsible for most of the glutamate uptake (Perego et al. 2000). These combined events contribute to excess glutamate present in the synaptic cleft and may activate receptors such as mGluR5 and GluN2B‐containing NMDARs. Relapse susceptibility is related to these long‐lasting effects on glutamate synaptic transmission, and given that astrocytes are important for glutamate clearance in the healthy brain, astrocyte dysfunction is likely to play a role in relapse (Scofield & Kalivas, 2014). Moreover excess extracellular glutamate may trigger effects known to modify spine structure and subsequent synaptic transmission (Halpain et al. 1998). In fact, cocaine self‐administration causes an increase in spine density in the two areas involved in addiction, NAc and PFC (Robinson TE, 2001). Restoration of the glutamate clearance capability of astrocytes in addiction disorders could help avoid relapse and aid recovery by regulating glutamatergic neurotransmission. Additionally, TNF‐α has been implicated in behavioural responses to drugs of abuse (Nakajima et al. 2004; Duseja et al. 2015), although the specific role of astrocyte‐secreted TNF‐α requires further research.
Epileptogenesis after injury
In injury‐induced epilepsy, enhanced excitatory connectivity in the neocortex and hippocampus play a role in epileptogenesis. Downregulation of expression or function of K+ channels in astrocytes has been connected to defective K+ buffering, which may contribute to this hyperexcitability (Carmignoto & Haydon, 2012). Additionally, there is strong support for a focal epilepsy model in which astrocytes exhibit large Ca2+ elevations, leading to neuronal hyperexcitability and production of focal seizure activity (Carmignoto & Haydon, 2012; Álvarez‐Ferradas et al. 2015; Crunelli et al. 2015). Of particular interest, given the known roles of astrocytes at the synapse, is the fact that an upregulation in expression of the Ca2+ channel α2δ1 subunit is seen in nerve and brain injury (Luo et al. 2001). α2δ1 is the receptor for TSP‐1, an astrocyte‐secreted factor known to play a prominent role in synaptogenesis during development. It is also the receptor for gabapentin, an antiepileptic drug that is known to inhibit TSP‐1‐induced excitatory synapse formation during development (Eroglu et al. 2009). Gabapentin prevents some of the excess excitatory connectivity seen in injury‐induced epileptogenesis (Li et al. 2012). Astrocytic TSP‐1 may therefore play a role in the development of epilepsy following CNS injury, and these data provide a mechanism for how gabapentin inhibits seizures.
Summary of other disorders
Synaptic dysfunction and alterations in glutamate reuptake by astrocytes have been implicated in many neurological disorders, including schizophrenia and addiction. Excess glutamate can contribute to excitotoxicity, impairing synaptic function and plasticity, and restoration of proper astrocytic reuptake of glutamate may be an avenue for addiction treatment. In addition, there is evidence that astrocyte‐secreted factors, such as TSP‐1 and TNF‐α, may play a role in the changes in plasticity seen with drug use and epilepsy. This suggests that synaptic dysfunction, characteristic of these disorders, might be alleviated by proper astrocytic protein expression and secretion. Further investigation into the roles and mechanisms of astrocytic factors in synaptic plasticity and the deficits seen in these disorders should help to uncover possible avenues of treatment.
Conclusions
As we begin to better understand the molecular basis for brain disorders and diseases, we are also beginning to uncover the roles that astrocytes play in these diseases (Table 1). Traditionally, astrocytes were merely considered to be filling the gaps between neurons, and therefore their study was neglected. However, new discoveries have put astrocytes in the spotlight of neuroscience research. Numerous current studies, like the ones mentioned in this review, are focusing their efforts on elucidating the role of astrocytes in neurological disorders. The dissection of astrocyte regulatory mechanisms will help to better understand a wide variety of neurological disorders and open new pathways for the development of innovative treatments.
Additional information
Competing interests
The authors declare no conflict of interests.
Author contributions
E.B.‐S., A.L.M.C. and N.J.A. conceived the structure and topic of the review. E.B.‐S. and A.L.M.C. co‐wrote the review and designed the figures. N.J.A. reviewed and edited the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NINDS grant R01 NS089791 to N.J.A.
Acknowledgements
We thank members of the Allen lab for critical reading of the manuscript.
Biographies
The Allen lab works to understand the role of astrocytes during the formation and development of neuronal circuits – in particular, how astrocyte‐secreted factors can influence the structure and function of the synapse. Nicola Allen carried out her PhD with David Attwell at UCL, and her Postdoc with Ben Barres at Stanford University. She has been an Assistant Professor at the Salk Institute for Biological Studies since 2012. Elena Blanco‐Suárez majored in Biology at the University of Oviedo. She received her PhD from the University of Bristol working in Biochemistry with Dr Jon Hanley. She is now a post‐doctoral researcher in the Allen lab at the Salk Institute.
Alison Caldwell completed her B.S. in Brain and Cognitive Science at MIT, and spent time as a research technologist at the Medical College of Wisconsin before joining the Neurosciences Graduate Program at UCSD, where she is currently a student in the Allen lab.
E. Blanco‐Suárez and A. L. M. Caldwell contributed equally to this work.
References
- Allen NJ (2014). Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol 30, 439–463. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ & Barres BA (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez VA & Sabatini BL (2007). Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci 30, 79–97. [DOI] [PubMed] [Google Scholar]
- Álvarez‐Ferradas C, Morales JC, Wellmann M, Nualart F, Roncagliolo M, Fuenzalida M & Bonansco C (2015). Enhanced astroglial Ca2+ signaling increases excitatory synaptic strength in the epileptic brain. Glia 63, 1507–1521. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U & Zoghbi HY (1999). Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nat Genet 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP & Haydon PG (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22, 208–215. [DOI] [PubMed] [Google Scholar]
- Araque A & Perea G (2004). Glial modulation of synaptic transmission in culture. Glia 47, 241–248. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C & Mandel G (2009). Non‐cell autonomous influence of MeCP2‐deficient glia on neuronal dendritic morphology. Nat Neurosci 12, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides‐Piccione R, Ballesteros‐Yanez I, de Lagran MM, Elston G, Estivill X, Fillat C, Defelipe J & Dierssen M (2004). On dendrites in Down syndrome and DS murine models: a spiny way to learn. Prog Neurobiol 74, 111–126. [DOI] [PubMed] [Google Scholar]
- Bernardinelli Y, Randall J, Janett E, Nikonenko I, König S, Jones EV, Flores CE, Murai KK, Bochet CG, Holtmaat A & Muller D (2014). Activity‐dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr Biol 24, 1679–1688. [DOI] [PubMed] [Google Scholar]
- Berry‐Kravis E (2014). Mechanism‐based treatments in neurodevelopmental disorders: fragile X syndrome. Pediatr Neurol 50, 297–302. [DOI] [PubMed] [Google Scholar]
- Blanco‐Suarez E & Hanley JG (2014). Distinct subunit‐specific α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptor trafficking mechanisms in cultured cortical and hippocampal neurons in response to oxygen and glucose deprivation. J Biol Chem 289, 4644–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson A, Boisseau S, Buisson A, Savasta M & Albrieux M (2015). Disruption of dopaminergic transmission remodels tripartite synapse morphology and astrocytic calcium activity within substantia nigra pars reticulata. Glia 63, 673–683. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Millar JK, Korth C, Sive H, Singh KK & Sawa A (2009). Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci 29, 12768–12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H & Yankner BA (2002). Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron 33, 677–688. [DOI] [PubMed] [Google Scholar]
- Calfa G, Percy AK & Pozzo‐Miller L (2011). Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp Biol Med (Maywood) 236, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ & Brandon NJ (2007). Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry 12, 74–86. [DOI] [PubMed] [Google Scholar]
- Carmignoto G & Haydon PG (2012). Astrocyte calcium signalling and epilepsy. Glia 60, 1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jiang P, Xue H, Peterson SE, Tran HT, McCann AE, Parast MM, Li S, Pleasure DE, Laurent LC, Loring JF, Liu Y & Deng W (2014). Role of astroglia in Down's syndrome revealed by patient‐derived human‐induced pluripotent stem cells. Nat Commun 5, 4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chever O, Djukic B, McCarthy KD & Amzica F (2010). Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial‐conditional Kir4.1 knock‐out mice. J Neurosci 30, 15769–15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P & Barres BA (2005). Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Carmignoto G & Steinhäuser C (2015). Novel astrocyte targets: new avenues for the therapeutic treatment of epilepsy. Neuroscientist 21, 62–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC (2001). Glutamate uptake. Prog Neurobiol 65, 1–105. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW & Surmeier DJ (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci 9, 251–259. [DOI] [PubMed] [Google Scholar]
- Dickson DW (2012). Parkinson's disease and Parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2, doi: 10.1101/cshperspect.a009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongen AMV. (ed) (2009). Biology of the NMDA Receptor. CRC Press/Taylor & Francis, Boca Raton. [Google Scholar]
- Duan S, Anderson CM, Stein BA & Swanson RA (1999). Glutamate induces rapid upregulation of astrocyte glutamate transport and cell‐surface expression of GLAST. J Neurosci 19, 10193–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duseja R, Heir R, Lewitus GM, Altimimi HF & Stellwagen D (2015). Astrocytic TNFα regulates the behavioral response to antidepressants. Brain Behav Immun 44, 187–194. [DOI] [PubMed] [Google Scholar]
- Eroglu Ç, Allen NJ, Susman MW, O'Rourke NA, Park CY, Özkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF & Barres BA (2009). Gabapentin receptor α2δ‐1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST & Hersch SM (1997). Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17, 1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I & Gullotta F (1990). Down's syndrome and Alzheimer's disease: dendritic spine counts in the hippocampus. Acta Neuropathol 79, 680–685. [DOI] [PubMed] [Google Scholar]
- Freitas BC, Trujillo CA, Carromeu C, Yusupova M, Herai RH & Muotri AR (2014). Stem cells and modeling of autism spectrum disorders. Exp Neurol 260, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia O, Torres M, Helguera P, Coskun P & Busciglio J (2010). A role for thrombospondin‐1 deficits in astrocyte‐mediated spine and synaptic pathology in Down's syndrome. PLoS One 5, e14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, Foust KD, Kaspar BK, Bird A & Mandel G (2013). Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci 33, 13612–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA & Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57, 65–73. [DOI] [PubMed] [Google Scholar]
- Gleichman AJ & Carmichael ST (2014). Astrocytic therapies for neuronal repair in stroke. Neurosci Lett 565, 47–52. [DOI] [PubMed] [Google Scholar]
- Graber E, Chacko E, Regelmann MO, Costin G & Rapaport R (2012). Down syndrome and thyroid function. Endocrinol Metab Clin North Am 41, 735–745. [DOI] [PubMed] [Google Scholar]
- Gu Y & Stornetta RL (2007). Synaptic plasticity, AMPA‐R trafficking, and Ras‐MAPK signaling. Acta Pharmacol Sin 28, 928–936. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K & Ramos O (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol 14, 471–479. [DOI] [PubMed] [Google Scholar]
- Halpain S, Hipolito A & Saffer L (1998). Regulation of F‐actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci 18, 9835–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H & Sheng M (2001). Dentritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2, 880–888. [DOI] [PubMed] [Google Scholar]
- Hibaoui Y, Grad I, Letourneau A, Sailani MR, Dahoun S, Santoni FA, Gimelli S, Guipponi M, Pelte MF, Béna F, Antonarakis SE & Feki A (2014). Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol Med 6, 259–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimori H, Morel L, Huth J, Lindemann L, Dulla C, Taylor A, Freeman M & Yang Y (2013). Astroglial FMRP‐dependent translational down‐regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum Mol Genet 22, 2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski, K & Rudelli RD (2015). Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet 41, 289–294. [DOI] [PubMed] [Google Scholar]
- Hotulainen P & Hoogenraad CC (2010). Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S & Malinow R (2006). AMPAR removal underlies Aβ‐induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R & Greenough WT (2000). Dendritic spine structural anomalies in fragile‐X mental retardation syndrome. Cereb Cortex 10, 1038–1054. [DOI] [PubMed] [Google Scholar]
- Jacobs S & Doering LC (2010. a). Astrocytes prevent abnormal neuronal development in the fragile X mouse. J Neurosci 30, 4508–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SNM & Doering LC (2010. b). Fragile X astrocytes induce developmental delays in dendrite maturation and synaptic protein expression. BMC Neurosci 11, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey LN, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi‐Buisson N, Leonard H, Bailey MES, Schanen NC, Zappella M, Renieri A, Huppke P, Percy AK; RettSearch Consortium (2010). Rett syndrome: Revised diagnostic criteria and nomenclature. Ann Neurol 68, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M, Njapo SA, Rastogi V, Hedna VS (2015). Taming glutamate excitotoxicity: strategic pathway modulation for neuroprotection. CNS Drugs 29, 153–162. [DOI] [PubMed] [Google Scholar]
- Jiang R, Diaz‐Castro B, Looger LL & Khakh BS (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington's disease model mice. J Neurosci 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EV, Bernardinelli Y, Tse YC, Chierzi S, Wong TP & Murai KK (2011). Astrocytes control glutamate receptor levels at developing synapses through SPARC–β‐integrin interactions. J Neurosci 31, 4154–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW (2009). The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10, 561–572. [DOI] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi‐Takagi A & Noguchi J (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci 33, 121–129. [DOI] [PubMed] [Google Scholar]
- Koleske AJ (2013). Molecular mechanisms of dendrite stability. Nat Rev Neurosci 14, 536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, Hokanson KC, Narayan AR, Dvornik J, Rooney GE, Rauen KA, Weiss LA, Rowitch DH & Ullian EM (2015). Dysregulation of astrocyte extracellular signaling in Costello syndrome. Sci Transl Med 7, 286ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R & Ullian EM (2013). A cellular star atlas: using astrocytes from human pluripotent stem cells for disease studies. Front Cell Neurosci 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA & Eroglu C (2011). Control of excitatory CNS synaptogenesis by astrocyte‐secreted proteins Hevin and SPARC. Proc Natl Acad Sci USA 108, E440–E449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H, MacDermott AB, Karayiorgou M & Gogos JA (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci USA 105, 7076–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Graber KD, Jin S, McDonald W, Barres BA & Prince DA (2012). Gabapentin decreases epileptiform discharges in a chronic model of neocortical trauma. Neurobiol Dis 48, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P & Murphy TH (2008). Two‐photon imaging during prolonged middle cerebral artery occlusion in mice reveals recovery of dendritic structure after reperfusion. J Neurosci 28, 11970–11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM & Selkoe DJ (2011). Soluble Aβ oligomers inhibit long‐term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B‐containing NMDA receptors. J Neurosci 31, 6627–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun G‐h, Percy M, Wildman‐Tobriner B, Bliss T, Guzman RG, Barres BA & Steinberg GK (2008). Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab 28, 1722–1732. [DOI] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N & Mandel G (2011). A role for glia in the progression of Rett's syndrome. Nature 475, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ & Zukin RS (2007). Ca2+‐permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci 30, 126–134. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME & Yaksh TL (2001). Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve‐injured rats. J Neurosci 21, 1868–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C & Malenka RC (2012). NMDA receptor‐dependent long‐term potentiation and long‐term depression (LTP/LTD). Cold Spring Harb Perspect Biol 4, doi: 10.1101/cshperspect.a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TM, Abazyan S, Abazyan B, Nomura J, Yang C, Seshadri S, Sawa A, Snyder SH & Pletnikov MV (2013). Pathogenic disruption of DISC1‐serine racemase binding elicits schizophrenia‐like behavior via D‐serine depletion. Mol Psychiatry 18, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC & Bear MF (2004). LTP and LTD: An embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Marin‐Padilla M (1976). Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. A Golgi study. J Comp Neurol 167, 63–81. [DOI] [PubMed] [Google Scholar]
- Matos M, Augusto E, Oliveira CR & Agostinho P (2008). Amyloid‐beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen‐activated protein kinase cascades. Neuroscience 156, 898–910. [DOI] [PubMed] [Google Scholar]
- McDuffie A, Thurman AJ, Hagerman RJ & Abbeduto L (2015). Symptoms of autism in males with fragile X syndrome: A comparison to nonsyndromic ASD using current ADI‐R scores. J Autism Dev Disord 45, 1925–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF & Lindemann L (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML (2004). Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol 33, 377–387. [DOI] [PubMed] [Google Scholar]
- Morales I, Sanchez A, Rodriguez‐Sabate C & Rodriguez M (2015). The degeneration of dopaminergic synapses in Parkinson's disease: A selective animal model. Behav Brain Res 289, 19–28. [DOI] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady Jr. RO, Lin DJ, Ferris CD, Rogawski MA & Snyder, SH (2000). d‐Serine is an endogenous ligand for the glycine site of the N‐methyl‐d‐aspartate receptor. Proceedings of the National Academy of Sciences 97, 4926–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S & Manahan‐Vaughan D (2013). Role of metabotropic glutamate receptors in persistent forms of hippocampal plasticity and learning. Neuropharmacology 66, 65–81. [DOI] [PubMed] [Google Scholar]
- Murmu RP, Li W, Holtmaat A & Li J‐Y (2013). Dendritic spine instability leads to progressive neocortical spine loss in a mouse model of Huntington's disease. J Neurosci 33, 12997–13009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima A, Yamada K, Nagai T, Uchiyama T, Miyamoto Y, Mamiya T, He J, Nitta A, Mizuno M, Tran MH, Seto A, Yoshimura M, Kitaichi K, Hasegawa T, Saito K, Yamada Y, Seishima M, Sekikawa K, Kim H‐C & Nabeshima T (2004). Role of tumor necrosis factor‐α in methamphetamine‐induced drug dependence and neurotoxicity. J Neurosci 24, 2212–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM & Svoboda K (2001). Abnormal development of dendritic spines in FMR1 knock‐out mice. J Neurosci 21, 5139–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte J (1999). The Human Brain. An Introduction to its Functional Anatomy. Mosby, St Louis. [Google Scholar]
- Okabe Y, Takahashi T, Mitsumasu C, Kosai K‐i, Tanaka E & Matsuishi T (2012). Alterations of gene expression and glutamate clearance in astrocytes derived from an MeCP2‐null mouse model of Rett syndrome. PLoS One 7, e35354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey LK & Doering LC (2007). Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia 55, 1601–1609. [DOI] [PubMed] [Google Scholar]
- Park CS, Zhong L & Tang S‐J (2009). Aberrant expression of synaptic plasticity‐related genes in the NF1+/− mouse hippocampus. J Neurosci Res 87, 3107–3119. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen J‐E & Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M & Araque A (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32, 421–431. [DOI] [PubMed] [Google Scholar]
- Perego C, Vanoni C, Bossi M, Massari S, Basudev H, Longhi R & Pietrini G (2000). The GLT‐1 and GLAST glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J Neurochem 75, 1076–1084. [DOI] [PubMed] [Google Scholar]
- Perez‐Alvarez A, Navarrete M, Covelo A, Martin ED & Araque A (2014). Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J Neurosci 34, 12738–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picchioni MM & Murray RM (2007). Schizophrenia BMJ 335, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao KVR, Curtis KM, Johnstone JT & Norenberg MD (2013). Amyloid‐β inhibits thrombospondin 1 release from cultured astrocytes: effects on synaptic protein expression. J Neuropathol Exp Neurol 72, 735–744. [DOI] [PubMed] [Google Scholar]
- Rauen KA (2013). The RASopathies. Annu Rev Genomics Hum Genet 14, 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Gorny G, Mitton E, Kolb B (2001). Cocaine self‐administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse 39, 257–266. [DOI] [PubMed] [Google Scholar]
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, Reilmann R, Unschuld PG, Wexler A, Margolis RL & Tabrizi SJ (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10, 204–216. [DOI] [PubMed] [Google Scholar]
- Ross WT & Olsen M (2014). Care of the adult patient with Down syndrome. South Med J 107, 715–721. [DOI] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure‐Kamionowska M, Connell F & Wisniewski HM (1985). Adult fragile X syndrome. Clinico‐neuropathologic findings. Acta Neuropathol 67, 289–295. [DOI] [PubMed] [Google Scholar]
- Scofield MD & Kalivas PW (2014). Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist 20, 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL & Südhof TC (2012). Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol 4, doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD & Huganir RL (2007). The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol 23, 613–643. [DOI] [PubMed] [Google Scholar]
- Shin J‐Y, Fang Z‐H, Yu Z‐X, Wang C‐E, Li S‐H & Li X‐J (2005). Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirao T & González‐Billault C (2013). Actin filaments and microtubules in dendritic spines. J Neurochem 126, 155–164. [DOI] [PubMed] [Google Scholar]
- Suetsugu M, Mehraein P (1980). Spine distribution along the apical dendrites of the pyramidal neurons in Down's syndrome. A quantitative Golgi study. Acta Neuropathol 50, 207–210. [DOI] [PubMed] [Google Scholar]
- Takashima S, Ieshima A, Nakamura H & Becker LE (1989). Dendrites, dementia and the Down syndrome. Brain Dev 11, 131–133. [DOI] [PubMed] [Google Scholar]
- Talantova M, Sanz‐Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S‐i, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang Y‐J, Tu S, Molokanova E, McKercher SR, Hires SA, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, Powers ET, Kelly JW, Roberts A, Tong G, Fang‐Newmeyer T, Parker J, Holland EA, Zhang D, Nakanishi N, Chen H‐SV, Wolosker H, Wang Y, Parsons LH, Ambasudhan R, Masliah E, Heinemann SF, Piña‐Crespo JC & Lipton SA (2013). Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA 110, E2518–E2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanahashi S, Yamamura S, Nakagawa M, Motomura E & Okada M (2012). Clozapine, but not haloperidol, enhances glial D‐serine and L‐glutamate release in rat frontal cortex and primary cultured astrocytes. Br J Pharmacol 165, 1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman WE & Rauen KA (2009). The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV & Khakh BS (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turovsky E, Karagiannis A, Abdala AP & Gourine AV (2015). Impaired CO2 sensitivity of astrocytes in a mouse model of Rett syndrome. J Physiol 593, 3159–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpelli C, Schmeisser M, Sala C & Boeckers T (2012). Scaffold proteins at the postsynaptic density In Synaptic Plasticity, ed. Kreutz MR. & Sala C, pp. 29–61. Springer, Vienna. [DOI] [PubMed] [Google Scholar]
- Villalba RM & Smith Y (2011). Neuroglial plasticity at striatal glutamatergic synapses in Parkinson's disease. Front Syst Neurosci 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EC, Zhong X, Mohamed A, Li R, Liu Y, Dong Q, Ananiev GE, Mok JC, Lin BR, Lu J, Chiao C, Cherney R, Li H, Zhang SC & Chang Q (2014). Mutant astrocytes differentiated from Rett syndrome patients‐specific iPSCs have adverse effects on wild‐type neurons. Hum Mol Genet 23, 2968–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Miller EC & Pozzo‐Miller L (2014). Dendritic spine dysgenesis in Rett syndrome. Front Neuroanat 8, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F & Gan W‐B (2009). Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara Y, De Roo M & Muller D (2009). Dendritic spine formation and stabilization. Curr Opin Neurobiol 19, 146–153. [DOI] [PubMed] [Google Scholar]
- Yudkin D, Hayward BE, Aladjem MI, Kumari D & Usdin K (2014). Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum Mol Genet 23, 2940–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]