

Abstract
Key points
When a skeletal muscle is stretched while it contracts, the muscle produces a relatively higher force than the force from an isometric contraction at the same length: a phenomenon referred to as residual force enhancement.
Residual force enhancement is puzzling because it cannot be directly explained by the classical force–length relationship and the sliding filament theory of contraction, the main paradigms in the muscle field.
We used custom‐built instruments to measure residual force enhancement in skeletal myofibrils, and, for the first time, in cardiac myofibrils.
Our data report that residual force enhancement is present in skeletal muscles, but not cardiac muscles, and is regulated by the different isoforms of the titin protein filaments.
Abstract
When a skeletal muscle contracts isometrically, the muscle produces a force that is relative to the final isometric sarcomere length (SL). However, when the same final SL is reached by stretching the muscle while it contracts, the muscle produces a relatively higher force: a phenomenon commonly referred to as residual force enhancement. In this study, we investigated residual force enhancement in rabbit skeletal psoas myofibrils and, for the first time, cardiac papillary myofibrils. A custom‐built atomic force microscope was used in experiments that stretched myofibrils before and after inhibiting myosin and actin interactions to determine whether the different cardiac and skeletal titin isoforms regulate residual force enhancement. At SLs ranging from 2.24 to 3.13 μm, the skeletal myofibrils enhanced the force by an average of 9.0%, and by 29.5% after hindering myosin and actin interactions. At SLs ranging from 1.80 to 2.29 μm, the cardiac myofibrils did not enhance the force before or after hindering myosin and actin interactions. We conclude that residual force enhancement is present only in skeletal muscles and is dependent on the titin isoforms.
Keywords: Blebbistatin, cardiac, residual force enhancement, myofibril, skeletal, static tension, titin
Key points
When a skeletal muscle is stretched while it contracts, the muscle produces a relatively higher force than the force from an isometric contraction at the same length: a phenomenon referred to as residual force enhancement.
Residual force enhancement is puzzling because it cannot be directly explained by the classical force–length relationship and the sliding filament theory of contraction, the main paradigms in the muscle field.
We used custom‐built instruments to measure residual force enhancement in skeletal myofibrils, and, for the first time, in cardiac myofibrils.
Our data report that residual force enhancement is present in skeletal muscles, but not cardiac muscles, and is regulated by the different isoforms of the titin protein filaments.
Abbreviations
- Custom‐AFM
custom‐built atomic force microscope
- E‐domains
glutamate domains
- Ig
immunoglobulin
- PEVK
proline, glutamate, valine and lysine
- SL
sarcomere length
Introduction
Isometric contraction of muscles produces a force that is related to the final isometric sarcomere length (SL) (Gordon et al. 1966). However, when the same isometric SL is reached by stretching a muscle while it contracts, the muscle maintains a relatively higher force until it is finally relaxed (Abbot & Aubert, 1952; Julian & Morgan, 1979; Edman et al. 1982; Sugi & Tsuchiya, 1988; Pinniger et al. 2006; Pun et al. 2010; Cornachione & Rassier, 2012; Rassier & Pavlov, 2012). This phenomenon of ‘residual force enhancement’ has been studied for over 60 years in skeletal muscles, but never directly investigated in cardiac muscles. Residual force enhancement is puzzling because it cannot be directly explained by the classical force–length relationship (Gordon et al. 1966) and the sliding filament theory of contraction (Huxley & Niedergerke, 1954; Huxley & Hanson, 1954), the main paradigms in the muscle field. The mechanisms responsible for residual force enhancement are still a matter of debate in the literature (Edman & Reggiani, 1984; Herzog & Leonard, 2002; Rassier et al. 2003a, b ; Pinniger et al. 2006; Minozzo & Rassier, 2010; Pun et al. 2010; Ranatunga et al. 2010; Cornachione & Rassier, 2012; Rassier & Pavlov, 2012; Cornachione et al. 2016). Recent studies suggest that residual force enhancement is regulated by the isoforms of the sarcomeric protein titin, the molecule mainly responsible for passive forces in muscles (Pinniger et al. 2006; Ranatunga et al. 2010; Cornachione & Rassier, 2012; Rassier & Pavlov, 2012; Cornachione et al. 2016). However, this hypothesis has not yet been subjected to rigorous experimental tests.
In this paper, we examined residual force enhancement and its relation to titin molecules in rabbit skeletal psoas myofibrils (N2A titin isoforms; Neagoe et al. 2003) and cardiac papillary myofibrils (predominantly N2B titin isoforms; Linke & Fernandez, 2002; Neagoe et al. 2003; Fujita et al. 2004; Lahmers et al. 2004). We used a custom‐built atomic force microscope (custom‐AFM) (Labuda et al. 2011) to synchronize the SLs of the myofibrils in different experimental conditions and directly compare the corresponding forces at well‐controlled lengths, a procedure not shown in the literature. Myofibrils are at the optimal size‐scale for studying how myosin, actin and titin cooperate while they stay in an ordered arrangement (Månsson et al. 2015). In fibres, the action of other cellular proteins can interfere with the measurements, leading to misinterpretation of the results. In isolated molecules, the cooperative action between myosin, actin and titin cannot be directly measured.
Methods
Ethical approval
New Zealand White rabbits were humanely treated as approved by the Animal Care Committee at McGill University and the Canadian Council on Animal Care (reference number: 20122–23). Female rabbits were obtained from Charles River Canada and weighed between 2.5 and 3.5 kg. They were fed ad libitum and killed at an age between 4 months and 1 year. The euthanasia protocol used exsanguination under general anaesthesia. Premedication was given via subcutaneous injection (in mg kg−1: 0.1 glycopyrrolate, 0.5 butorphanol, 0.75 acepromazine) and anaesthetics were administered via intramuscular injection (in mg kg−1: 35 ketamine, 5 xylazine). The anaesthetic depth was determined by pedal reflex.
Preparation of the myofibrils and solutions
Myofibrils were prepared, isolated, and tested based on previously documented procedures and solutions for the skeletal myofibrils (Rassier et al. 2003a; Rassier, 2008; Minozzo & Rassier, 2010; Cornachione & Rassier, 2012) and the cardiac myofibrils (Linke et al. 1993). Table 1 summarizes the different solutions used in the experiments.
Table 1.
Chemical composition, pH and pCa of six solutions used in preparing and testing myofibrils
| Cardiac rigor | Skeletal rigor | Activation | Relaxation | Blebbistatin + relaxation | ||
|---|---|---|---|---|---|---|
| pH (adjusted by HCl or NaOH) | 7.1 | 7.0 | 7.0 | 7.0 | 7.0 | |
| pCa | – | – | 4.5 | 9.0 | 9.0 | |
| Chemicals (mm) | KCl | 5.00 | 100.60 | 52.31 | 68.68 | 68.68 |
| MgCl2.6H2O | 1.00 | 1.97 | 5.41 | 5.41 | 5.41 | |
| EGTA | 5.00 | 1.05 | 7.20 | 7.20 | 7.20 | |
| Tris | – | 50.02 | – | – | – | |
| Tes | 10.00 | – | – | – | – | |
| Glucose | 7.00 | – | – | – | – | |
| NaCl | 132.00 | – | – | – | – | |
| Imidazole | – | – | 20.27 | 20.27 | 20.27 | |
| ATP | – | – | 6.17 | 6.10 | 6.10 | |
| PCr | – | – | 18.97 | 18.97 | 18.97 | |
| CaCl2 | – | – | 7.00 | 0.014 | 0.014 | |
| Blebbistatin (+/−) | – | – | – | – | 0.08 |
Note that the chemicals in the cardiac rigor solution are different from those of the skeletal rigor solution in an attempt to increase the yield of isolated cardiac myofibrils, as previously documented (Linke et al. 1993). A software program calculated the concentration of chemicals in the rest of the solutions (Fabiato, 1988).
After the rabbits were killed, pieces were obtained from their skeletal psoas muscles (2–3 cm long), and cardiac papillary muscles (4–6 mm long). The pieces were placed at 0°C for 4 h with a mixture of skeletal rigor solution or cardiac rigor solution for each of the respective muscles, and protease inhibitors to avoid protein degradation (Protease Inhibitor Cocktail Tablets, Roche Diagnostics, Indianapolis, IN, USA). The mixtures were then refreshed and an equal amount of glycerol solution was added for 15 h at 0°C. Afterwards, the solutions and glycerol were refreshed again and stored for at least 1 week at −20°C. This procedure chemically permeabilized the muscles.
On the day of the experiments, skeletal psoas muscles were placed in the skeletal rigor solution and were defrosted at 4°C for at least an hour. Pieces of muscle 2–3 mm long were then excised and homogenized three times at 12,000 r.p.m. for 5 s, and thee times at 28,000 r.p.m. for 3 s (VWR Power AHS250 homogenizer with a 5 mm generator, Radnor, PA, USA). In the case of the cardiac papillary muscles, 2–3 mm pieces were excised and defrosted for 30 min at 4°C in a mixture of cardiac rigor solution, protease inhibitors and 0.05% of Triton X‐100 detergent. After defrosting, the muscles were moved to cardiac rigor solution for approximately 30 min before they were homogenized three times at 26,000 r.p.m. for 7 s. Triton X‐100 and the faster homogenization protocol increased the yield of isolated myofibrils. Triton X‐100 was used at a concentration at least 10 times lower than previously documented (Linke et al. 1993) to reduce any damage to the muscles. One of the advantages of using myofibrils that are isolated after muscle bundles are permeabilized is that they can be activated by an increase of Ca2+ concentration in the media, without the need for electrical stimulation.
During experiments, rigor solution was completely washed out using relaxation solution with a high concentration of ATP and a low concentration of Ca2+. Two other solutions were used during the experiments: activation solution containing a high Ca2+ concentration and ATP, and Blebbistatin solution previously mixed with relaxation solution. Dimethylformamide was initially used to dissolve Blebbistatin to a concentration of 160 μm, and then the mixture was diluted with relaxation solution to a final concentration of 80 μm. Blebbistatin is a photosensitive chemical that inhibits myosin II from interacting with actin and is used in the dark or with a red light filter (Sakamoto et al. 2005; Minozzo & Rassier, 2010; Cornachione & Rassier, 2012).
Instrumentation
Figure 1 shows a schematic illustration of the different components of the custom‐AFM (Labuda et al. 2011). Isolated myofibrils were placed in a thermally controlled chamber filled with relaxation solution and maintained at 10°C. An inverted microscope with phase‐contrast illumination was used to identify myofibrils that were not damaged and to measure the initial sarcomere length of the myofibrils, based on the contrast between the dark bands of myosin (A‐bands) and the light bands of actin (I‐bands). A myofibril or a small bundle of myofibrils was then glued (3145 RTV MIL‐A‐46146 Adhesive, Dow Corning, Auburn, MI, USA) and suspended between the tips of a rigid glass micro‐needle and a silicon atomic force cantilever (ATEC‐CONTPt‐20, Nanosensors, Watsonville, CA, USA). A laser‐tracking system monitored the deflection of the cantilevers in response to the force exerted by the myofibrils. The motion of the micro‐needle was controlled by a piezo‐electric controller. The micro‐needle motion and the cantilever deflection were used to calculate changes in the sarcomere length and the force produced by the myofibrils. Finally, the myofibrils were exposed to different chemical solutions using a micro‐fluidic perfusion system connected to a double‐barrel pipette (Labuda et al. 2011).
Figure 1. Custom‐built atomic force microscope components and basic functioning principles.
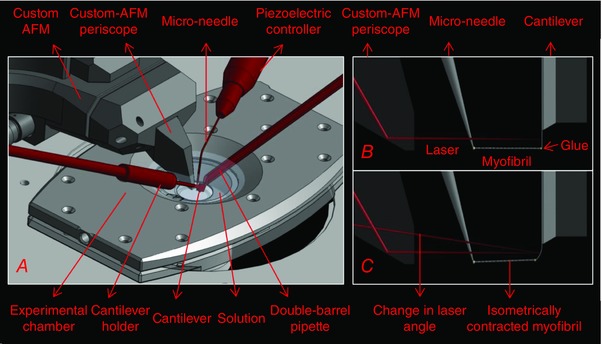
A, schematic diagram of the position of the micro‐needle, cantilever and double‐barrel pipette within the custom‐AFM. B, side‐view showing a myofibril attached between the micro‐needle and cantilever with the laser aligned onto the cantilever surface. C, side‐view showing a change in laser angle as a myofibril contracts isometrically and the cantilever can no longer bend.
Force and sarcomere length calculations
The force applied by a single sarcomere is equivalent to that of a single myofibril and was calculated using
| (1) |
where is the force per myofibril as a function of time (t), is the number of myofibrils in a tested bundle, is the displacement of the cantilever tip (sampling rate: 0.2 MHz averaged over 1280 data points), and k C is the stiffness of the silicon cantilever. The cantilever stiffness was determined either by measuring the cantilever dimensions through scanning electron microscope images (Hopcroft et al. 2010; Lübbe et al. 2012), or by constructing force‐bending curves using cantilevers previously calibrated with the scanning electron microscope images (Lübbe et al. 2012). Four different cantilevers were used in this study (stiffness: 46.5, 66.2, 130.4 and 40.3 nN μm−1).
The sarcomere length was calculated using
| (2) |
where represents the average sarcomere length as a function of time and captures the central tendency of the sarcomeres given any non‐uniformity in the sarcomere length behaviour. When we refer to sarcomere length in this paper, we imply the average sarcomere length of the different sarcomeres in the myofibril. Moreover, M(0) is the myofibril length at rest, c(0) is the initial position of the cantilever tip, is the input motion of the micro‐needle, n(0) is the initial position of the micro‐needle, and is the number of sarcomeres within the myofibril.
The random errors in and were minimized in four ways. First, cantilever displacement was calibrated before the start of each experimental set using the laser‐tracking system. Second, the micro‐needle was at least 20 times thicker than the cantilever and did not deflect during the experiments. Third, the micro‐needle was controlled by a piezoelectric controller with resolution greater than the noise of the system and did not limit the detectability of the myofibril response. Fourth, myofibrils with a large number of sarcomeres were studied so that the SL could better represent the central tendencies of the sarcomeres, despite any non‐uniformity in behaviour.
Experimental protocol
Residual force enhancement was measured using three consecutive tests on the same myofibrils. In all three tests, myofibrils were first placed in relaxation solution and then held taut at a resting position with no tensile force. In the first test (Test 1), myofibrils were stretched (10 μm s−1) to a desired SL and then fully activated. Consequently, the myofibrils contracted and were held isometrically as the force from the myofibrils could no longer bend the cantilever. Accordingly, the SL dropped to a point referred to in this study as the ‘synchronization SL’. The myofibrils were then relaxed and finally shortened back to the resting position. In the second test (Test 2), the myofibrils were stretched (10 μm s−1) to a shorter SL than that of the first test, then fully activated. After an isometric contraction and while still activating, the myofibrils were stretched (450 μm s−1) to bring the SL to the synchronization SL. The myofibrils were then relaxed and shortened back to the resting position. In the third test (Test 3), the myofibrils were stretched (10 μm s−1) to bring the SL to the synchronization SL and then shortened back to the resting position. Forces were compared between the three tests when the myofibrils stabilized at the synchronization SL.
We chose to use synchronization SLs around 2.85 μm for the skeletal myofibrils and around 2.00 μm for the cardiac myofibrils because at these lengths the passive forces start to develop and are directly associated with titin. In cardiac muscles, the passive force is determined mainly by titin at short‐to‐intermediate physiological SLs, like the ones we investigated in this study, whereas extracellular collagen fibres dominate the stiffness and passive force development at higher SLs (Linke et al. 1994; Granzier & Irving, 1995; Wu et al. 2000; Linke & Fernandez, 2002). Therefore, we used a length in both muscles in which titin starts to play a role, without invoking other components that can develop passive force (as indicated by the passive forces produced by the myofibrils during passive stretches that we show in the Results section). Furthermore, when we stretched activated myofibrils to very long lengths (e.g. sarcomere lengths beyond 2.4–2.5 μm for the cardiac myofibrils), they were easily damaged and could compromise the reproducibility of the results.
To identify if titin regulates residual force enhancement, the same myofibrils were treated with Blebbistatin and relaxation solution for approximately 40 min under a red light filter and then the three previous tests were repeated in the same order (Tests 4–6). Blebbistatin hindered myosin and actin interactions and prevented myofibril contraction.
Procedure to identify titin isoforms
To identify the titin isoforms of cardiac and skeletal muscles, an electrophoresis agarose gel was performed, as previously described (Warren et al. 2003; Hudson et al. 2011). Briefly, cardiac papillary and skeletal psoas muscles were incubated for 1 h at 4°C in two solutions: relaxation solution and Blebbistatin mixed with relaxation solution (80 μm). The muscles were pulverized and the yield was solubilized at 60°C in a solution that mixed protease inhibitors (Protease Inhibitor Cocktail Tablets, Roche Diagnostics, indianapolis, IN, USA), urea buffer (in mm: 8000 urea, 2000 thiourea, 104 SDS, 75 DTT, 50 Tris‐HCl), and an equal amount of glycerol. Next, 1% agarose gels were performed at 15 mA per gel for 3.2 h at 4°C using a Standard Vertical Unit (Hoefer SE 600, Hoefer Inc., Holliston, MA, USA). The gels were then stained using Coomassie brilliant blue.
Statistical analysis
Forces per myofibril and SLs from the different conditions were grouped then compared using a one‐way analysis of variance (ANOVA) for repeated measures and a post hoc analysis using a Holm–Sidak test (statistical package: SigmaPlot 13, Systat Software Inc., San Jose, CA, USA). A significance value of 5% (P < 0.05) was adopted in all comparisons. In some cases, data sets were summarized using box plots showing the median ± interquartile range (box), the ± range (whiskers), and the outliers as values greater than 1.5 times the length of the box.
Results
Test conditions
Using myofibril bundles or single myofibrils when possible, experiments were conducted on 8 skeletal psoas samples and 10 cardiac papillary samples. Figure 2 shows an example and summary of the initial conditions for all the tested myofibrils. Note that multiple images were used to determine the initial conditions and each of these images focused on different parts of the myofibrils. Figure 3 summarizes the most important SL conditions used during the experiments. Notably, the SLs are within the physiological range that invokes the titin molecules for the skeletal myofibrils and cardiac myofibrils. Furthermore, the absolute error in synchronizing SLs was kept to a minimum and experiments with larger deviations than those shown were discarded.
Figure 2. Summary of initial conditions in the experiments with myofibrils.
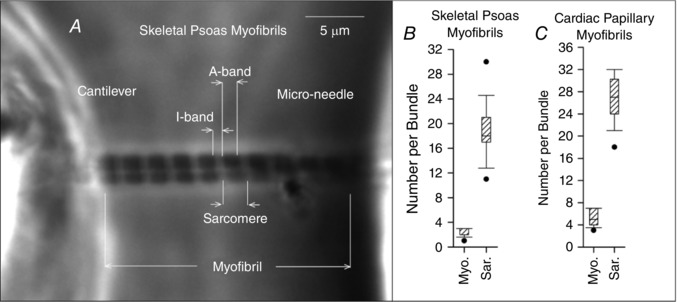
A, example of a bundle of 2 skeletal psoas myofibrils suspended between a cantilever and micro‐needle with a resting SL of 2.16 μm. To the right of the image, box plots summarize the number of myofibrils and sarcomeres per tested bundle for the skeletal psoas myofibrils (B) and the cardiac papillary myofibrils (C).
Figure 3. Box plots of the skeletal psoas myofibrils (A) and cardiac papillary myofibrils (B) summarizing the resting SL, the synchronization SL, the amount of stretch during activation, and the amount the SL deviated from the synchronization SL after the stretch during activation.
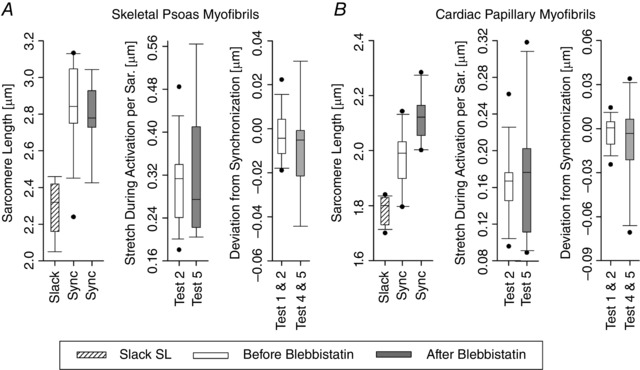
Note that 14 skeletal and 10 cardiac experimental sets were performed. After treating the myofibril with Blebbistatin, 9 skeletal and 10 cardiac experimental sets were performed.
Forces and sarcomere lengths
Plots representing the first three experiments measuring residual force enhancement are shown in Fig. 4. In these examples, myofibrils were first set to the same resting SL (annotated by ‘a’ in the SL plots). In Test 1, the myofibrils were stretched to a pre‐determined SL (b: skeletal SL of 3.18 μm and a cardiac SL of 2.17 μm), then fully activated. Consequently, the myofibrils isometrically contracted to the synchronization SL (c: skeletal SL of 2.80 μm and cardiac SL of 1.91 μm). In Test 2, the myofibrils were first stretched to a pre‐determined SL (d: skeletal SL of 2.87 μm and a cardiac SL of 1.97 μm), then activated (e: skeletal SL of 2.55 μm and cardiac SL of 1.72 μm). As the myofibrils contracted and while still activated, the myofibrils were stretched to the same synchronization SL (c) from Test 1, and then held isometrically once again. At the synchronization SL (c), the steady‐state force in the skeletal myofibrils was 3.62% higher relative to the force produced during the isometric contraction from Test 1. The cardiac myofibrils showed no increase in force. In Test 3, the myofibrils were stretched to the same synchronization SL (c) to measure the passive forces of the myofibrils.
Figure 4. Plots representing the first three tests on a skeletal psoas myofibril (A) and a cardiac papillary myofibril (B).
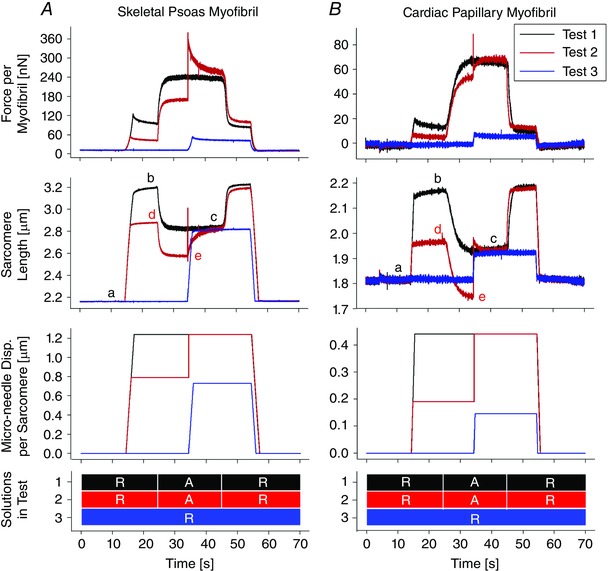
The SL plots are annotated with letters a–e with the synchronization SL (c) denoting the period when forces were compared. The micro‐needle input and solutions exchanged are plotted to clarify the time period they were applied. ‘R’ and ‘A’ indicate the use of relaxation solution and activation solution, respectively.
Plots representing the next three experiments to identify if titin regulates residual force enhancement are shown in Fig. 5. In these examples, myofibrils were first set to the same resting SL from the first three tests and then treated with Blebbistatin for approximately 40 min to hinder myosin and actin interactions. In Test 4, the myofibrils were stretched to a pre‐determined SL (b: skeletal SL of 2.91 μm and a cardiac SL of 2.16 μm), then fully activated. The myofibrils experienced minimal to no change in SL at the synchronization SL (c). In Test 5, the myofibrils were stretched to a pre‐determined SL (d: skeletal SL of 2.64 μm and a cardiac SL of 1.97 μm), then fully activated. The myofibrils again experienced minimal to no change in SL (e). While still activating, the myofibrils were stretched to the synchronization SL (c), and then held isometrically. At the synchronization SL (c), the steady‐state force in the skeletal myofibrils was 30.13% higher relative to the force produced during full activation from Test 4. The cardiac myofibrils showed no such increase in force. In Test 6, the myofibrils were stretched to the same synchronized SL (c) to again verify the passive forces of the myofibrils.
Figure 5. Plots representing the additional three tests after inhibiting myosin and actin interactions on a skeletal psoas myofibril (A) and a cardiac papillary myofibril (B).

The SL plots are annotated with letters a–e with the synchronization SL (c) denoting the period when all forces were compared. The micro‐needle input and solutions exchanged are plotted to clarify the time period they were applied. ‘R’ and ‘A’ indicate the use of relaxation solution and activation solution, respectively.
The synchronization SL and the corresponding forces were statistically compared for all the tested myofibrils. Tests 1 and 2 were individually grouped and compared and, similarly, Tests 4 and 5 were grouped and compared (Fig. 6).
Figure 6. Means ± SEM of the force per myofibril and the SL at the synchronization SL for all tested skeletal psoas myofibrils (A) and cardiac papillary myofibrils (B).
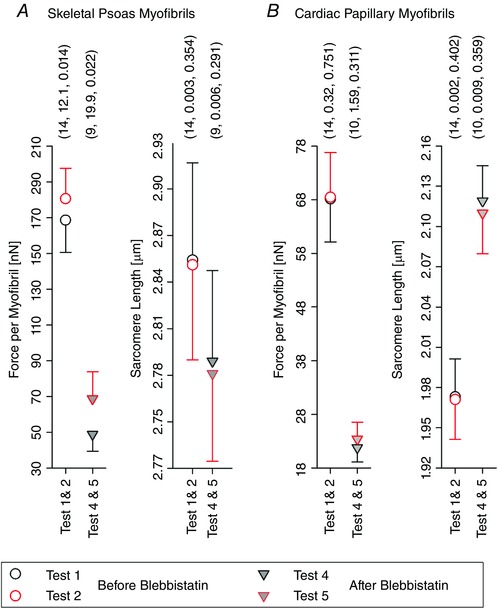
The error bars were plotted by showing the upper half for the upper symbol and the lower half for the lower symbol. Three parenthesized values are shown above each group – sequentially: number of observations, difference of mean values, and P value arising from the one‐way ANOVA for repeated measures.
In all conditions, the synchronization SLs were not statistically different. However, the forces after stretch were statistically different for the skeletal myofibrils, but not the cardiac myofibrils. Skeletal myofibrils showed an average increase of 12.1 nN (9.0%) before the Blebbistatin treatment and 19.9 nN (29.5%) after the Blebbistatin treatment.
Myofibril fatigue and effectiveness of the Blebbistatin treatment
Experiments were further analysed to determine if the myofibrils fatigued during the experiments. The forces and SLs were extracted from any passive stretches (stretches during relaxation) or from any activation (contraction without stretching). Using the passive and activation responses, a force–SL relationship for each myofibril was constructed then grouped into responses before and after the treatment with Blebbistatin. Each group was fitted with one of two linear regression models. The first model correlated the force per myofibril due to passive stretching, F P, with the SL after the application of the passive stretch, S P, using
| (3) |
where k P is the passive stiffness of the myofibril, S P(0) is the SL at rest, and F P(0) is the passive force at the resting SL (set to zero as the myofibrils did not experience any forces at rest). The second model correlated the force per myofibril due to activation, F A, to the SL after the activation, S A, using
| (4) |
where k A is the activation stiffness of the myofibril and F A(0) is the force per myofibril corresponding to the activation that would occur at the resting SL, S P(0). Figure 7 shows typical examples of the force–SL relationship and models from a skeletal and cardiac myofibril. In both cases the passive responses were not different before and after Blebbistatin treatment, the activation responses had an almost complete drop in force after the Blebbistatin treatment, and all passive and activation responses did not significantly decrease in force with an increase in SL.
Figure 7. Plots representing the force–SL relationship and the linear regression models for a skeletal psoas myofibril (A) and a cardiac papillary myofibril (B).
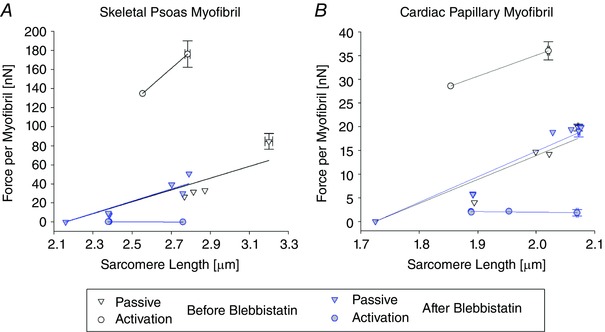
The passive and activation responses are shown before Blebbistatin treatment, Tests 1–3, and after Blebbistatin treatment, Tests 4–6. In some cases, the means ± SEM are given instead of a single data point when a test was repeated without modifying the input motion of the micro‐needle.
The model parameters depicted in eqns (3) and (4) were grouped for all tested myofibrils, and were compared before and after the Blebbistatin treatment (Fig. 8).
Figure 8. Means ± SEM of the passive stiffness (k P), activation stiffness (k A), and the activation force per myofibril expected at the resting SL (F A(0)) from all tested skeletal psoas myofibrils (A) and cardiac papillary myofibrils (B).
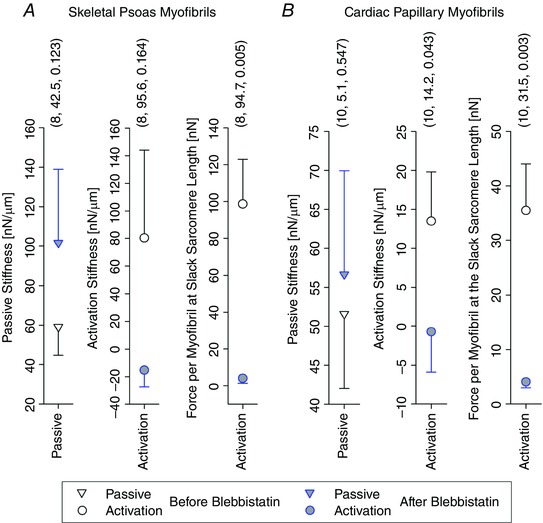
These values were grouped for all tests before the Blebbistatin treatment, Tests 1–3, and all tests after the Blebbistatin treatment, Tests 4–6. The error bars were plotted by showing the upper half for the upper symbol and the lower half for the lower symbol. Three parenthesized values are shown above each group – sequentially: number of observations, difference of mean values, and P value arising from the one‐way ANOVA for repeated‐measures.
The passive stiffness was not statistically different before and after the Blebbistatin treatment. The activation stiffness and the expected force at the resting SL were statistically different and showed an almost complete drop in force after the Blebbistatin treatment.
Synchronization of the sarcomere lengths
Forces can be meaningfully compared when the corresponding SLs between tests are accurately synchronized. However, this can be challenging because changes in SL can vary between myofibrils from the same muscle sample and can even vary between tests on the same myofibril. Furthermore, changes in SL depend on the condition of the myofibrils after multiple cycles of testing. Three techniques were used to overcome these challenges. First, the synchronized length was determined only after measuring the response from the activation (Tests 1 and 4 from both Figs 4 and 5). Second, if the myofibrils did not respond as expected to micro‐needle motion, the micro‐needle input was adjusted and the tests were repeated until the SLs synchronized. Third, if the myofibrils were fatigued, Tests 1 or 4 were repeated and a new synchronization SL was established as a reference for the remaining tests. This ensured that results at different levels of myofibril fatigue were never compared to each other.
Figure 9 demonstrates the process of synchronization and the consequences of mismatching synchronization. In these examples, Test 4 was used as the synchronization SL at (c). Test 5‐II was then conducted and did not match well to the synchronization SL. The test was then repeated in Test 5‐I after adjusting the micro‐needle input and matching the synchronization SL. In the skeletal myofibril, the test that well synchronized (Test 5‐I) deviated by −3.1% from the synchronization SL and had a 28.1% increase in steady‐state force. In contrast, the test that did not synchronize (Test 5‐II) deviated by −10.1% from the synchronization SL and had a significant difference in force. In the cardiac myofibril, the test that did not synchronize (Test 5‐II) deviated by −19.7% from the synchronization SL and had a 27.5% decrease in steady‐state force. In contrast, the test that well synchronized (Test 5‐I) deviated by 1.75% from the synchronization SL and had no significant difference in force. Hence, in both cases, mismatches in the synchronization SL could have wrongly identified the amount of residual force enhancement.
Figure 9. Examples of experiments repeated to synchronize SLs between tests for a skeletal psoas myofibril (A) and a cardiac papillary myofibril (B).

The SL plots are annotated with letters a–e with the synchronization SL (c) denoting the period when all forces were compared. Test 5‐I (red) synchronized with Test 4 at ‘c’, unlike Test 5‐II (green). ‘R’ and ‘A’ indicate the use of relaxation solution and activation solution, respectively. Note that the micro‐needle input at ‘e’ was increased to synchronize the SLs.
Titin isoforms
Using the electrophoresis agarose gel, titin isoform bands were identified for the skeletal psoas and cardiac papillary muscles with reference to previous studies (Linke & Fernandez, 2002; Neagoe et al. 2003; Fujita et al. 2004; Lahmers et al. 2004) (Fig. 10). The cardiac titin isoforms (predominantly N2B) had lower molecular weight and higher mobility through the gel relative to the skeletal isoforms (N2A). Furthermore, the treatment with Blebbistatin did not change the mobility of the titin isoforms through the gel.
Figure 10. Relevant sections of the electrophoresis agarose gel for each of the skeletal psoas muscles and cardiac papillary muscles.
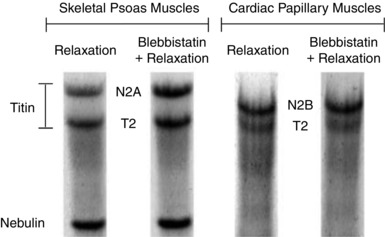
Each muscle was incubated in two columns with different solutions: a relaxation solution and a Blebbistatin solution mixed with relaxation solution. The gel shows the skeletal titin isoform (N2A) and the cardiac titin isoform (N2B). Note that the T2 bands result from normal degradation of titin (Linke & Fernandez, 2002; Neagoe et al. 2003; Fujita et al. 2004; Lahmers et al. 2004). Furthermore, the nebulin bands are below the T2 bands in the skeletal psoas columns, but not in the cardiac papillary columns.
Discussion
The results of this study are consistent with the claim that residual force enhancement is present and is regulated by titin in skeletal psoas myofibrils, but not cardiac papillary myofibrils. SLs were quantitatively synchronized between tests before comparing forces and reported a 0.009 μm maximum difference in the mean synchronization SL for all the tests. The force–SL relationships in our study confirmed that Blebbistatin almost completely hindered myosin and actin interactions and did not affect the passive force associated with titin, as further supported by the results of the gel electrophoresis experiments (Fig. 10).
Comparisons to other studies
When the skeletal psoas myofibrils were activated then stretched (stretches ranging from 0.18 to 0.48 μm per sarcomere), the force increased by an average of 12.1 nN (9.0%) relative to an isometric contraction at the same SL (SLs ranging from 2.24 to 3.13 μm). A similar increase in force was observed in previous studies, which used myofibrils from psoas muscles of rabbits (Pun et al. 2010; Rassier & Pavlov, 2012), psoas fibres of rabbits (Cornachione & Rassier, 2012), tibialis anterior fibres of frogs (Abbot & Aubert, 1952; Julian & Morgan, 1979; Edman et al. 1982; Sugi & Tsuchiya, 1988; Edman & Tsuchiya, 1996), skeletal fibres of dogfish (Abbot & Aubert, 1952), and flexor hallucis brevis fibres of rats (Pinniger et al. 2006). When the cardiac papillary myofibrils were activated then stretched (stretches ranging from 0.10 to 0.26 μm per sarcomere), the force was similar to that from an isometric contraction at the same SL (SLs ranging from 1.80 to 2.14 μm). No other studies with cardiac muscles were available to compare to our study. However, since our results with skeletal psoas myofibrils agree with the literature, we are confident that the results with cardiac papillary myofibrils are reproducible. It is important to note that, although the synchronization SLs used for the skeletal and cardiac papillary myofibrils are different, the comparisons are valid because both muscles are operating at a SL in which the passive force starts to develop. Comparing the muscles at similar absolute SLs would not be correct, as myofibrils would be operating in very distinct functional SLs, given the intrinsic differences in these two muscles.
After inhibiting myosin and actin interactions, the skeletal psoas myofibrils (N2A titin isoforms) were activated then stretched (stretches ranging from 0.22 to 0.56 μm per sarcomere) and the force of the myofibrils increased by an average of 19.9 nN (29.5%) relative to complete activation at the same SL (SLs ranging from 2.43 to 3.04 μm). A similar increase in force was observed in previous studies which used psoas myofibrils of rabbits (Cornachione et al. 2016), soleus myofibrils of rabbits (Cornachione et al. 2016), psoas fibres of rabbits (Cornachione & Rassier, 2012), soleus fibres of mice (Labeit et al. 2003), and flexor hallucis brevis fibres of rats (Pinniger et al. 2006). When the cardiac papillary myofibrils (predominantly N2B titin isoforms) were activated then stretched (stretches ranging from 0.09 to 0.32 μm per sarcomere), the force was similar to that from a complete activation at the same SL (SLs ranging from 2.00 to 2.29 μm). Similar force behaviour was observed in previous studies using ventricle myofibrils of rabbits (Cornachione et al. 2016) and trabeculae fibres of rat (Fujita et al. 2004). Conversely, a study of bovine left atrium fibres (Fujita et al. 2004) did show an increase in force. However, larger animals such as bovine are predominantly composed of N2BA isoforms of titin which are different from the predominantly N2B isoforms of titin in rats and rabbit (rabbit muscles were used in this study) (Linke & Fernandez, 2002; Neagoe et al. 2003; Fujita et al. 2004; Lahmers et al. 2004).
Potential mechanism of residual force enhancement
The results suggest that activation affects N2A titin isoforms of skeletal psoas myofibrils causing residual force enhancement after stretch, but not N2B titin isoforms of cardiac papillary myofibrils. When the myofibrils are activated, the released Ca2+ binds to the I‐band region of titin containing proline, glutamate, valine and lysine (PEVK) which are rich with glutamate domains (E‐domains) (Labeit et al. 2003). Consequently, titin would reduce its persistence length (Takahashi et al. 1992; Kolmerer et al. 1996; Tatsumi et al. 1996, 2001), increase its stiffness (Labeit et al. 2003), and produce more force when stretched (Bagni et al. 1994, 2002). The N2A titin isoforms of the skeletal psoas myofibrils have larger PEVK segments with more E‐domains than the N2B titin isoforms of the cardiac papillary myofibrils (Labeit & Kolmerer, 1995; Bang et al. 2001; Labeit et al. 2003; Fujita et al. 2004). Therefore, skeletal psoas myofibrils are more likely to show an increase of titin force during activation than cardiac papillary myofibrils.
This mechanism aligns with recent studies that show static tension and directly associate it with residual force enhancement in skeletal muscles (Pinniger et al. 2006; Cornachione & Rassier, 2012; Cornachione et al. 2016), but not in cardiac muscles (Cornachione et al. 2016). The studies show that N2A titin isoforms of skeletal muscles induced static tension in titin molecules of mice (Labeit et al. 2003), myofibrils of rabbits (Cornachione et al. 2016), fibres of rabbits (Cornachione & Rassier, 2012), and fibres of rats (Pinniger et al. 2006), but not in N2B titin isoforms of cardiac muscles from myofibrils of rabbits (Cornachione et al. 2016) and fibres of rats (Fujita et al. 2004).
There is also the possibility that Ca2+ increases the binding between actin and titin, which would increase the sarcomere stiffness and consequently cause the residual force enhancement after stretch. It is known that titin binds to actin (Kellermayer & Granzier, 1996a, b ; Linke et al. 1997, 2002; Trombitas et al. 1997; Stuyvers et al. 1998; Kulke et al. 2001; Yamasaki et al. 2001) in PEVK segments (Yamasaki et al. 2001) and that Ca2+ regulates the binding (Kellermayer & Granzier, 1996b; Kulke et al. 2001; Yamasaki et al. 2001; Linke et al. 2002; Stuyvers et al. 2002). In vitro motility assays show that actin motility over myosin molecules is reduced after actin binds to titin in the presence of Ca2+ (Kellermayer & Granzier, 1996b; Yamasaki et al. 2001), which suggests an increase in the stiffness. However, several studies contradict these findings and showed an increase in actin motility over myosin in the presence of titin and Ca2+ (Stuyvers et al. 1998; Kulke et al. 2001). Another study showed that S100A1 (a soluble Ca2+‐binding protein highly concentrated in striated muscles; Kato & Kimura, 1985) hindered actin from interacting with PEVK segments of titin (Yamasaki et al. 2001), which would prevent an increase of stiffness. Ultimately, the effect of Ca2+ on titin binding to actin seems to be inconclusive and requires further investigation.
Finally, a study using molecular dynamics suggested that Ca2+ reduces the persistence length of titin by binding to immunoglobulin (Ig) segments spliced within the titin‐I‐band region (Lu et al. 1998). However, single molecule experiments did not correlate Ca2+ to a reduction in persistence length of Ig segments (Watanabe et al. 2002).
There are other potential mechanisms to explain residual force enhancement that are independent of titin. Some studies suggest that residual force enhancement is regulated by non‐uniformity in SL. According to this hypothesis, when muscles are activated and then stretched, weaker sarcomeres are passively overstretched and stronger sarcomeres contract to a shorter length and produce more force than expected (Julian & Morgan, 1979; Edman & Reggiani, 1984). However, studies have shown that non‐uniformity in SL behaviour cannot solely explain residual force enhancement (Rassier et al. 2003b).
Finally, some studies suggested that residual force enhancement is caused by an increase in the number of attached cross‐bridges between actin and myosin (Minozzo & Rassier, 2010; Ranatunga et al. 2010). However, studies with skeletal muscles that hindered myosin and actin interactions, such as this study, still observed residual force enhancement (Pinniger et al. 2006; Cornachione & Rassier, 2012).
Additional information
Competing interests
None declared.
Authors contributions
The research was performed at the Muscle Physiology and Biophysics Laboratory of McGill University. N.S. assembled, improved, and validated the custom‐built atomic force microscope, helped design the study and draft the manuscript, performed the experiments and data analyses; A.C. assisted with the experiments with the cardiac myofibrils and helped draft the manuscript. F.L. performed the electrophoresis agarose gel and helped draft the manuscript. S.V. co‐supervised the study and helped design it and draft the manuscript; D.E.R. conceived and co‐supervised the study and helped design the experiments and draft the manuscript. All authors gave approval for publication, agreed to be accountable for all aspects of the work, and are qualified for authorship. All those who qualify for authorship are listed.
Funding
The Canadian Government funded the research through the Canadian Institutes of Health Research (no. 125898 to D.E.R.), the Natural Sciences and Engineering Research Council of Canada (no. 2016‐05317 to D.E.R.) and the Canada Research Chairs program (no. 241488 to D.E.R.). F.S.L. was a recipient of a scholarship from CNPq (Brazil).
Acknowledgements
The authors thank Tiberius Brastaviceanu for collaborating with the development of new software for the experiments, and Dr Fabio Minozzo and Albert Kalganov for advising and assisting throughout the project.
References
- Abbot BC & Aubert XM (1952). The force exerted by active striated muscle during and after change of length. J Physiol 117, 77–86. [PMC free article] [PubMed] [Google Scholar]
- Bagni MA, Cecchi G, Colombini B & Colomo F (2002). A non‐cross‐bridge stiffness in activated frog muscle fibers. Biophys J 82, 3118–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni MA, Cecchi G, Colomo F & Garzella P (1994). Development of stiffness precedes cross‐bridge attachment during the early tension rise in single frog muscle fibres. J Physiol 481, 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H & Labeit S (2001). The complete gene sequence of titin, expression of an unusual approximately 700‐kDa titin isoform, and its interaction with obscurin identify a novel Z‐line to I‐band linking system. Circ Res 89, 1065–1072. [DOI] [PubMed] [Google Scholar]
- Cornachione AS, Leite F, Bagni MA & Rassier DE (2016). The increase in non‐cross‐bridge forces after stretch of activated striated muscle is related to titin isoforms. Am J Physiol Cell Physiol 310, C19–C26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornachione AS & Rassier DE (2012). A non‐cross‐bridge, static tension is present in permeabilized skeletal muscle fibers after active force inhibition or actin extraction. Am J Physiol Cell Physiol 302, C566–C574. [DOI] [PubMed] [Google Scholar]
- Edman KAP, Elzinga G & Noble MIM (1982). Residual force enhancement after stretch of contracting frog single muscle fibers. J Gen Physiol 80, 769–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman KAP & Reggiani C (1984). Redistribution of sarcomere length during isometric contraction of frog muscle fibres and its relation to tension creep. J Physiol 351, 169–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman KAP & Tsuchiya T (1996). Strain of passive elements during force enhancement by stretch in frog muscle fibres. J Physiol 490, 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A (1988). Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol 157, 378–417. [DOI] [PubMed] [Google Scholar]
- Fujita H, Labeit D, Gerull B, Labeit S & Granzier HL (2004). Titin isoform‐dependent effect of calcium on passive myocardial tension. Am J Physiol Heart Circ Physiol 287, H2528–H2534. [DOI] [PubMed] [Google Scholar]
- Gordon AM, Huxley AF & Julian FJ (1966). The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol 184, 170–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier HL & Irving TC (1995). Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68, 1027–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog W & Leonard TR (2002). Force enhancement following stretching of skeletal muscle: a new mechanism. J Exp Biol 205, 1275–1283. [DOI] [PubMed] [Google Scholar]
- Hopcroft MA, Nix WD & Kenny TW (2010). What is the Young's modulus of silicon? J Microelectromech Syst 19, 229–238. [Google Scholar]
- Hudson B, Hidalgo C, Saripalli C & Granzier H (2011). Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction‐induced diastolic dysfunction. Circ Res 109, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley AF & Niedergerke R (1954). Structural changes in muscle during contraction: interference microscopy of living muscle fibres. Nature 173, 971–973. [DOI] [PubMed] [Google Scholar]
- Huxley H & Hanson J (1954). Changes in the cross‐striations of muscle during contraction and stretch and their structural interpretation. Nature 173, 973–976. [DOI] [PubMed] [Google Scholar]
- Julian FJ & Morgan DL (1979). The effect on tension of non‐uniform distribution of length changes applied to frog muscle fibres. J Physiol 293, 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K & Kimura S (1985). S100ao (αα) protein is mainly located in the heart and striated muscles. Biochim Biophys Acta 842, 146–150. [DOI] [PubMed] [Google Scholar]
- Kellermayer MSZ & Granzier HL (1996a). Elastic properties of single titin molecules made visible through fluorescent F‐actin binding. Biochem Biophys Res Commun 221, 491–497. [DOI] [PubMed] [Google Scholar]
- Kellermayer MSZ & Granzier HL (1996b). Calcium‐dependent inhibition of in vitro thin‐filament motility by native titin. FEBS Lett 380, 281–286. [DOI] [PubMed] [Google Scholar]
- Kolmerer B, Olivieri N, Witt CC, Herrmann BG & Labeit S (1996). Genomic organization of M line titin and its tissue‐specific expression in two distinct isoforms. J Mol Biol 256, 556–563. [DOI] [PubMed] [Google Scholar]
- Kulke M, Fujita‐Becker S, Rostkova E, Neagoe C, Labeit D, Manstein DJ, Gautel M & Linke WA (2001). Interaction between PEVK‐titin and actin filaments: origin of a viscous force component in cardiac myofibrils. Circ Res 89, 874–881. [DOI] [PubMed] [Google Scholar]
- Labeit D, Watanabe K, Witt C, Fujita H, Wu Y, Lahmers S, Funck T, Labeit S & Granzier H (2003). Calcium‐dependent molecular spring elements in the giant protein titin. Proc Natl Acad Sci USA 100, 13716–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit S & Kolmerer B (1995). Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270, 293–296. [DOI] [PubMed] [Google Scholar]
- Labuda A, Brastaviceanu T, Pavlov I, Paul W & Rassier DE (2011). Optical detection system for probing cantilever deflections parallel to a sample surface. Rev Sci Instrum 82, 013701. [DOI] [PubMed] [Google Scholar]
- Lahmers S, Wu Y, Call DR, Labeit S & Granzier H (2004). Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 94, 505–513. [DOI] [PubMed] [Google Scholar]
- Linke WA, Bartoo ML & Pollack GH (1993). Spontaneous sarcomeric oscillations at intermediate activation levels in single isolated cardiac myofibrils. Circ Res 73, 724–734. [DOI] [PubMed] [Google Scholar]
- Linke WA & Fernandez JM (2002). Cardiac titin: molecular basis of elasticity and cellular contribution to elastic and viscous stiffness components in myocardium. Muscle Res Cell Motil 23, 483–497. [DOI] [PubMed] [Google Scholar]
- Linke WA, Ivemeyer M, Labeit S, Hinssen H, Ruegg JC & Gautel M (1997). Actin‐titin interaction in cardiac myofibrils: probing a physiological role. Biophys J 73, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke WA, Kulke M, Li H, Fujita‐Becker S, Neagoe C, Manstein DJ, Gautel M & Fernandez JM (2002). PEVK domain of titin: an entropic spring with actin‐binding properties. J Struct Biol 137, 194–205. [DOI] [PubMed] [Google Scholar]
- Linke WA, Popov VI & Pollack GH (1994). Passive and active tension in single cardiac myofibrils. Biophys J 67, 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Isralewitz B, Krammer A, Vogel V & Schulten K (1998). Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys J 75, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübbe J, Doering L & Reichling M (2012). Precise determination of force microscopy cantilever stiffness from dimensions and eigenfrequencies. Meas Sci Technol 23, 045401. [Google Scholar]
- Månsson A, Rassier D & Tsiavaliaris G (2015). Poorly understood aspects of striated muscle contraction. Biomed Res Int 2015, 245154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minozzo FC & Rassier DE (2010). Effects of blebbistatin and Ca2+ concentration on force produced during stretch of skeletal muscle fibers. Am J Physiol Cell Physiol 299, C1127–C1135. [DOI] [PubMed] [Google Scholar]
- Neagoe C, Opitz CA, Makarenko I & Linke WA (2003). Gigantic variety: expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J Muscle Res Cell Motil 24, 175–189. [DOI] [PubMed] [Google Scholar]
- Pinniger GJ, Ranatunga KW & Offer GW (2006). Crossbridge and non‐crossbridge contributions to tension in lengthening rat muscle: force‐induced reversal of the power stroke. J Physiol 573, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun C, Syed A & Rassier DE (2010). History‐dependent properties of skeletal muscle myofibrils contracting along the ascending limb of the force‐length relationship. Proc Biol Sci 277, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranatunga KW, Roots H, Pinniger GJ & Offer GW (2010). Crossbridge and non‐crossbridge contributions to force in shortening and lengthening muscle. Adv Exp Med Biol 682, 207–221. [DOI] [PubMed] [Google Scholar]
- Rassier DE (2008). Pre‐power stroke cross bridges contribute to force during stretch of skeletal muscle myofibrils. Proc Biol Sci 275, 2577–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassier DE, Herzog W & Pollack GH (2003a). Dynamics of individual sarcomeres during and after stretch in activated single myofibrils. Proc Biol Sci 270, 1735–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassier DE, Herzog W, Wakeling J & Syme DA (2003b). Stretch‐induced, steady‐state force enhancement in single skeletal muscle fibers exceeds the isometric force at optimum fiber length. J Biomech 36, 1309–1316. [DOI] [PubMed] [Google Scholar]
- Rassier DE & Pavlov I (2012). Force produced by isolated sarcomeres and half‐sarcomeres after an imposed stretch. Am J Physiol Cell Physiol 302, C240–C248. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Limouze J, Combs CA, Straight AF & Sellers JR (2005). Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry 44, 584–588. [DOI] [PubMed] [Google Scholar]
- Stuyvers BD, McCulloch AD, Guo JQ, Duff HJ & ter Keurs HEDJ (2002). Effect of stimulation rate, sarcomere length and Ca2+ on force generation by mouse cardiac muscle. J Physiol 544, 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuyvers BD, Miura M, Jin J‐P & ter Keurs HEDJ (1998). Ca2+‐dependence of diastolic properties of cardiac sarcomeres: involvement of titin. Prog Biophys Mol Biol 69, 425–443. [DOI] [PubMed] [Google Scholar]
- Sugi H & Tsuchiya T (1988). Stiffness changes during enhancement and deficit of isometric force by slow length changes in frog skeletal muscle fibres. J Physiol 407, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Hattori A, Tatsumi R & Takai K (1992). Calcium‐induced splitting of connectin filaments into β‐connectin and a 1,200‐kDa subfragment. J Biochem 111, 778–782. [DOI] [PubMed] [Google Scholar]
- Tatsumi R, Hattori A & Takahashi K (1996). Splitting of connectin/titin filaments into β‐connectin/T2 and a 1,200‐kDa subfragment by 0.1 mm calcium ions. Adv Biophys 33, 65–77. [PubMed] [Google Scholar]
- Tatsumi R, Maeda K, Hattori A & Takahashi K (2001). Calcium binding to an elastic portion of connectin/titin filaments. J Muscle Res Cell Motil 22, 149–162. [DOI] [PubMed] [Google Scholar]
- Trombitas K, Greaser ML & Pollack GH (1997). Interaction between titin and thin filaments in intact cardiac muscle. J Muscle Res Cell Motil 18, 345–351. [DOI] [PubMed] [Google Scholar]
- Warren CM, Krzesinski PR & Greaser ML (2003). Vertical agarose gel electrophoresis and electroblotting of high‐molecular‐weight proteins. Electrophoresis 24, 1695–1702. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Muhle‐Goll C, Kellermayer MSZ, Labeit S & Granzier H (2002). Different molecular mechanics displayed by titin's constitutively and differentially expressed tandem Ig segments. J Struct Biol 137, 248–258. [DOI] [PubMed] [Google Scholar]
- Wu Y, Cazorla O, Labeit D, Labeit S & Granzier H (2000). Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J Mol Cell Cardiol 32, 2151–2161. [DOI] [PubMed] [Google Scholar]
- Yamasaki R, Berri M, Wu Y, Trombitás K, McNabb M, Kellermayer MSZ, Witt C, Labeit D, Labeit S, Greaser M & Granzier H (2001). Titin–actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys J 81, 2297–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]