

Abstract
Key points
Hypercapnia or parafacial respiratory group (pFRG) disinhibition at normocapnia evokes active expiration in rats by recruitment of pFRG late‐expiratory (late‐E) neurons.
We show that hypercapnia simultaneously evoked active expiration and exaggerated glottal dilatation by late‐E synaptic excitation of abdominal, hypoglossal and laryngeal motoneurons. Simultaneous rhythmic expiratory activity in previously silent pFRG late‐E neurons, which did not express the marker of ventral medullary CO2‐sensitive neurons (transcription factor Phox2b), was also evoked by hypercapnia.
Hypercapnia‐evoked active expiration, neural and neuronal late‐E activities were eliminated by pFRG inhibition, but not after blockade of synaptic excitation.
Hypercapnia produces disinhibition of non‐chemosensitive pFRG late‐E neurons to evoke active expiration and concomitant cranial motor respiratory responses controlling the oropharyngeal and upper airway patency.
Abstract
Hypercapnia produces active expiration in rats and the recruitment of late‐expiratory (late‐E) neurons located in the parafacial respiratory group (pFRG) of the ventral medullary brainstem. We tested the hypothesis that hypercapnia produces active expiration and concomitant cranial respiratory motor responses controlling the oropharyngeal and upper airway patency by disinhibition of pFRG late‐E neurons, but not via synaptic excitation. Phrenic nerve, abdominal nerve (AbN), cranial respiratory motor nerves, subglottal pressure, and medullary and spinal neurons/motoneurons were recorded in in situ preparations of juvenile rats. Hypercapnia evoked AbN active expiration, exaggerated late‐E discharges in cranial respiratory motor outflows, and glottal dilatation via late‐E synaptic excitation of abdominal, hypoglossal and laryngeal motoneurons. Simultaneous rhythmic late‐E activity in previously silent pFRG neurons, which did not express the marker of ventral medullary CO2‐sensitive neurons (transcription factor Phox2b), was also evoked by hypercapnia. In addition, hypercapnia‐evoked AbN active expiration, neural and neuronal late‐E activities were eliminated by pFRG inhibition, but not after blockade of synaptic excitation. On the other hand, pFRG inhibition did not affect either hypercapnia‐induced inspiratory increases in respiratory motor outflows or CO2 sensitivity of the more medial Phox2b‐positive neurons in the retrotrapezoid nucleus (RTN). Our data suggest that neither RTN Phox2b‐positive nor other CO2‐sensitive brainstem neurons activate Phox2b‐negative pFRG late‐E neurons under hypercapnia to produce AbN active expiration and concomitant cranial motor respiratory responses controlling the oropharyngeal and upper airway patency. Hypercapnia produces disinhibition of non‐chemosensitive pFRG late‐E neurons in in situ preparations of juvenile rats to activate abdominal, hypoglossal and laryngeal motoneurons.
Keywords: abdominal active expiration, airway patency, late‐expiratory neurons, parafacial respiratory group, respiratory motoneurons
Key points
Hypercapnia or parafacial respiratory group (pFRG) disinhibition at normocapnia evokes active expiration in rats by recruitment of pFRG late‐expiratory (late‐E) neurons.
We show that hypercapnia simultaneously evoked active expiration and exaggerated glottal dilatation by late‐E synaptic excitation of abdominal, hypoglossal and laryngeal motoneurons. Simultaneous rhythmic expiratory activity in previously silent pFRG late‐E neurons, which did not express the marker of ventral medullary CO2‐sensitive neurons (transcription factor Phox2b), was also evoked by hypercapnia.
Hypercapnia‐evoked active expiration, neural and neuronal late‐E activities were eliminated by pFRG inhibition, but not after blockade of synaptic excitation.
Hypercapnia produces disinhibition of non‐chemosensitive pFRG late‐E neurons to evoke active expiration and concomitant cranial motor respiratory responses controlling the oropharyngeal and upper airway patency.
Abbreviations
- AbN
abdominal nerve
- aug‐E
augmenting‐expiratory
- BIC
bicuculline
- BötC
Bötzinger complex
- cNA
caudal nucleus ambiguus
- DE
duration of expiration
- DI
duration of inspiration
- E2 phase
second‐half of expiration
- GABAA
GABA receptor type A
- GLY
glycine
- HN
hypoglossal nerve
- KYN
kynurenic acid
- late‐E
late‐expiratory
- MUS
muscimol
- PFA
paraformaldehyde
- pFRG
parafacial respiratory group
- Phox2b
paired‐like homeobox 2b
- PN
phrenic nerve
- pre‐BötC
pre‐Bötzinger complex
- RLN
recurrent laryngeal nerve
- RTN
retrotrapezoid nucleus
- sEPSC
spontaneous excitatory post‐synaptic currents
- SGP
subglottal pressure
- SLN
superior laryngeal nerve
- Sp5
spinal trigeminal tract
- STRY
strychnine
- VRG
ventral respiratory group
Introduction
During baseline breathing, rats exhibit minimal expiratory motor activity (passive expiration) (Iizuka, 2011; Andrews & Pagliardini, 2015). Hypercapnia produces active expiration, i.e. a forced expulsion of air in breathing, via stimulation of abdominal expiratory motor activity (Abdala et al. 2009; Molkov et al. 2011; Huckstepp et al. 2015). In the ventral medullary brainstem, the transition from passive to active expiration in juvenile or adult rats seems to be determined by the rhythmic activity of the so‐called parafacial respiratory group (pFRG) late‐expiratory (late‐E) neurons located more laterally to, or overlapping with, the retrotrapezoid nucleus (RTN), producing late‐E discharges in the abdominal expiratory muscles (Abdala et al. 2009; Pagliardini et al. 2011; Moraes et al. 2012 a, 2014 a; Huckstepp et al. 2016). It has been proposed that the pFRG late‐E neurons, driving active expiration, sense increases in CO2/[H+] concentration (Marina et al. 2010) or are activated by the central chemoreceptors during hypercapnia (Molkov et al. 2011, 2014). On the other hand, pFRG disinhibition also produces active expiration at normocapnia (Pagliardini et al. 2011; Huckstepp et al. 2015), suggesting that pFRG late‐E neurons are actively suppressed via GABA and/or glycine neurotransmission at baseline breathing. Therefore, the mechanisms by which the pFRG late‐E neuronal activity induces active expiration during hypercapnia and how these discharges affect respiratory motor outflows are not fully understood.
Hypoglossal (HN) and superior laryngeal (SLN) nerves were also found to exhibit late‐E discharges during hypercapnia that coincide with abdominal late‐E activity (Abdala et al. 2009), suggesting that the observed late‐E discharges in abdominal nerve (AbN), HN and SLN originate from the same generator, i.e. pFRG late‐E neuronal activity (Molkov et al. 2010). This would serve to advance the control of oropharyngeal and upper airway patency, thereby timing it with AbN active expiration when respiratory demands increase due to changes in blood gases. The RTN is proposed as a major site for central CO2 chemoreception because it contains Phox2b‐positive neurons intrinsically activated by CO2/[H+] (Stornetta et al. 2006; Guyenet & Bayliss, 2015) and projects to several expiratory and inspiratory brainstem nuclei (Rosin et al. 2006). However, the brainstem (Pagliardini et al. 2011) and spinal projections of pFRG late‐E neurons to respiratory neurons/motoneurons and the physiological role of these projections have been little explored.
Using in situ perfused preparations of juvenile rats (Paton, 1996), we tested the hypothesis that hypercapnia produces AbN active expiration and concomitant cranial respiratory motor responses controlling oropharyngeal and upper airway patency by disinhibition of pFRG late‐E neurons, but not via synaptic excitation. Hypercapnia evoked AbN active expiration, exaggerated late‐E discharges in cranial respiratory motor outflows and glottal dilatation by late‐E synaptic excitation of abdominal, hypoglossal and laryngeal motoneurons. Simultaneous rhythmic late‐E activity in non‐chemosensitive (Phox2b‐negative) pFRG neurons was also evoked by hypercapnia. In addition, hypercapnia‐evoked AbN active expiration, neural and neuronal late‐E activities were eliminated by pFRG inhibition, but not after blockade of synaptic excitation. Our data suggest that, under hypercapnia, neither Phox2b‐positive RTN nor other CO2‐sensitive brainstem neurons activate Phox2b‐negative pFRG late‐E neurons in in situ preparations of juvenile rats. Instead, hypercapnia induces disinhibition of non‐chemosensitive pFRG late‐E neurons to produce active expiration and concomitant cranial respiratory motor responses controlling the oropharyngeal and upper airway patency.
Methods
Animals and ethical approval
Experiments were performed on male Wistar juvenile rats (100–150 g) from the Animal Care of the University of São Paulo, Campus of Ribeirão Preto, Brazil. The animals were maintained in standard environmental conditions (23 ± 2°C; 12/12 h dark/light cycle) with water and chow available ad libitum. All experimental protocols were approved by the Ethical Committee on Animal Experimentation of the School of Medicine of Ribeirão Preto, University of São Paulo (protocol 064/2010).
Ventral and dorsal approaches of perfused brainstem preparation
Rats were prepared for in situ perfused brainstem preparations. The animals were deeply anaesthetized with halothane (AstraZeneca do Brasil Ltda, Cotia, SP, Brazil), transected caudal to the diaphragm, exsanguinated and submerged in a cooled Ringer's solution (in mm: NaCl, 125; NaHCO3, 24; KCl, 3; CaCl2, 2.5; MgSO4, 1.25; KH2PO4, 1.25; dextrose, 10). Rats were then immediately decerebrated at the precollicular level, and thus rendered insentient, and skinned. The carotid sinus nerves were bilaterally cut to disrupt the carotid chemoreceptor afferents (McBryde et al. 2013). The efficacy of peripheral chemoreceptor denervation was confirmed by the absence of respiratory motor responses to peripheral chemoreflex activation using potassium cyanide (0.05%, 50 μl; Merck, Darmstadt, Germany). Using the ventral approach of perfused brainstem preparation (Moraes et al. 2013), the ventral medullary surface was exposed for neuronal recordings in the caudal nucleus ambiguus (cNA), RTN or pFRG and simultaneous microinjections into pFRG. In some preparations, a ventral spinal cord laminectomy was also performed for motoneuron recordings in ventral aspect of the thoracic spinal cord. Using the dorsal approach of perfused brainstem preparation (Paton, 1996), the dorsal medullary surface was exposed for motoneuron recordings in the hypoglossal nucleus and simultaneous microinjections into the pFRG. All preparations were transferred to a recording chamber and the descending aorta was cannulated and perfused with Ringer's solution containing an oncotic agent (MW 20,000; 1.25% polyethylene glycol; Sigma, St Louis, MO, USA), and continuously gassed with 5% CO2 and 95% O2 using a 520S peristaltic pump (Watson‐Marlow, Falmouth, UK). The perfusate was warmed to 31°C and filtered using a nylon mesh (pore size: 25 μm; Millipore, Billerica, MA, USA). In preparations in which we performed neural and neuronal recordings, a neuromuscular blocker was administered (vecuronium bromide, 3–4 μg ml−1; Cristália Produtos Químicos Farmacêuticos Ltda, São Paulo, Brazil).
Electrophysiological data acquisition
Respiratory motor nerves were isolated and recorded using bipolar glass suction electrodes. Left phrenic nerve (PN) activity was recorded from its central end. The SLN and HN were isolated in the cervical region, cut distally and their activity recorded. The lower thoracic spinal segment of AbN (T12) was isolated from the abdominal external oblique muscle, cut distally and its activity recorded. Recurrent laryngeal nerve (RLN) was isolated for stimulation (see below). All nerves were recorded in absolute units (μV) and analyses were performed off‐line in rectified and integrated (time constant: 50 ms) signals. Baseline PN activity was assessed by their burst frequency and duration (duration of inspiration; DI). The time intervals between consecutive PN bursts were considered the duration of expiration (DE). The duration of the late‐E component of SLN and HN preceding the PN were analysed by its relationship to the beginning of PN burst. The duration of SLN post‐inspiratory activity was also quantified. The changes in the PN and HN inspiratory amplitude were expressed as percentage values in relation to basal values. Active expiration was determined when the AbN late‐E activity at the end of the second half of expiration (E2 phase) was greater (μV) than the AbN post‐inspiratory activity.
Single‐unit extracellular recordings of RTN and pFRG late‐E neurons, respiratory medullary and spinal motoneurons (Duo 773 Electrometer; World Precision Instruments, Sarasota, FL, USA) were made using glass microelectrodes (10–30 MΩ) filled with 0.5 m sodium acetate and 1.5% biocytin (Molecular Probes, Grand Island, NY, USA) to juxtacellular labelling of recorded neurons/motoneurons (200 ms pulses of 1.0–4.0 nA at 2.5 Hz for 1–5 min) (Schreihofer & Guyenet, 1997). Blind whole cell patch clamp recordings were also performed to study the effects of hypercapnia on membrane potential trajectories and on spontaneous excitatory post‐synaptic currents (sEPSCs) to spinal and medullary respiratory motoneurons. Electrodes were filled with a solution containing (in mm): potassium gluconate, 130; MgCl2, 4.5; trisphosphocreatine, 14; Hepes, 10; EGTA, 5; Na‐ATP, 4; Na‐GTP, 0.3; biocytin, 0.2%; pH 7.4 and osmolality 300 mosmol kg–1 and had resistances of 3–4.5 MΩ when tested in bath solution. Electrodes were mounted on a micromanipulator (PatchStar; Scientifica, Uckfield, UK) and positioned onto the ventral or dorsal medullary surfaces or ventral surface of spinal cord under visual control (microscope; Seiler, St Louis, MO, USA) using surface landmarks (trapezoid body, rootlets of the HN, basilar artery, area postrema or calamus scriptorius). Voltage (−70 mV) and current clamp experiments were performed using an Axopatch‐200B integrating amplifier (Molecular Devices, Sunnyvale, CA, USA). A liquid junction potential of 15 mV was corrected off‐line. Gigaseals (>1 GΩ) were formed and whole cell configuration was obtained by suction. sEPSCs were isolated by locally applying 50 μm of the GABAA and the glycinergic receptor antagonists bicuculline (BIC; Sigma) and strychnine (STRY; Sigma), respectively. Motoneurons in the cNA, hypoglossal nucleus and ventral aspect of thoracic spinal cord were mapped by searching for antidromic field potentials following stimulation of the RLN, HN or AbN (5–10 V, 1 Hz, 0.2 ms pulse width), respectively. pFRG late‐E neurons were identified 0.3‐0.5 mm caudal to the trapezoid body, 1.8–2.0 mm lateral to the midline and 30–70 μm beneath the ventral surface (Moraes et al. 2012 a, 2014 a,b). RTN CO2‐sensitive neurons were identified 0.6–0.7 mm caudal to the trapezoid body, 1.5–1.7 mm lateral to the midline and 10–70 μm beneath the ventral surface. All signals recorded were amplified, band‐pass filtered (50 Hz – 2 kHz) and acquired (5–20 KHz) with an A/D converter (CED 1401; Cambridge Electronic Design, CED, UK), using Spike 2 software (Cambridge Electronic Design).
Microinjections into pFRG
The coordinates used for microinjections of BIC/STRY, vehicle, muscimol/glycine (MUS/GLY; 1 mm; Sigma) or kynurenic acid [KYN; 200 mm; Sigma (Moraes et al. 2012 b)] into pFRG were determined in accordance with the location of late‐E neurons: (i) 0.3–0.5 mm caudal to the trapezoid body, 1.8–2.0 mm lateral to the midline and 30–70 μm beneath the ventral surface using the ventral approach of in situ preparation; or (ii) 1.5–1.7 mm rostral to calamus scriptorius, 1.8–2.0 mm lateral to the midline and 3–3.2 mm ventral to the dorsal surface of medulla using the dorsal approach of in situ preparation. Drugs were applied bilaterally via a glass micropipette connected to a picopump (Picospritzer II; Parker Instruments, Cleveland, OH, USA) and the injected volume was approximately 20 nl. We included a 1.5% dilatation of fluorescent microbeads (Lumafluor, New York, NY, USA) with all drug application. The drugs were freshly dissolved in saline (0.9%) and the pH was adjusted to 7.4. The pH was determined using a pH indicator (Spezialindikator; Merck).
Measuring subglottal pressure
Changes in upper airway patency were evaluated by direct measurement of subglottal pressure (SGP) (Moraes & Machado, 2015). The SGP was recorded using a pressure transducer (Model PT 300; Grass, West Warwick, RI, USA). Increases and decreases in SGP were indicative of constriction (adduction) and dilatation (abduction) of the glottis, respectively, thereby giving an index of the dynamic changes in upper airway patency during the respiratory cycle. We measured the time between the decrease of SGP and inspiratory activity (PN) over 30 s periods. Mean values for these 30 s periods were determined.
Hypercapnic stimulus
Using a gas mix device (Gas mixer Pegas 4000, Columbus Instruments, Columbus, OH, USA), the proportion of the gases in the perfusate was altered to increase CO2. For hypercapnic stimulus, the concentration was 10% CO2/90% O2.
Histology
At the end of the experiments, the preparations were perfused first with PBS (0.1 m) and then with 4% paraformaldehyde (PFA) and brainstems or spinal cord were removed and post‐fixed in 4% PFA for 24 h. Transverse sections (30 μm thickness) were cut through the brainstem or spinal cord. Immunofluorescence staining was performed with free‐floating sections. Sections were blocked and permeabilized in PBS containing 10% normal horse serum and 0.5% Triton X‐100 for 1 h at room temperature. After three PBS washes, the sections were incubated in primary antibody rabbit anti‐Phox2b (1:800; gift from J.‐F. Brunet, Ecole Normale Supèrieure, Paris, France) for 24 h at 4°C. After three PBS washes, the sections were incubated in secondary antibodies Alexa 488‐conjugated streptavidin (1:1000; Molecular Probes) and Alexa 647‐conjugated goat anti‐rabbit (1:500; Molecular Probes) for 1 h at room temperature. Sections were washed three times in PBS and mounted in Fluoromount (Sigma). Images were collected on a Leica TCS SP5 confocal microscope (Buffalo Grove, IL, USA) equipped with 488 and 633 nm laser lines and tunable emission wavelength detection.
Statistical analyses
Results are expressed as mean ± SEM and compared using Student's paired t test or one‐way ANOVA with Bonferroni post‐tests (GraphPad Prism 4, La Jolla, CA, USA), in accordance with the requirements of each experimental protocol. Differences were considered significant at P < 0.05.
Results
We first investigated whether in situ preparations of juvenile rats did not generate AbN active expiration, and thus enhanced late‐E activity in cranial motor respiratory outflows controlling oropharyngeal and upper airway patency at normocapnia due to pFRG suppression by synaptic inhibition. Consistent with this hypothesis and previous studies using in vivo anesthetized preparations (Pagliardini et al. 2011; Huckstepp et al. 2015), bilateral microinjections into pFRG (Fig. 1 Aa and Ab) of antagonists for GABAergic and glycinergic receptors (BIC/STRY) evoked active expiration, as indicated by bursts of AbN at the late part of E2 phase (Fig. 1 B, Ca and Cb). Late‐E HN (0.43 ± 0.01 vs. 0.19 ± 0.09 s; P < 0.0001; n = 11) and SLN (0.44 ± 0.02 vs. 0.16 ± 0.006 s; P < 0.0001; n = 11) activities increased after bilateral microinjections of BIC/STRY into pFRG (Fig. 1 B–Db), while the duration of SLN post‐inspiratory activity was not affected (P > 0.05; Fig. 1 Ca and Cb). Measuring SGP revealed that pFRG disinhibition also exaggerated glottal dilatation at the end of E2 phase (0.36 ± 0.02 vs. 0.09 ± 0.07 s; P = 0.0001; n = 6; Fig. 2 Aa–Ac). During AbN active expiration, DE was 4.28 ± 0.05 s and significantly longer compared with the baseline condition (3.35 ± 0.07 s; P < 0.0001). With a decrease in DI (0.54 ± 0.01 vs. 1.26 ± 0.03 s; P < 0.0001), PN frequency did not change after BIC/STRY (P > 0.05; n = 12; Fig. 1 B, Ca, Cb and Dc). The ratio of the frequency of AbN active expiration to the frequency of PN bursts increased stepwise from about 1:4/1:3 after unilateral disinhibition of pFRG, to 1:1 after bilateral disinhibition (Fig. 1 B). These effects were in contrast to those for bilateral vehicle microinjections into pFRG that produced neither AbN active expiration nor any significant change in cranial and spinal motor respiratory outflows (P > 0.05; n = 7; Fig. 3 A, Ba and Bb).
Figure 1. pFRG disinhibition evoked active expiration and concomitant responses in cranial and spinal respiratory motor outflows in in situ preparations of rats.
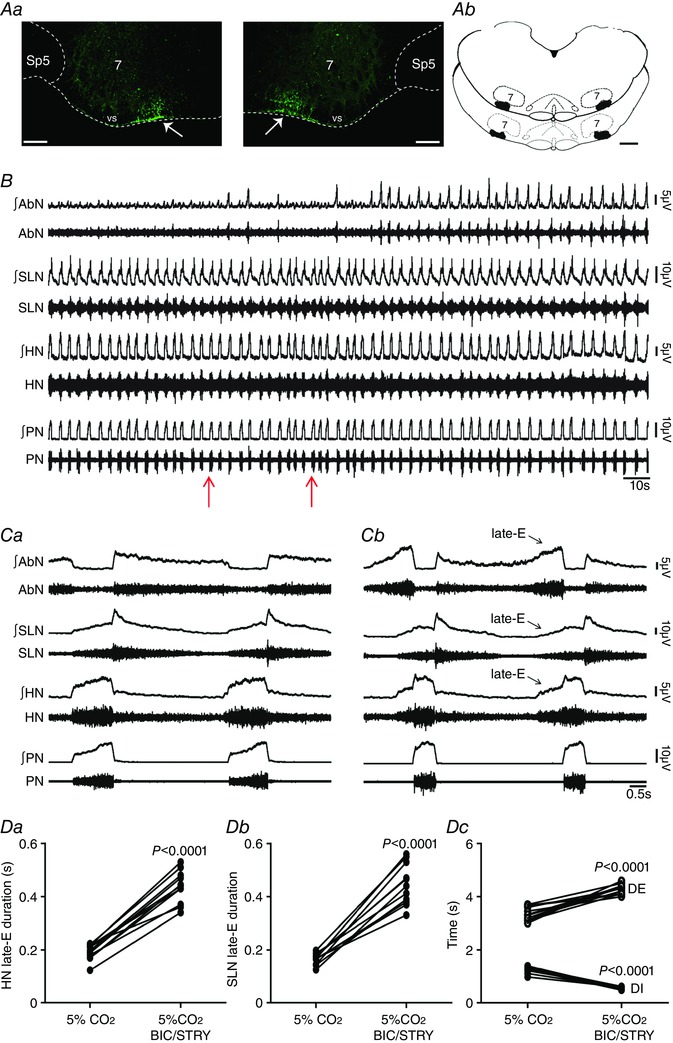
Aa, photomicrographs showing typical sites of bilateral microinjections in the pFRG (at bregma level −11.40 mm; 200 μm rostral to the caudal pole of facial nucleus). The fluorescent microbeads are located in the ventral medullary surface (vs; arrows) ∼ 500 μm medial to the spinal trigeminal tract (Sp5). Scale bars: 100 μm; 7, motor facial nucleus. Ab, schematic drawings of coronal sections of the brainstem showing the sites of bilateral microinjections into the pFRG of all in situ preparations of rats used in the present study (at bregma level between –11.30 mm and −11.50 mm; 100–300 μm rostral to the caudal pole of facial nucleus). Scale bar: 500 μm. B, raw and integrated (∫) records of AbN, SLN, HN and PN activities from one in situ preparation of rat under normocapnia before and after bilateral microinjections (arrows) of BIC/STRY into pFRG. C, magnification of two respiratory cycles from the same in situ preparation of rat before (Ca) and after (Cb) bilateral microinjections of BIC/STRY into pFRG. Note that pFRG disinhibition evoked AbN active expiration, HN and SLN late‐E activities, decreased DI, increased DE, but did not affect the PN frequency. D, grouped data comparing the duration of HN (Da) and SLN (Db) late‐E activity, DI and DE (Dc) under normocapnia before and after bilateral microinjections of BIC/STRY into pFRG. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2. Hypercapnia or pFRG disinhibition exaggerated late‐E glottal dilatation in in situ preparations of rats.
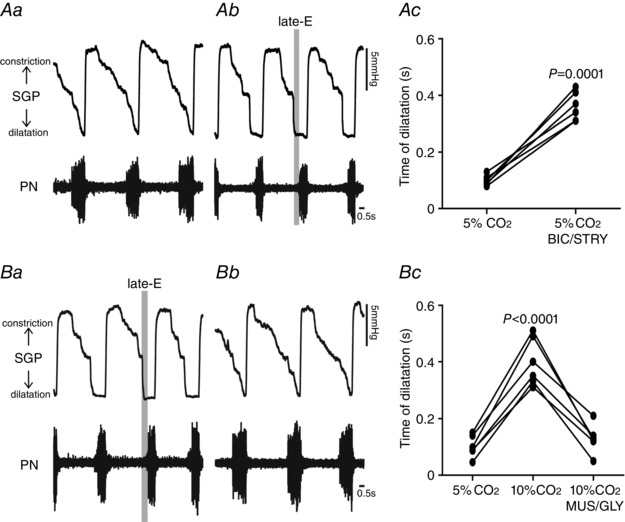
Representative tracings of dynamic changes in glottis during the respiratory cycle, including dilatation during inspiration (decrease in SGP) and constriction following inspiration (increase in SGP) from one in situ preparation of rat under normocapnia before (Aa) and after bilateral microinjections of BIC/STRY into pFRG (Ab). Ac, grouped data comparing the time of glottal dilatation in relation to inspiratory activity (PN) before and after bilateral microinjections of BIC/STRY into pFRG of rats under normocapnia. Note that pFRG disinhibition exaggerated late‐E glottal dilatation. B, representative tracings of dynamic changes in glottal resistance during the respiratory cycle from one in situ preparation of rat under hypercapnia before (Ba) and after bilateral microinjections of MUS/GLY into pFRG (Bb). Bc, grouped data comparing the time of glottal dilatation in relation to inspiratory activity (PN) before and after bilateral microinjections of MUS/GLY into pFRG of rats under hypercapnia. Note that pFRG inhibition normalized the exaggerated late‐E glottal dilatation evoked by hypercapnia.
Figure 3. Bilateral microinjections of vehicle into pFRG did not affect the respiratory pattern in in situ preparations of rats.
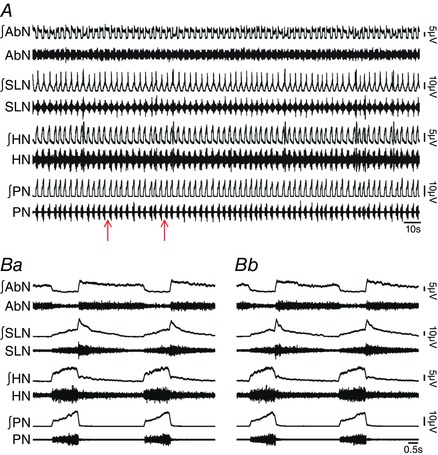
A, raw and integrated (∫) records of AbN, SLN, HN and PN activities from one in situ preparation of rat under normocapnia before and after bilateral microinjections (arrows) of vehicle into pFRG. B, magnification of two respiratory cycles from the same in situ preparation of rat before (Ba) and after (Bb) bilateral microinjections of vehicle into pFRG. Note the absence of changes in the respiratory pattern after vehicle microinjections into pFRG. [Color figure can be viewed at wileyonlinelibrary.com]
Hypercapnia also produced marked changes in the cranial and spinal respiratory motor outflows in in situ preparations of rats (Fig. 4 A). These changes were very similar to those observed after bilateral microinjections of BIC/STRY into pFRG. Expiratory AbN activity was significantly enhanced, with the presence of a novel high‐amplitude burst peaking at the end of E2 phase (late‐E). PN burst exhibited a reduced duration during inspiration (DI; 0.51 ± 0.01 vs. 1.23 ± 0.03 s; P < 0.0001), DE increased (4.57 ± 0.11 vs. 3.47 ± 0.03 s; P < 0.0001) with no changes in PN frequency (P > 0.05; n = 12; Fig. 4 A, Ba, Bb and Cc). Late‐E HN (0.41 ± 0.01 vs. 0.18 ± 0.006 s; P < 0.0001; n = 12) and SLN (0.27 ± 0.01 vs. 0.1 ± 0.008 s; P < 0.0001; n = 12) activities increased (Fig. 4 A–Cb), as well as glottal dilatation at the end of E2 phase (0.39 ± 0.03 vs. 0.1 ± 0.01 s; n = 6; P < 0.0001; Fig. 2 Ba and Bc), showing an enhancement of expiratory control of oropharyngeal and upper airway patency, phase‐locked with the occurrence of AbN active expiration. On the other hand, hypercapnia increased the amplitude of inspiratory PN (93.5 ± 3.8%) and HN (103 ± 2.3%) activities and reduced the duration of SLN post‐inspiratory activity (2.75 ± 0.06 vs. 3.36 ± 0.1 s; P < 0.0001; Fig. 4 A, Ba and Bb), which were not observed after bilateral microinjections of BIC/STRY into pFRG (P > 0.05; Fig. 1 B, Ca and Cb).
Figure 4. pFRG disinhibition did not affect hypercapnia‐evoked active expiration and concomitant responses in cranial and spinal respiratory motor outflows in in situ preparations of rats.
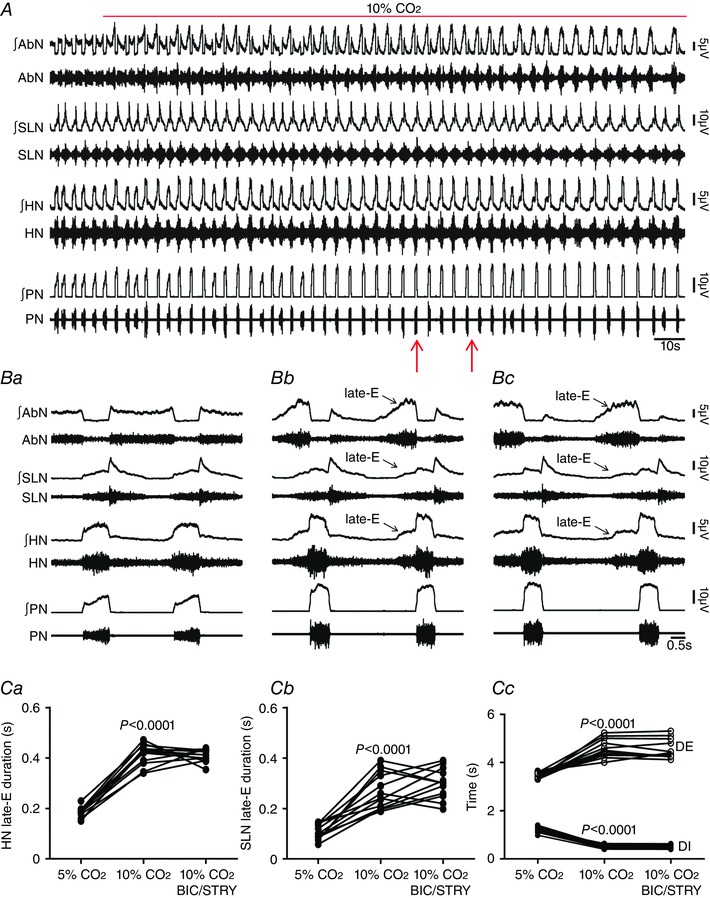
A, raw and integrated (∫) records of AbN, SLN, HN and PN activities from one in situ preparation of rat under normocapnia, hypercapnia and after bilateral microinjections (arrows) of BIC/STRY into pFRG during hypercapnia. B, magnification of two respiratory cycles from the same in situ preparation of rat under normocapnia (Ba), hypercapnia (Bb) and after bilateral microinjections of BIC/STRY into pFRG during hypercapnia (Bc). Note that hypercapnia evoked AbN active expiration, HN and SLN late‐E activities, decreased DI, increased DE, but did not affect the PN frequency. pFRG disinhibition was not able to produce any additional change in the AbN and in cranial and spinal respiratory motor outflows during hypercapnia. C, grouped data comparing the duration of HN (Ca) and SLN (Cb) late‐E activity, DI and DE (Cc) under normocapnia, hypercapnia and after bilateral microinjections of BIC/STRY into pFRG during hypercapnia. [Color figure can be viewed at wileyonlinelibrary.com]
pFRG disinhibition in in situ preparations of rats during hypercapnia was not able to produce any additional change in AbN active expiration, late‐E HN and SLN activities, or DI and DE (P > 0.05; n = 12; Fig. 4 A–Cc), suggesting that hypercapnia eliminated the synaptic inhibition to pFRG. In this regard, activation of GABAergic and glycinergic receptors in the pFRG, using MUS/GLY, eliminated the hypercapnia‐evoked AbN active expiration, HN (0.23 ± 0.02 vs. 0.22 ± 0.01 s; n = 9) and SLN (0.12 ± 0.01 vs. 0.11 ± 0.01 s; n = 9) late‐E activities, normalized DE (3.47 ± 0.09 vs. 3.37 ± 0.08 s) and DI (1.04 ± 0.05 vs. 1.15 ± 0.05 s; n = 9; Fig. 5 A–Cc) and glottal dilatation at the end of E2 phase (0.12 ± 0.02 vs. 0.1 ± 0.01 s; n = 6; Fig. 2 Ba and Bc) in relation to normocapnia. However, activation of GABAergic and glycinergic receptors in the pFRG did not affect the hypercapnia‐evoked increases of PN and HN inspiratory amplitude (P > 0.05; n = 9; Fig. 5 A–Bb). In addition, blockade of ionotropic glutamatergic receptors using KYN did not affect the hypercapnia‐evoked changes in the cranial and spinal respiratory motor outflows (P > 0.05; n = 8; Fig. 6 A–Bb).
Figure 5. pFRG inhibition eliminated hypercapnia‐evoked active expiration and concomitant responses in cranial and spinal respiratory motor outflows in in situ preparations of rats.
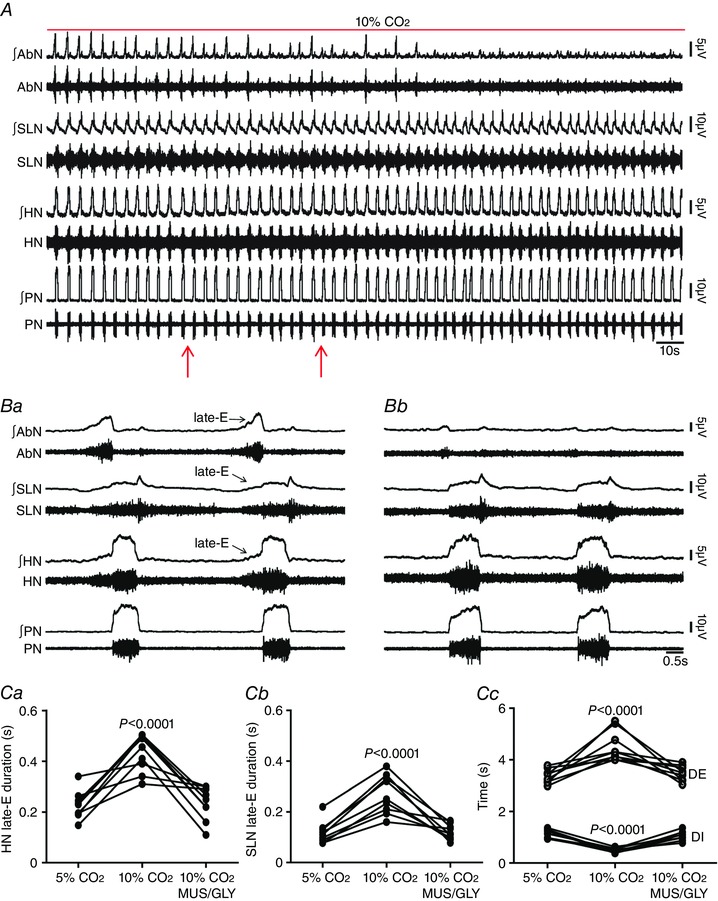
A, raw and integrated (∫) records of AbN, SLN, HN and PN activities from one in situ preparation of rat under hypercapnia before and after bilateral microinjections (arrows) of MUS/GLY into pFRG. B, magnification of two respiratory cycles from the same in situ preparation of rat under hypercapnia before (Ba) and after (Bc) bilateral microinjections of MUS/GLY into pFRG. Note that hypercapnia evoked AbN active expiration, HN and SLN late‐E activities, decreased DI and increased DE. pFRG inhibition eliminated hypercapnia‐evoked active expiration and concomitant responses in cranial and spinal respiratory motor outflows. C, grouped data comparing the duration of HN (Ca) and SLN (Cb) late‐E activity, DI and DE (Cc) under hypercapnia before and after bilateral microinjections of MUS/GLY into pFRG. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 6. Blockade of ionotropic glutamatergic receptors in pFRG did not affect hypercapnia‐evoked active expiration and concomitant responses in cranial and spinal respiratory motor outflows in in situ preparations of rats.
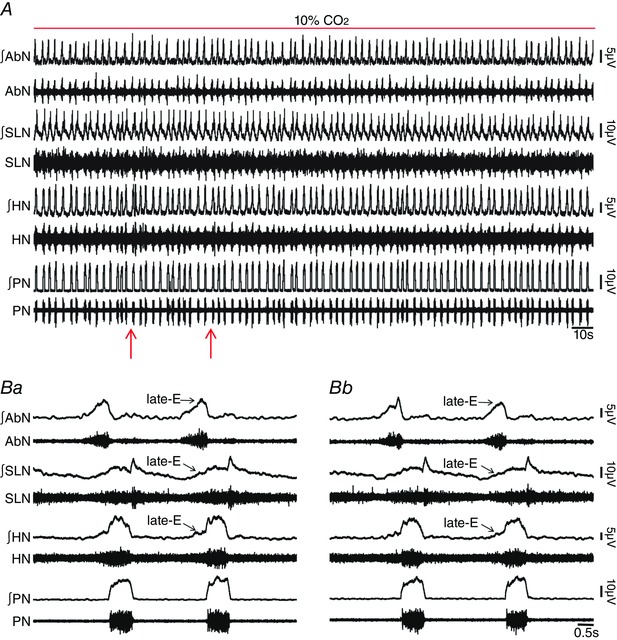
A, raw and integrated (∫) records of AbN, SLN, HN and PN activities from one in situ preparation of rat under hypercapnia before and after bilateral microinjections (arrows) of KYN into pFRG. B, magnification of two respiratory cycles from the same in situ preparation of rat under hypercapnia before (Ba) and after (Bc) bilateral microinjections of KYN into pFRG. Note that hypercapnia evoked AbN active expiration, HN and SLN late‐E activity, decreased DI and increased DE. Blockade of synaptic excitation in the pFRG did not affect these responses. [Color figure can be viewed at wileyonlinelibrary.com]
To determine the effects of hypercapnia, inhibition, disinhibition or blockade of synaptic excitation in the pFRG on the medullary and spinal neuronal activity, we performed neuronal extracellular and whole cell patch clamp recordings using ventral and dorsal approaches of in situ preparations of rats, while injecting MUS/GLY, BIC/STRY or KYN into pFRG.
Spinal abdominal aug‐E motoneurons
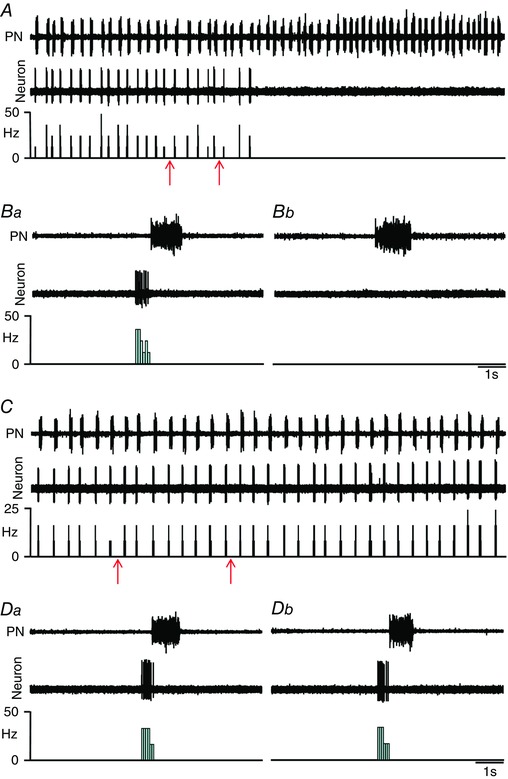
The ventral aspect of the thoracic spinal cord region contains abdominal aug‐E motoneurons that drive expiratory abdominal muscles (Iizuka, 2011). We tested whether hypercapnia‐evoked AbN active expiration is due to an increase in firing frequency of abdominal aug‐E motoneurons and whether this response is determined by pFRG. Abdominal aug‐E motoneurons were antidromically activated by T12 AbN stimulations (Fig. 7 A), extracellularly recorded ventrally in the thoracic region of the spinal cord (Fig. 7 B), and showed increasing firing frequency at E2 phase (Fig. 7 C and Da). Bilateral microinjections of BIC/STRY into pFRG evoked AbN active expiration and increased the firing frequency of abdominal aug‐E motoneurons at the end of E2 phase (109 ± 6.74 vs. 26.38 ± 2.98 Hz; P < 0.0001; n = 6; Fig. 7 C–Db) to values similar to those recorded during hypercapnia (102.3 ± 6.52 Hz; n = 7; Fig. 8 A and Ba) in in situ preparations of rats. In addition, activation of GABAergic and glycinergic receptors in the pFRG of in situ preparations during hypercapnia (n = 7), using MUS/GLY, simultaneously eliminated the AbN active expiration and normalized the firing frequency of abdominal aug‐E motoneurons in relation to motoneurons recorded during normocapnia (28.61 ± 3.76 vs. 26.38 ± 2.98 Hz; n = 13; Figs 7 and 8 A–Bb).
Figure 7. pFRG disinhibition increased the firing frequency of spinal abdominal aug‐E motoneurons in in situ preparations of rats.
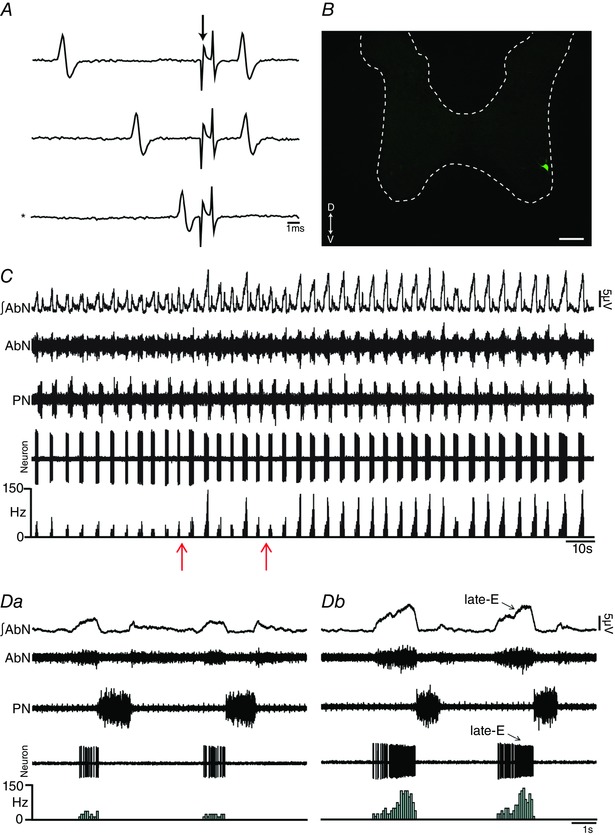
A, representative spinal abdominal aug‐E motoneuron that was antidromically activated from the T12 AbN. Asterisk indicates sweep when the antidromic spike was absent as the result of a collision with a spontaneous spike used to trigger the stimulus (stimulus artifact at arrow). B, spinal abdominal aug‐E motoneuron labelled in situ with biocytin (Alexa 488, green) located in the ventral aspect of spinal cord. Scale bar: 100 μm. C, raw and integrated (∫) extracellular records of AbN, PN and spinal abdominal aug‐E motoneuron from one in situ preparation of rat under normocapnia before and after bilateral microinjections (arrows) of BIC/STRY into pFRG. D, magnification of two respiratory cycles from the same in situ preparation of rat under normocapnia before (Da) and after (Dc) bilateral microinjections of BIC/STRY into pFRG. Note that pFRG disinhibition evoked AbN active expiration and increased the firing frequency of spinal abdominal aug‐E motoneurons at the end of E2 phase. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 8. pFRG inhibition blockade the effects of hypercapnia on the firing frequency of spinal abdominal aug‐E motoneurons in in situ preparations of rats.
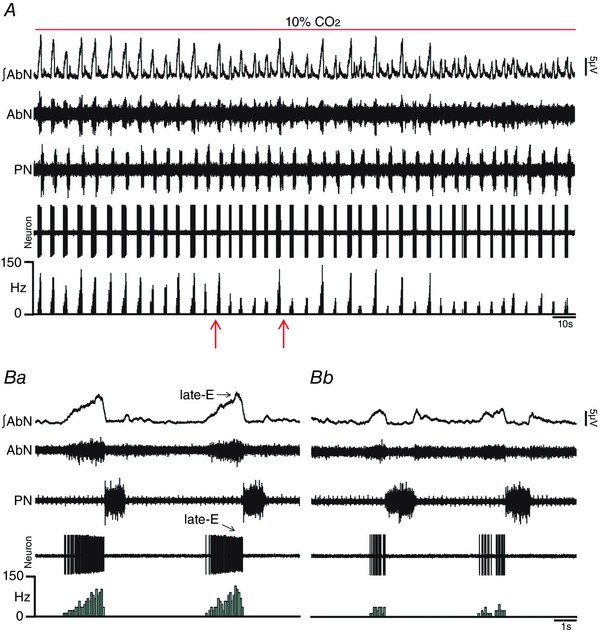
A, raw and integrated (∫) extracellular records of AbN, PN and spinal abdominal aug‐E motoneurons from one in situ preparation of rat under hypercapnia before and after bilateral microinjections (arrows) of MUS/GLY into pFRG. B, magnification of two respiratory cycles from the same in situ preparation of rat under hypercapnia before (Ba) and after (Bc) bilateral microinjections of MUS/GLY into pFRG. Note that pFRG inhibition eliminated AbN active expiration and normalized the increased firing frequency of spinal abdominal aug‐E motoneurons, at the end of E2 phase, induced by hypercapnia. [Color figure can be viewed at wileyonlinelibrary.com]
Whole cell patch current and voltage clamp recordings were performed to evaluate whether hypercapnia affects membrane potential trajectories, frequency and amplitude of sEPSCs to spinal abdominal aug‐E motoneurons. In this regard, hypercapnia did not affect the membrane potential of abdominal aug‐E motoneurons either during inspiration (P > 0.05) or following inspiration (P > 0.05; Fig. 9 A and B). On the other hand, hypercapnia selectively increased the frequency (121.4 ± 5.2 vs. 58.69 ± 5.92 Hz; P < 0.0001; n = 7), but not the amplitude (P > 0.05), of sEPSCs to abdominal aug‐E motoneurons at the end of E2 phase (Fig. 9 C and D). These data suggest a possible functional connectivity between pFRG and spinal abdominal aug‐E motoneurons for generating AbN active expiration.
Figure 9. Hypercapnia increases synaptic excitation to spinal abdominal aug‐E motoneurons in in situ preparations of rats.
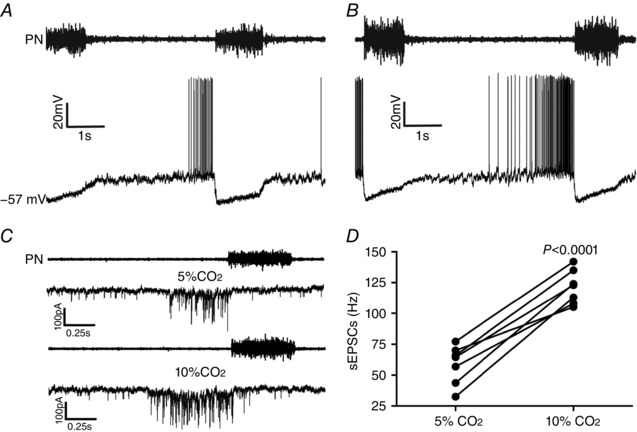
A and B, raw records of PN and whole cell patch current clamp records of spinal abdominal aug‐E motoneurons from one in situ preparation of rat under normocapnia (A) and hypercapnia (B). C, raw records of PN and whole cell patch voltage clamp (−70 mV) records of spinal abdominal aug‐E motoneurons from one in situ preparation of rat under normocapnia and hypercapnia. Note that hypercapnia increased the spinal abdominal aug‐E motoneuron firing frequency and the frequency, but not the amplitude, of sEPSCs at the end of E2 phase. D, grouped data comparing the frequency of sEPSCs to spinal abdominal aug‐E motoneurons at the end of E2 phase under normocapnia and hypercapnia.
Hypoglossal and laryngeal inspiratory motoneurons
Late‐E HN and SLN activities and control of oropharyngeal and upper airway patency at the end of E2 phase are determined by inspiratory hypoglossal (Gestreau et al. 2005) and laryngeal (Moraes & Machado, 2015) motoneurons, respectively. We tested whether the hypercapnia‐evoked enhancement of late‐E HN and SLN activities is due to increases in firing frequency of inspiratory motoneurons in hypoglossal nucleus and in cNA, respectively, and whether these responses are determined by pFRG. Medullary respiratory motoneurons were antidromically activated from the ipsilateral HN (Fig. 10 A) or RLN (Fig. 11 A) stimulations and were located in the hypoglossal nucleus (Fig. 10 B) or cNA (Fig. 11 B), respectively. Inspiratory hypoglossal (Fig. 10 C) and laryngeal motoneurons in in situ preparations of rats during normocapnia showed a ramping pattern of activities with maximal firing frequency during inspiration. Bilateral microinjections of BIC/STRY into pFRG evoked AbN active expiration and increased the firing frequency of inspiratory hypoglossal (26.16 ± 2.05 vs. 4.28 ± 0.98 Hz; n = 8; P < 0.0001; Fig. 10 C–Db) and laryngeal (26.95 ± 2.27 vs. 3.38 ± 0.93 Hz; n = 6; P = 0.0002) motoneurons, only at the end of E2 phase, to values similar to those found during hypercapnia [(hypoglossal: 26.58 ± 4.24 Hz; n = 5) (laryngeal: 23.67 ± 2.83 Hz; n = 6; Fig. 11 C and Da)]. Activation of GABAergic and glycinergic receptors in the pFRG of in situ preparations of rats during hypercapnia, using MUS/GLY, simultaneously eliminated AbN active expiration and normalized the firing frequency of inspiratory hypoglossal (4.16 ± 0.83 vs. 4.28 ± 0.98 Hz; n = 13) and laryngeal (3.6 ± 0.72 vs. 3.38 ± 0.93 Hz; n = 12; Fig. 11 C–Db) motoneurons at the end of E2 phase, suggesting that pFRG drives hypoglossal and laryngeal motoneurons to generate late‐E control of oropharyngeal and upper airway patency. Hypercapnia also increased the firing frequency of hypoglossal (42.96 ± 1.17 vs. 34.61 ± 1.72 Hz; P = 0.01) and laryngeal (44.33 ± 1.68 vs. 32.45 ± 1.7 Hz; P = 0.002) motoneurons during inspiration, which were not affected by pFRG inhibition using MUS/GLY (P > 0.05; Fig. 11 C–Db).
Figure 10. pFRG disinhibition increased the firing frequency of hypoglossal inspiratory motoneurons in in situ preparations of rats.
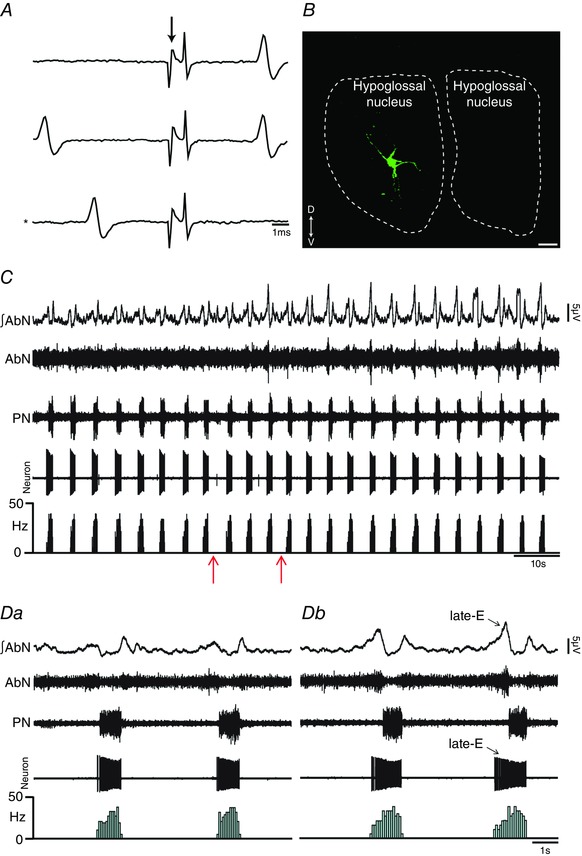
A, representative hypoglossal inspiratory motoneuron that was antidromically activated from HN. Asterisk indicates sweep when the antidromic spike was absent as the result of a collision with a spontaneous spike used to trigger the stimulus (stimulus artifact at arrow). B, hypoglossal inspiratory motoneuron labelled in situ with biocytin (Alexa 488, green) located in the hypoglossal nucleus. Scale bar: 25 μm. C, raw and integrated (∫) extracellular records of AbN, PN and hypoglossal inspiratory motoneurons from one in situ preparation of rat under normocapnia before and after bilateral microinjections (arrows) of BIC/STRY into pFRG. D, magnification of two respiratory cycles from the same in situ preparation of rat under normocapnia before (Da) and after (Db) bilateral microinjections of BIC/STRY into pFRG. Note that pFRG disinhibition evoked AbN active expiration and increased the firing frequency of hypoglossal inspiratory motoneurons at the end of E2 phase. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 11. pFRG inhibition blockade the effects of hypercapnia on the firing frequency of laryngeal motoneurons in in situ preparations of rats.
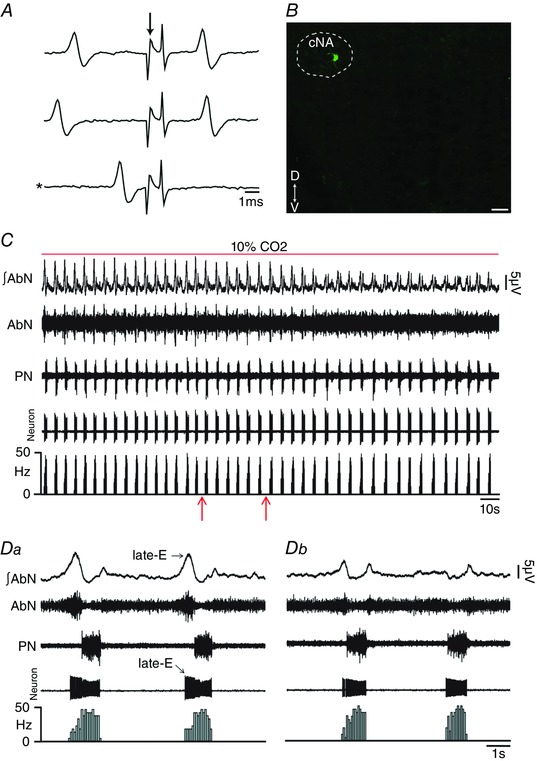
A, representative laryngeal inspiratory motoneuron that was antidromically activated from RLN. Asterisk indicates sweep when the antidromic spike was absent as the result of a collision with a spontaneous spike used to trigger the stimulus (stimulus artifact at arrow). B, laryngeal inspiratory motoneuron labelled in situ with biocytin (Alexa 488, green) located in the cNA. Scale bar: 50 μm. C, raw and integrated (∫) extracellular records of AbN, PN and laryngeal inspiratory motoneurons from one in situ preparation of rat under hypercapnia before and after bilateral microinjections (arrows) of MUS/GLY into pFRG. D, magnification of two respiratory cycles from the same in situ preparation of rat under hypercapnia before (Da) and after (Db) bilateral microinjections of MUS/GLY into pFRG. Note that pFRG inhibition eliminated AbN active expiration and normalized the increased firing frequency of laryngeal inspiratory motoneurons at the end of E2 phase induced by hypercapnia. [Color figure can be viewed at wileyonlinelibrary.com]
Whole cell patch current and voltage clamp recordings were also performed to evaluate whether hypercapnia affects membrane potential trajectories, frequency and amplitude of sEPSCs to inspiratory hypoglossal and laryngeal motoneurons. Hypercapnia did not affect the membrane potential of inspiratory hypoglossal (Fig. 12 Aa and Ab) or laryngeal (Fig. 12 Da and Db) motoneurons either after inspiration (P > 0.05) or during E2 phase (P > 0.05). Hypercapnia selectively increased the frequency, but not the amplitude (P > 0.05), of sEPSCs to both hypoglossal and laryngeal motoneurons not only at the end of E2 phase [(hypoglossal: 29.87 ± 2.57 vs. 3.83 ± 0.59 Hz; P = 0.0002) (laryngeal: 31.8 ± 3.03 vs. 2.62 ± 0.4 Hz; P = 0.0007)], but also during inspiration [(hypoglossal: 71.17 ± 2.86 vs. 37.37 ± 2.98 Hz; P = 0.0004; n = 6) (laryngeal: 69.5 ± 2.63 vs. 42.58 ± 4.04 Hz; P = 0.005; n = 5), Fig. 12 Ba–Cb and Ea‐Fb].
Figure 12. Hypercapnia increases synaptic excitation to hypoglossal and laryngeal inspiratory motoneurons in in situ preparations of rats.
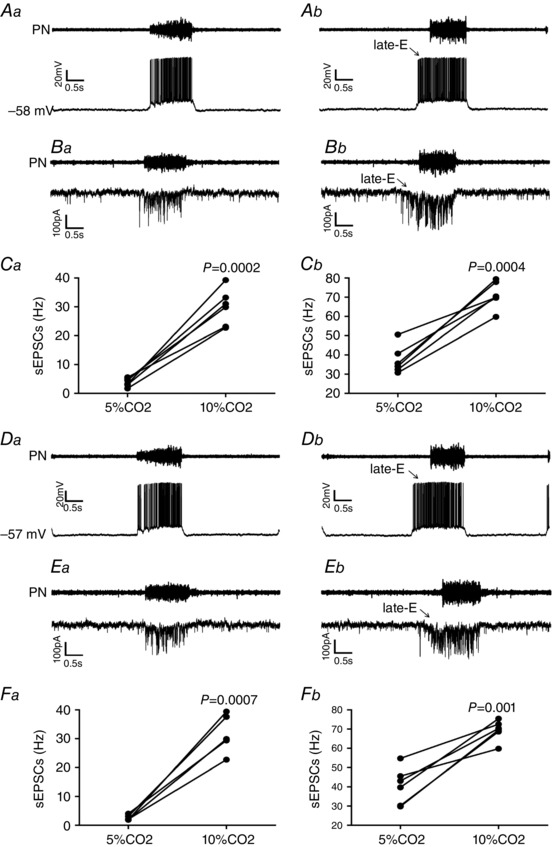
A, raw records of PN and whole cell patch current clamp records of hypoglossal inspiratory motoneurons from one in situ preparation of rat under normocapnia (Aa) and hypercapnia (Ab). B, raw records of PN and whole cell patch voltage clamp (−70 mV) records of hypoglossal inspiratory motoneurons from one in situ preparation of rat under normocapnia (Ba) and hypercapnia (Bb). C, grouped data comparing the frequency of sEPSCs to hypoglossal inspiratory motoneurons under normocapnia and hypercapnia at the end of E2 phase (Ca) and during inspiration (Cb). D, raw records of PN and whole cell patch current clamp records of laryngeal inspiratory motoneurons from one in situ preparation of rat under normocapnia (Da) and hypercapnia (Db). E, raw records of PN and whole cell patch voltage clamp (−70 mV) records of laryngeal inspiratory motoneurons from one in situ preparation of rat under normocapnia (Ea) and hypercapnia (Eb). F, grouped data comparing the frequency of sEPSCs to laryngeal inspiratory motoneurons under normocapnia and hypercapnia at the end of E2 phase (Fa) and during inspiration (Fb). Note that hypercapnia increased the firing frequency of hypoglossal and laryngeal inspiratory motoneurons, and the frequency, but not the amplitude, of sEPSCs not only at the end of E2 phase but also during inspiration.
pFRG late‐E neurons
We performed extracellular recordings of late‐E neurons in the pFRG of in situ preparations of rats during hypercapnia to test whether these neurons are the common source of synaptic excitation to spinal abdominal aug‐E, hypoglossal and laryngeal inspiratory motoneurons at the end of E2 phase. Under hypercapnia, we found Phox2b‐negative (Fig. 13 Aa–Ac) expiratory rhythmic neurons (late‐E neurons; 31.19 ± 2.19 Hz), more laterally to RTN, with a decreasing pattern at the end of E2 phase in the pFRG of in situ preparations of rats, well correlated with AbN active expiration and HN late‐E activity (Fig. 13 B). Switching from hypercapnia to normocapnia, pFRG late‐E neurons became silent in all in situ preparations (Fig. 13 C) and bilateral microinjections of BIC/STRY into pFRG again activated these late‐E neurons (27.26 ± 2.94 Hz; n = 7; Fig. 13 D), suggesting that pFRG late‐E neurons are the source of synaptic excitation to spinal and medullary respiratory motoneurons at hypercapnia. In addition, activation of GABAergic and glycinergic receptors using MUS/GLY (Fig. 14 A–Bb), but not blockade of ionotropic glutamatergic receptors (29.9 ± 3.57 vs. 28.57 ± 2.79 Hz; n = 7; Fig. 14 C–Db), in the pFRG of in situ preparations of rats during hypercapnia eliminated the pFRG late‐E neuronal activity, consistent with the idea that pFRG late‐E neuronal disinhibition drives late‐E discharges in cranial and spinal respiratory motor outflows under hypercapnia.
Figure 13. Hypercapnia or pFRG disinhibition evoked Phox2b‐negative late‐E neuronal activity in in situ preparations of rats.
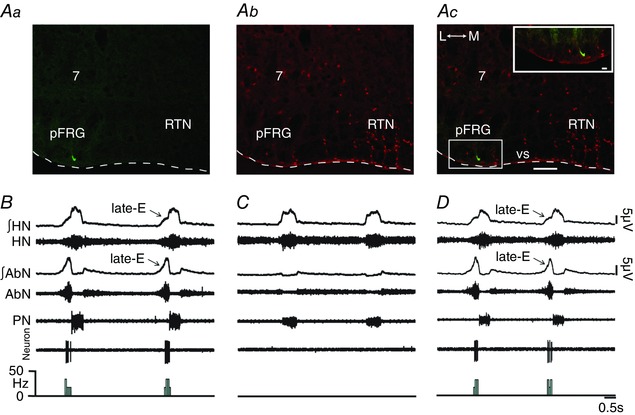
A, late‐E neuron labelled in situ with biocytin (Alexa 488, green; at bregma level −11.40 mm) located in the ventral medullary surface (vs) in pFRG more laterally to the RTN (Aa), Phox2b immunofluorescence revealed with Alexa 647 (red) (Ab) and the overlay (Ac). Scale bar: 100 μm; 7, motor facial nucleus; L, lateral; M, medial. Note the absence of Phox2b immunostaining in the labelled pFRG late‐E neuron in the enlargement of the corresponding box in Ac. Scale bar: 20 μm. B, raw and integrated (∫) extracellular records of HN, AbN, PN and pFRG late‐E neurons from one in situ preparation of rat under hypercapnia. Note that hypercapnia evoked AbN active expiration and simultaneous late‐E activity in HN and in pFRG neurons. C, normocapnia simultaneously eliminated AbN active expiration, HN and pFRG late‐E activity. D, bilateral microinjections of BIC/STRY into pFRG again evoked pFRG late‐E neuronal activity in the same in situ preparation of rat. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 14. pFRG inhibition, but not blockade of synaptic excitation, eliminated hypercapnia‐evoked pFRG late‐E neuronal activity in in situ preparations of rats.
A, raw extracellular records of PN and pFRG late‐E neurons from one in situ preparation of rat under hypercapnia before and after bilateral microinjections of MUS/GLY (arrows) into pFRG. B, magnification of one respiratory cycle from the same in situ preparation under hypercapnia before (Ba) and after (Bb) bilateral microinjections of MUS/GLY into pFRG. Note that pFRG inhibition eliminated pFRG late‐E neuronal activity induced by hypercapnia. C, raw extracellular records of PN and pFRG late‐E neurons from another in situ preparation of rat under hypercapnia before and after bilateral microinjections of KYN (arrows) into pFRG. D, magnification of one respiratory cycle from the same in situ preparation under hypercapnia before (Da) and after (Db) bilateral microinjections of KYN into pFRG. Note the absence of changes in late‐E neuronal activity after blockade of ionotropic glutamatergic receptors in the pFRG. [Color figure can be viewed at wileyonlinelibrary.com]
RTN neurons
To evaluate the effects of pFRG inhibition on the CO2 sensitivity of Phox2b‐positive RTN neurons, we performed extracellular recordings of RTN neurons under hypercapnia while MUS/GLY was bilateral microinjected into pFRG. We found Phox2b‐positive (Fig. 15 Aa–Ac) tonically firing neurons in the RTN that were CO2‐sensitive (12.82 ± 0.67 vs. 4.97 ± 0.71 Hz; P < 0.0001; Fig. 15 B). Bilateral microinjections of MUS/GLY into pFRG under hypercapnia did not affect the CO2‐induced increase in the RTN neurons firing frequency (13.04 ± 0.62 vs. 12.82 ± 0.67 Hz; n = 5), despite the elimination of AbN active expiration (Fig. 15 B). These data suggest that the CO2 sensitivity of Phox2b‐positive RTN neurons does not depend on the integrity of pFRG late‐E neurons.
Figure 15. pFRG inhibition did not affect CO2 sensitivity of Phox2b‐positive RTN neurons in in situ preparations of rats.
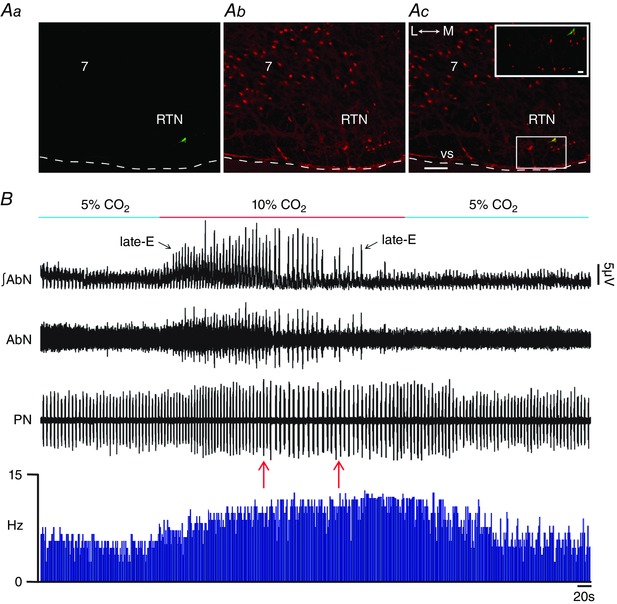
Aa, RTN CO2‐sensitive neuron labelled in situ with biocytin (Alexa 488, green; at bregma level −11.60 mm), Phox2b immunofluorescence (Ab) revealed with Alexa 647 (red) and the overlay (Ac). Scale bar: 100 μm; 7, motor facial nucleus; L, lateral; M, medial. Note the Phox2b immunostaining in the nucleus of a labelled RTN neuron in the enlargement of the corresponding box in Ac. Scale bar: 20 μm. B, raw and integrated (∫) extracellular records of AbN, PN and RTN Phox2b‐positive neurons from one in situ preparation of rat under normocapnia, hypercapnia, before and after bilateral microinjections of MUS/GLY (arrows) into pFRG. Note that hypercapnia evoked AbN active expiration and increased the Phox2b‐positive RTN neuron firing frequency. Bilateral inhibition of pFRG did not affect the CO2 sensitivity of Phox2b‐positive RTN neurons, despite elimination of AbN active expiration. Normocapnia reduced the RTN Phox2b‐positive neuron firing frequency. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
The present study has extended our current knowledge of the control of expiration by pFRG late‐E neurons, and their interactions with spinal and medullary respiratory motoneurons, under hypercapnia. Our data suggest that hypercapnia evokes disinhibition of non‐chemosensitive (Phox2b‐negative) pFRG late‐E neurons, but not synaptic excitation. In addition, under hypercapnia, pFRG late‐E neurons seem to be the common source for generating AbN active expiration and its concomitant cranial respiratory motor responses that control the oropharyngeal and upper airway patency, at the end of E2 phase, via synaptic excitation of spinal abdominal aug‐E, hypoglossal and laryngeal inspiratory motoneurons.
Expiration becomes actively involved in breathing when pFRG late‐E neurons are disinhibited or photo‐activated, suggesting that pFRG is silent at rest due to synaptic inhibition (Pagliardini et al. 2011; Huckstepp et al. 2015). We have confirmed our hypothesis that hypercapnia also produces active expiration by disinhibition of pFRG late‐E neurons, but not via synaptic excitation. Disinhibition of pFRG evokes late‐E activity in pFRG neurons and AbN active expiration in in situ preparations of juvenile rats, as shown before in adult anaesthetized rats (Pagliardini et al. 2011; Huckstepp et al. 2015). Switching to hypercapnic conditions in in situ preparations of rats also evoked well‐expressed expiratory discharges in pFRG late‐E neurons and AbN active expiration, which were identical to those described previously by others (Abdala et al. 2009; Moraes et al. 2012 a, 2014 a; Huckstepp et al. 2015). Additional changes in AbN active expiration were not observed following disinhibition of pFRG under hypercapnia. On the other hand, activation of GABAergic and glycinergic receptors, but not blockade of ionotropic glutamatergic receptors, eliminated late‐E neuronal activity in the pFRG and AbN active expiration. We conclude that pFRG late‐E neurons are silent at normocapnia due to post‐synaptic inhibition mediated by GABAergic and/or glycinergic receptors (Pagliardini et al. 2011) and hypercapnia sufficiently excites these neurons by synaptic disinhibition, which triggers active expiration.
Synaptic inhibition in the brainstem seems to be essential for generating stable respiratory patterns and breathing movements (Smith et al. 2013; Abdala et al. 2015; Marchenko et al. 2016). According to different models, this may involve reciprocal inhibition between different expiratory and inspiratory neuronal populations in the Bötzinger (BötC) and pre‐Bötzinger (pre‐BötC) complexes (Molkov et al. 2011; Smith et al. 2013). The present study also suggests that synaptic inhibition to pFRG late‐E neurons is important to control the generation of AbN active expiration and concomitant responses in the cranial respiratory motor outflows under hypercapnia. Synaptic inhibition to pFRG late‐E neurons may originate in respiratory GABAergic and glycinergic neurons from ventral respiratory group (VRG) (Smith et al. 2013), particularly from the BötC post‐inspiratory ones. In this regard, hypercapnia, but not pFRG disinhibition, reduced the duration of SLN following inspiration. It has also been shown that hypercapnia hyperpolarizes VRG post‐inspiratory neurons (Kawai et al. 1996), which may reduce the synaptic inhibition following inspiration to pFRG late‐E neurons, increasing the remaining expiratory time and allowing late‐E neurons to fire at the end of E2 phase. On the other hand, we cannot rule out the possible contribution of tonic synaptic inhibition to pFRG for the control of late‐E neuron excitability at baseline breathing. Inputs from the nucleus tractus solitarius inhibitory neurons (Takakura et al. 2006) could be a source of tonic synaptic inhibition to pFRG, which may control membrane potential subthreshold oscillations of the presumably rhythmic intrinsic bursting late‐E neurons (Abdala et al. 2009) during baseline breathing, avoiding neuronal action potential at the end of E2 phase. Hypercapnia could directly reduce the respiratory‐related or tonic synaptic inhibition to pFRG late‐E neurons presynaptically, allowing late‐E neurons to fire at the end of E2 phase. Therefore, new electrophysiological experiments are needed to evaluate membrane potential trajectories, rhythmic intrinsic bursting properties and inhibitory post‐synaptic currents to pFRG late‐E neurons at different phases of the respiratory cycle under normocapnia and hypercapnia for a better understanding of synaptic mechanisms determining the excitability of pFRG late‐E neurons. It is also important to determine whether synaptic disinhibition contributes to the activation of pFRG late‐E neuronal oscillations under other physiological conditions in which active expiration is recruited, such as peripheral chemoreflex activation (Moraes et al. 2012 a), exercise (Abraham et al. 2002) and sleep‐associated irregular breathing (Andrews & Pagliardini, 2015). Network dysfunctions in these circuit mechanisms may also underlie the presence of AbN active expiration, associated with the emergence of pathogenic processes, such as those observed in rats subjected to chronic intermittent hypoxia (Zoccal et al. 2008; Moraes et al. 2013) or sustained hypoxia (Moraes et al. 2014 a) and in spontaneously hypertensive rats (Moraes et al. 2014 b).
It has been proposed that CO2 chemoreception involves Phox2b‐positive neurons located in RTN, which play an important role in respiratory responses to central chemoreceptor activation (Stornetta et al. 2006; Marina et al. 2010; Takakura et al. 2014; Guyenet & Bayliss, 2015). RTN CO2‐sensitive neurons are glutamatergic and establish connections with several medullary respiratory neurons and motoneurons (Rosin et al. 2006). Our experimental study suggests that hypercapnia‐evoked AbN active expiration, HN and SLN late‐E activities, and exaggerated glottal dilatation at the end of E2 phase are determined by disinhibition of Phox2b‐negative pFRG late‐E neurons located more laterally to the RTN. On the other hand, pFRG inhibition did not affect the inspiratory increases (amplitude) in cranial and spinal respiratory motor outflows in response to hypercapnia in in situ preparations of rats, which may involve connections between RTN CO2‐sensitive and inspiratory neurons/motoneurons of pre‐BötC, hypoglossal nucleus and cNA (Rosin et al. 2006). The pFRG contains glutamatergic neurons in neonatal rats (Onimaru et al. 2008) and pFRG late‐E neurons seem to exert excitatory activity not only on spinal abdominal aug‐E motoneurons, possibly also involving bulbospinal caudal VRG aug‐E neurons (Iizuka, 2011; Silva et al. 2016), but also on hypoglossal (Pagliardini et al. 2011) and laryngeal inspiratory motoneurons, culminating in AbN active expiration and late‐E increases in HN and SLN discharges at the end of E2 phase. The last two may be important for the control of oropharyngeal and upper airway patency at the end of expiration when respiratory demands increase due to changes in blood gases.
In the present study we used a very small and well‐defined injection volume (20 nl) into pFRG, around 300–500 μm medial to the spinal trigeminal tract (Sp5), in the same region in which we previously recorded ventral medullary late‐E neurons (Moraes et al. 2012 a, 2014 a,b). Therefore, it is not surprising that all pFRG microinjections provided consistent responses in terms of recruitment (pFRG disinhibition) or elimination (pFRG inhibition) of AbN active expiration, and concomitant cranial respiratory motor responses. Previous studies have demonstrated that disinhibition of ventral medullary neurons [using large injection volumes (50–200 nl) of BIC/STRY] in the lateral edge of the facial nucleus, juxtaposed to the Sp5, was also able to evoke AbN active expiration (Pagliardini et al. 2011; Huckstepp et al. 2015). It is conceivable that neurons located medial (300–500 μm medial to the Sp5) to the centre of their injection sites could have been affected to various degrees by injecting large volumes juxtaposed to the SP5. Therefore, it is possible that pFRG late‐E neurons are distributed in the ventral (up to 500 μm medial to the Sp5) and lateral edges (juxtaposed to the Sp5) of the facial nucleus in the ventral medullary surface. Although we cannot exclude the possibility of caudal spreading of drugs after our small volume microinjections into the pFRG, especially toward more medial RTN CO2‐sensitive neurons (Stornetta et al. 2006), we recorded late‐E neurons in the pFRG more laterally to the RTN (Fig. 13), concurrent with AbN active expiration and late‐E activity in HN. MUS/GLY microinjections into pFRG eliminated AbN active expiration and neuronal late‐E activity. This limitation attributed to microinjection spread was also addressed in parallel experiments with inactivation of pFRG and simultaneous records of RTN CO2‐sensitive neurons, showing that MUS/GLY into pFRG eliminated AbN active expiration, but did not affect the CO2 sensitivity of more medial Phox2b‐positive RTN neurons.
Several recent studies have suggested that pFRG neurons present characteristics consistent with a late‐E oscillator, producing AbN active expiration in juvenile/adult rats only under conditions of high central respiratory drive (Abdala et al. 2009; Moraes et al. 2012 a, 2014 a,b; Molkov et al. 2014; Huckstepp et al. 2015). At normocapnia, in the absence of active expiration, pFRG neurons in juvenile/adult animals are silent (Abdala et al. 2009; Pagliardini et al. 2011; Moraes et al. 2012 a, 2014 a,b). When disinhibited or photo‐activated, they induce active expiration and become late‐E modulated (Pagliardini et al. 2011). Previous studies also suggest that RTN Phox2b‐positive neurons drive active expiration (Marina et al. 2010; Abbott et al. 2011) by sensing high levels of CO2/[H+] and influencing the oscillatory activity of the pFRG late‐E neurons to drive active expiration (Molkov et al. 2010, 2011, 2014). However, there is no data showing that Phox2b‐positive RTN CO2‐sensitive neurons present late‐E modulation to influence pFRG late‐E neurons under hypercapnia in juvenile/adult rats. Given that the blockade of ionotropic glutamatergic receptors did not affect either AbN active expiration or pFRG late‐E oscillations, the present study suggests that RTN Phox2b‐positive neurons, or other brainstem CO2‐sensitive neurons, do not activate pFRG late‐E neurons under hypercapnia to produce AbN active expiration. On the other hand, we cannot rule out the possibility that pFRG late‐E neurons present intrinsic CO2/[H+] sensitivity and do not need synaptic excitation to produce AbN active expiration under hypercapnia, despite the absence of immunostaining for Phox2b. We believe that pFRG Phox2b‐negative late‐E neurons are distinct from the more medial Phox2b‐positive RTN CO2‐sensitive neurons and that both populations are recruited under conditions of enhanced respiratory drive (hypercapnia) to accelerate expiration and inspiration, respectively, as proposed by Huckstepp et al. (2015). However, additional studies are needed to test the intrinsic CO2/[H+] sensitivity of pFRG late‐E neurons in juvenile/adults rats.
In conclusion, by evoking non‐chemosensitive (Phox2b‐negative) pFRG late‐E neurons via synaptic disinhibition, hypercapnia promotes synaptic excitation of spinal and medullary motoneurons and generates AbN active expiration and concomitant responses in cranial respiratory motor outflows that control oropharyngeal and upper airway patency in in situ preparations of juvenile rats. We postulate that CO2‐sensitive brainstem neurons do not activate pFRG late‐E neurons under hypercapnia to produce cranial and spinal motor responses at expiration; rather, hypercapnia produces disinhibition of the conditional pFRG late‐E neuronal oscillator.
Additional information
Competing interests
None.
Author contributions
A.A.B. and D.J.A.M. contributed to experiments involving nerve recordings and central microinjections. D.J.A.M. contributed to experiments involving neuronal and subglottal pressure recordings. A.A.B. and D.J.A.M. contributed to the conception and experimental design, data analyses and interpretation of the findings and preparation of the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by grant from ‘Fundação de Amparo à Pesquisa do Estado de São Paulo’ (FAPESP; Young Investigator Project, 2013/10484‐5).
Acknowledgements
We thank Professor Benedito H. Machado and Professor Julian F. R. Paton for their very important support with all in situ studies. We gratefully acknowledge J. F. Brunet (Departement de Biologie, Ecole Normale Superieure, Paris, France) for the Phox2b antibody.
References
- Abbott SB, Stornetta RL, Coates MB & Guyenet PG (2011). Phox2b‐expressing neurons of the parafacial region regulate breathing rate, inspiration, and expiration in conscious rats. J Neurosci 31, 16410–16422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Paton JF & Smith JC (2015). Defining inhibitory neurone function in respiratory circuits: opportunities with optogenetics? J Physiol 593, 3033–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Rybak IA, Smith JC & Paton JF (2009). Abdominal expiratory activity in the rat brainstem‐spinal cord in situ: patterns, origins and implications for respiratory rhythm generation. J Physiol 587, 3539–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham KA, Feingold H, Fuller DD, Jenkins M, Mateika JH & Fregosi RF (2002). Respiratory‐related activation of human abdominal muscles during exercise. J Physiol 541, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews CG & Pagliardini S (2015). Expiratory activation of abdominal muscle is associated with improved respiratory stability and an increase in minute ventilation in REM epochs of adult rats. J Appl Physiol (1985) 119, 968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestreau C, Dutschmann M, Obled S & Bianchi AL (2005). Activation of XII motoneurons and premotor neurons during various oropharyngeal behaviors. Respir Physiol Neurobiol 147, 159–176. [DOI] [PubMed] [Google Scholar]
- Guyenet PG & Bayliss DA (2015). Neural control of breathing and CO2 homeostasis. Neuron 87, 946–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Cardoza KP, Henderson LE & Feldman JL (2015). Role of parafacial nuclei in control of breathing in adult rats. J Neurosci 35, 1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Henderson LE, Cardoza KP & Feldman JL (2016). Interactions between respiratory oscillators in adult rats. Elife 5, e14203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M (2011). Respiration‐related control of abdominal motoneurons. Respir Physiol Neurobiol 179, 80–88. [DOI] [PubMed] [Google Scholar]
- Kawai A, Ballantyne D, Muckenhoff K & Scheid P (1996). Chemosensitive medullary neurones in the brainstem–spinal cord preparation of the neonatal rat. J Physiol 492, 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko V, Koizumi H, Mosher B, Koshiya N, Tariq MF, Bezdudnaya TG, Zhang R, Molkov YI, Rybak IA & Smith JC (2016). Perturbations of respiratory rhythm and pattern by disrupting synaptic inhibition within pre‐Botzinger and Botzinger complexes. eNeuro 3, ENEURO.0011‐16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Abdala AP, Trapp S, Li A, Nattie EE, Hewinson J, Smith JC, Paton JF & Gourine AV (2010). Essential role of Phox2b‐expressing ventrolateral brainstem neurons in the chemosensory control of inspiration and expiration. J Neurosci 30, 12466–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P, Moraes DJ, Sobotka PA & Paton JF (2013). The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4, 2395. [DOI] [PubMed] [Google Scholar]
- Molkov YI, Abdala AP, Bacak BJ, Smith JC, Paton JF & Rybak IA (2010). Late‐expiratory activity: emergence and interactions with the respiratory CpG. J Neurophysiol 104, 2713–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkov YI, Shevtsova NA, Park C, Ben‐Tal A, Smith JC, Rubin JE & Rybak IA (2014). A closed‐loop model of the respiratory system: focus on hypercapnia and active expiration. PLoS One 9, e109894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkov YI, Zoccal DB, Moraes DJ, Paton JF, Machado BH & Rybak IA (2011). Intermittent hypoxia‐induced sensitization of central chemoreceptors contributes to sympathetic nerve activity during late expiration in rats. J Neurophysiol 105, 3080–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes DJ, Bonagamba LG, Costa KM, Costa‐Silva JH, Zoccal DB & Machado BH (2014. a). Short‐term sustained hypoxia induces changes in the coupling of sympathetic and respiratory activities in rats. J Physiol 592, 2013–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes DJ, da Silva MP, Bonagamba LG, Mecawi AS, Zoccal DB, Antunes‐Rodrigues J, Varanda WA & Machado BH (2013). Electrophysiological properties of rostral ventrolateral medulla presympathetic neurons modulated by the respiratory network in rats. J Neurosci 33, 19223–19237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes DJ, Dias MB, Cavalcanti‐Kwiatkoski R, Machado BH & Zoccal DB (2012. a). Contribution of the retrotrapezoid nucleus/parafacial respiratory region to the expiratory‐sympathetic coupling in response to peripheral chemoreflex in rats. J Neurophysiol 108, 882–890. [DOI] [PubMed] [Google Scholar]
- Moraes DJ & Machado BH (2015). Electrophysiological properties of laryngeal motoneurones in rats submitted to chronic intermittent hypoxia. J Physiol 593, 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes DJ, Machado BH & Paton JF (2014. b). Specific respiratory neuron types have increased excitability that drive presympathetic neurones in neurogenic hypertension. Hypertension 63, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Moraes DJ, Zoccal DB & Machado BH (2012. b). Sympathoexcitation during chemoreflex active expiration is mediated by l‐glutamate in the RVLM/Botzinger complex of rats. J Neurophysiol 108, 610–623. [DOI] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K & Kawakami K (2008). CO2‐sensitive preinspiratory neurons of the parafacial respiratory group express Phox2b in the neonatal rat. J Neurosci 28, 12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliardini S, Janczewski WA, Tan W, Dickson CT, Deisseroth K & Feldman JL (2011). Active expiration induced by excitation of ventral medulla in adult anesthetized rats. J Neurosci 31, 2895–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF (1996). A working heart‐brainstem preparation of the mouse. J Neurosci Methods 65, 63–68. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Chang DA & Guyenet PG (2006). Afferent and efferent connections of the rat retrotrapezoid nucleus. J Comp Neurol 499, 64–89. [DOI] [PubMed] [Google Scholar]
- Schreihofer AM & Guyenet PG (1997). Identification of C1 presympathetic neurons in rat rostral ventrolateral medulla by juxtacellular labeling in vivo . J Comp Neurol 387, 524–536. [DOI] [PubMed] [Google Scholar]
- Silva JN, Tanabe FM, Moreira TS & Takakura AC (2016). Neuroanatomical and physiological evidence that the retrotrapezoid nucleus/parafacial region regulates expiration in adult rats. Respir Physiol Neurobiol 227, 9–22. [DOI] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Borgmann A, Rybak IA & Paton JF (2013). Brainstem respiratory networks: building blocks and microcircuits. Trends Neurosci 36, 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA & Guyenet PG (2006). Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci 26, 10305–10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Barna BF, Cruz JC, Colombari E & Moreira TS (2014). Phox2b‐expressing retrotrapezoid neurons and the integration of central and peripheral chemosensory control of breathing in conscious rats. Exp Physiol 99, 571–585. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL & Guyenet PG (2006). Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2‐sensitive neurons in rats. J Physiol 572, 503–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccal DB, Simms AE, Bonagamba LG, Braga VA, Pickering AE, Paton JF & Machado BH (2008). Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol 586, 3253–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]