

Abstract
Astrocytes are the most common glial cells in the brain with fine processes and endfeet that intimately contact both neuronal synapses and the cerebral vasculature. They play an important role in mediating neurovascular coupling (NVC) via several astrocytic Ca2+‐dependent signalling pathways such as K+ release through BK channels, and the production and release of arachidonic acid metabolites. They are also involved in maintaining the resting tone of the cerebral vessels by releasing ATP and COX‐1 derivatives. Evidence also supports a role for astrocytes in maintaining blood pressure‐dependent change in cerebrovascular tone, and perhaps also in blood vessel‐to‐neuron signalling as posited by the ‘hemo‐neural hypothesis’. Thus, astrocytes are emerging as new stars in preserving the intricate balance between the high energy demand of active neurons and the supply of oxygen and nutrients from the blood by maintaining both resting blood flow and activity‐evoked changes therein. Following neuropathology, astrocytes become reactive and many of their key signalling mechanisms are altered, including those involved in NVC. Furthermore, as they can respond to changes in vascular pressure, cardiovascular diseases might exert previously unknown effects on the central nervous system by altering astrocyte function. This review discusses the role of astrocytes in neurovascular signalling in both physiology and pathology, and the impact of these findings on understanding BOLD‐fMRI signals.
Keywords: astrocytes, cerebral blood flow, functional hyperemia, hemo‐neural hypothesis, neuroglial signalling, neurovascular coupling
Abbreviations
- AA
arachidonic acid
- AD
Alzheimer's disease
- AMPA
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid
- ATP
adenosine triphosphate
- BAPTA
1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid tetrakis(acetoxymethyl ester)
- BOLD fMRI
blood oxygenation level‐dependent functional magnetic resonance imaging
- COX
cyclooxygenase
- CNS
central nervous system
- CYP
cytochrome P450
- EETs
epoxyeicosatrienoic acids
- fNIRS
functional near‐infrared spectroscopy
- GABA
γ‐aminobutyric acid
- GECI
genetically encoded calcium indicator
- 20‐HETE
hydroxyeicosatetraenoic acid
- mGluR
metabotropic glutamate receptor
- NMDA
N‐methyl‐d‐aspartate
- NO
nitric oxide
- NOS
nitric oxide synthase
- NVC
neurovascular coupling
- PET
positron emission tomography
- PGE2
prostaglandin E2
- RNA
ribonucleic acid
- TRP
transient receptor potential
- VSMC
vascular smooth muscle cell
Introduction
Astrocytes are a major type of glial cell in the central nervous system (CNS). They uniformly tile the entire nervous system and occupy distinct minimally overlapping territories in the neuropil, referred to as astrocyte domains. These domains encompass thousands of synapses from many neurons (Bushong et al. 2002; Ogata & Kosaka, 2002; Bushong et al. 2004), suggesting that each astrocyte can sense the activity of a large number of neurons. Furthermore, these cells are coupled by gap junctions that allow direct cell‐to‐cell communication (Theis & Giaume, 2012), forming the astrocyte syncytium (Kirchhoff et al. 2001). This syncytium increases the spatial buffering capacity for K+ to help regulate neuronal excitability (Kofuji & Newman, 2004). It may also allow astrocytes to convey glucose from blood vessels to neurons (Rouach et al. 2008), and to integrate information over large areas or act as a diffuse conduit to pass information to distant neurons (Araque et al. 2014) and, perhaps, to the vasculature to control cerebral blood flow.
Astrocytes have historically thought to be passive housekeeping cells, but research over the last few decades has revealed their multifaceted role in the development and maintenance of a healthy brain. They are necessary for synapse formation and maturation during development, a function likely to be carried over into adulthood, particularly in the context of learning, and of repair after injury (Pfrieger & Barres, 1997; Ullian et al. 2001). They respond to neuronal activity by releasing gliotransmitters such as glutamate, ATP and d‐serine to modulate synaptic properties (Parpura et al. 1994; Henneberger et al. 2010; Panatier et al. 2011; Araque et al. 2014), although the mechanism underlying this release remains highly controversial (Hamilton & Attwell, 2010; Bazargani & Attwell, 2016). They maintain the temporal and spatial precision of synaptic transmission by taking up excess neurotransmitters (Barbour et al. 1988; Chatton et al. 2003) and buffering K+ from the extracellular space (Newman et al. 1984). It is also becoming increasingly clear that astrocytes contribute significantly to the regulation of, and may even respond to changes in, cerebral blood flow (Moore & Cao, 2008; Attwell et al. 2010).
Cerebral blood flow
The brain has a very high energy demand compared to the rest of the body – although it comprises only 2% of the body weight, it commands 20% of the body's resting state energy usage (Kety, 1957; Sokoloff, 1960; Attwell & Laughlin, 2001; Harris et al. 2012). This high energy demand is supplied by an extensive cerebral vasculature. Large pial arteries, covered by multiple layers of vascular smooth muscle cells (VSMCs), lie on the surface of the cortex and branch into penetrating arterioles, ensheathed by a single layer of VSMCs, which enter the cortical parenchyma. These parenchymal arterioles further branch into the capillary network and are drained by ascending venules into the pial veins (Blinder et al. 2013).
To ensure sufficient supplies of oxygen and nutrients to the brain in the face of varying systemic blood pressure, cerebral blood flow is tightly controlled via a process termed autoregulation (Tzeng & Ainslie, 2014). Despite this regulatory mechanism, and due to a lack of major energy stores in the brain (besides some glycogen granules: Holmes & Holmes, 1926), increases in neuronal activity demand a further increase in energy supply, which is delivered by a corresponding increase in local blood flow. This coupling between neuronal activity and cerebral blood flow, termed functional hyperaemia or neurovascular coupling (NVC), was first described by Mosso in 1880, and further characterised by Roy and Sherrington a decade later (1890). NVC has now come to be accepted as a fundamental aspect of brain function and many non‐invasive brain imaging techniques used in both clinical and research settings, such as blood oxygenation level dependent functional magnetic resonance imaging (BOLD fMRI), functional near‐infrared spectroscopy (fNIRS) and some forms of positron emission tomography (PET), exploit these changes in blood flow as a proxy measure of neuronal activity. Although the significance of neurovascular coupling for healthy brain function is widely accepted, the cellular mechanisms underlying this phenomenon have not yet been fully characterised.
Mechanisms of neurovascular signalling
Much of the initial efforts in NVC research focused on defining direct signalling from neurons to VSMCs. In many brain regions, glutamatergic activation of principal neurons results in the downstream production and release of nitric oxide (NO) and prostaglandins, vasoactive substances that can induce arteriole dilation (Meng et al. 1995 a, b ; Yang et al. 2000; Lacroix et al. 2015). The intracortical vasculature is also innervated by nerve terminals from subcortical regions that can release vasoactive neurotransmitters such as acetylcholine, serotonin, dopamine and noradrenaline (Krimer et al. 1998; Hamel, 2004, 2006). Adding another layer of complexity, pial arteries also receive nerve terminals from the peripheral nervous system which can release agents to dilate or constrict them (Hamel, 2006). However, this vascular innervation from subcortical and peripheral axons is distributed widely throughout the brain and pial vasculature, respectively, suggesting that this level of regulation will change vascular tone over large regions rather than mediate local NVC. A subset of subcortical neuronal inputs may also activate local interneurons as intermediaries in signalling to the vasculature (Cauli et al. 2004). Physiological activation of a cortical region would also activate the interneurons therein, and signalling through these interneurons may be one possible mechanism by which local NVC control can be gained. Indeed, interneurons are the primary neurons that express neuronal nitric oxide synthase (NOS) in the brain (Cauli et al. 2004; Jaglin et al. 2012). However, the contribution of interneurons to activity‐evoked NVC is not completely understood and both dilatory and constrictory effects have been reported (Hamel, 2006). A detailed report of the neurogenic signals involved in regulating the cerebrovasculature can be found elsewhere (Hamel, 2006; Attwell et al. 2010; Cauli & Hamel, 2010). This review will focus instead on the regulation of cerebral blood flow by astrocyte‐mediated signalling mechanisms.
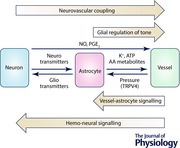
The role of astrocytes in mediating NVC
Numerous studies in the last few decades have suggested a role for astrocytes in regulating cerebral blood flow. Astrocytes are characterised by a dense cloud of fine processes (Bushong et al. 2004) which contact hundreds of synapses (Ventura & Harris, 1999), while specialised astrocyte processes called endfeet enwrap the vascular network in the brain parenchyma (Simard et al. 2003; Mathiisen et al. 2010), making them ideally positioned to mediate NVC (Fig. 1). Based on observations of the astrocyte morphology, Ramón y Cajal speculated, as early as 1895, that they might regulate vascular diameter (Ramón y Cajal, 1895). More than a hundred years later, we now know that astrocytes can indeed respond to neuronal activity with rises in intracellular Ca2+ concentration (Cornell‐Bell et al. 1990; Zonta et al. 2003; Hirase et al. 2004; Nimmerjahn et al. 2009) and, in turn, release various gliotransmitters (Parpura et al. 1994; Parri et al. 2001; Henneberger et al. 2010; Panatier et al. 2011; reviewed in Araque et al. 2014) and other factors into the microenvironment, some of which lead to the alteration of vascular tone.
Figure 1. Astrocyte endfeet processes enwrap blood vessels in the central nervous system.
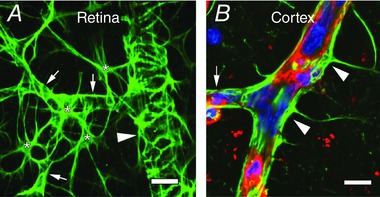
Inner surface of a whole mount retina (A) and a cortical slice (B) immunolabelled for glial fibrillary acidic protein (in green) showing astrocytes with their endfeet around blood vessels. Stars indicate astrocyte somata, arrowheads point to endfeet surrounding large vessels, arrows point to endfeet surrounding capillaries. The cortical section is also stained with DAPI (blue) for nuclei and isolectin B4 (red) for the vascular basement membrane. Scale bars = 10 μm. (Image by A. Mishra and Y. Chen.)
One of the first hypotheses regarding astrocyte control of vascular diameter came from Paulson and Newman in 1987 (Paulson & Newman, 1987). Neuronal activity results in an increase in extracellular [K+] ([K+]e), which is taken up by astrocytes to buffer [K+]e and thus regulate the excitability of nearby neurons. It was proposed that glial cells might release this K+ in a directed manner via their endfeet processes, which are enriched in K+ channels, thus raising the [K+]e around the vessels and resulting in VSMC hyperpolarisation (via the unusual dependence of VSMC inward rectifier K+ channels on [K+]e; Longden & Nelson, 2015) and thus dilation of blood vessels (Paulson & Newman, 1987). This K+ siphoning hypothesis was later disproved by experiments showing that mice lacking in Kir 4.1 channels, the primary K+ channels on glial endfeet around vessels, did not affect NVC (Metea et al. 2007). However, an alternative mechanism of NVC dependent on K+ release from astrocytes was brought forward by Nelson and colleagues (Filosa et al. 2006). Neuronal activity leads to a rise in astrocyte [Ca2+]i, which, in turn, can activate large conductance Ca2+‐activated K+ (BK) channels on astrocyte endfeet, leading to an efflux of K+ onto the vasculature. This rise in [K+]e results in smooth muscle hyperpolarisation and dilation of arterioles (Filosa et al. 2006).
A separate mechanism of astrocyte‐mediated NVC was proposed in 1998, which depends on the synthesis and release of vasoactive agents from astrocytes (Harder et al. 1998). Astrocytes can metabolise membrane phospholipids to produce arachidonic acid (AA) (Stella et al. 1994), which can be used to synthesise vasodilatory substances such as prostaglandins and epoxyeicosatrienoic acids (EETs) within astrocytes (Amruthesh et al. 1993; Alkayed et al. 1996 b), or it can be released onto VSMCs where it can be metabolised to the vasoconstrictor 20‐hydroxyeicosatetraenoic acid (20‐HETE) (Gebremedhin et al. 2000; Roman, 2002). Furthermore, altering the AA metabolism pathway was shown to reduce functional hyperaemia in vivo (Alkayed et al. 1996 a). It was thus suggested that neuronal activation may lead to the release of astrocyte‐derived AA metabolites to mediate NVC.
Evidence soon emerged from in vitro slice experiments that neuronal stimulation‐evoked vasodilation of cortical arterioles was dependent on prostaglandin E2 production and associated with rises in astrocyte [Ca2+]i (Zonta et al. 2003). In hippocampal slices, direct activation of astrocytes, by Ca2+ uncaging or activation of metabotropic glutamate receptors (mGluRs), was shown to evoke 20‐HETE‐dependent arteriole vasoconstriction (Mulligan & Macvicar, 2004). Interestingly, in the retina, light‐evoked neuronal activation led to both dilation (by EETs) and constriction (by 20‐HETE) of retinal arterioles in a manner dependent on glial activation (Metea & Newman, 2006). Around the same time, an in vivo study reported that cortical arterioles dilate in response to both neuronal and glial activation in a prostaglandin‐dependent manner (Takano et al. 2006). Furthermore, spontaneous glial Ca2+ waves in the retina were also observed to evoke vasoconstriction when they propagated across an arteriole (Kurth‐Nelson et al. 2009), although the molecular mechanism for this is as yet unknown.
The fact that glial activation evoked dilations in some studies but constrictions in others opened up the possibility of bidirectional control of the vasculature by astrocytes and raised new questions regarding the conditions that define the polarity of the neurovascular response. These studies also suggested possible regional differences in the molecular signals mediating NVC, for example EETs might mediate dilation in the retina but prostaglandin E2 might be the predominant dilating agent in the brain (Attwell et al. 2010; Macvicar & Newman, 2015). More recent studies, however, have also found a role for glial‐derived PGE2 in mediating retinal arteriole dilation (Mishra & Newman, 2010) and EETs in mediating cortical arteriole dilation (Peng et al. 2002, 2004; Lecrux et al. 2011; Liu et al. 2011).
To further establish the role of astrocytes in NVC, some researchers have also used models of glial ablation. In one such study, increases in cerebral blood flow produced by basal forebrain stimulation were reduced by ∼50% when the gliotoxins fluorocitrate and fluoroacetate (Paulsen et al. 1987) were used to damage astrocytes in the cortex (Lecrux et al. 2011). Based on pharmacology, the authors concluded that basal forebrain stimulation evokes the release of EETs onto blood vessels by activating astrocytes (Lecrux et al. 2011). Another study found that ablation of glial cells with L‐2‐aminoadipic acid, another potent gliotoxin (Huck et al. 1984), significantly reduced light‐evoked retinal blood flow without altering electroretinograms in the cat retina in vivo (Song et al. 2015). Although these studies support the idea of astrocyte‐mediated NVC, they should be interpreted with caution as ablation of glial cells will also undoubtedly have some indirect effects on neurons due to changes in homeostatic mechanisms such as glutamate–glutamine cycling (Hassel et al. 1992), K+ buffering (Lian & Stringer, 2004 a,b) and extracellular pH regulation (Stringer & Aribi, 2003). These neuronal effects, however, are likely to be minimal or only present at high doses (Paulsen et al. 1987; Virgili et al. 1991; Hassel et al. 1992), which is also demonstrated by the maintenance of the electroretinogram in the study by Song et al. (2015).
Taken together, these findings have established astrocytes as being important players in regulating cerebrovascular blood flow (Fig. 2) and lent credence to the concept of neuro‐glio‐vascular coupling, as originally posited by Cohen et al. (1997). However, they also raise a number of new questions. (i) How is the polarity of the astrocyte‐mediated NVC determined? (ii) How is the balance between dilation vs. constriction regulated to achieve the desired response? (iii) What is the relative contribution of BK channels vs. AA metabolites to astrocyte‐mediated NVC? (iv) What is the precise role of astrocyte [Ca2+]i? (v) What is the relative contribution of neurons vs astrocytes in the haemodynamic response?
Figure 2. Pathways mediating NVC.
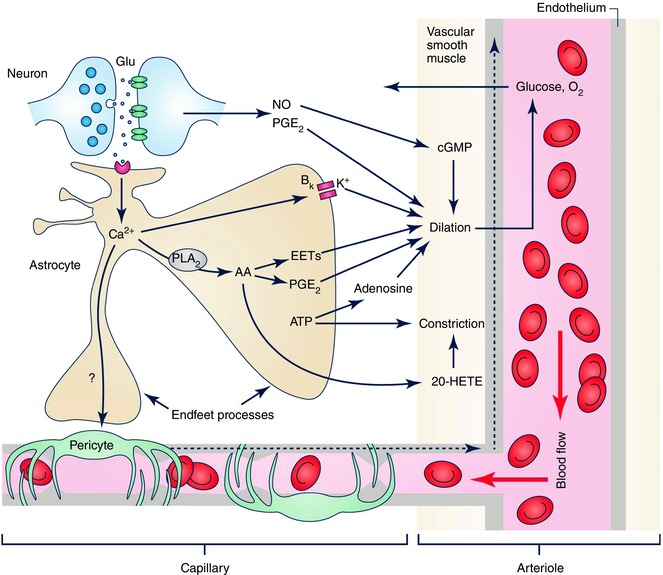
Neuronal activity results in synaptic release of glutamate, activating both postsynaptic neurons and astrocytes. Neuronal messengers such as NO and PGE2 may directly dilate cerebral arterioles. Activation of astrocytes results in a cascade of several signalling pathways. Ca2+‐dependent activation of BK channels can release K+ to dilate vessels. Ca2+ can also activate phospholipase A2, leading to the synthesis and release of AA metabolites such as EETs and PGE2 to dilate vessels (both arterioles via VSMCs, and capillaries via pericytes). AA can also be released from astrocytes onto vascular smooth muscle cells, where it can be metabolised to the vasoconstrictor 20‐HETE. Release of ATP from astrocytes can directly cause constriction of the vessels, or it can be metabolised by ectonucleotidases to raise the level of adenosine, a vasodilator. A dilation evoked at the capillary level may propagate to upstream arterioles via gap junctions between endothelial cells (dashed arrows). Some of the astrocyte‐generated vasoactive factors (e.g. ATP and PGE2) may contribute to the maintenance of basal tone, whereas others (EETs, PGE2, 20‐HETE, K+ efflux from BK channels) are involved in activity‐evoked changes in vascular diameter, which lead to increases in blood flow to bring O2 and glucose to supply the energy demand of active neurons. (Image by A. Mishra.)
Additionally, it is worth noting that although functional hyperaemia has been traditionally thought to be mediated exclusively at the arteriole level, evidence now suggests that pericyte‐mediated dilation of capillaries also contributes to this response (Peppiatt et al. 2006; Hall et al. 2014; Kornfield & Newman, 2014). Given the large relative resistance of capillaries in the vascular network in vivo (Blinder et al. 2013), they may contribute significantly to neurovascular coupling at the local level compared to arterioles alone (Hall et al. 2014). The contribution of astrocytes to capillary blood flow regulation has yet to be explored in more detail.
Bidirectional control of blood vessel diameter by astrocytes
Why brain activity should sometimes result in vasoconstriction as observed by several groups (Zonta et al. 2003; Mulligan & Macvicar, 2004; Metea & Newman, 2006), conceivably producing a reduction in energy supply, became a point of contention in the field. Some of these findings might be due to the variables of slice work. In the retina, light‐evoked dilation of retinal arterioles was enhanced and constriction was suppressed when the NO concentration in the tissue was lowered (Metea & Newman, 2006). This seems counterintuitive at first because NO is an established vasodilating agent, having been initially identified as the endothelium‐derived relaxing factor (Palmer et al. 1987). However, it can be explained when the effect of NO on AA metabolism is considered. The enzymes that metabolise AA (epoxygenases which produce EETs, cyclooxygenases which produce prostaglandins and ω‐hydroxylases which produce 20‐HETE) are susceptible to modulation by NO (Roman, 2002; Attwell et al. 2010). It is possible that damage caused during dissection induces an increase in the activity of nitric oxide synthases (NOS) in the retina, resulting in abnormal NO levels and altering the relative balance of AA metabolites produced.
Furthermore, these aforementioned studies were conducted in 95% O2‐bubbled perfusate, a widely accepted practice in neuroscience, which results in the abnormally high tissue [O2] of > 500 mmHg (Mishra et al. 2011) compared to physiological measurements ranging between ∼15–20 mmHg in the cortex (Metzger et al. 1971), 12.7–64.4 mmHg in the cerebellum (Offenhauser et al. 2005) and ∼5–40 mmHg in the retina (Cringle & Yu, 2010). When slices are exposed to 20% O2, tissue [O2] ranges between 10–20 mmHg at depths of 50–90 μm in the hippocampus (Gordon et al. 2008) and 30–60 mmHg in the retina (Mishra et al. 2011), approximating physiological concentrations. When neurovascular coupling experiments were repeated under these conditions mimicking physiological [O2], astrocyte stimulation led to dilation of hippocampal arterioles (Gordon et al. 2008) while also inducing an increase in glycolytic metabolism and raising the concentration of lactate in the tissue. Lactate inhibits the PGE2 transporter (Chan et al. 2002) and thus raises the extracellular PGE2 concentration, leading to dilations (Gordon et al. 2008). On the other hand, O2 is also a strong modulator of haem enzymes (see Attwell et al. 2010 for review) and when experiments were conducted in 20% O2, 20‐HETE‐mediated constrictions were suppressed and a prostaglandin‐mediated dilation component was revealed in the retina (Mishra et al. 2011). This is probably because of the different O2 affinities of the enzymes synthesising 20‐HETE and prostaglandins (Attwell et al. 2010). Thus, it appears that O2 can modulate NVC by changing the metabolic state of the tissue and altering the AA metabolites synthesised. However, high levels of inspired O2 in vivo did not alter the neurovascular response in either the retina (Mishra et al. 2011) or the cortex (Lindauer et al. 2010), perhaps because autoregulatory mechanisms constrict the vasculature and reduce cerebral blood flow upon exposure to high [O2] in order to maintain tissue within the physiological range despite high arterial (Torbati et al. 1978; Omae et al. 1998; Floyd et al. 2003; Lu et al. 2009; Mishra et al. 2011).
Given these effects of NO and O2 on NVC, it is noteworthy that the NOS enzymes themselves are also sensitive to [O2] (Stuehr et al. 2004) and that NO can act as either a mediator (in the cerebellum: Akgören et al. 1996; Yang et al. 2000) or a modulator of NVC (in the cortex: Lindauer et al. 1999), depending on the brain region being studied. We must also consider the possibility that other factors that are altered in slice preparations bathed with artificial cerebrospinal fluid, such as glucose and other metabolic products (Yamanishi et al. 2006; Puro, 2007), may also alter NVC. Even more importantly, vessels in brain slices completely lack perfusion pressure and physiological tone. A few studies have overcome this limitation by artificially perfusing the vasculature in slices via cannulation (Lovick et al. 2005; Kim & Filosa, 2012); however, due to the difficult nature of this technique, it is not widely practiced. Thus, although slice experiments are a necessity for the ease of carrying out pharmacological experiments and observing active dilation of the vasculature (as opposed to passive dilation resulting from an upstream increase in blood flow), the pitfalls and confounds of in vitro experiments should be taken into account in data interpretation.
Bidirectional modulation of NVC may also be explained, in part, by the extracellular [K+] that the vascular smooth muscle cells are exposed to. Although moderate increases in [K+]e induces VSMC hyperpolarisation and thus dilation (Filosa et al. 2006), concentrations above 20 mm can result in VSMC depolarisation and constriction of cerebral arterioles (Knot et al. 1996; Horiuchi et al. 2002), which is expected from the [K+]e dependence of the current that flows through VSMC inwardly rectifying K+ channels (Farr & David, 2011; Longden & Nelson, 2015). Following neuronal stimulation or astrocyte Ca2+ uncaging, moderate increases in astrocyte [Ca2+]i correlate with dilation but large increases correlate with vasoconstriction (Girouard et al. 2010), suggesting that the magnitude of the astrocyte [Ca2+]i signal may determine the polarity of vasomotor response, possibly by altering [K+] in the extracellular space surrounding the vessels. It has also been suggested that vasoconstriction might be physiologically relevant in certain instances in vivo, whereby increases in blood flow to active brain regions may be assisted by the active constriction of blood vessels in the surround or the contralateral hemisphere, a hypothesis further supported by the observation of negative BOLD signals in the periphery of activated regions (Devor et al. 2007).
The controversies surrounding the role of astrocytes in NVC
Astrocyte‐mediated mechanisms of NVC are believed to be dependent on astrocyte Ca2+ signalling. Activation of neurons results in the synaptic release of neurotransmitters such as glutamate which, in addition to acting on postsynaptic receptors, can also activate astrocytic group I metabotropic glutamate receptors (mGluR1 and mGluR5), leading to inositol trisphosphate (IP3)‐dependent Ca2+ release from internal stores (Panatier & Robitaille, 2015). Until recently, this was thought to be the main neuron‐to‐astrocyte signalling mechanism responsible for the downstream release of vasoactive agents. However, recent studies have questioned this conclusion. Whisker stimulation‐evoked haemodynamic responses were found to be independent of mGluR5 activation (Calcinaghi et al. 2011), a finding supported by the lack of group I mGluR expression in adult astrocytes (Sun et al. 2013). Furthermore, mice lacking IP3R2, the predominant IP3 receptor in astrocytes, were shown to have normal NVC (Takata et al. 2013; Bonder & McCarthy, 2014) and fMRI responses (Jego et al. 2014), challenging the idea that astrocytes are required for NVC. There is also controversy regarding whether astrocyte Ca2+ signals, when they do occur, are fast enough (Nizar et al. 2013) or reliable enough (Winship et al. 2007) to generate functional hyperaemia.
However, it is important to consider the possibility that astrocyte Ca2+ signals important for neurovascular signalling might be mediated by pathways other than mGluR‐mediated IP3 release, or may not have been detected using traditional methods of imaging using bulk loading of Ca2+ indicator dyes. In addition, most previous reports investigated [Ca2+]i changes within astrocyte cell bodies alone, but the signals that are important for neuroglial communication and NVC probably occur in their fine processes near synapses and/or endfeet. Indeed, astrocyte fine processes display Ca2+ transients and localised process‐wide Ca2+ waves that are not always reflected by soma measurements (Di Castro et al. 2011; Panatier et al. 2011; Shigetomi et al. 2013 a) and are poorly detected by bulk loaded indicators (Reeves et al. 2011). Observations made in vivo have achieved some degree of success in demonstrating stimulation‐evoked rapid [Ca2+]i rises in astrocyte cell bodies and endfeet in the somatosensory cortex (Lind et al. 2013). The development of genetically encoded calcium indicators (GECIs) such as GCaMP has further aided the detection of these astrocyte Ca2+ signals. Using astrocyte specific expression of GCaMP6f, it was recently shown that although large Ca2+ fluctuations in astrocyte soma are absent in the IP3R2 knockout mouse, Ca2+ transients in astrocyte processes are still present (Srinivasan et al. 2015), indicating that alternative mechanisms that raise astrocyte [Ca2+]i must exist. Similarly, using GCaMP3 expression, Otsu et al. (2015) also detected the presence of mGluR‐independent [Ca2+] signals in astrocyte processes in the olfactory bulb of adult mice and found them to precede neuronally evoked vessel dilation (Otsu et al. 2015). In contrast, another study using GCaMP6f found that neuronal stimulation‐evoked arteriole dilations were preceded by a [Ca2+]i rise in astrocyte processes only when the stimulation was above a certain threshold (Institoris et al. 2015), implying that NVC may be mediated by astrocytes in conditions of high activity but is generated perhaps by neurons directly following mild activation. It has also been proposed that the initiation and maintenance of the NVC response might be mediated differentially, whereby neuronal or Ca2+‐independent astrocyte signalling pathways may initiate vascular dilation and Ca2+‐dependent astrocyte signalling component may be important only in the maintenance phase of the response (Martindale et al. 2005; Calcinaghi et al. 2011; Rosenegger & Gordon, 2015). A computational model of neuro‐glio‐vascular coupling also suggested an astrocytic contribution only during high levels of neuronal activity (Blanchard et al. 2016), supporting this hypothesis. However, this study only included astrocytic neurotransmitter uptake in their model. Incorporating astrocytic mechanisms involved in NVC such as K+ buffering and AA production/release into such models may offer more significant insights into this process.
In addition, astrocyte [Ca2+]i signalling can be driven by other mechanisms that do not require a dependence on IP3, including AMPA receptors (Seifert & Steinhauser, 1995), NMDA receptors (Lalo et al. 2006), purinergic receptors (Lalo et al. 2008), TRPA1 channels (Shigetomi et al. 2013 b), Na+–Ca2+ exchangers and ryanodine receptors (Kirischuk et al. 1997). These mechanisms should be considered as possible alternatives when investigating neuroglial communication. In particular, ATP has long been known to raise astrocyte [Ca2+]i (Fellin et al. 2006; Newman, 2006) and is often used as a positive control in experiments studying astrocyte [Ca2+]i (Sun et al. 2013; Otsu et al. 2015). Glial Ca2+ waves are primarily propagated by ATP release from glial cells acting on purinoceptors on neighbouring cells (Newman, 2001; Bowser & Khakh, 2007), and ATP also plays a role in direct neuroglial communication, as demonstrated in the retina (Newman, 2006), cerebellum (Piet & Jahr, 2007) and the cortex (Ase et al. 2010; Lalo et al. 2011). Recently, overexpression of an ectonucleotidase to break down extracellular ATP was shown to significantly decrease the BOLD signal evoked by electrical forepaw stimulation (Wells et al. 2015), suggesting that purinergic signalling may be involved in NVC. However, the source(s) of endogenously released ATP, and the specific receptor(s) involved in mediating its possible neurovascular effects, have yet to be defined.
It is also important to keep in mind that NVC may involve Ca2+‐independent astrocyte signalling. Due to their electrically passive nature, the biology of astrocytes was largely unexplored until the middle of the twentieth century when researchers started characterising them electrophysiologically (Hild et al. 1958; Tasaki & Chang, 1958; Kuffler & Potter, 1964), but the field really bloomed only in the 90s when Ca2+ indicators were used to show astrocyte Ca2+ waves in response to glutamate and neuronal stimulation (Cornell‐Bell et al. 1990; Dani et al. 1992). Measuring Ca2+ is, to date, the best method we have to study astrocyte activity, but this should not lead us to believe that this is the only way that signalling within or via astrocytes can be achieved. The development of new methods to study astrocytes is therefore required to improve our understanding of their function, both Ca2+‐dependent and ‐independent.
Astrocyte control of pial arterioles
Although pial vascular tone is largely mediated by endothelial and myogenic factors, astrocytes have also been implicated in neurovascular signalling to pial arteries. Neuronally evoked dilation of pial arterioles is partly dependent on purinergic signalling from the glia limitans, a layer of specialised astrocyte processes that form a barrier around the brain and abut pial vessels (Xu et al. 2008). In this study, interrupting the signalling along the astrocytic syncytium, by damaging the glia limitans with the gliotoxin l‐2‐aminoadipic acid, reduced neuronally evoked pial arteriole dilation, but endothelial damage induced by photoactivation of an intravascular dye had little effect (Xu et al. 2008). In contrast, a more recent study reported that damaging endothelial cells locally using a similar photoactivated dye method stopped the propagation of dilation along pial arterioles beyond the region of damage and, when the damage is induced over a broader region, caused a reduction in the haemodynamic response (Chen et al. 2014). Perhaps the different observations reported by these two studies can be explained by differences in stimulation protocol or the anaesthesia regime, but, at this point in time, the relative contribution of astrocytes and the endothelium in propagating dilations to upstream arterioles is still unclear and a role for astrocytes cannot be ruled out. In addition, factors other than ATP have also been suggested in astrocyte signalling to pial arterioles, for example AA metabolites (Ellis et al. 1990), and have yet to be investigated in more detail.
Astrocytes and resting vessel tone
Cerebrovascular tone is generated largely by autoregulatory means, including pressure‐induced (Faraci et al. 1989) and flow‐induced (Pohl et al. 1991) myogenic mechanisms. Regulation of cerebral vessels by perivascular nerve terminals (both peripheral, in the case of extracerebral pial vessels, and subcortical, in the case of parenchymal vessels) also contributes to neurogenic vascular tone (Hamel, 2006). Recently, however, a role for astrocytes in maintaining the resting tone of arterioles has also been unveiled. In the in vivo retina, increasing extracellular ATP constricted arterioles while breakdown of extracellular ATP, inhibition of purinergic receptors and application of the gliotoxin fluorocitrate all dilated arterioles (Kur & Newman, 2014). A purinergic signalling mechanism also contributed to the development of pressure‐evoked vasomotor tone in the cortex (Kim et al. 2015). The development and maintenance of this vascular tone was found to be dependent on TRPV4‐mediated astrocytic [Ca2+]i rises both in vivo and in vitro, presumably leading to ATP release from astrocytes to constrict vessels (Kim et al. 2015).
Rosenegger et al. (2015) reported that Ca2+‐dependent mechanisms in astrocytes contribute to COX1‐mediated tonic dilation of cortical arterioles. When they used the Ca2+ chelator BAPTA to arrest Ca2+ fluctuations within the astrocyte syncytium surrounding arterioles, the resting diameter of the arterioles decreased significantly (Rosenegger et al. 2015). However, another group found that similarly arresting astrocyte Ca2+ with BAPTA‐filling from a patch pipette led to a decrease in pressure‐induced vascular tone, i.e. dilation (Kim et al. 2015). It is highly probable that these opposing mechanisms (COX1‐mediated dilation and purinergic constriction) may operate together to maintain the right balance of constrictory and dilatory factors to stabilise vascular tone in the face of changing microenvironment (Fig. 2), both within the brain and the blood, an effect that is well known elsewhere in the vascular system (Meininger & Davis, 1992) and in extracerebral pial arteries (Faraci et al. 1989).
Vascular signalling to astrocytes
Cortical astrocytes have been reported to respond with a [Ca2+]i rise to pressure changes in arterioles both in vitro and in vivo (Kim et al. 2015). This suggests two interesting possibilities: (i) the nervous system can sense vascular changes and may be able to adjust them in turn; and (ii) the cardiovascular system may be able to alter neuronal function. In the Kim et al. study, changes in vascular pressure raised [Ca2+]i in astrocytes, which contributed to the maintenance of pressure‐evoked cerebrovascular tone, thus providing some support to the first conjecture (Kim et al. 2015). There is also evidence that astrocytes in the brain stem sense blood pH to activate chemoreceptor neurons and alter breathing rate (Gourine et al. 2010), and those in the subfornical organ sense blood Na+ levels to control salt intake behaviour (Shimizu et al. 2007). Similarly, it is possible that astrocyte sensing of vascular pressure may, directly or via astrocyte‐mediated neuronal activation, lead to the regulation of cardiovascular function. On the other hand, signals from blood vessels might also be altering neuronal function. An example of direct signalling from blood vessels to neurons via NO production and release has been demonstrated in the optic nerve (Garthwaite et al. 2006). As Ca2+‐dependent signalling within astrocytes can regulate neuronal activity by releasing gliotransmitters (Parpura et al. 1994; Henneberger et al. 2010; Panatier et al. 2011; Araque et al. 2014), it is plausible that vascular‐evoked changes in astrocyte Ca2+ may modulate neuronal function, as proposed by the ‘hemo‐neural hypothesis’ (Moore & Cao, 2008; Kim et al. 2015). All these possibilities ought to be further explored, especially in the context of altered vascular tone in cardiovascular diseases such as hypertension that accompany, or may lead to, neurodegenerative disorders (Bloch et al. 2015).
Astrocytes and cerebrovascular dysfunction in CNS pathology
Astrocytes respond to almost all forms of CNS dysfunction with a process termed reactive astrogliosis (Barres, 2008; Anderson et al. 2014), during which their morphology and protein expression patterns change drastically (Hamby et al. 2012; Zamanian et al. 2012). This suggests that the function of astrocytes may evolve during disease, although it is unclear whether these changes contribute to the progression of pathology or exist as protective mechanisms. Challenging cultured astrocytes with inflammatory mediators alters the expression of many proteins involved in neuron‐to‐astrocyte signalling and, accordingly, astrocyte Ca2+ signalling is markedly altered (Hamby et al. 2012). Reactive astrocytes change their expression of neurotransmitter receptors and transporters such as mGluRs (Aronica et al. 2000) and GLT‐1 (Rothstein et al. 1995; Bramlett & Dietrich, 2004). They also alter the expression of the inwardly rectifying K+ channels, altering their ability to buffer extracellular K+ (Bordey et al. 2000, 2001; Tong et al. 2014). Such changes in astrocyte physiology, particularly in their Ca2+ dynamics and K+ buffering ability, are expected to alter their role in the regulation of cerebral blood flow with possible negative effects on neuronal function. The need to address the neurovascular unit as a therapeutic target in the context of diseases with a vascular component, including Alzheimer's disease (AD), cortical spreading depression, traumatic brain injury and hypertension, has been recently highlighted (Calcinaghi et al. 2013; Iadecola, 2013; Bloch et al. 2015; Lok et al. 2015; Ostergaard et al. 2015). Astrogliosis‐induced changes in NVC deserve further attention in this regard. Indeed, a recent report showed that an angiotensin II type 1 receptor blocker can improve the cognitive and cerebrovascular deficits in AD by reducing astrogliosis and inflammation, while leaving Aβ pathology intact (Ongali et al. 2014). Another study reported that glioma cells invade the space between astrocyte endfeet and the vasculature, disrupting NVC (Watkins et al. 2014). Such structural changes are likely to alter the generation of vasoactive signals as well as the response of the neurovascular unit to these signals and perhaps also lead to blood–brain barrier dysfunction, although these ideas have not yet been investigated. Together, these findings imply that CNS pathology may partly comprise a deficit in gliovascular coupling and commands more attention.
In diabetic patients, a reduction in functional hyperaemia precedes clinical retinopathy (Garhofer et al. 2004; Mandecka et al. 2007). In a rodent model of diabetes, a similar deficit in NVC was reported. This seemed to be mediated by altered glial AA metabolism due to high NO levels produced by increased inducible NOS (iNOS) expression (Mishra & Newman, 2010). This deficit could be pharmacologically reversed in diabetic animals by acute i.v. or chronic oral administration of an iNOS blocker (Mishra & Newman, 2011). As iNOS upregulation is associated with inflammatory responses that occur after injury throughout the body, including in the brain following stroke (Iadecola et al. 1995; Garry et al. 2015), and can alter glial derived vascular messengers (Metea & Newman, 2007; Mishra & Newman, 2010), its role in glial‐mediated NVC in neuropathology needs to be further explored.
Another factor to consider in disease is the tissue O2 level. Although high levels of O2 exposure have no effect on NVC responses in the healthy brain in vivo (Lindauer et al. 2010; Mishra et al. 2011) because of autoregulatory mechanisms that maintain tissue within an acceptable range (Torbati et al. 1978; Omae et al. 1998; Floyd et al. 2003; Lu et al. 2009; Mishra et al. 2011), this might be altered in disease states. In injuries such as ischaemic stroke, subarachnoid haemorrhage and traumatic brain injury, a breakdown of the blood–brain barrier often ensues (Doczi, 1985; Germano et al. 2000; Price et al. 2016; Ueno et al. 2016), and indeed is being recognised as another common hallmark of CNS pathology (Zhao et al. 2015). It is plausible that this breakdown, combined with a lack of autoregulatory compensation, exposes the brain to pathological levels of O2 and contributes to the observed reduction (Fordsmann et al. 2013) or inversion (Koide et al. 2012; Pappas et al. 2015) of the neurovascular response. In light of the astrogliosis following brain injury and the ensuing changes in their [Ca2+]i dynamics (Hamby et al. 2012) and K+ uptake (Bordey et al. 2000), astrocyte‐mediated neurovascular pathways such as those dependent on BK channels (Pappas et al. 2015) or 20‐HETE synthesis (Fordsmann et al. 2013) might be altered in disease. This is a particularly attractive hypothesis in the context of reperfusion injury, where reinstatement of blood flow after a stroke beyond a limited therapeutic time window results in further damage to the nervous tissue (Bai & Lyden, 2015; Marshall, 2015). The sudden rise in tissue oxygenation produced by the return of blood, especially in the absence of a healthy blood–brain barrier, is known to induce oxidative stress (Bai & Lyden, 2015) but it might also result in the production of glial‐derived vasoconstricting factors (Fordsmann et al. 2013), limiting the supply of oxygen and glucose to neurons and resulting in further injury.
Furthermore, autoregulation and NVC are both dependent on the resting tone of cerebral vessels (Aaslid et al. 1989; Blanco et al. 2008). Given that astrocytes play a role in both these processes and that the resting tone of vessels is altered or disrupted in CNS pathologies such as subarachnoid haemorrhage (Terpolilli et al. 2015), glioma (Watkins et al. 2014) and cortical spreading depression (Ayata & Lauritzen, 2015), understanding the role of astrocytes in cerebrovascular function in both healthy and pathological conditions is essential for unravelling disease mechanisms and developing novel therapeutic targets.
Development of the neuro‐glio‐vascular unit
Although neurogenesis is largely complete before birth in rodents, their connections take longer to mature. Astrocytes and the vasculature, integral members of the neurovascular unit, begin development late in the embryo and continue for the first few weeks of life (Caley & Maxwell, 1970; Bayraktar et al. 2015). Astrocyte density and morphology in the rat matures to adult levels differentially in different brain regions: at about postnatal day 8 in cortical layer I (Stichel et al. 1991), 2–3 weeks in the hippocampus (Nixdorf‐Bergweiler et al. 1994) and cortical layer VI (Stichel et al. 1991), and approximately 6 weeks in cortical layers II–V (Stichel et al. 1991). This is reflected in the K+ buffering capacity of the brain, which does not reach mature levels until 4 weeks after birth (Nixdorf‐Bergweiler et al. 1994). The diffuse cloud of fine astrocyte processes mature and form astrocyte territories during the third week after birth (Bushong et al. 2004) and synthesis of vasoactive mediators such as AA metabolites in the brain also reaches adult levels at the same time, probably as a result of the establishment of adult levels of enzyme expression in the astrocytes (Seregi et al. 1987). Existence of the morphologically complete neurovascular unit, comprising the vasculature, neurons and the fine processes and endfeet of astrocytes, is only apparent 3 weeks after birth in the cortex (Caley & Maxwell, 1970). Accordingly, hindpaw stimulation in rodents younger than 2 weeks (mimicking neonatal age in humans) evokes a decrease in blood flow and pial arterial constrictions in vivo (Zehendner et al. 2013; Kozberg & Hillman, 2016). This response evolves to display stimulation‐evoked localised increases in blood flow and pial dilations in a time course parallel to the structural development of the neurovascular unit, starting at around 3 weeks of age (Zehendner et al. 2013; Kozberg & Hillman, 2016). This early inverted vascular response is thought to be an important factor in the development of a healthy, mature cerebrovascular architecture and perhaps also the blood–brain barrier (reviewed in Lacoste & Gu, 2015).
Besides the inversion of the neurovascular response, other age‐related factors may also contribute to the discrepancies found in NVC research. Studies that use in vitro experiments to investigate NVC are largely conducted on tissue from young animals, while in vivo studies primarily use adult animals, presenting a co‐confound of age and experimental preparation. For example, mGluR‐dependent astrocyte‐mediated NVC was demonstrated in slices from young animals (P9–21; Zonta et al. 2003; Mulligan & Macvicar, 2004; Gordon et al. 2008) but, perhaps owing to the developmental downregulation of mGluR I expression in astrocytes (Sun et al. 2013), this mechanism did not appear to play a role in adults animals in vivo (Calcinaghi et al. 2011). Therefore, the age of animals should be carefully considered in the design and interpretation of NVC studies.
Implications of astrocyte control of NVC for BOLD signals
The BOLD signal, used in most functional imaging studies as a proxy measure of brain activity, measures the relative concentration of deoxyhaemoglobin, which rises during activity due to oxygen consumption by active neurons but falls as the increase in blood flow brings in more oxyhaemoglobin. Because the blood flow response is much greater than the fall in deoxyhaemoglobin, BOLD signals primarily reflect the increase in blood flow evoked by neuronal activation (Attwell & Iadecola, 2002). As outlined in this review, astrocytes play a complex role in generating the neurovascular response as well as maintaining the baseline tone of the cerebral vasculature, which influences the magnitude of the BOLD response. This has important implications for the interpretation of functional imaging studies, particularly with reference to different arousal states, disease and development stages. Subcortical neuronal mechanisms that are responsible for modulating brain activity during sleep or arousal states also exert control of the cerebral parenchymal arterioles over large regions of the cerebrum (Hamel, 2004, 2006) and appear to be at least partly mediated by glial cells (Cauli & Hamel, 2010; Lecrux et al. 2011). Depending on the arousal state of the subject, the relationship between neuronal activity, astrocyte activity and vascular responses might be differentially modulated by these subcortical nerve terminals, and therefore impact the BOLD signal.
In most CNS diseases, neuronal activity is noticeably altered, but less perceptible are the changes that occur in astrocytes and vascular reactivity. This is partly due to our lack of complete understanding of the disease processes and partly because these components of the nervous system have not been studied in as much detail as neurons. It is very likely that neurovascular signalling pathways might be altered in disease due to altered signalling from reactive astrocytes (as discussed above) or a change in vascular rigidity or reactivity (Hamel, 2015; Tong & Hamel, 2015), leading to a reduction in the coupling or even a complete uncoupling of neuronal activity from vascular responses. Furthermore, conditions like tissue damage and oedema might change the water component of the tissue, which can also alter the BOLD signal (Krings et al. 2002; Kim & Ogawa, 2012). BOLD may still be a powerful tool in identifying a disease or measuring signals that correlate with symptoms, but it would be impractical to use BOLD signals as a measure of neuronal activity per se under these conditions.
The use of BOLD signals to study developmental processes also comes with severe limitations (Harris et al. 2011; Lacoste & Gu, 2015). As discussed above, the development of the neuro‐glio‐vascular unit, which is essential for the proper execution of neurovascular signals that give rise to activity‐evoked blood flow changes, continues for weeks after birth in rodents. In humans, gliogenesis is essentially complete within the first few months after birth (Roessmann & Gambetti, 1986), but synaptogenesis and synaptic pruning continue into the teenage years while myelination can last into the twenties in humans (reviewed in Semple et al. 2013). Anatomical studies suggest that although the human brain is 95% of its adult size by age 6, it is still developing into the teenage years (Lenroot & Giedd, 2006). The ‘mature’ neuro‐glio‐vascular unit in the rat is only apparent after 3 weeks of age and continues to develop until at least 6 weeks of age (Caley & Maxwell, 1970). Using a simplistic model to compare the developmental stages of rodent and human brains, this suggests a minimum age of 1.5 years before the neurovascular response can be expected to mature in humans (http://www.translatingtime.net). Indeed, activity‐evoked blood flow and BOLD responses are inverted in young rodents and neonatal humans (Anderson et al. 2001; Born et al. 2002; Zehendner et al. 2013; Kozberg & Hillman, 2016), probably reflecting the large oxygen consumption of the developing brain combined with a decrease in blood flow (Lacoste & Gu, 2015). This developmental negative relationship between neuronal activity and blood flow is thought to be important in patterning the cerebrovasculature and, ultimately, brain maturation (Lacoste & Gu, 2015). Given these developmental changes, our ability to effectively decipher BOLD signals during childhood and adolescence is severely hampered by a lack of systematic studies investigating the neurovascular relationship during the course of early life in humans (further discussed in Harris et al. 2011) and should be carefully considered in interpreting data from these populations.
The transcriptome as a resource for future directions
Many aspects of the astrocytic regulation of cerebral blood flow are as yet uncharted territory. One possible strategy to develop new hypotheses and guide future experiments would be to use published transcriptome analyses (Cahoy et al. 2008; Hamby et al. 2012; Zamanian et al. 2012; Zhang et al. 2014). A quick study of the genes expressed at a > 15‐fold higher level in astrocytes than in all other cell types in the transcriptome published from the Barres lab (Zhang et al. 2014) identified the adenosine receptor A2b, mGluR3, small and intermediate conductance K+ channels, many enzymes (of particular interest were phospholipases, epoxide hydrolases and CYP enzymes) and a large number of transporters including those for glutamate, GABA, glucose, zinc and cationic amino acids. Many of these proteins, such as mGluRs, adenosine receptors, K+ channels and enzymes involved in AA metabolism, are already known to be expressed in astrocytes and play important roles in maintaining homeostasis of the extracellular environment and NVC, as discussed in this review. Many others probably play an important role in astrocyte‐specific functions and would be of interest to future neuroglial and neurovascular studies. Furthermore, astrocyte‐specific genes that help them associate with the vasculature, such as vascular cell adhesion molecule 1, or play a role in their response to disease, such as pentraxin 3 and fibroblast growth factors (particularly FGF8 and FGF3 and their respective receptors) (Rosenman et al. 1995; Ravizza et al. 2001; Rubio et al. 2010; Kang et al. 2014), might be of particular interest with respect to alterations in cerebrovascular tone and NVC following disease. Although the transcriptome provides a measure of the level of RNA transcribed and not protein expression, it may prove to be an important starting point in understanding the functions of astrocytes and other glial cells.
Conclusion
The evidence undeniably favours a multifaceted role for astrocytes in cerebrovascular regulation. NVC occurs to supply the energy demand of active neurons and, therefore, it is indisputable that neurons initiate the response, but increasing evidence supports a role for astrocytes as mediators of neurovascular signals. However, controversies still exist regarding the relative contribution of vasoactive signals arising directly from neurons (such as NO and prostaglandins) versus those released by astrocytes (AA metabolites, K+ release), as well as the location and timing of astrocytic Ca2+ signalling. The pattern of astrocyte Ca2+ signalling (for example, localised transients in processes compared to global intracellular waves) may also be important in defining the vascular response generated. We must endeavour to develop better experimental designs and [Ca2+] detection techniques to resolve these outstanding questions. Astrocytes also play a role in regulating the resting tone of arterioles by releasing both dilating and constricting agents. They can respond to alterations in cardiovascular factors such as pressure and flow which, given their role in neuronal homeostasis, may alter neuronal activity. This suggests a novel binaural role for astrocytes whereby they listen to signals from both neurons and vessels and, accordingly, orchestrate signalling to the cerebral vasculature to maintain a healthy blood supply to the brain. Their role in maintaining this intricate balance in healthy brains, and how it is altered in pathology, must be explored further with a view to developing new therapeutic targets for diseases of the CNS where the cerebrovasculature is affected.
Additional information
Competing interests
The author declares no competing interests.
Acknowledgements
I thank David Attwell, Grant R. Gordon, Renaud B. Jolivet, Steven J. Sullivan and Yang Chen for their help in the preparation of this manuscript.
Biography
Anusha Mishra started her research career in Kristen Harris's lab at the Medical College of Georgia, studying electron micrographs of the brain. She then did her PhD with Eric Newman at the University of Minnesota where she investigated changes in retinal neurovascular coupling in pathology and discovered a drug that reverses the loss of this response in diabetic animals. She is currently doing her postdoctoral training in David Attwell's lab at University College London, where she has been studying capillary level neurovascular coupling in health and disease and the role of astrocytes in mediating this response.

References
- Aaslid R, Lindegaard KF, Sorteberg W & Nornes H (1989). Cerebral autoregulation dynamics in humans. Stroke 20, 45–52. [DOI] [PubMed] [Google Scholar]
- Akgören N, Dalgaard P & Lauritzen M (1996). Cerebral blood flow increases evoked by electrical stimulation of rat cerebellar cortex: relation to excitatory synaptic activity and nitric oxide synthesis. Brain Res 710, 204–214. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Birks EK, Hudetz AG, Roman RJ, Henderson L & Harder DR (1996. a). Inhibition of brain P‐450 arachidonic acid epoxygenase decreases baseline cerebral blood flow. Am J Physiol Heart Circ Physiol 271, H1541–H1546. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ & Harder DR (1996. b). Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke 27, 971–979. [DOI] [PubMed] [Google Scholar]
- Amruthesh SC, Boerschel MF, McKinney JS, Willoughby KA & Ellis EF (1993). Metabolism of arachidonic acid to epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and prostaglandins in cultured rat hippocampal astrocytes. J Neurochem 61, 150–159. [DOI] [PubMed] [Google Scholar]
- Anderson AW, Marois R, Colson ER, Peterson BS, Duncan CC, Ehrenkranz RA, Schneider KC, Gore JC & Ment LR (2001). Neonatal auditory activation detected by functional magnetic resonance imaging. Magn Reson Imaging 19, 1–5. [DOI] [PubMed] [Google Scholar]
- Anderson MA, Ao Y & Sofroniew MV (2014). Heterogeneity of reactive astrocytes. Neurosci Lett 565, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R & Volterra A (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH & Gorter JA (2000). Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci 12, 2333–2344. [DOI] [PubMed] [Google Scholar]
- Ase AR, Bernier LP, Blais D, Pankratov Y & Seguela P (2010). Modulation of heteromeric P2X1/5 receptors by phosphoinositides in astrocytes depends on the P2X1 subunit. J Neurochem 113, 1676–1684. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA & Newman EA (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D & Iadecola C (2002). The neural basis of functional brain imaging signals. Trends Neurosci 25, 621–625. [DOI] [PubMed] [Google Scholar]
- Attwell D & Laughlin SB (2001). An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21, 1133–1145. [DOI] [PubMed] [Google Scholar]
- Ayata C & Lauritzen M (2015). Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95, 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J & Lyden PD (2015). Revisiting cerebral postischemic reperfusion injury: new insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke 10, 143–152. [DOI] [PubMed] [Google Scholar]
- Barbour B, Brew H & Attwell D (1988). Electrogenic glutamate uptake in glial cells is activated by intracellular potassium. Nature 335, 433–435. [DOI] [PubMed] [Google Scholar]
- Barres BA (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. [DOI] [PubMed] [Google Scholar]
- Bayraktar OA, Fuentealba LC, Alvarez‐Buylla A & Rowitch DH (2015). Astrocyte development and heterogeneity. Cold Spring Harb Perspect Biol 7, a020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazargani N & Attwell D (2016). Astrocyte calcium signaling: the third wave. Nat Neurosci 19, 182–189. [DOI] [PubMed] [Google Scholar]
- Blanchard S, Saillet S, Ivanov A, Benquet P, Benar CG, Pelegrini‐Issac M, Benali H & Wendling F (2016). A new computational model for neuro‐glio‐vascular coupling: astrocyte activation can explain cerebral blood flow nonlinear response to interictal events. PloS One 11, e0147292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco VM, Stern JE & Filosa JA (2008). Tone‐dependent vascular responses to astrocyte‐derived signals. Am J Physiol Heart Circ Physiol 294, H2855–H2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinder P, Tsai PS, Kaufhold JP, Knutsen PM, Suhl H & Kleinfeld D (2013). The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat Neurosci 16, 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch S, Obari D & Girouard H (2015). Angiotensin and neurovascular coupling: beyond hypertension. Microcirculation 22, 159–167. [DOI] [PubMed] [Google Scholar]
- Bonder DE & McCarthy KD (2014). Astrocytic Gq‐GPCR‐linked IP3R‐dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo . J Neurosci 34, 13139–13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, Hablitz JJ & Sontheimer H (2000). Reactive astrocytes show enhanced inwardly rectifying K+ currents in situ . Neuroreport 11, 3151–3155. [DOI] [PubMed] [Google Scholar]
- Bordey A, Lyons SA, Hablitz JJ & Sontheimer H (2001). Electrophysiological characteristics of reactive astrocytes in experimental cortical dysplasia. J Neurophysiol 85, 1719–1731. [DOI] [PubMed] [Google Scholar]
- Born AP, Rostrup E, Miranda MJ, Larsson HB & Lou HC (2002). Visual cortex reactivity in sedated children examined with perfusion MRI (FAIR). Magn Reson Imaging 20, 199–205. [DOI] [PubMed] [Google Scholar]
- Bowser DN & Khakh BS (2007). Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J Gen Physiol 129, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlett HM & Dietrich WD (2004). Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab 24, 133–150. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME & Ellisman MH (2004). Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. In J Dev Neurosci 22, 73–86. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ & Ellisman MH (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ & Barres BA (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcinaghi N, Jolivet R, Wyss MT, Ametamey SM, Gasparini F, Buck A & Weber B (2011). Metabotropic glutamate receptor mGluR5 is not involved in the early hemodynamic response. J Cereb Blood Flow Metab 31, e1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcinaghi N, Wyss MT, Jolivet R, Singh A, Keller AL, Winnik S, Fritschy JM, Buck A, Matter CM & Weber B (2013). Multimodal imaging in rats reveals impaired neurovascular coupling in sustained hypertension. Stroke 44, 1957–1964. [DOI] [PubMed] [Google Scholar]
- Caley DW & Maxwell DS (1970). Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J Comp Neurol 138, 31–47. [DOI] [PubMed] [Google Scholar]
- Cauli B & Hamel E (2010). Revisiting the role of neurons in neurovascular coupling. Front Neuroenergetics 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J & Hamel E (2004). Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci 24, 8940–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan BS, Endo S, Kanai N & Schuster VL (2002). Identification of lactate as a driving force for prostanoid transport by prostaglandin transporter PGT. Am J Physiol Renal Physiol 282, F1097–F1102. [DOI] [PubMed] [Google Scholar]
- Chatton JY, Pellerin L & Magistretti PJ (2003). GABA uptake into astrocytes is not associated with significant metabolic cost: implications for brain imaging of inhibitory transmission. Proc Natl Acad Sci USA 100, 12456–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BR, Kozberg MG, Bouchard MB, Shaik MA & Hillman EM (2014). A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc 3, e000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Z, Molinatti G & Hamel E (1997). Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab 17, 894–904. [DOI] [PubMed] [Google Scholar]
- Cornell‐Bell AH, Finkbeiner SM, Cooper MS & Smith SJ (1990). Glutamate induces calcium waves in cultured astrocytes: long‐range glial signaling. Science 247, 470–473. [DOI] [PubMed] [Google Scholar]
- Cringle SJ & Yu DY (2010). Oxygen supply and consumption in the retina: implications for studies of retinopathy of prematurity. Doc Ophthalmol 120, 99–109. [DOI] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A & Smith SJ (1992). Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 8, 429–440. [DOI] [PubMed] [Google Scholar]
- Devor A, Tian P, Nishimura N, Teng IC, Hillman EM, Narayanan SN, Ulbert I, Boas DA, Kleinfeld D & Dale AM (2007). Suppressed neuronal activity and concurrent arteriolar vasoconstriction may explain negative blood oxygenation level‐dependent signal. J Neurosci 27, 4452–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, Tiret P & Volterra A (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci 14, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Doczi T (1985). The pathogenetic and prognostic significance of blood–brain barrier damage at the acute stage of aneurysmal subarachnoid haemorrhage. Clinical and experimental studies. Acta Neurochir 77, 110–132. [DOI] [PubMed] [Google Scholar]
- Ellis EF, Police RJ, Yancey L, McKinney JS & Amruthesh SC (1990). Dilation of cerebral arterioles by cytochrome P‐450 metabolites of arachidonic acid. Am J Physio l Heart Circ Physiol 259, H1171–H1177. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Baumbach GL & Heistad DD (1989). Myogenic mechanisms in the cerebral circulation. J Hypertens Suppl 7, S61–64; discussion S65. [PubMed] [Google Scholar]
- Farr H & David T (2011). Models of neurovascular coupling via potassium and EET signalling. J Theoret Biol 286, 13–23. [DOI] [PubMed] [Google Scholar]
- Fellin T, Sul JY, D'Ascenzo M, Takano H, Pascual O & Haydon PG (2006). Bidirectional astrocyte–neuron communication: the many roles of glutamate and ATP. Novartis Found Symp 276, 208–217; discussion 217–221, 233–207, 275–281. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW & Nelson MT (2006). Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9, 1397–1403. [DOI] [PubMed] [Google Scholar]
- Floyd TF, Clark JM, Gelfand R, Detre JA, Ratcliffe S, Guvakov D, Lambertsen CJ & Eckenhoff RG (2003). Independent cerebral vasoconstrictive effects of hyperoxa and accompanying arterial hypocapnia at 1 ATA. J Appl Physiol 95, 2453–2461. [DOI] [PubMed] [Google Scholar]
- Fordsmann JC, Ko RW, Choi HB, Thomsen K, Witgen BM, Mathiesen C, Lonstrup M, Piilgaard H, MacVicar BA & Lauritzen M (2013). Increased 20‐HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci 33, 2562–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garhofer G, Zawinka C, Resch H, Kothy P, Schmetterer L & Dorner GT (2004). Reduced response of retinal vessel diameters to flicker stimulation in patients with diabetes. Br J Ophthalmol 88, 887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry PS, Ezra M, Rowland MJ, Westbrook J & Pattinson KT (2015). The role of the nitric oxide pathway in brain injury and its treatment – from bench to bedside. Exp Neurol 263, 235–243. [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Bartus K, Malcolm D, Goodwin D, Kollb–Sielecka M, Dooldeniya C & Garthwaite J (2006). Signaling from blood vessels to CNS axons through nitric oxide. J Neurosci 26, 7730–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, Narayanan J, Falck JR, Okamoto H, Roman RJ, Nithipatikom K, Campbell WB & Harder DR (2000). Production of 20‐HETE and its role in autoregulation of cerebral blood flow. Circ Res 87, 60–65. [DOI] [PubMed] [Google Scholar]
- Germano A, d'Avella D, Imperatore C, Caruso G & Tomasello F (2000). Time‐course of blood–brain barrier permeability changes after experimental subarachnoid haemorrhage. Acta Neurochir 142, 575–580; discussion 580–571. [DOI] [PubMed] [Google Scholar]
- Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW & Nelson MT (2010). Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci USA 107, 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis‐Davies GC & Macvicar BA (2008). Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K & Kasparov S (2010). Astrocytes control breathing through pH‐dependent release of ATP. Science 329, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M & Attwell D (2014). Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS & Sofroniew MV (2012). Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G‐protein‐coupled receptors. J Neurosci 32, 14489–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E (2004). Cholinergic modulation of the cortical microvascular bed. Prog Brain Res 145, 171–178. [DOI] [PubMed] [Google Scholar]
- Hamel E (2006). Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol 100, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Hamel E (2015). Cerebral circulation: function and dysfunction in Alzheimer's disease. J Cardiovasc Pharmacol 65, 317–324. [DOI] [PubMed] [Google Scholar]
- Hamilton NB & Attwell D (2010). Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11, 227–238. [DOI] [PubMed] [Google Scholar]
- Harder DR, Alkayed NJ, Lange AR, Gebremedhin D & Roman RJ (1998). Functional hyperemia in the brain: hypothesis for astrocyte‐derived vasodilator metabolites. Stroke 29, 229–234. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R & Attwell D (2012). Synaptic energy use and supply. Neuron 75, 762–777. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Reynell C & Attwell D (2011). The physiology of developmental changes in BOLD functional imaging signals. Dev Cogn Neurosci 1, 199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Paulsen RE, Johnsen A & Fonnum F (1992). Selective inhibition of glial cell metabolism in vivo by fluorocitrate. Brain Res 576, 120–124. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH & Rusakov DA (2010). Long‐term potentiation depends on release of d‐serine from astrocytes. Nature 463, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hild W, Chang JJ & Tasaki I (1958). Electrical responses of astrocytic glia from the mammalian central nervous system cultivated in vitro . Experientia 14, 220–221. [DOI] [PubMed] [Google Scholar]
- Hirase H, Qian L, Bartho P & Buzsaki G (2004). Calcium dynamics of cortical astrocytic networks in vivo . PLoS Biol 2, E96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EG & Holmes BE (1926). Contributions to the study of brain metabolism: carbohydrate metabolism relationship of glycogen and lactic acid. Biochem J 20, 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T, Dietrich HH, Hongo K & Dacey RG Jr (2002). Mechanism of extracellular K+‐induced local and conducted responses in cerebral penetrating arterioles. Stroke 33, 2692–2699. [DOI] [PubMed] [Google Scholar]
- Huck S, Grass F & Hatten ME (1984). Gliotoxic effects of alpha‐aminoadipic acid on monolayer cultures of dissociated postnatal mouse cerebellum. Neuroscience 12, 783–791. [DOI] [PubMed] [Google Scholar]
- Iadecola C (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Xu S, Casey R & Ross ME (1995). Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 15, 378–384. [DOI] [PubMed] [Google Scholar]
- Institoris A, Rosenegger DG & Gordon GR (2015). Arteriole dilation to synaptic activation that is sub‐threshold to astrocyte endfoot Ca2+ transients. J Cereb Blood Flow Metab 35, 1411–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaglin XH, Hjerling‐Leffler J, Fishell G & Batista‐Brito R (2012). The origin of neocortical nitric oxide synthase‐expressing inhibitory neurons. Front Neural Circuits 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego P, Pacheco‐Torres J, Araque A & Canals S (2014). Functional MRI in mice lacking IP3‐dependent calcium signaling in astrocytes. J Cereb Blood Flow Metab 34, 1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K, Lee SW, Han JE, Choi JW & Song MR (2014). The complex morphology of reactive astrocytes controlled by fibroblast growth factor signaling. Glia 62, 1328–1344. [DOI] [PubMed] [Google Scholar]
- Kety SS (1957). The general metabolism of the brain in vivo In Metabolism of the Nervous System, ed. Richter D, pp. 221–237. Pergamon, London. [Google Scholar]
- Kim KJ & Filosa JA (2012). Advanced in vitro approach to study neurovascular coupling mechanisms in the brain microcirculation. J Physiol 590, 1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Iddings JA, Stern JE, Blanco VM, Croom D, Kirov SA & Filosa JA (2015). Astrocyte contributions to flow/pressure‐evoked parenchymal arteriole vasoconstriction. J Neurosci 35, 8245–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG & Ogawa S (2012). Biophysical and physiological origins of blood oxygenation level‐dependent fMRI signals. J Cereb Blood Flow Metab 32, 1188–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff F, Dringen R & Giaume C (2001). Pathways of neuron–astrocyte interactions and their possible role in neuroprotection. Eur Arch Psychiatry Clin Neurosci 251, 159–169. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Kettenmann H & Verkhratsky A (1997). Na+/Ca2+ exchanger modulates kainate‐triggered Ca2+ signaling in Bergmann glial cells in situ . FASEB J 11, 566–572. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA & Nelson MT (1996). Extracellular K+‐induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J Physiol 492, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P & Newman EA (2004). Potassium buffering in the central nervous system. Neuroscience 129, 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide M, Bonev AD, Nelson MT & Wellman GC (2012). Inversion of neurovascular coupling by subarachnoid blood depends on large‐conductance Ca2+‐activated K+ (BK) channels. Proc Natl Acad Sci USA 109, E1387–E1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfield TE & Newman EA (2014). Regulation of blood flow in the retinal trilaminar vascular network. J Neurosci 34, 11504–11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozberg M & Hillman E (2016). Neurovascular coupling and energy metabolism in the developing brain. Prog Brain Res 225, 213–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimer LS, Muly EC 3rd, Williams GV & Goldman‐Rakic PS (1998). Dopaminergic regulation of cerebral cortical microcirculation. Nat Neurosci 1, 286–289. [DOI] [PubMed] [Google Scholar]
- Krings T, Reinges MH, Willmes K, Nuerk HC, Meister IG, Gilsbach JM & Thron A (2002). Factors related to the magnitude of T2* MR signal changes during functional imaging. Neuroradiology 44, 459–466. [DOI] [PubMed] [Google Scholar]
- Kuffler SW & Potter DD (1964). Glia in the leech central nervous system: physiological properties and neuron–glia relationship. J Neurophysiol 27, 290–320. [DOI] [PubMed] [Google Scholar]
- Kur J & Newman EA (2014). Purinergic control of vascular tone in the retina. J Physiol 592, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth‐Nelson ZL, Mishra A & Newman EA (2009). Spontaneous glial calcium waves in the retina develop over early adulthood. J Neurosci 29, 11339–11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste B & Gu C (2015). Control of cerebrovascular patterning by neural activity during postnatal development. Mech Dev 138, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix A, Toussay X, Anenberg E, Lecrux C, Ferreiros N, Karagiannis A, Plaisier F, Chausson P, Jarlier F, Burgess SA, Hillman EM, Tegeder I, Murphy TH, Hamel E & Cauli B (2015). COX‐2‐derived prostaglandin E2 produced by pyramidal neurons contributes to neurovascular coupling in the rodent cerebral cortex. J Neurosci 35, 11791–11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA & Verkhratsky A (2006). NMDA receptors mediate neuron‐to‐glia signaling in mouse cortical astrocytes. J Neurosci 26, 2673–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Wichert SP, Rossner MJ, North RA, Kirchhoff F & Verkhratsky A (2008). P2X1 and P2X5 subunits form the functional P2X receptor in mouse cortical astrocytes. J Neurosci 28, 5473–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Verkhratsky A & Pankratov Y (2011). Ionotropic ATP receptors in neuronal–glial communication. Semin Cell Dev Biol 22, 220–228. [DOI] [PubMed] [Google Scholar]
- Lecrux C, Toussay X, Kocharyan A, Fernandes P, Neupane S, Levesque M, Plaisier F, Shmuel A, Cauli B & Hamel E (2011). Pyramidal neurons are "neurogenic hubs" in the neurovascular coupling response to whisker stimulation. J Neurosci 31, 9836–9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenroot RK & Giedd JN (2006). Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci Biobehav Rev 30, 718–729. [DOI] [PubMed] [Google Scholar]
- Lian XY & Stringer JL (2004. a). Astrocytes contribute to regulation of extracellular calcium and potassium in the rat cerebral cortex during spreading depression. Brain Res 1012, 177–184. [DOI] [PubMed] [Google Scholar]
- Lian XY & Stringer JL (2004. b). Energy failure in astrocytes increases the vulnerability of neurons to spreading depression. Eur J Neurosci 19, 2446–2454. [DOI] [PubMed] [Google Scholar]
- Lind BL, Brazhe AR, Jessen SB, Tan FC & Lauritzen MJ (2013). Rapid stimulus‐evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo . Proc Natl Acad Sci USA 110, E4678–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindauer U, Leithner C, Kaasch H, Rohrer B, Foddis M, Fuchtemeier M, Offenhauser N, Steinbrink J, Royl G, Kohl‐Bareis M & Dirnagl U (2010). Neurovascular coupling in rat brain operates independent of hemoglobin deoxygenation. J Cereb Blood Flow Metab 30, 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindauer U, Megow D, Matsuda H & Dirnagl U (1999). Nitric oxide: a modulator, but not a mediator, of neurovascular coupling in rat somatosensory cortex. Am J Physiol Heart Circ Physiol 277, H799–H811. [DOI] [PubMed] [Google Scholar]
- Liu X, Li C, Gebremedhin D, Hwang SH, Hammock BD, Falck JR, Roman RJ, Harder DR & Koehler RC (2011). Epoxyeicosatrienoic acid‐dependent cerebral vasodilation evoked by metabotropic glutamate receptor activation in vivo . Am J Physiol Heart Circ Physiol 301, H373–H381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok J, Wang XS, Xing CH, Maki TK, Wu LM, Guo SZ, Noviski N, Arai K, Whalen MJ, Lo EH & Wang XY (2015). Targeting the neurovascular unit in brain trauma. CNS Neurosci Ther 21, 304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden TA & Nelson MT (2015). Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 22, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovick TA, Brown LA & Key BJ (2005). Neuronal activity‐related coupling in cortical arterioles: involvement of astrocyte‐derived factors. Exp Physiol 90, 131–140. [DOI] [PubMed] [Google Scholar]
- Lu J, Dai G, Egi Y, Huang S, Kwon SJ, Lo EH & Kim YR (2009). Characterization of cerebrovascular responses to hyperoxia and hypercapnia using MRI in rat. Neuro Image 45, 1126–1134. [DOI] [PubMed] [Google Scholar]
- Macvicar BA & Newman EA (2015). Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect Biol 7, a020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandecka A, Dawczynski J, Blum M, Muller N, Kloos C, Wolf G, Vilser W, Hoyer H & Muller UA (2007). Influence of flickering light on the retinal vessels in diabetic patients. Diabetes Care 30, 3048–3052. [DOI] [PubMed] [Google Scholar]
- Marshall RS (2015). Progress in intravenous thrombolytic therapy for acute stroke. JAMA Neurol 72, 928–934. [DOI] [PubMed] [Google Scholar]
- Martindale J, Berwick J, Martin C, Kong Y, Zheng Y & Mayhew J (2005). Long duration stimuli and nonlinearities in the neural–haemodynamic coupling. J Cereb Blood Flow Metab 25, 651–661. [DOI] [PubMed] [Google Scholar]
- Mathiisen TM, Lehre KP, Danbolt NC & Ottersen OP (2010). The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58, 1094–1103. [DOI] [PubMed] [Google Scholar]
- Meininger GA & Davis MJ (1992). Cellular mechanisms involved in the vascular myogenic response. Am J Physiol Heart Circ Physiol 263, H647–H659. [DOI] [PubMed] [Google Scholar]
- Meng W, Colonna DM, Tobin JR & Busija DW (1995. a). Nitric oxide and prostaglandins interact to mediate arteriolar dilation during cortical spreading depression. Am J Physiol Heart Circ Physiol 269, H176–H181. [DOI] [PubMed] [Google Scholar]
- Meng W, Tobin JR & Busija DW (1995. b). Glutamate‐induced cerebral vasodilation is mediated by nitric oxide through N‐methyl‐D‐aspartate receptors. Stroke 26, 857–862. [DOI] [PubMed] [Google Scholar]
- Metea MR, Kofuji P & Newman EA (2007). Neurovascular coupling is not mediated by potassium siphoning from glial cells. J Neurosci 27, 2468–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea MR & Newman EA (2006). Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci 26, 2862–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea MR & Newman EA (2007). Signalling within the neurovascular unit in the mammalian retina. Exp Physiol 92, 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger H, Erdmann W & Thews G (1971). Effect of short periods of hypoxia, hyperoxia, and hypercapnia on brain O2 supply. J Appl Physiol 31, 751–759. [DOI] [PubMed] [Google Scholar]
- Mishra A, Hamid A & Newman EA (2011). Oxygen modulation of neurovascular coupling in the retina. Proc Natl Acad Sci USA 108, 17827–17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A & Newman EA (2010). Inhibition of inducible nitric oxide synthase reverses the loss of functional hyperemia in diabetic retinopathy. Glia 58, 1996–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A & Newman EA (2011). Aminoguanidine reverses the loss of functional hyperemia in a rat model of diabetic retinopathy. Front Neuroenergetics 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CI & Cao R (2008). The hemo‐neural hypothesis: on the role of blood flow in information processing. J Neurophysiol 99, 2035–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosso A (1880). Sulla circolazione del sangue nel cervello dell'uomo. [On the circulation of blood in the human brain]. Rend Accad Lincei 5, 122. [Google Scholar]
- Mulligan SJ & Macvicar BA (2004). Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199. [DOI] [PubMed] [Google Scholar]
- Newman EA (2001). Propagation of intercellular calcium waves in retinal astrocytes and Muller cells. J Neurosci 21, 2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA (2006). A purinergic dialogue between glia and neurons in the retina. Novartis Found Symp 276, 193–202. [PubMed] [Google Scholar]
- Newman EA, Frambach DA & Odette LL (1984). Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 225, 1174–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Mukamel EA & Schnitzer MJ (2009). Motor behavior activates Bergmann glial networks. Neuron 62, 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixdorf–Bergweiler BE, Albrecht D & Heinemann U (1994). Developmental changes in the number, size, and orientation of GFAP‐positive cells in the CA1 region of rat hippocampus. Glia 12, 180–195. [DOI] [PubMed] [Google Scholar]
- Nizar K, Uhlirova H, Tian P, Saisan PA, Cheng Q, Reznichenko L, Weldy KL, Steed TC, Sridhar VB, MacDonald CL, Cui J, Gratiy SL, Sakadzic S, Boas DA, Beka TI, Einevoll GT, Chen J, Masliah E, Dale AM, Silva GA & Devor A (2013). In vivo stimulus‐induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J Neurosci 33, 8411–8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offenhauser N, Thomsen K, Caesar K & Lauritzen M (2005). Activity‐induced tissue oxygenation changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. J Physiol 565, 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata K & Kosaka T (2002). Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neurosci 113, 221–233. [DOI] [PubMed] [Google Scholar]
- Omae T, Ibayashi S, Kusuda K, Nakamura H, Yagi H & Fujishima M (1998). Effects of high atmospheric pressure and oxygen on middle cerebral blood flow velocity in humans measured by transcranial Doppler. Stroke 29, 94–97. [DOI] [PubMed] [Google Scholar]
- Ongali B, Nicolakakis N, Tong XK, Aboulkassim T, Papadopoulos P, Rosa–Neto P, Lecrux C, Imboden H & Hamel E (2014). Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer's disease model. Neurobiol Dis 68, 126–136. [DOI] [PubMed] [Google Scholar]
- Ostergaard L, Dreier JP, Hadjikhani N, Jespersen SN, Dirnagl U & Dalkara T (2015). Neurovascular coupling during cortical spreading depolarization and ‐depression. Stroke 46, 1392–1401. [DOI] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE & Charpak S (2015). Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci 18, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG & Moncada S (1987). Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature 327, 524–526. [DOI] [PubMed] [Google Scholar]
- Panatier A & Robitaille R (2015). Astrocytic mGluR5 and the tripartite synapse. Neurosci 323, 29–34. [DOI] [PubMed] [Google Scholar]
- Panatier A, Vallee J, Haber M, Murai KK, Lacaille JC & Robitaille R (2011). Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146, 785–798. [DOI] [PubMed] [Google Scholar]
- Pappas AC, Koide M & Wellman GC (2015). Astrocyte Ca2+ signaling drives inversion of neurovascular coupling after subarachnoid hemorrhage. J Neurosci 35, 13375–13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S & Haydon PG (1994). Glutamate‐mediated astrocyte–neuron signalling. Nature 369, 744–747. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM & Crunelli V (2001). Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR‐mediated neuronal excitation. Nat Neurosci 4, 803–812. [DOI] [PubMed] [Google Scholar]
- Paulsen RE, Contestabile A, Villani L & Fonnum F (1987). An in vivo model for studying function of brain tissue temporarily devoid of glial cell metabolism: the use of fluorocitrate. J Neurochem 48, 1377–1385. [DOI] [PubMed] [Google Scholar]
- Paulson OB & Newman EA (1987). Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science 237, 896–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, Traystman RJ & Koehler RC (2002). Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. Am J Physiol Heart Circ Physiol 283, H2029–H2037. [DOI] [PubMed] [Google Scholar]
- Peng X, Zhang C, Alkayed NJ, Harder DR & Koehler RC (2004). Dependency of cortical functional hyperemia to forepaw stimulation on epoxygenase and nitric oxide synthase activities in rats. J Cereb Blood Flow Metab 24, 509–517. [DOI] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P & Attwell D (2006). Bidirectional control of CNS capillary diameter by pericytes. Nature 443, 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW & Barres BA (1997). Synaptic efficacy enhanced by glial cells in vitro . Science 277, 1684–1687. [DOI] [PubMed] [Google Scholar]
- Piet R & Jahr CE (2007). Glutamatergic and purinergic receptor‐mediated calcium transients in Bergmann glial cells. J Neurosci 27, 4027–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl U, Herlan K, Huang A & Bassenge E (1991). EDRF‐mediated shear‐induced dilation opposes myogenic vasoconstriction in small rabbit arteries. Am J Physiol Heart Circ Physiol 261, H2016–H2023. [DOI] [PubMed] [Google Scholar]
- Price L, Wilson C & Grant G (2016). Blood–brain barrier pathophysiology following traumatic brain injury In Translational Research in Traumatic Brain Injury, eds Laskowitz D. & Grant G. CRC Press/Taylor and Francis Group, Boca Raton, FL, U: SA. [PubMed] [Google Scholar]
- Puro DG (2007). Physiology and pathobiology of the pericyte‐containing retinal microvasculature: new developments. Microcirculation 14, 1–10. [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S (1895). Algunas conjeturas sobre el mecanismo anatómico de la ideación, asociación y atención. [Some conjectures on the anatomical mechanism of ideation, association and attention]. Rev Med Cirugía Prácticas 19, 12. [Google Scholar]
- Ravizza T, Moneta D, Bottazzi B, Peri G, Garlanda C, Hirsch E, Richards GJ, Mantovani A & Vezzani A (2001). Dynamic induction of the long pentraxin PTX3 in the CNS after limbic seizures: evidence for a protective role in seizure‐induced neurodegeneration. Neurosci 105, 43–53. [DOI] [PubMed] [Google Scholar]
- Reeves AM, Shigetomi E & Khakh BS (2011). Bulk loading of calcium indicator dyes to study astrocyte physiology: key limitations and improvements using morphological maps. J Neurosci 31, 9353–9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessmann U & Gambetti P (1986). Astrocytes in the developing human brain. An immunohistochemical study. Acta Neuropathol 70, 308–313. [DOI] [PubMed] [Google Scholar]
- Roman RJ (2002). P‐450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82, 131–185. [DOI] [PubMed] [Google Scholar]
- Rosenegger DG & Gordon GR (2015). A slow or modulatory role of astrocytes in neurovascular coupling. Microcirculation 22, 197–203. [DOI] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CH, Wamsteeker Cusulin JI & Gordon GR (2015). Tonic local brain blood flow control by astrocytes independent of phasic neurovascular coupling. J Neurosci 35, 13463–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenman SJ, Shrikant P, Dubb L, Benveniste EN & Ransohoff RM (1995). Cytokine‐induced expression of vascular cell adhesion molecule‐1 (VCAM‐1) by astrocytes and astrocytoma cell lines. J Immunol 154, 1888–1899. [PubMed] [Google Scholar]
- Rothstein JD, Van KM, Levey AI, Martin LJ & Kuncl RW (1995). Selective loss of glial glutamate transporter GLT‐1 in amyotrophic lateral sclerosis. Ann Neurol 38, 73–84. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K & Giaume C (2008). Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322, 1551–1555. [DOI] [PubMed] [Google Scholar]
- Roy CS & Sherrington CS (1890). On the Regulation of the blood‐supply of the brain. J Physiol 11, 85–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio N, Sanz–Rodriguez F & Arevalo MA (2010). Up‐regulation of the vascular cell adhesion molecule‐1 (VCAM‐1) induced by Theiler's murine encephalomyelitis virus infection of murine brain astrocytes. Cell Commun Adhes 17, 57–68. [DOI] [PubMed] [Google Scholar]
- Seifert G & Steinhauser C (1995). Glial cells in the mouse hippocampus express AMPA receptors with an intermediate Ca2+ permeability. Eur J Neurosci 7, 1872–1881. [DOI] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM & Noble–Haeusslein LJ (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 106–107, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seregi A, Keller M & Hertting G (1987). Are cerebral prostanoids of astroglial origin? Studies on the prostanoid forming system in developing rat brain and primary cultures of rat astrocytes. Brain Res 404, 113–120. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson‐Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH & Khakh BS (2013. a). Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno‐associated viruses. J Gen Physiol 141, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Jackson‐Weaver O, Huckstepp RT, O'Dell TJ & Khakh BS (2013. b). TRPA1 channels are regulators of astrocyte basal calcium levels and long‐term potentiation via constitutive D‐serine release. J Neurosci 33, 10143–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Watanabe E, Hiyama TY, Nagakura A, Fujikawa A, Okado H, Yanagawa Y, Obata K & Noda M (2007). Glial Nax channels control lactate signaling to neurons for brain [Na+] sensing. Neuron 54, 59–72. [DOI] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS & Nedergaard M (2003). Signaling at the gliovascular interface. J Neurosci 23, 9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L (1960). The metabolism of the central nervous system in vivo In Handbook of Physiology, Section I, Neurophysiology, eds Field J, Magoun HW, Hall VE, pp. 1843–1864. American Physiological Society, Washington DC. [Google Scholar]
- Song Y, Nagaoka T, Yoshioka T, Nakabayashi S, Tani T & Yoshida A (2015). Role of glial cells in regulating retinal blood flow during flicker‐induced hyperemia in cats. Invest Ophthalmol Vis Sci 56, 7551–7559. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P & Khakh BS (2015). Ca2+ signaling in astrocytes from Ip3r2−/− mice in brain slices and during startle responses in vivo . Nat Neurosci 18, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Tence M, Glowinski J & Premont J (1994). Glutamate‐evoked release of arachidonic acid from mouse brain astrocytes. J Neurosci 14, 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stichel CC, Muller CM & Zilles K (1991). Distribution of glial fibrillary acidic protein and vimentin immunoreactivity during rat visual cortex development. J Neurocytol 20, 97–108. [DOI] [PubMed] [Google Scholar]
- Stringer JL & Aribi AM (2003). Effects of glial toxins on extracellular acidification in the hippocampal CA1 region in vivo . Epilepsy Res 54, 163–170. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Santolini J, Wang ZQ, Wei CC & Adak S (2004). Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem 279, 36167–36170. [DOI] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y & Nedergaard M (2013). Glutamate‐dependent neuroglial calcium signaling differs between young and adult brain. Science 339, 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X & Nedergaard M (2006). Astrocyte‐mediated control of cerebral blood flow. Nat Neurosci 9, 260–267. [DOI] [PubMed] [Google Scholar]
- Takata N, Nagai T, Ozawa K, Oe Y, Mikoshiba K & Hirase H (2013). Cerebral blood flow modulation by basal forebrain or whisker stimulation can occur independently of large cytosolic Ca2+ signaling in astrocytes. PloS One 8, e66525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki I & Chang JJ (1958). Electric response of glia cells in cat brain. Science 128, 1209–1210. [DOI] [PubMed] [Google Scholar]
- Terpolilli NA, Feiler S, Dienel A, Muller F, Heumos N, Friedrich B, Stover J, Thal S, Scholler K & Plesnila N (2015). Nitric oxide inhalation reduces brain damage, prevents mortality, and improves neurological outcome after subarachnoid hemorrhage by resolving early pial microvasospasms. J Cereb Blood Flow Metab DOI: 10.1177/0271678X15605848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M & Giaume C (2012). Connexin‐based intercellular communication and astrocyte heterogeneity. Brain Res 1487, 88–98. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV & Khakh BS (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XK & Hamel E (2015). Simvastatin restored vascular reactivity, endothelial function and reduced string vessel pathology in a mouse model of cerebrovascular disease. J Cereb Blood Flow Metab 35, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbati D, Parolla D & Lavy S (1978). Blood flow in rat brain during exposure to high oxygen pressure. Aviat Space Environ Med 49, 963–967. [PubMed] [Google Scholar]
- Tzeng YC & Ainslie PN (2014). Blood pressure regulation IX: cerebral autoregulation under blood pressure challenges. Eur J Appl Physiol 114, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Chiba Y, Murakami R, Matsumoto K, Kawauchi M & Fujihara R (2016). Blood–brain barrier and blood–cerebrospinal fluid barrier in normal and pathological conditions. Brain Tumor Pathol 33, 89–96. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS & Barres BA (2001). Control of synapse number by glia. Science 291, 657–661. [DOI] [PubMed] [Google Scholar]
- Ventura R & Harris KM (1999). Three‐dimensional relationships between hippocampal synapses and astrocytes. J Neurosci 19, 6897–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgili M, Paulsen R, Villani L, Contestabile A & Fonnum F (1991). Temporary impairment of Muller cell metabolism in the rat retina by intravitreal injection of fluorocitrate. Exp Eye Res 53, 115–122. [DOI] [PubMed] [Google Scholar]
- Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis–Davies G & Sontheimer H (2014). Disruption of astrocyte–vascular coupling and the blood–brain barrier by invading glioma cells. Nat Commun 5, 4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, Christie IN, Hosford PS, Huckstepp RT, Angelova PR, Vihko P, Cork SC, Abramov AY, Teschemacher AG, Kasparov S, Lythgoe MF & Gourine AV (2015). A critical role for purinergic signalling in the mechanisms underlying generation of BOLD fMRI responses. J Neurosci 35, 5284–5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winship IR, Plaa N & Murphy TH (2007). Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo . J Neurosci 27, 6268–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HL, Mao L, Ye S, Paisansathan C, Vetri F & Pelligrino DA (2008). Astrocytes are a key conduit for upstream signaling of vasodilation during cerebral cortical neuronal activation in vivo . Am J Physiol Heart Circ Physiol 294, H622–H632. [DOI] [PubMed] [Google Scholar]
- Yamanishi S, Katsumura K, Kobayashi T & Puro DG (2006). Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am J Physiol Heart Circ Physiol 290, H925–H934. [DOI] [PubMed] [Google Scholar]
- Yang G, Huard JM, Beitz AJ, Ross ME & Iadecola C (2000). Stellate neurons mediate functional hyperemia in the cerebellar molecular layer. J Neurosci 20, 6968–6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG & Barres BA (2012). Genomic analysis of reactive astrogliosis. J Neurosci 32, 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehendner CM, Tsohataridis S, Luhmann HJ & Yang JW (2013). Developmental switch in neurovascular coupling in the immature rodent barrel cortex. PloS One 8, e80749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA & Wu JQ (2014). An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C & Zlokovic BV (2015). Establishment and dysfunction of the blood–brain barrier. Cell 163, 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T & Carmignoto G (2003). Neuron‐to‐astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6, 43–50. [DOI] [PubMed] [Google Scholar]