

Abstract
Small heat shock proteins (sHsps) are an evolutionary conserved class of ATP‐independent chaperones that protect cells against proteotoxic stress. sHsps form assemblies with aggregation‐prone misfolded proteins, which facilitates subsequent substrate solubilization and refolding by ATP‐dependent Hsp70 and Hsp100 chaperones. Substrate solubilization requires disruption of sHsp association with trapped misfolded proteins. Here, we unravel a specific interplay between Hsp70 and sHsps at the initial step of the solubilization process. We show that Hsp70 displaces surface‐bound sHsps from sHsp–substrate assemblies. This Hsp70 activity is unique among chaperones and highly sensitive to alterations in Hsp70 concentrations. The Hsp70 activity is reflected in the organization of sHsp–substrate assemblies, including an outer dynamic sHsp shell that is removed by Hsp70 and a stable core comprised mainly of aggregated substrates. Binding of Hsp70 to the sHsp/substrate core protects the core from aggregation and directs sequestered substrates towards refolding pathway. The sHsp/Hsp70 interplay has major impact on protein homeostasis as it sensitizes substrate release towards cellular Hsp70 availability ensuring efficient refolding of damaged proteins under favourable folding conditions.
Keywords: Hsp100 disaggregase, Hsp70, protein aggregation, protein refolding, sHsps
Subject Categories: Protein Biosynthesis & Quality Control
Introduction
Exposure of cells to stress leads to increased formation of misfolded, aggregation‐prone proteins and results in the breakdown of protein homeostasis (Tyedmers et al, 2010; Hartl et al, 2011; Labbadia & Morimoto, 2015). Appearance of misfolded protein species in the cytosol not only results in a loss of active proteins, but also promotes the trapping and aggregation of other proteins (Richter et al, 2010; Kim et al, 2013). The problem of protein aggregation in response to environmental stress is particularly severe in case of unicellular and sessile organisms. A conserved network of three molecular chaperone systems has evolved early in evolution to counteract protein aggregation in the cell. This system comprises small heat shock proteins (sHsps: IbpA and IbpB in Escherichia coli and Hsp26 or Hsp42 in Saccharomyces cerevisiae) and two ATP‐driven chaperones: Hsp70 (DnaK in E. coli and Ssa1 in S. cerevisiae) and an Hsp100 disaggregase (ClpB in E. coli and Hsp104 in S. cerevisiae), which together efficiently rescue and refold polypeptides trapped in aggregates (Mogk et al, 2003a; Cashikar et al, 2005; Haslbeck et al, 2005; Liberek et al, 2008; Doyle et al, 2013). The refolding of proteins from aggregates is important for the cell since severe heat stress conditions impair both transcription and translation (Yost & Lindquist, 1986; Panniers, 1994). In agreement with a crucial function of sHsps as part of a chaperone triad, deletion of sHsps gene in Synechocystis sp. (Lee et al, 2000) and Neurospora crassa (Plesofsky‐Vig & Brambl, 1995) results in temperature‐sensitive growth phenotypes, while deletion of sHsps in E. coli decreases cell viability during prolonged growth at high temperature (Kuczynska‐Wisnik et al, 2002) or under conditions of limiting DnaK levels (Mogk et al, 2003a). Similarly, deletion of Hsp26 or Hsp42 in yeast S. cerevisiae results in a dramatic change in cell morphology under heat shock conditions pointing to a general protective function of sHsps for proteome homeostasis in yeast (Haslbeck et al, 2004).
Small Hsps, protecting the cell from irreversible protein aggregation, are widely distributed in both prokaryotes and eukaryotes (Haslbeck & Vierling, 2015). Bacteria, Archaea and unicellular eukaryotes usually possess one or two genes coding for sHsps. The number of genes encoding sHsps is higher in multicellular eukaryotes, reaching 19 in Arabidopsis thaliana and 10 in human (Haslbeck & Vierling, 2015). Members of this family are characterized by low molecular mass (12–43 kDa) and a conserved β‐sandwich α‐crystallin domain of ~90 amino acid residues (Haslbeck & Vierling, 2015; Treweek et al, 2015). A striking feature of most sHsps is their ability to assemble into oligomers in which dynamic exchange of subunits takes place (Giese & Vierling, 2002; Lentze et al, 2004; Painter et al, 2008; Benesch et al, 2010; Aquilina et al, 2013; Delbecq & Klevit, 2013; Shi et al, 2013). The oligomeric forms possess limited ability to interact with substrates (Ecroyd et al, 2007; Ahmad et al, 2008; Peschek et al, 2013; Treweek et al, 2015). Many sHsp family members become activated by increase in temperature, which shifts the equilibrium from oligomeric forms towards smaller species, usually dimers, which readily bind partially misfolded proteins exposing hydrophobic patches (Cheng et al, 2008; Stengel et al, 2010; Basha et al, 2013). The interaction between a single sHsp dimer and a substrate is dynamic; however, the oligomeric sHsp–substrate assemblies are quite stable, suggesting that substrates are tethered at multiple sites (Mogk et al, 2003b; Cheng et al, 2008).
Small Hsps constitute the first line of defence against protein aggregation. Under heat stress, the cytoplasmic levels of sHsps rapidly increase. Heat‐activated sHsps bind misfolded substrates in an ATP‐independent manner to form assemblies containing both misfolded substrate proteins and sHsps (Ehrnsperger et al, 1997; Lee et al, 1997; Ratajczak et al, 2009). These assemblies are not only morphologically different from protein aggregates formed in the absence of sHsps but also substantially smaller in size. Although these assemblies have decreased size, the molecular mass still typically exceeds 1 MDa (Ratajczak et al, 2009; Stengel et al, 2010, 2012). More importantly, trapped polypeptides are rescued from these assemblies more efficiently by the Hsp70–Hsp100‐dependent disaggregation and refolding systems, which reduces the need for resynthesis of essential proteins required for stress recovery (Veinger et al, 1998; Mogk et al, 2003b; Matuszewska et al, 2005).
An intimate cooperation between Hsp70 and Hsp100 is needed for protein disaggregate and refolding (Glover & Lindquist, 1998). Hsp70 binds first to protein aggregates, recruiting and activating the cooperating Hsp100 hexamers in a second step through direct physical interactions (Miot et al, 2011; Seyffer et al, 2012; Lee et al, 2013; Rosenzweig et al, 2013). Activated Hsp100 extracts trapped polypeptides by threading the polypeptides through its central channel (Weibezahn et al, 2004). Hsp70 activity is regulated by J‐proteins (DnaJ in E. coli and Ydj1/Sis1 in S. cerevisiae) and nucleotide exchange factors (NEFs; GrpE in E. coli, Sse1/Fes1 in S. cerevisiae). J‐protein binding and concomitant interaction with the substrate trigger ATP hydrolysis in Hsp70, allowing stable substrate interaction. Substrate release from Hsp70 is facilitated by the action of NEFs, which release ADP, allowing the rebinding of ATP to complete the cycle (Langer et al, 1992; Schmid et al, 1994; Laufen et al, 1999; Kampinga & Craig, 2010).
Here, we analysed the mechanism of extraction and refolding of misfolded proteins from sHsp–substrate assemblies. The initial binding of sHsps to substrates is beneficial by preventing the formation of large aggregates and creating smaller assemblies possessing greater accessible surface for Hsp70–Hsp100 action. At the same time, sHsp–substrate interactions might also hamper subsequent Hsp70/Hsp100 association and have to be broken to allow for substrate refolding. How this Janus‐faced sHsp feature is circumvented has not been addressed so far. Here, we demonstrate a step‐by‐step mechanism during which the sHsp–substrate assemblies are solubilized and the substrates reactivated. We show that sHsp–substrate assemblies are composed of two substructures, a stable core and a dynamic outer shell exclusively composed of sHsps. Substrate recovery starts with the displacement of sHsps from the assembly surface by Hsp70. Hsp70 outcompetes dynamic sHsp molecules for rebinding to the assemblies. Hsp70–substrate interaction at this point preserves the beneficial properties of the assembly core and protects it from aggregation. Hsp70 next recruits Hsp100, ensuring efficient substrate solubilization and targeting to refolding pathways. The revealed mechanism explains the phenomenon of efficient refolding of aggregated proteins assembled with sHsps.
Results
Dual role of Hsp70 in disaggregating IbpAB–substrate assemblies
The extraction of misfolded protein molecules from assemblies with sHsps requires accessibility of these substrates on the surface of the assemblies and an ability of the disaggregating chaperones to separate the sHsp–substrate associations. We therefore first analysed the size of sHsp–substrate assemblies, which determines the potential chaperone accessible surface. Thermal denaturation of substrates in the presence of sHsps results in formation of sHsp–substrate assemblies that are morphologically different from protein aggregates formed in their absence. Electron microscopy studies of thermolabile luciferase denatured in the presence of the E. coli sHsps IbpA and IbpB (IbpAB) revealed the formation of small, uniform and nearly globular structures that are distinct from luciferase aggregates formed in the absence of sHsps (Ratajczak et al, 2009). Size determination by dynamic light scattering (DLS) revealed that IbpAB–luciferase assemblies are 30 times smaller (average diameter: 58 nm) compared with luciferase aggregates (average diameter: 2,000 nm; Fig 1A). For additional characterization, we sedimented both the IbpAB–luciferase assemblies and luciferase aggregates in a glycerol gradient. The assemblies sedimented to the middle of the gradient in contrast to aggregates, which were found at the bottom following sedimentation (Fig EV1A). Based on SDS–PAGE followed by SYPRO Ruby staining, the assemblies isolated from the gradient showed an IbpA:IbpB:luciferase stoichiometry of ~1:0.8:1 ratio (Fig EV1A and B). DLS measurements estimated the molecular mass of the assemblies to be ~9 MDa. Based on molecular mass and stoichiometry estimation, each assembly on average contains ~90 luciferase molecules and a similar number of both IbpA and IbpB molecules.
Figure 1. IbpAB–luciferase assemblies differ in biophysical and biochemical properties from luciferase aggregates.
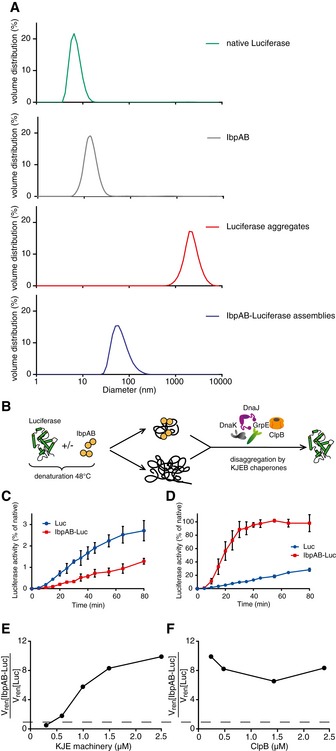
-
ADynamic light scattering graphs depicting the size distribution of the following entities: native luciferase (green), IbpAB (grey), luciferase aggregates (red), IbpAB–luciferase assemblies (blue), shown as volume distribution of analysed species. Luciferase was present at 1.5 μM, IbpA at 3 μM, IbpB at 7 μM concentration.
-
BScheme of the disaggregation experiment.
-
C, DRenaturation kinetics for luciferase denatured in the presence or absence of IbpAB. Luciferase (1.5 μM) was denatured in the presence of IbpA (3 μM) and IbpB (7 μM), diluted to 40 nM and refolded at limited (0.3 μM; C) or elevated (2.4 μM; D) KJE machinery concentrations and standard (1.5 μM) ClpB concentration. The KJE machinery concentration is given with respect to DnaK with a constant DnaK:DnaJ:GrpE ratio 1:0.3:0.3. Data are the mean ± SD of three independent experiments.
-
EA comparison of renaturation rates for luciferase denatured in the presence or absence of IbpAB in changing KJE and constant ClpB concentrations. The KJE machinery concentration is given with respect to DnaK with a constant DnaK:DnaJ:GrpE ratio 1:0.3:0.3.
-
FA comparison of renaturation rates for luciferase denatured in the presence or absence of IbpAB in changing ClpB and constant KJE concentrations. The dashed line indicates the theoretical scenario that luciferase refolding from aggregates and sHsp assemblies proceeds with identical refolding rates.
Figure EV1. Sedimentational analysis of IbpAB–luciferase assemblies.
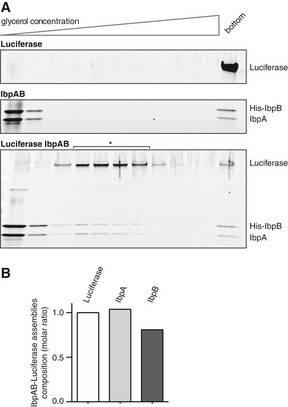
- Comparison of sedimentational properties of IbpAB–luciferase assemblies and luciferase aggregates. Luciferase (1.5 μM)(upper gel), IbpA (3 μM) and IbpB (7 μM)(middle gel) or luciferase (1.5 μM), IbpA (3 μM) and IbpB (7 μM)(lower gel) were incubated at 48°C for 10 min and subjected to glycerol gradient sedimentation (SW 60 Ti rotor, 40,000 rpm, 1 h, 10°C). Fractions were collected from the top of the gradient. Proteins in the fractions were visualized by SYPRO Ruby staining following SDS–PAGE.
- Stoichiometry of the proteins in the IbpAB–luciferase assemblies. Fractions marked with an asterisk containing IbpAB–luciferase assemblies were pooled and following SDS–PAGE and SYPRO Ruby staining, a densitometric analysis was performed. The relative molar ratio of proteins in assemblies was calculated by taking into account the respective molecular masses.
Source data are available online for this figure.
As a consequence of sHsp/substrate assembly formation, the potentially accessible surface of the same amount of misfolded luciferase is much larger in IbpAB assemblies compared with its aggregated state. This may facilitate Hsp70–Hsp100‐dependent disaggregation and refolding by offering more chaperone binding sites for initiating disaggregation. On the other hand, IbpAB present on the surface of the IbpAB–luciferase assemblies potentially also restricts chaperone binding to misfolded luciferase. Furthermore, substrate refolding requires disruption of sHsp–substrate interactions. To analyse how sHsp–substrate interactions influence subsequent Hsp70–Hsp100‐dependent refolding, we compared the dependencies of luciferase reactivation from IbpAB–luciferase assemblies and luciferase aggregates on varying Hsp70 and Hsp100 chaperone concentrations (Fig 1B).
We started with an unexpected observation: when luciferase was denatured in the presence of IbpAB to form smaller assemblies, its reactivation by Hsp70–Hsp100 was less efficient compared with luciferase aggregates when using limiting concentrations of the Hsp70 system (KJE: 0.3 μM DnaK, 0.1 μM DnaJ and 0.1 μM GrpE; Fig 1C). While the yield of luciferase refolding was low under such conditions, significant and reproducible differences in refolding kinetics were determined for both substrate types: the assemblies were refolded at a twofold lower rate than the aggregates (Fig 1C). In contrast, eight times higher KJE concentrations (2.4 μM DnaK, 0.8 μM DnaJ and 0.8 μM GrpE) allowed for 10 times more efficient and faster protein refolding from IbpAB–luciferase assemblies as compared to luciferase aggregates (Fig 1D). Thus, IbpAB plays an inhibitory role in substrate disaggregation and refolding at the limiting KJE concentration, while it becomes beneficial at elevated KJE level.
We next compared the initial rates of luciferase refolding from either IbpAB–luciferase assemblies or luciferase aggregates over a wide range of both KJE and ClpB concentrations. In this set‐up, the level of KJE system was varied, while the Hsp100 chaperone (e.g. ClpB) was kept at saturating concentration. Again, we observed that the relative refolding rate of IbpAB‐bound luciferase was highly dependent on KJE concentrations (Fig 1E). Increasing KJE concentrations allowed for superior refolding of luciferase from IbpAB–luciferase assemblies. In contrast, at saturating KJE concentration, ClpB, under all concentrations tested, stimulated luciferase reactivation from IbpAB–luciferase assemblies ~eightfold as compared to luciferase aggregates (Fig 1F).
These findings reveal that IbpAB–luciferase assemblies require much higher concentration of the Hsp70 chaperone system for efficient disaggregation and refolding than luciferase aggregates. At the same time, Hsp100 results in a similar degree of stimulation of the disaggregation of assemblies in comparison to aggregates over a range of Hsp104 concentrations used. This suggests that the Hsp70 chaperone system, in addition to its role in Hsp100 activation and docking to aggregates, has a novel function critical to sHsp–substrate assembly dissociation.
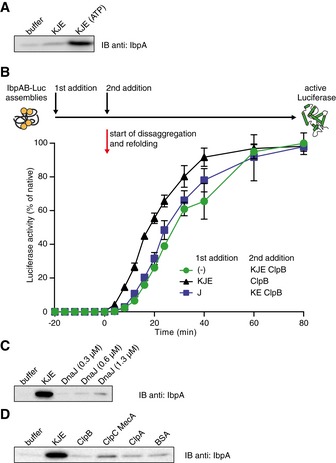
Hsp70–Hsp100‐dependent disaggregation and refolding of aggregated proteins are preceded by a delay time (Zietkiewicz et al, 2004). We noticed that the delay time during luciferase reactivation from IbpAB–luciferase assemblies is significantly longer than the delay observed under analogous conditions for luciferase aggregates (Appendix Fig S1A and B). This observation, together with the higher demand for Hsp70 in luciferase rescuing from IbpAB–luciferase assemblies, led us to perform order of addition experiments to analyse which step of the disaggregation and refolding process requires elevated Hsp70 concentration. IbpAB–luciferase assemblies were initially incubated with specific components of the KJE–ClpB bichaperone machinery and the missing components were added at a later step to initiate disaggregation. The final yield of refolded luciferase was almost identical for all reactions. When IbpAB–luciferase assemblies were first incubated with KJE and ATP, the refolding reaction started earlier, as compared to the reactions in which these assemblies were initially incubated with either KJE in the absence of ATP or ClpB and ATP or without any chaperones (Fig 2). Additionally, initial incubation with KJE and ATP resulted in a 1.6‐fold increased refolding rate, underlining an ClpB‐independent activity of DnaK towards IbpAB–luciferase assemblies.
Figure 2. KJE acts prior to the disaggregation step of IbpAB–luciferase assemblies.
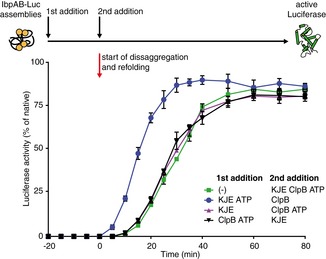
An order of addition experiment. Luciferase (1.5 μM) was denatured in the presence of IbpA (3 μM) and IbpB (7 μM), diluted to 40 nM and incubated first for 20 min with the components listed in the legend and then complemented with the missing components of the functional ClpB‐KJE bichaperone system. The DnaK, DnaJ, GrpE, ClpB concentrations used were as follows: 1, 0.3, 0.3 and 1.5 μM. Data are the mean ± SD of three independent experiments.
To generalize our kinetics observations, we used thermolabile malate dehydrogenase (MDH) as an alternative substrate. MDH was denatured in the presence of IbpAB, and Hsp70–Hsp100‐dependent MDH refolding was performed. As observed earlier for IbpAB–luciferase assemblies, initial incubation of IbpAB–MDH with KJE (+ATP) resulted in a faster initiation of MDH reactivation but did neither influence the rate of MDH reactivation nor the final refolding yield (Fig EV2).
Figure EV2. KJE acts prior to disaggregation of IbpAB–MDH assemblies.
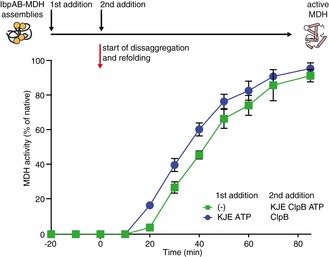
An order of addition experiment. MDH (2 μM) was denatured in the presence of IbpA (3 μM) and IbpB (7 μM) at 47°C for 30 min. IbpAB–MDH assemblies were diluted fourfold and first incubated with the components listed in the legend and then complemented with the missing components of the functional ClpB‐KJE bichaperone system. The DnaK, DnaJ, GrpE, ClpB concentrations used were as follows: 1, 0.3, 0.3 and 1.5 μM. Data are the mean ± SD of three independent experiments.
Summarized, we observed that assembly formation between sHsps and substrates causes a delay in substrate refolding and at low Hsp70 concentrations negatively affects substrate refolding. Both effects are overcome by initially treating sHsp–substrate assemblies with higher Hsp70 levels, suggesting that Hsp70 is playing an additional, so far non‐recognized role upstream of Hsp70–Hsp100‐mediated protein disaggregation.
Hsp70 releases sHsp‐dependent inhibition of disaggregation
Our kinetic experiments (Fig 2) suggested that the role of Hsp70 in processing of sHsp–substrate assemblies is not limited to recruitment and activation of Hsp100 during protein disaggregation, but that Hsp70 possesses an additional activity at the initial step of the process. However, it is problematic to directly investigate an independent Hsp70 activity in the processing of sHsp–substrate assemblies, since substrate disaggregation and reactivation require both Hsp70 and Hsp100. To divide this process into discrete steps depending exclusively on one of these chaperone systems, we took advantage of the properties of a hyperactive D484K variant of the yeast Hsp104 disaggregase (Lipinska et al, 2013). This variant is able to recover aggregated substrates in the absence of Hsp70 as it does not require recruitment and activation by the Hsp70 partner chaperone.
We speculated that the determined Hsp70 activity is related to the release of an inhibitory sHsp impact on the disaggregation process. In order to investigate this hypothesis, we used aggregated GFP as model substrate. GFP was initially aggregated at 85°C and the preformed GFP aggregates were subsequently incubated at 48°C with IbpAB to trigger sHsp binding to the aggregates (Appendix Fig S2A). As a result, the size of GFP aggregates and GFP aggregates coated with IbpAB (IbpAB–GFP assemblies) is similar (Appendix Fig S2B). Any observable changes in GFP reactivation by chaperones can therefore be directly related to IbpAB association with the aggregate surface. Similar to the findings obtained for IbpAB–luciferase assemblies, rescuing of GFP from IbpAB–GFP assemblies by KJE–ClpB proceeded at a slower rate compared with aggregated GFP when KJE was used at low concentration (Appendix Fig S2C). This inhibitory effect of IbpAB was again overcome by increasing KJE concentration (Appendix Fig S2D).
The comparison of GFP refolding efficiencies from IbpAB–GFP assemblies and GFP aggregates by Hsp104‐D484K revealed that IbpAB presence strongly inhibits disaggregase activity (Fig 3A). In a control experiment, GFP aggregates and IbpAB were incubated separately at 48°C and mixed after cooling. This treatment does not allow for IbpAB binding to the surface of GFP aggregates. Under these conditions, no inhibition of Hsp104‐D484K‐mediated GFP refolding was observed (Fig 3A), demonstrating that the association of IbpAB with GFP aggregates but not the mere presence of IbpAB inhibits the hyperactive Hsp104 variant.
Figure 3. Hsp70, independently of Hsp100, releases IbpAB‐dependent inhibition of disaggregation.
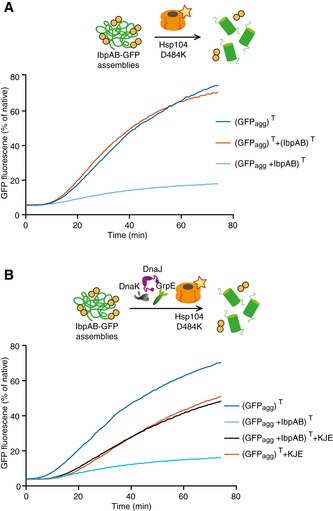
- IbpAB association with GFP aggregates blocks substrate translocation through the central Hsp100 pore. Aggregated GFP (1.5 μM) was incubated for 10 min at 48°C (marked “T” in the Figure) alone (blue) or with IbpA and IbpB (3 μM and 7 μM, respectively; sky blue) or aggregated GFP and IbpAB were incubated separately at 48°C and then mixed (orange). Hsp104 D484K hyperactive variant (1 μM) was added, and GFP reactivation was monitored.
- KJE machinery allows to overcome the IbpAB‐dependent inhibition of IbpAB–GFP disaggregation by hyperactive Hsp104 D484K. Aggregated GFP was incubated alone at 48°C (blue and orange) or with IbpAB at 48°C (sky blue and black). After addition of the Hsp104 D484K hyperactive mutant (all plots) and KJE machinery (black and magenta), GFP reactivation was monitored. DnaK, DnaJ and GrpE were present at 1 μM, 0.3 μM and 0.3 μM. Aggregated GFP, IbpAB and Hsp104 D484K were in concentrations as in (A).
We speculated that IbpAB association restricts Hsp104‐D484K access to the binding sites at the aggregate surface, thereby reducing its disaggregation activity. This model predicts that the additional role of KJE in substrate reactivation from IbpAB–substrate assemblies observed in the kinetic and order of addition experiments (Figs 1C and 2) is required to overcome this inhibitory IbpAB effect. To test this, we added KJE chaperones to the reactivation reaction containing IbpAB–GFP assemblies and Hsp104‐D484K. It is important to note that the bacterial Hsp70 system does not cooperate with the yeast Hsp104 disaggregase (Glover & Lindquist, 1998; Krzewska et al, 2001), neither does it efficiently refold denatured GFP on its own (Appendix Fig S3). Therefore, any changes in GFP reactivation noticed upon KJE addition can be attributed to its activity at the initial step of disaggregation. We observed a substantial increase in GFP reactivation from the IbpAB–GFP assemblies by Hsp104‐D484K in the presence of KJE (Fig 3B). This means that an Hsp100‐independent activity of KJE resulted in the release of an IbpAB‐dependent inhibition of GFP reactivation. In a control experiment, we showed that the addition of KJE chaperones to a reaction containing Hsp104‐D484K and GFP aggregates lacking IbpAB did not stimulate, but partially inhibited GFP disaggregation (Fig 3B), excluding that the beneficial effect of KJE observed in case of IbpAB–GFP assemblies stems from cooperation with Hsp104‐D484K. The noticed inhibition can be explained by competition between KJE and Hsp104‐D484K for binding to GFP aggregates. We conclude that KJE releases the IbpAB‐dependent inhibitory effect in a process not involving cooperation with the Hsp100 disaggregase. This Hsp70 activity is only required when IbpAB is present in substrate assemblies but not for protein aggregates lacking sHsps.
Hsp70 is required for dissociation of sHsps from assemblies
The exact nature of KJE activity in releasing the sHsp‐dependent inhibition of disaggregation is unknown. KJE activity may result in rearrangement of sHsp–substrate assemblies facilitating subsequent refolding reactions. Alternatively, KJE might cause the release of sHsps from the substrate assembly. To distinguish between these two possibilities, we isolated IbpAB–luciferase assemblies by sedimentation in a glycerol gradient. The assemblies were incubated for different periods of time in the presence of varying chaperone combinations and tested for integrity by subjecting them to a second round of sedimentation (Fig 4A). Assemblies incubated without chaperones remained stable. Cosedimentation of IbpAB and luciferase in the middle of the gradient, indicative for assembly presence, was confirmed by Western blot (Fig 4B). Incubation of isolated IbpAB–luciferase assemblies with KJE for 30 min resulted in the release of over 40% of IbpAB from the assemblies in an ATP‐dependent manner (Figs 4B and EV3A). The released IbpAB molecules were found in the top fractions of the gradient. IbpAB dissociation from the assemblies was further increased to 80% upon longer incubation (180 min) with KJE chaperones. Notably, a small fraction of IbpAB remained associated with luciferase and could not be displaced (Fig 4B). These findings define the existence of two distinct IbpAB populations in their assemblies with substrates. We hypothesize that the majority of IbpAB molecules is surface‐exposed and can be displaced by KJE activity, whereas a second, smaller population forms an integral part of the assembly. The KJE‐mediated release of IbpAB from the assemblies was not linked to a regain of luciferase activity. Accordingly, luciferase species migrated to nearly the same position in the glycerol gradient showing that no major change in sedimentation properties of assemblies took place following KJE‐mediated release of IbpAB.
Figure 4. Hsp70 releases sHsps from sHsp–substrate assemblies in vitro .
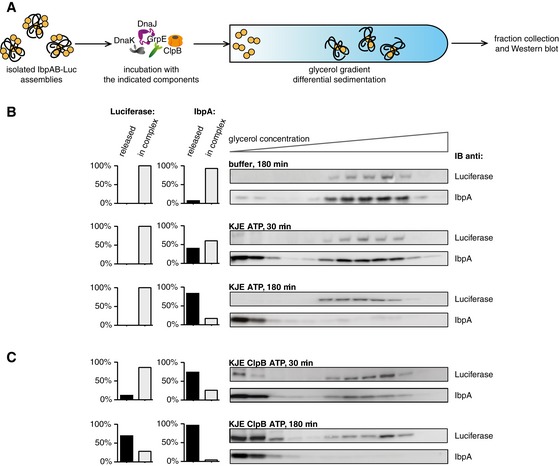
- Experimental scheme. Purified IbpAB–luciferase assemblies were incubated with indicated components for the indicated time and resubjected to glycerol gradient sedimentation.
- KJE releases IbpAB from IbpAB–luciferase assemblies. Fractions were collected from the top of the gradient, and luciferase and IbpA were visualized by Western blot following SDS–PAGE. The band intensities of luciferase and IbpA retained in the core assembly (fractions 5–12) or released from the assembly (fractions 1–3) were quantified and plotted on the graphs on the left. IbpA–IbpB–luciferase assemblies (approximate stoichiometry 1:0.8:1), DnaK, DnaJ and GrpE were present at 100 nM (calculated for luciferase monomer), 1 μM, 0.3 μM and 0.3 μM concentrations, respectively.
- Concerted action of ClpB‐KJE results in release of IbpAB and luciferase from IbpAB–luciferase assemblies. Experiments were performed and analysed as in (B). ClpB was present at 1.5 μM concentration.
Source data are available online for this figure.
Figure EV3. Hsp70 specifically displaces sHsps sHsp–substrate assemblies.
- KJE requires ATP to release IbpAB from IbpAB–luciferase assemblies. The isolated assemblies were incubated with the indicated components for 60 min and resubjected to glycerol gradient sedimentation as in Fig 4B, but instead of showing the entire gradients, the fractions corresponding to the released material (1–3) were pooled together and IbpA was visualized by Western blot following SDS–PAGE.
- An order of addition experiment as in Fig 2. IbpAB–luciferase assemblies were first incubated with the components listed in the legend and then complemented with the missing components of the functional ClpB‐KJE bichaperone system. The DnaK, DnaJ, GrpE and ClpB concentrations used were as follows: 1, 0.6, 0.3 and 1.5 μM. Data are the mean ± SD of three independent experiments.
- Incubation of the IbpAB–luciferase assemblies in the presence of DnaJ alone does not lead to IbpAB release. Experiment performed as in (A).
- Incubation of the IbpAB–luciferase assemblies in the presence of Hsp100 family members does not lead to IbpAB release. Experiments were performed as in (A). ClpB, ClpC, MecA, ClpA and BSA were present at 1 μM concentration.
Source data are available online for this figure.
Next, we carried out a set of analogous sedimentation experiments additionally including ClpB. Incubation of the assemblies with both KJE and ClpB for 30 min resulted in a release of 70% of IbpAB species and also led to the appearance of monomeric, active luciferase in the top of the gradient (Fig 4C). Prolonged incubation for 180 min substantially enriched the top fractions of the gradient with native luciferase and resulted in dissociation of all IbpAB molecules from the assemblies (Fig 4C). It is worth noticing that the release of IbpAB from assemblies (30 min) preceded the formation of substantial amounts of active luciferase (180 min).
To verify whether the determined, KJE‐dependent release of sHsps from sHsp–substrate assemblies is not related to one particular substrate, luciferase, similar experiments were performed using IbpAB–MDH as alternative assemblies. As observed for IbpAB–luciferase assemblies, the majority of IbpAB but not MDH was released following incubation with KJE and ATP (Fig EV4A and B). Native MDH only appeared in the gradient top fractions when ClpB was additionally present (Fig EV4B). These findings confirm that KJE releases surface‐located sHsps without affecting the integrity of the core of the sHsp–substrate assembly.
Figure EV4. KJE dissociates IbpAB from IbpAB–MDH assemblies in vitro .
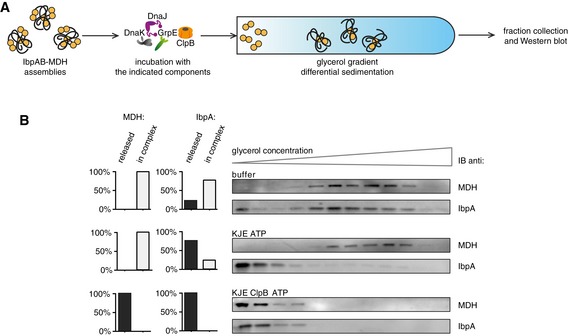
- Experimental scheme. IbpAB–MDH assemblies, isolated from unbound IbpAB via sedimentation in glycerol gradient, were incubated with the indicated components for 120 min and resubjected to a second round of sedimentation.
- KJE dissociates IbpAB from IbpAB–MDH assemblies. Fractions were collected from the top of the gradient, and MDH and IbpA were visualized by Western blot following SDS–PAGE. IbpA and MDH levels in each fraction were determined by quantitative immunoblot analysis using IbpA‐ and MDH‐specific antibodies, respectively. The band intensities of IbpA and MDH retained in the core assembly (fractions 5–12) or released from the assembly (fractions 1–3) were quantified and plotted on the graphs on the left. IbpAB–MDH assemblies (approximate stoichiometry 1:0.5:1), DnaK, DnaJ, GrpE and ClpB were present at 400 nM (calculated for MDH monomers), 1 μM, 0.3 μM, 0.3 μM and 1.5 μM concentrations, respectively.
Source data are available online for this figure.
Hsp70 binding displaces dynamic sHsps from the assembly
We considered two possible modes of KJE‐mediated release of sHsps from the assemblies. The first mode, defined as an active one, involves remodelling of sHsp–substrate assemblies by Hsp70 in cycles of ATP hydrolysis leading to IbpAB release. The second mode suggests that binding of Hsp70 to the sHsp–substrate assembly is sufficient to displace sHsp molecules. This latter mode of DnaK action in releasing sHsp molecules from assemblies demands for dynamic interactions between sHsps and substrates. Constant dissociation and re‐association of sHsps with sHsp–substrate assemblies has indeed been reported (Friedrich et al, 2004).
To test for spontaneous dissociation of IbpAB from assemblies, we performed an experiment in which we blocked the ability of dissociated IbpA molecules to re‐associate with substrate assemblies. This was achieved by adding an excess of the peptide pC (PEAKKPRRRIEIN), which corresponds to the twelve C‐terminal residues of IbpA. Peptide pC efficiently inhibits IbpA ability to bind to substrates (Strozecka et al, 2012). As control, the inactive peptide variant pC‐AEA was used, in which the two conserved isoleucines were replaced with alanines. Isolated IbpAB–luciferase assemblies were incubated with pC or pC‐AEA peptide and tested for integrity via sedimentation. The presence of pC but not pC‐AEA peptide resulted in partial release of IbpAB from the assemblies as evident from appearance of IbpAB in the top fractions of the gradient (Fig 5). These results are in line with dynamic dissociation and re‐association of sHsps from sHsp–substrate assemblies, with the rebinding step being inhibited by peptide pC.
Figure 5. sHsps spontaneously dissociate from and re‐associate with sHsp–substrate assemblies.

Surface‐exposed IbpAB are dynamically binding and dissociating from the assemblies. Purified IbpAB–luciferase assemblies (100 nM; calculated for luciferase monomer; approximate stoichiometry 1:0.8:1) were incubated with the indicated peptides (1 mM each) for 60 min and subjected to glycerol gradient sedimentation. Fractions were collected from the top of the gradient, and luciferase and IbpA were visualized by Western blot following SDS–PAGE. Source data are available online for this figure.
Notably, incubation of IbpAB–luciferase assemblies with peptide pC also strongly increased the size of assemblies, which were now found in the bottom fraction of the gradient (Fig 5). This shows that the pC‐mediated displacement of surface‐located IbpAB causes aggregation of the core assemblies, including luciferase and some IbpAB. Core assembly aggregation is not observed upon IbpAB dissociation by Hsp70 (Fig 4), indicating that Hsp70 preserves the size of the sHsp–substrate assembly core. This suggests stable association of Hsp70 with the IbpAB–luciferase core assemblies following IbpAB displacement. To analyse this, IbpAB–luciferase assemblies were incubated with the KJE chaperone system and, following sedimentation, the association of DnaK with IbpAB–luciferase assemblies was investigated by Western blot analysis. DnaK cosedimented with luciferase following KJE‐mediated dissociation of IbpAB from assemblies and was present in analogous fractions to IbpAB–luciferase assemblies (Fig 6A). This indicates that DnaK replaces surface‐located dynamic IbpAB, thereby preserving assembly size and preventing its aggregation.
Figure 6. Hsp70 remains associated with the assembly following sHsp displacement.
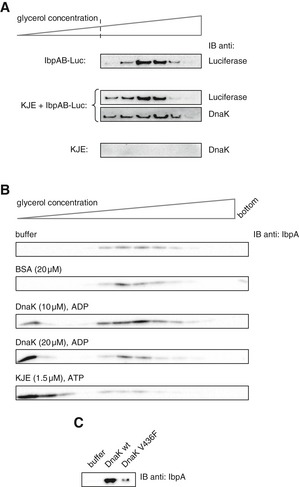
- Purified IbpAB–luciferase assemblies were incubated with buffer or KJE and 2 mM ATP at 25°C for 60 min and resubjected to glycerol gradient sedimentation. Fractions were collected from the top of the gradient, and luciferase and DnaK were visualized by Western blot analysis. To avoid visualizing dominant, unbound DnaK, only the lower, IbpAB–luciferase assembly‐containing fractions of the gradient are shown. Sedimentation analysis of KJE only is provided as a control. IbpAB–luciferase assemblies (approximate stoichiometry 1:0.8:1), DnaK, DnaJ and GrpE were present at 100 nM (calculated for luciferase monomer), 1 μM, 0.3 μM and 0.3 μM concentrations, respectively.
- DnaK binding to IbpAB–luciferase assemblies is sufficient to release IbpAB. Purified IbpAB–luciferase assemblies (100 nM) were incubated with the indicated components for 60 min and subjected to glycerol gradient sedimentation. Fractions were collected from the top of the gradient and IbpA was visualized by Western blot following SDS–PAGE.
- Purified IbpAB–luciferase assemblies (100 nM) were incubated for 60 min with buffer or DnaK (10 μM) or DnaK V436F (10 μM) and subjected to glycerol gradient sedimentation as in Fig 4B, but instead of showing the entire gradients, the fractions corresponding to the released material (1–3) were pooled together and IbpA was visualized by Western blot following SDS–PAGE.
Source data are available online for this figure.
In an alternative approach to support a competitive dissociation mechanism of IbpAB by DnaK, we took advantage of the ability of DnaK to bind substrates independent of DnaJ if present at high concentration in the presence of ADP (Liberek et al, 1991). Under such conditions, DnaK displays no refolding activity. We performed sedimentation experiments in which DnaK, at 12‐fold higher concentration compared with the standard KJE–ATP reaction, and ADP were added to the isolated IbpAB–luciferase assemblies. Indeed, high concentrations of DnaK caused release of IbpAB (Fig 6B), suggesting that DnaK binding is sufficient to displace IbpAB from the assemblies. As control, we incubated the IbpAB–luciferase assemblies with the DnaK‐V436F variant, which possess 20‐fold lower association rate for binding substrate proteins (Laufen et al, 1999). The amount of IbpAB released upon incubation with DnaK‐V436F and ADP was substantially lower (Fig 6C). This supports the model that the mere binding of DnaK is sufficient to efficiently outcompete dynamic, surface‐located IbpAB from the assemblies.
Hsp70 has unique ability to displace sHsps
We next analysed whether chaperones other than DnaK can also displace sHsps from sHsp–substrate assemblies. We tested for activity of DnaJ, as it binds misfolded proteins and delivers them to DnaK (Hartl et al, 2011). We first analysed if DnaJ alone is able to release the inhibitory influence of sHsps on disaggregation by performing order of addition experiments. When IbpAB–luciferase assemblies were initially incubated with DnaJ and the missing components of the KJE–ClpB machinery were added at a later step, no change in the start of refolding reaction was observed (Fig EV3B). DnaJ is therefore not sufficient to shorten the delay time. Next, a series of sedimentation experiments was performed to directly probe for the ability of DnaJ to dissociate IbpAB from IbpAB–luciferase assemblies (Fig EV3C). High DnaJ concentrations released only a small amount of IbpAB that was marginal compared with equivalent DnaK concentrations (Fig EV3C). These results confirm DnaK as the crucial component of the KJE chaperone system that is displacing IbpAB from IbpAB–luciferase assemblies.
Next we investigated whether other chaperones that act on protein aggregates are able to release IbpAB from assemblies. We focussed on AAA+ subunits of ATP‐dependent proteases, ClpA and ClpC (with its adaptor MecA), which possess disaggregation activity in vitro (Dougan et al, 2002; Schlothauer et al, 2003). None of the AAA+ chaperones caused substantial dissociation of IbpAB from the IbpAB–luciferase assemblies as shown by sedimentation experiments (Fig EV3D). These findings further demonstrate the unique ability of KJE to act on sHsp–substrate assemblies.
Hsp70 releases sHsps from aggregates in bacterial cells
All our in vitro experimental approaches indicate that IbpAB chaperones, in an Hsp70‐dependent process, must dissociate from the misfolded substrate prior to substrate disaggregation and reactivation. To test whether our in vitro observations are relevant in the cell and whether dissociation of sHsps from aggregates takes place prior to disaggregation in vivo, we made use of E. coli ΔclpB cells, in which aggregates are stably maintained (Mogk et al, 1999). Additionally, the native heat shock promoter of the dnaK dnaJ operon was replaced by an IPTG‐inducible Lac promoter, which allows manipulating DnaK and DnaJ levels on demand. This experimental set‐up enabled us to uncouple Hsp100‐dependent and Hsp100‐independent Hsp70 activities and to test whether Hsp70‐mediated release of sHsp is observed in vivo in the absence of ongoing disaggregation. ΔclpB PIPTG dnaKJ cells were first grown in the absence of IPTG, and formation of protein aggregates was induced by a 30‐min heat shock at 46°C (Fig 7A). Next, the bacterial culture was transferred to 30°C and divided into two flasks. IPTG was added to one of them to induce the synthesis of DnaK and DnaJ. Following 2 h from induction, the level of DnaK was similar to the level of heat‐shocked wild‐type cells (Fig 7E). Protein aggregates were isolated from both cultures during the recovery phase, and the levels of IbpAB associated with aggregates were analysed. As expected, the fraction of aggregated proteins remained stable, regardless of DnaK and DnaJ induction, due to the absence of ClpB (Fig 7B and D). However, induction of DnaK and DnaJ led to a substantial decrease in the amount of IbpAB associated with aggregates (Fig 7C and D). The Hsp70 system is therefore able to dissociate IbpAB from protein aggregates in vivo, in agreement with the in vitro results. IbpAB dissociation proceeded with slow kinetics under the given experimental conditions. However, one should take into account that DnaK, a major and highly abundant bacterial chaperone, was depleted to ~5% of its physiological level before and during heat shock. In combination with the clpB deletion, this resulted in massive protein aggregation. Moreover, DnaK levels corresponding to the heat shock‐induced levels were only achieved 2 h after addition of IPTG, further rationalizing the delayed release of IbpAB.
Figure 7. Hsp70 releases sHsps from the fraction of aggregated proteins in vivo .
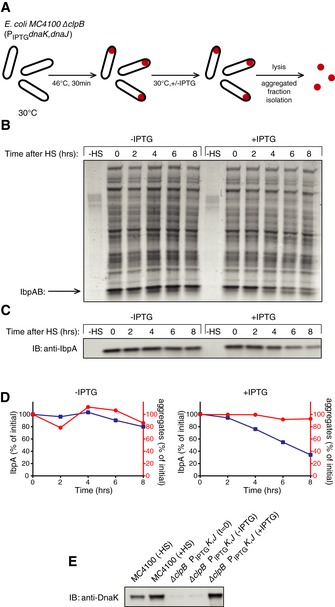
- Experimental scheme. BB6410 (PA1lacO‐1) dnaK, dnaJ, lacIq, ΔclpB cells were grown at 30°C in LB medium until late log phase, heat‐shocked at 46°C for 30 min, then the culture was separated into two flasks and IPTG was added at 1 mM. At indicated time points, aliquots were taken, cells were lysed and protein aggregates were isolated.
- The isolated fraction of aggregates was analysed by SDS–PAGE followed by staining with Coomassie brilliant blue at the indicated time points.
- Levels of IbpA present in aggregates at the indicated time points were determined by quantitative immunoblot analysis using IbpA‐specific antibodies.
- Quantification of (B) and (C).
- DnaK cellular levels were determined by quantitative immunoblot analysis using DnaK‐specific antibodies.
Source data are available online for this figure.
Yeast Hsp70 releases Hsp26 from substrate assemblies
We finally tested whether the determined novel Hsp70 activity is evolutionary conserved by repeating key experiments with the respective S. cerevisiae chaperones (sHsp: Hsp26; Hsp70 system: Ssa1, Ydj1, Sse1 and Hsp104 disaggregase) and MDH as substrate. Order of addition experiments showed that initial incubation of Hsp26–MDH assemblies with the yeast Hsp70 system (SYS: Ssa1, Ydj1, Sse1) resulted in a faster start of MDH refolding following addition of Hsp104 (Fig 8A), analogous to the results obtained for the bacterial chaperones (Figs 2 and EV2). Assemblies composed of misfolded MDH and Hsp26 were also isolated using sedimentation. Such isolated assemblies were incubated for 180 min in the presence of SYS, SYS and Hsp104 or with buffer only, and subjected to a second round of sedimentation (Fig 8B and C). Assemblies composed of Hsp26 and misfolded MDH were less stable than those comprising bacterial IbpAB and denatured substrates. Incubation of Hsp26–MDH assemblies with buffer only followed by recentrifugation caused dissociation of almost 40% of Hsp26 molecules, suggesting increased dynamics of Hsp26 binding to Hsp26/MDH core assemblies. Incubation of Hsp26–MDH assemblies with the yeast Hsp70 chaperone system, however, increased the fraction of released Hsp26 to 80%, indicating a conserved Hsp70 function in sHsp displacement (Fig 8C); 20% of Hsp26 remained associated with MDH assemblies suggesting that this fraction of Hsp26 is not surface‐exposed but is an integral part of the assembly. This indicates that Hsp26–MDH assemblies are also composed of a stable Hsp26–MDH core structure and a dynamic Hsp26 outer shell. Hsp26 displacement by SYS did not result in release of free MDH, which still sedimented at almost the same position in the gradient as observed before for bacterial chaperones. Substrate solubilization and reactivation required addition of Hsp104, as seen by quantitative movement of MDH to the top fractions of the gradient indicative for formation of active MDH (Fig 8C). Accordingly, under these experimental conditions, all Hsp26 was found in the top fractions of the gradient. We finally showed that the bacterial KJE system also efficiently releases Hsp26 from Hsp26–MDH assemblies (Fig EV5A and B), indicating that Hsp70–sHsp interplay does not involve species‐specific cooperation consistent with a mechanism based on the direct binding of Hsp70 to sequestered substrates. Together, these experiments show that the Hsp70 system harbours an evolutionary conserved activity to displace sHsps from substrate assemblies, preparing the assemblies for efficient substrate reactivation.
Figure 8. Eukaryotic Hsp70 dissociates eukaryotic sHsps from sHsp–substrate assemblies in vitro .
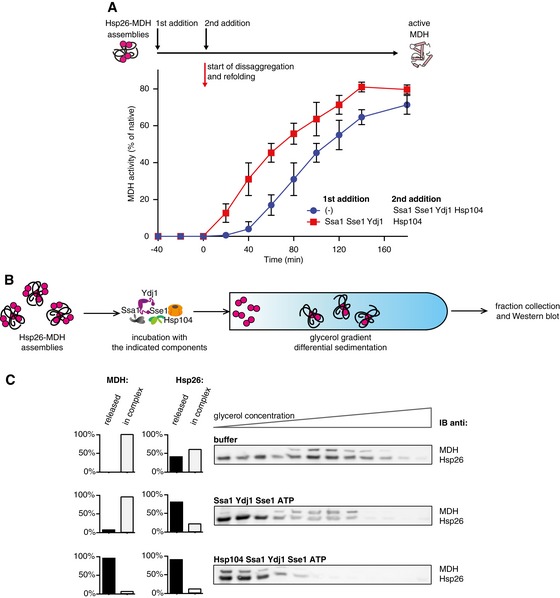
- Yeast Hsp70 system acts prior to disaggregation of Hsp26–MDH assemblies. An order of addition experiment. MDH (5 μM) denatured in the presence of Hsp26 (5 μM) was diluted 10‐fold and first incubated with the components listed in the legend and then complemented with the missing components of the functional Hsp104‐SYS (Ssa1, Ydj1, Sse1) bichaperone system present at 1.5 μM, 4 μM, 2 μM and 0.15 μM concentrations, respectively. Data are the mean ± SD of three independent experiments.
- Experimental scheme. Purified Hsp26–MDH assemblies were incubated with indicated components and subjected to glycerol gradient sedimentation.
- SYS (Ssa1, Ydj1, Sse1) dissociates Hsp26 from Hsp26–MDH assemblies. Fractions were collected from the top of the gradient, and MDH and Hsp26 were visualized by Western blot following SDS–PAGE. The band intensities of MDH and Hsp26 retained in the core assembly (fractions 5–12) or released from the assembly (fractions 1–3) were quantified and presented on the graphs on the left. Hsp26–MDH assemblies (approximate stoichiometry 1.25:1), Ssa1, Ydj1, Sse1 and Hsp104 were present at 400 nM (calculated for MDH monomers), 4 μM, 2 μM, 0.15 μM and 1.5 μM concentrations, respectively.
Source data are available online for this figure.
Figure EV5. Prokaryotic Hsp70 dissociates eukaryotic sHsps from sHsp–substrate assemblies in vitro .
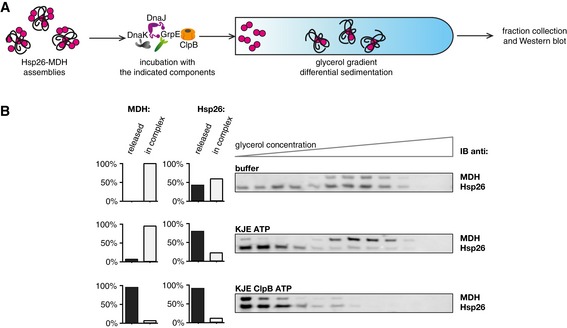
- Experimental scheme. Hsp26–MDH assemblies isolated from unbound Hsp26 via sedimentation in a glycerol gradient were incubated with the indicated components for 120 min and resubjected to a second round of sedimentation.
- DnaK, DnaJ, GrpE dissociate Hsp26 from Hsp26–MDH assemblies. Fractions were collected from the top of the gradient and MDH and Hsp26 were visualized by Western blot following SDS–PAGE. MDH and Hsp26 levels in each fraction were determined by quantitative immunoblot analysis using MDH‐ and Hsp26‐specific antibodies, respectively. The band intensities of Hsp26 and MDH retained in the core assembly (fractions 5–12) or released from the assembly (fractions 1–3) were quantified and presented on the graphs on the left. Hsp26–MDH assemblies (approximate stoichiometry 1.25:1), DnaK, DnaJ, GrpE and ClpB were present at 400 nM (calculated for MDH monomers), 1 μM, 0.3 μM, 0.3 μM and 1.5 μM concentrations, respectively.
Source data are available online for this figure.
Discussion
sHsps act as the first line of defence during protein misfolding stress and are key factors in modifying protein aggregation. Their presence results in the formation of small‐sized sHsp–substrate assemblies as compared to protein aggregates formed in sHsp absence. This assembly formation has beneficial effects on rates and yields of Hsp70–Hsp100‐mediated protein disaggregation and reactivation. This positive impact can be explained by an altered conformational state of the bound substrate and an increased surface offering more substrate binding sites for chaperone association. However, here we demonstrate that sHsps have ambivalent effects as their presence also causes a delay in substrate recovery and even inhibits the disaggregation and refolding processes at limited Hsp70 concentrations. Thus, while increasing the binding‐competent surface of the sHsp–substrate assemblies, sHsps also seem to reduce chaperone binding, presumably through direct competition. Additionally, binding of sHsps to substrates is likely also confining the fate of the bound substrate to the refolding pathway.
In this work, we unravel a novel, evolutionary conserved function of Hsp70 chaperones, which directly affects the ambivalent influence of sHsps on the disaggregation process. We show that Hsp70 binding allows for efficient, permanent dissociation of surface‐located sHsps from sHsp–substrate assemblies at the initial phase of disaggregation (Fig 9). This Hsp70 activity is unique and is not executed by other chaperones that either act on protein aggregates or can bind misfolded proteins (Fig EV3). It also does not involve cooperation with the Hsp100 partner protein. This has important evolutionary implications as the Hsp100 disaggregase is absent in metazoa, which encode for more potent Hsp70 chaperone systems that exhibit robust disaggregation activity (Nillegoda et al, 2015). The conserved key role of Hsp70 in initiating protein disaggregation strongly suggests that the activity of Hsp70 towards sHsp–substrate assemblies established here for bacterial and yeast chaperones is conserved in all kingdoms of life.
Figure 9. Schematic model of the Hsp100‐Hsp70 bichaperone system action on sHsp–substrate assemblies.
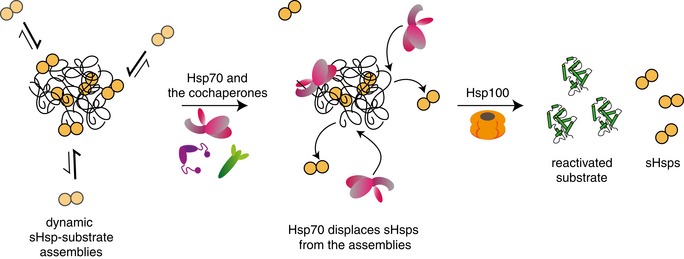
sHsp–substrate assemblies consist of a stable sHsp–substrate core and an dynamic outer shell, comprising the majority of sHsps. Surface‐exposed sHsps are dynamically binding and dissociating from the sHsp–substrate assemblies, with the equilibrium shifted towards the bound state. Hsp70 outcompetes sHsps in binding to the assembly surface, protecting the sHsp–substrate core and rendering sHsps in the unbound state. Once the sHsps are displaced from the surface, the misfolded polypeptides are extracted from the assemblies and refolded by a concerted action of the Hsp100‐Hsp70 bichaperone system.
We provide evidence that Hsp70 displaces sHsps from the assemblies by a competitive dissociation mechanism as mere binding of Hsp70 to the assemblies is sufficient to cause sHsp release. The competition model that we propose predicts that direct interactions between Hsp70 and sHsps are not necessary for sHsp displacement by Hsp70. Indeed, species‐specific cooperation between Hsp70 and sHsps in disaggregation reactions was neither observed here nor in former studies and direct interactions between both chaperones have not been reported (Giese & Vierling, 2002; Mogk et al, 2003b; Friedrich et al, 2004; Bepperling et al, 2012). The efficient dissociation of sHsps from the assemblies is dependent on a functional Hsp70 chaperone cycle. Hsp70 has to be activated for binding to the substrate by J‐proteins (e.g. DnaJ) to compete with sHsps for binding to the assemblies. DnaJ itself, however, is not able to dissociate sHsps from assemblies. We speculate that the interaction between DnaJ and substrate is too transient to efficiently compete with dynamic IbpAB. Alternatively DnaJ may recognize distinct substrate segments compared with IbpAB allowing for their simultaneous binding at the complex surface.
The proposed competition model demands for a dynamic behaviour of sHsps. Our work defines two distinct sHsps populations within sHsp–substrate assemblies forming specific substructures with diverse dynamics. The first and less abundant population of sHsps forms an integral, immobile part of sHsp–substrate assemblies. This population of sHsps is only liberated during Hsp100 disaggregase activity, which results in the complete disintegration of the assemblies. The second and more abundant sHsp fraction is surface‐exposed and dynamically interacts with the assembly. The presence of two sHsp populations within sHsp–substrate assemblies is supported by kinetic studies revealing biphasic kinetics of sHsp–substrate interactions: substrates are rapidly bound by a small number of sHsps, and extra sHsps are subsequently recruited to the assembly (Stengel et al, 2010). We suggest that the first of these two populations represents the core assembly population defined in this work and the latter represents the surface‐exposed dynamic fraction that can be displaced by Hsp70. We consider two scenarios for the displacement of surface‐exposed sHsps by Hsp70, given that sHsps associate with substrates via multiple binding sites (Jaya et al, 2009; Fu et al, 2013). First, sHsps completely dissociate from the assemblies and are prevented from re‐association by Hsp70 binding. Second, sHsps only partially dissociate from the assembly by loosing some substrate interactions while retaining others. Binding of Hsp70 to the core assembly at a site that becomes vacant upon partial sHsp release might then ultimately cause sHsp dissociation.
We also predict that the outer shell of dynamic sHsps has various important physiological functions. First, it maintains size and solubility of the core assembly by shielding hydrophobic sites within the substrate that are exposed at the surface of the assemblies. Second, it forms a selective barrier that is only penetrated by the Hsp70 chaperone system but not by, for example, AAA+ proteases, which have disaggregation potential and are linked to proteolysis. The sHsp shell thereby indirectly targets sequestered substrates of the core structure to refolding pathways. Third, it increases the demand for Hsp70 chaperone activity, as sHsp–substrate assemblies require higher Hsp70 concentrations for disaggregation compared with aggregated substrates. We suggest that such mechanism allows to protect sequestered substrates under unfavourable folding conditions such as chronic stress, which reduces the availability of free Hsp70 chaperones.
Taken together, our work unravels a novel function of Hsp70 in protein disaggregation, namely the displacement of surface‐located sHsps from sHsp–substrate assemblies. This Hsp70 activity directs sequestered substrates to refolding pathways. The Hsp70 function is independent from the cooperating Hsp100 disaggregase and is therefore expected to also play crucial roles in metazoan protein quality control systems.
Materials and Methods
Proteins
Purifications of the proteins were performed as described previously: DnaK, DnaJ, GrpE, ClpB, IbpA, His‐IbpB (N‐terminal His‐tag) and GFP (Ratajczak et al, 2009), and Ssa1, Sse1, Ydj1, Hsp104 wt, Hsp104 D484K and Hsp26 (Seyffer et al, 2012; Lipinska et al, 2013). Pyruvate kinase and creatine kinase were purchased from Sigma, pig heart muscle MDH from Roche and firefly luciferase from Promega. Protein concentrations were determined with the Bio‐Rad Bradford assay using BSA as standard. Protein concentrations refer to monomers.
Dynamic light scattering
Particle sizing was performed using Malvern Instruments ZetaSizer Nano S dynamic light scattering instrument. Measurements were taken in buffer F (50 mM Tris, pH 7.4, 150 mM KCl, 20 mM Mg acetate, 2 mM DTT). Luciferase was present at 1.5 μM, IbpA at 3 μM, IbpB at 7 μM concentration. Conditions were as follows: measurement volume—40 μl, scattering angle—173°, wavelength—633 nm, temperature—25°C. For every measurement, minimum five 10‐s runs were averaged and particle size distribution was calculated by fitting to 70 size bins between 0.4 and 10,000 nm. Results are shown as particle distribution by volume distribution of analysed species.
Luciferase denaturation and refolding experiments
Luciferase (1.5 μM) was denatured at 48°C for 10 min in buffer A (50 mM Tris, pH 7.5, 150 mM KCl, 20 mM MgCl2, 2 mM DTT) either with or without IbpA (3 μM) and His‐IbpB (7 μM). Protein refolding was started by 40‐fold dilution of denatured luciferase in the chaperone cocktail. Unless noted otherwise, the chaperone concentrations used were as follows: DnaK 1 μM, DnaJ 0.3 μM, GrpE 0.3 μM, ClpB 1.5 μM. All assays were performed in the presence of an ATP‐regenerating system (3 mM phosphoenol pyruvate, 20 μg/ml pyruvate kinase, 2 mM ATP). The disaggregation reaction was carried out at 25°C. Luciferase activities were measured using a Sirius Luminometer (Berthold). Refolding rates were calculated from the linear range of increase in luciferase activity.
GFP denaturation and refolding experiments
The experiments were performed essentially as described previously (Ratajczak et al, 2009). GFP (150 μM) was thermally inactivated in buffer B (40 mM Tris (pH 7.5), 10% (v/v) glycerol, 150 mM NaCl and 100 mM 2‐mercaptoethanol) by incubation for 10 min at 85°C. After denaturation, GFP aggregates were diluted to a final concentration of 1.5 μM in buffer A supplemented with sHsps (3 μM IbpA and 7 μM IbpB) as indicated, and incubation proceeded for 10 min at 48°C. Reactivation was initiated by threefold dilution of GFP in buffer C (40 mM Tris (pH 7.8), 85 mM potassium glutamate, 20 mM magnesium acetate, 10% (v/v) glycerol and 5 mM 2‐mercaptoethanol) supplemented with 10 mM ATP and an ATP regeneration system comprising creatine phosphate (120 mM) and creatine kinase (0.2 mg/ml) and chaperone proteins (DnaK, DnaJ, GrpE, and Hsp104 D484K at final concentrations of 1, 0.3, 0.3, and 1 μM, respectively). The disaggregation reaction was carried out at 25°C in 400 μl final volume in a spectrofluorometric cuvette. The fluorescence of refolded GFP was measured in a PerkinElmer LS50B spectrofluorimeter with excitation at 395 nm and emission at 510 nm.
MDH denaturation and refolding experiments
MDH (5 μM) was denatured in the presence of Hsp26 (5 μM) in buffer D (50 mM HEPES, pH 7.5, 50 mM KCl, 5 mM MgCl2, 2 mM DTT). Protein refolding was started by diluting aggregated MDH to 500 nM in the chaperone cocktail: Ssa1 4 μM, Ydj1 2 μM, Sse1 0.15 μM, Hsp104 1.5 μM. All assays were performed in the presence of an ATP‐regenerating system (3 mM phosphoenol pyruvate; 20 μg/ml pyruvate kinase; 2 mM ATP). The disaggregation reaction was carried out at 30°C. Determination of enzymatic activities followed published protocols (Mogk et al, 2003a).
SDS–PAGE, immunoblotting and protein quantifications
Gel electrophoresis was performed using 4–20% ExpressPlus PAGE Gels (Genescript) and stained with Coomassie brilliant blue. Immunoblotting was performed according to the standard procedures, using rabbit anti‐sera specific for IbpA, DnaK, Hsp26, MDH or luciferase as primary antibody, and developed either with a Vistra ECF fluorescence immunoblotting kit (Amersham) using alkaline phosphatase‐conjugated anti‐rabbit IgG as secondary antibodies (Vector Laboratories) or, alternatively SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific), using (H+L) HRP‐conjugated anti‐rabbit IgG (Bio‐Rad) as secondary antibodies. Developed immunoblots were scanned using a fluoroimager ImageQuant LAS 4000 or ChemiDoc MP Imaging System (Bio‐Rad) and quantified using ImageJ software.
sHsp–substrate assembly isolation and sedimentation analysis
Luciferase (1.5 μM) was denatured at 48°C for 10 min in buffer A in the presence of IbpA (3 μM) and His‐IbpB (7 μM). To isolate the IbpAB–luciferase assemblies from unbound IbpAB, 600 μl of preformed assemblies was applied on a 3.2 ml 10–60% glycerol gradient in buffer A. Samples were centrifuged at 4°C in a Beckman SW 60 rotor at 160,000 g for 1.5 h; fractions were collected from the top. Protein distribution in each fraction was verified by SDS–PAGE followed by immunoblotting. Fractions containing IbpA‐IbpB‐luciferase assemblies (approximate stoichiometry 1:0.8:1) were pooled together, and the substrate concentration was determined by optical densitometry following SDS–PAGE as 1 μM (luciferase monomer). To analyse the effect of chaperones on isolated assemblies, the assemblies were diluted 1:10 in chaperone cocktail (DnaK 1 μM, DnaJ 0.3 μM, GrpE 0.3 μM, ClpB 1.5 μM) and subjected to a second round of sedimentation under conditions listed above.
MDH (5 μM) was denatured at 47°C for 30 min in buffer D in the presence of Hsp26 (15 μM). To isolate the Hsp26–MDH assemblies from unbound sHsps, 600 μl of the assemblies was applied on a 3.2 ml 10–60% glycerol gradient in buffer D. Samples were centrifuged at 4°C in a Beckman SW 60 rotor at 210,000 g for 80 min; fractions were collected from the top. Protein distribution in each fraction was verified by SDS–PAGE followed by immunoblotting. Fractions containing Hsp26–MDH assembly (approximate stoichiometry 1.25:1) were pooled together, and substrate concentration was determined by optical densitometry following SDS–PAGE as 1.6 μM (MDH monomer). To analyse the effect of chaperones on isolated assemblies, the assemblies were diluted 1:4 in chaperone cocktail (Ssa1 4 μM, Ydj1 2 μM, Sse1 0.15 μM, Hsp104 1.5 μM or, alternatively, DnaK 1 μM, DnaJ 0.3 μM, GrpE 0.3 μM, ClpB 1.5 μM) and subjected to a second round of sedimentation under conditions listed above.
Isolation of aggregated proteins in vivo
The E. coli strain used was a derivative of MC4100 and is described in Mogk et al (2003a). The strain was cultured at 30°C in Luria broth (LB). Chloramphenicol and kanamycin were used at final concentrations of 10 and 20 μg/ml, respectively. Temperature‐shift experiments were performed in orbital shaking water baths. The cells were grown overnight to OD = 0.7, heat‐shocked at 46°C for 30 min and shifted back to 30°C. To induce synthesis of DnaK and DnaJ, IPTG was added to 1 mM. Four hours after the heat shock spectinomycin was added to 50 μg/ml to the cultures to prevent the residual accumulation of DnaK and DnaJ synthetized due to PA1/lacO‐1 promoter leakage in IPTG non‐induced culture. At indicated time points, 10‐ml aliquots were collected and the aggregated protein fraction was isolated as described in Tomoyasu et al (2001).
Author contributions
SZw, AK, NBN and IO performed the experiments. SZw, AK, NBN, SZi, BB, AM and KL designed the experiments. AP performed and analysed the DLS experiments. SZw, AM and KL wrote the manuscript. SZw, AM, BB and KL conceived the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
This work was supported by a grant of the Polish National Science Centre (2013/08/A/NZ1/00683) to K.L. and a grant of the Deutsche Forschungsgemeinschaft (SFB1036 project A8) to A.M. and B.B. S.Z. was supported by Foundation for Polish Science International Ph.D. Projects Grant MPD/2010/5 cofinanced by the European Union European Regional Development Fund, Operational Program Innovative Economy 2007–2013. S.Z. was also a recipient of EMBO short‐term fellowship (ASTF 550‐2014). A.K. is a recipient of “Diamentowy Grant” from the Ministry of Science and Higher Education. We thank Dr. Matthias Mayer for discussions.
The EMBO Journal (2017) 36: 783–796
References
- Ahmad MF, Raman B, Ramakrishna T, Rao CHM (2008) Effect of phosphorylation on alpha B‐crystallin: differences in stability, subunit exchange and chaperone activity of homo and mixed oligomers of alpha B‐crystallin and its phosphorylation‐mimicking mutant. J Mol Biol 375: 1040–1051 [DOI] [PubMed] [Google Scholar]
- Aquilina JA, Shrestha S, Morris AM, Ecroyd H (2013) Structural and functional aspects of hetero‐oligomers formed by the small heat shock proteins alphaB‐crystallin and HSP27. J Biol Chem 288: 13602–13609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha E, Jones C, Blackwell AE, Cheng G, Waters ER, Samsel KA, Siddique M, Pett V, Wysocki V, Vierling E (2013) An unusual dimeric small heat shock protein provides insight into the mechanism of this class of chaperones. J Mol Biol 425: 1683–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch JL, Aquilina JA, Baldwin AJ, Rekas A, Stengel F, Lindner RA, Basha E, Devlin GL, Horwitz J, Vierling E, Carver JA, Robinson CV (2010) The quaternary organization and dynamics of the molecular chaperone HSP26 are thermally regulated. Chem Biol 17: 1008–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepperling A, Alte F, Kriehuber T, Braun N, Weinkauf S, Groll M, Haslbeck M, Buchner J (2012) Alternative bacterial two‐component small heat shock protein systems. Proc Natl Acad Sci USA 109: 20407–20412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashikar AG, Duennwald M, Lindquist SL (2005) A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J Biol Chem 280: 23869–23875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Basha E, Wysocki VH, Vierling E (2008) Insights into small heat shock protein and substrate structure during chaperone action derived from hydrogen/deuterium exchange and mass spectrometry. J Biol Chem 283: 26634–26642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbecq SP, Klevit RE (2013) One size does not fit all: the oligomeric states of alphaB crystallin. FEBS Lett 587: 1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan DA, Reid BG, Horwich AL, Bukau B (2002) ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9: 673–683 [DOI] [PubMed] [Google Scholar]
- Doyle SM, Genest O, Wickner S (2013) Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol 14: 617–629 [DOI] [PubMed] [Google Scholar]
- Ecroyd H, Meehan S, Horwitz J, Aquilina JA, Benesch JL, Robinson CV, Macphee CE, Carver JA (2007) Mimicking phosphorylation of alphaB‐crystallin affects its chaperone activity. Biochem J 401: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnsperger M, Graber S, Gaestel M, Buchner J (1997) Binding of non‐native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J 16: 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich KL, Giese KC, Buan NR, Vierling E (2004) Interactions between small heat shock protein subunits and substrate in small heat shock protein‐substrate complexes. J Biol Chem 279: 1080–1089 [DOI] [PubMed] [Google Scholar]
- Fu X, Shi X, Yin L, Liu J, Joo K, Lee J, Chang Z (2013) Small heat shock protein IbpB acts as a robust chaperone in living cells by hierarchically activating its multi‐type substrate‐binding residues. J Biol Chem 288: 11897–11906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KC, Vierling E (2002) Changes in oligomerization are essential for the chaperone activity of a small heat shock protein in vivo and in vitro . J Biol Chem 277: 46310–46318 [DOI] [PubMed] [Google Scholar]
- Glover JR, Lindquist S (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94: 73–82 [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer‐Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475: 324–332 [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Braun N, Stromer T, Richter B, Model N, Weinkauf S, Buchner J (2004) Hsp42 is the general small heat shock protein in the cytosol of Saccharomyces cerevisiae . EMBO J 23: 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Miess A, Stromer T, Walter S, Buchner J (2005) Disassembling protein aggregates in the yeast cytosol. The cooperation of Hsp26 with Ssa1 and Hsp104. J Biol Chem 280: 23861–23868 [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Vierling E (2015) A first line of stress defense: small heat shock proteins and their function in protein homeostasis. J Mol Biol 427: 1537–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaya N, Garcia V, Vierling E (2009) Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc Natl Acad Sci USA 106: 15604–15609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11: 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer‐Hartl M, Hartl FU (2013) Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82: 323–355 [DOI] [PubMed] [Google Scholar]
- Krzewska J, Langer T, Liberek K (2001) Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett 489: 92–96 [DOI] [PubMed] [Google Scholar]
- Kuczynska‐Wisnik D, Kedzierska S, Matuszewska E, Lund P, Taylor A, Lipinska B, Laskowska E (2002) The Escherichia coli small heat‐shock proteins IbpA and IbpB prevent the aggregation of endogenous proteins denatured in vivo during extreme heat shock. Microbiology 148: 1757–1765 [DOI] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU (1992) Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone‐mediated protein folding. Nature 356: 683–689 [DOI] [PubMed] [Google Scholar]
- Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B (1999) Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci USA 96: 5452–5457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GJ, Roseman AM, Saibil HR, Vierling E (1997) A small heat shock protein stably binds heat‐denatured model substrates and can maintain a substrate in a folding‐competent state. EMBO J 16: 659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Owen HA, Prochaska DJ, Barnum SR (2000) HSP16.6 is involved in the development of thermotolerance and thylakoid stability in the unicellular cyanobacterium, Synechocystis sp. PCC 6803. Curr Microbiol 40: 283–287 [DOI] [PubMed] [Google Scholar]
- Lee J, Kim JH, Biter AB, Sielaff B, Lee S, Tsai FT (2013) Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc Natl Acad Sci USA 110: 8513–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentze N, Aquilina JA, Lindbauer M, Robinson CV, Narberhaus F (2004) Temperature and concentration‐controlled dynamics of rhizobial small heat shock proteins. Eur J Biochem 271: 2494–2503 [DOI] [PubMed] [Google Scholar]
- Liberek K, Skowyra D, Zylicz M, Johnson C, Georgopoulos C (1991) The Escherichia coli DnaK chaperone, the 70‐kDa heat shock protein eukaryotic equivalent, changes conformation upon ATP hydrolysis, thus triggering its dissociation from a bound target protein. J Biol Chem 266: 14491–14496 [PubMed] [Google Scholar]
- Liberek K, Lewandowska A, Zietkiewicz S (2008) Chaperones in control of protein disaggregation. EMBO J 27: 328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinska N, Zietkiewicz S, Sobczak A, Jurczyk A, Potocki W, Morawiec E, Wawrzycka A, Gumowski K, Slusarz M, Rodziewicz‐Motowidlo S, Chrusciel E, Liberek K (2013) Disruption of ionic interactions between the nucleotide binding domain 1 (NBD1) and middle (M) domain in Hsp100 disaggregase unleashes toxic hyperactivity and partial independence from Hsp70. J Biol Chem 288: 2857–2869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuszewska M, Kuczynska‐Wisnik D, Laskowska E, Liberek K (2005) The small heat shock protein IbpA of Escherichia coli cooperates with IbpB in stabilization of thermally aggregated proteins in a disaggregation competent state. J Biol Chem 280: 12292–12298 [DOI] [PubMed] [Google Scholar]
- Miot M, Reidy M, Doyle SM, Hoskins JR, Johnston DM, Genest O, Vitery MC, Masison DC, Wickner S (2011) Species‐specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proc Natl Acad Sci USA 108: 6915–6920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Tomoyasu T, Goloubinoff P, Rudiger S, Roder D, Langen H, Bukau B (1999) Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J 18: 6934–6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Deuerling E, Vorderwulbecke S, Vierling E, Bukau B (2003a) Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol Microbiol 50: 585–595 [DOI] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Friedrich KL, Schonfeld HJ, Vierling E, Bukau B (2003b) Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem 278: 31033–31042 [DOI] [PubMed] [Google Scholar]
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R, Guilbride DL, Wade RC, Morimoto RI, Mayer MP, Bukau B (2015) Crucial HSP70 co‐chaperone complex unlocks metazoan protein disaggregation. Nature 524: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter AJ, Jaya N, Basha E, Vierling E, Robinson CV, Benesch JL (2008) Real‐time monitoring of protein complexes reveals their quaternary organization and dynamics. Chem Biol 15: 246–253 [DOI] [PubMed] [Google Scholar]
- Panniers R (1994) Translational control during heat shock. Biochimie 76: 737–747 [DOI] [PubMed] [Google Scholar]
- Peschek J, Braun N, Rohrberg J, Back KC, Kriehuber T, Kastenmuller A, Weinkauf S, Buchner J (2013) Regulated structural transitions unleash the chaperone activity of alphaB‐crystallin. Proc Natl Acad Sci USA 110: E3780–E3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesofsky‐Vig N, Brambl R (1995) Disruption of the gene for hsp30, an alpha‐crystallin‐related heat shock protein of Neurospora crassa, causes defects in thermotolerance. Proc Natl Acad Sci USA 92: 5032–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak E, Zietkiewicz S, Liberek K (2009) Distinct activities of Escherichia coli small heat shock proteins IbpA and IbpB promote efficient protein disaggregation. J Mol Biol 386: 178–189 [DOI] [PubMed] [Google Scholar]
- Richter K, Haslbeck M, Buchner J (2010) The heat shock response: life on the verge of death. Mol Cell 40: 253–266 [DOI] [PubMed] [Google Scholar]
- Rosenzweig R, Moradi S, Zarrine‐Afsar A, Glover JR, Kay LE (2013) Unraveling the mechanism of protein disaggregation through a ClpB‐DnaK interaction. Science 339: 1080–1083 [DOI] [PubMed] [Google Scholar]
- Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K (2003) MecA, an adaptor protein necessary for ClpC chaperone activity. Proc Natl Acad Sci USA 100: 2306–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D, Baici A, Gehring H, Christen P (1994) Kinetics of molecular chaperone action. Science 263: 971–973 [DOI] [PubMed] [Google Scholar]
- Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, Zahn R, Sourjik V, Bukau B, Mogk A (2012) Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol 19: 1347–1355 [DOI] [PubMed] [Google Scholar]
- Shi J, Koteiche HA, McDonald ET, Fox TL, Stewart PL, McHaourab HS (2013) Cryoelectron microscopy analysis of small heat shock protein 16.5 (Hsp16.5) complexes with T4 lysozyme reveals the structural basis of multimode binding. J Biol Chem 288: 4819–4830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel F, Baldwin AJ, Painter AJ, Jaya N, Basha E, Kay LE, Vierling E, Robinson CV, Benesch JL (2010) Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc Natl Acad Sci USA 107: 2007–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel F, Baldwin AJ, Bush MF, Hilton GR, Lioe H, Basha E, Jaya N, Vierling E, Benesch JL (2012) Dissecting heterogeneous molecular chaperone complexes using a mass spectrum deconvolution approach. Chem Biol 19: 599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strozecka J, Chrusciel E, Gorna E, Szymanska A, Zietkiewicz S, Liberek K (2012) Importance of N‐ and C‐terminal regions of IbpA, Escherichia coli small heat shock protein, for chaperone function and oligomerization. J Biol Chem 287: 2843–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoyasu T, Mogk A, Langen H, Goloubinoff P, Bukau B (2001) Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol Microbiol 40: 397–413 [DOI] [PubMed] [Google Scholar]
- Treweek TM, Meehan S, Ecroyd H, Carver JA (2015) Small heat‐shock proteins: important players in regulating cellular proteostasis. Cell Mol Life Sci 72: 429–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B (2010) Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 11: 777–788 [DOI] [PubMed] [Google Scholar]
- Veinger L, Diamant S, Buchner J, Goloubinoff P (1998) The small heat‐shock protein IbpB from Escherichia coli stabilizes stress‐denatured proteins for subsequent refolding by a multichaperone network. J Biol Chem 273: 11032–11037 [DOI] [PubMed] [Google Scholar]
- Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber‐Ban EU, Dougan DA, Tsai FT, Mogk A, Bukau B (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119: 653–665 [DOI] [PubMed] [Google Scholar]
- Yost HJ, Lindquist S (1986) RNA splicing is interrupted by heat shock and is rescued by heat shock protein synthesis. Cell 45: 185–193 [DOI] [PubMed] [Google Scholar]
- Zietkiewicz S, Krzewska J, Liberek K (2004) Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. J Biol Chem 279: 44376–44383 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8