

Abstract
PAX5 is a tumor suppressor in B‐ALL, while the role of PAX5 fusion proteins in B‐ALL development is largely unknown. Here, we studied the function of PAX5‐ETV6 and PAX5‐FOXP1 in mice expressing these proteins from the Pax5 locus. Both proteins arrested B‐lymphopoiesis at the pro‐B to pre‐B‐cell transition and, contrary to their proposed dominant‐negative role, did not interfere with the expression of most regulated Pax5 target genes. Pax5‐Etv6, but not Pax5‐Foxp1, cooperated with loss of the Cdkna2a/b tumor suppressors in promoting B‐ALL development. Regulated Pax5‐Etv6 target genes identified in these B‐ALLs encode proteins implicated in pre‐B‐cell receptor (BCR) signaling and migration/adhesion, which could contribute to the proliferation, survival, and tissue infiltration of leukemic B cells. Together with similar observations made in human PAX5‐ETV6+ B‐ALLs, these data identified PAX5‐ETV6 as a potent oncoprotein that drives B‐cell leukemia development.
Keywords: B‐cell leukemia, CDKN2A/B cooperation, mouse model, PAX5‐ETV6, regulated target genes
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; Immunology
Introduction
The transcription factor Pax5 is an essential regulator of B‐cell commitment and development, which is exclusively expressed in the B‐lymphoid lineage of the hematopoietic system (Nutt et al, 1999; Horcher et al, 2001; Medvedovic et al, 2011). Pax5 acts as a transcriptional repressor to suppress B‐lineage‐inappropriate genes (Delogu et al, 2006) and functions as an activator to induce gene expression required for B‐cell development and function (Schebesta et al, 2007; Revilla‐i‐Domingo et al, 2012). Notably, mature B cells undergoing complete loss of Pax5 dedifferentiate to uncommitted progenitor cells and give rise to an aggressive progenitor cell leukemia, demonstrating that Pax5 maintains B‐cell identity throughout B‐lymphopoiesis and functions as a tumor suppressor in the B‐lymphoid lineage (Cobaleda et al, 2007).
PAX5 was identified as a haploinsufficient tumor suppressor gene in human B‐cell precursor acute lymphoblastic leukemia (B‐ALL), as heterozygous deletions and loss‐of‐function mutations are present in one‐third of all B‐ALLs (Mullighan et al, 2007). In mouse models, Pax5 heterozygosity cooperates with constitutive activation of STAT5, JAK1, or JAK3 in promoting B‐ALL development, which demonstrated that Pax5 functions as a haploinsufficient tumor suppressor in leukemogenesis (Heltemes‐Harris et al, 2011; Dang et al, 2015). Importantly, these mouse B‐ALL tumors require continuous Pax5 deficiency for their maintenance, as restoration of normal Pax5 expression allows the leukemic cells to further differentiate along the B‐cell pathway, leading to disease remission (Liu et al, 2014). Hence, heterozygous loss of the tumor suppressor Pax5 contributes to B‐ALL development, at least in part, by arresting B‐cell development in the oncogenic tumor setting.
PAX5 translocations occur at a frequency of 2–3% in human B‐ALLs and involve several fusion partner genes, generating novel chimeric PAX5 transcription factors (Nebral et al, 2009; Coyaud et al, 2010). All PAX5 fusion proteins contain the N‐terminal DNA‐binding paired domain of PAX5 and lack C‐terminal PAX5 sequences including the potent transactivation domain (Nebral et al, 2009; Coyaud et al, 2010). PAX5 fusion proteins are able to interact with Pax5‐binding sites at target genes, but may possess different transcriptional activities depending on the C‐terminal sequences provided by their partner proteins. PAX5‐ETV6 was identified as the first and most frequently described PAX5 translocation, which fuses the PAX5 paired domain to almost the entire ETV6 transcription factor (Cazzaniga et al, 2001). Although detected in fewer B‐ALL cases, PAX5‐FOXP1 is also a recurrent translocation that links the N‐terminal PAX5 sequences to almost the whole FOXP1 transcription factor (Mullighan et al, 2007; Coyaud et al, 2010). Chimeric PAX5 fusion proteins are complex multidomain transcription factors, as exemplified by PAX5‐ETV6 and PAX5‐FOXP1. Both proteins contain a second DNA‐binding domain (Ets domain of ETV6 or forkhead domain of FOXP1) as well as an oligomerization motif (pointed domain of ETV6 or coiled‐coil domain of FOXP1) (Cazzaniga et al, 2001; Mullighan et al, 2007). Consequently, PAX5‐ETV6 and other PAX5 fusion proteins were shown to oligomerize (Kawamata et al, 2012; Fortschegger et al, 2014). Notably, several PAX5 fusion proteins, including PAX5‐ETV6 and PAX5‐FOXP1, are able to inhibit the transcriptional activity of wild‐type PAX5 in transient transfection assays (Bousquet et al, 2007; Mullighan et al, 2007; Kurahashi et al, 2011; Kawamata et al, 2012). Hence, these chimeric PAX5 transcription factors are thought to act as dominant‐negative proteins to interfere with the function of PAX5 that is expressed from the retained wild‐type allele. The function of PAX5‐ETV6 was further investigated by retroviral overexpression in cultured mouse pro‐B cells, which resulted in moderate expression changes of some B‐cell‐specific genes, suggesting a role for PAX5‐ETV6 in controlling cell survival and oncogenic STAT5 signaling (Fazio et al, 2008, 2013; Cazzaniga et al, 2015). However, this overexpression system could neither recapitulate the correct relative expression levels of PAX5‐ETV6 and PAX5 seen in B‐ALL nor provide information about the developmental and oncogenic function of PAX5‐ETV6. Hence, little is known to date about the role of PAX5 fusion proteins in B‐ALL development.
Here, we have generated two mouse models that express PAX5‐ETV6 or PAX5‐FOXP1 under the control of the Pax5 locus. By analyzing the Pax5 Etv6/+ and Pax5 Foxp1/+ mice, we could demonstrate that Pax5‐Etv6 and Pax5‐Foxp1 impaired B‐cell development already at the pro‐B to pre‐B‐cell transition, but did not interfere with the expression of most regulated Pax5 target genes. Heterozygous loss of the Cdkn2a/b tumor suppressor locus cooperated with Pax5‐Etv6, but not with Pax5‐Foxp1, in promoting B‐ALL development in Cdkn2ab +/− Pax5 Etv6/+ mice. The identification of regulated Pax5‐Etv6 target genes revealed several Pax5‐Etv6‐controlled pathways, including pre‐B‐cell receptor (pre‐BCR) signaling and migration/adhesion, which likely contributed to the proliferation, survival, and tissue infiltration of the leukemic B cells. Moreover, several of these target genes were similarly regulated in human PAX5‐ETV6+ B‐ALLs. Hence, these data provided novel insight into the molecular role of PAX5‐ETV6 as potent oncoprotein in driving B‐ALL development.
Results
Normal B‐cell development in heterozygous Pax5 mutant mice
As Pax5 is an essential regulator of B‐cell development (Medvedovic et al, 2011) and PAX5 heterozygosity is frequently associated with human B‐ALL (Mullighan et al, 2007), it is assumed that inactivation of one PAX5 allele contributes to leukemia formation by inducing a B‐cell developmental arrest. To test this hypothesis, we compared B‐cell development in Pax5 +/+ and Pax5 +/− mice by flow cytometry. As shown in Fig 1A, similar numbers of each B‐cell type were present in the bone marrow and spleen of Pax5 +/+ and Pax5 +/− mice. As competitive bone marrow transplantation is best suited for detecting minor differences in the developmental fitness of competing cells, we reconstituted lethally irradiated Pax5 +/+ (Ly5.1) mice with a 1:1 mixture of Pax5 +/− (Ly5.2) and Pax5 +/+ (Ly5.1) bone marrow cells. Flow cytometric analysis of the bone marrow of a chimeric mouse 12 weeks after transplantation demonstrated that short‐lived granulocytes, reflecting the activity of hematopoietic stem cells, consisted of 59% Pax5 +/− and 39% Pax5 +/+ cells and that this ratio was minimally increased in favor of the Pax5 +/− genotype in pre‐B and immature B cells (Fig 1B). Analysis of four reconstituted mice revealed that the average ratio of the two genotypes was close to 50% for granulocytes, macrophages, and the different B‐cell subsets analyzed (Fig 1C). RNA‐sequencing (RNA‐seq) comparison of ex vivo sorted Pax5 +/+ and Pax5 +/− pro‐B cells furthermore revealed only one upregulated and 16 downregulated genes in Pax5 +/− pro‐B cells (Fig 1D and Appendix Fig S1A). Nine of these genes were previously identified as direct Pax5 target genes (Revilla‐i‐Domingo et al, 2012), thus indicating that these target genes were already deregulated in response to a twofold reduction of Pax5 expression (Appendix Fig S1A). In summary, we conclude that heterozygous loss of Pax5 expression does not impair B‐cell development under steady‐state conditions in the mouse.
Figure 1. Normal B‐cell development in heterozygous Pax5 mutant mice.
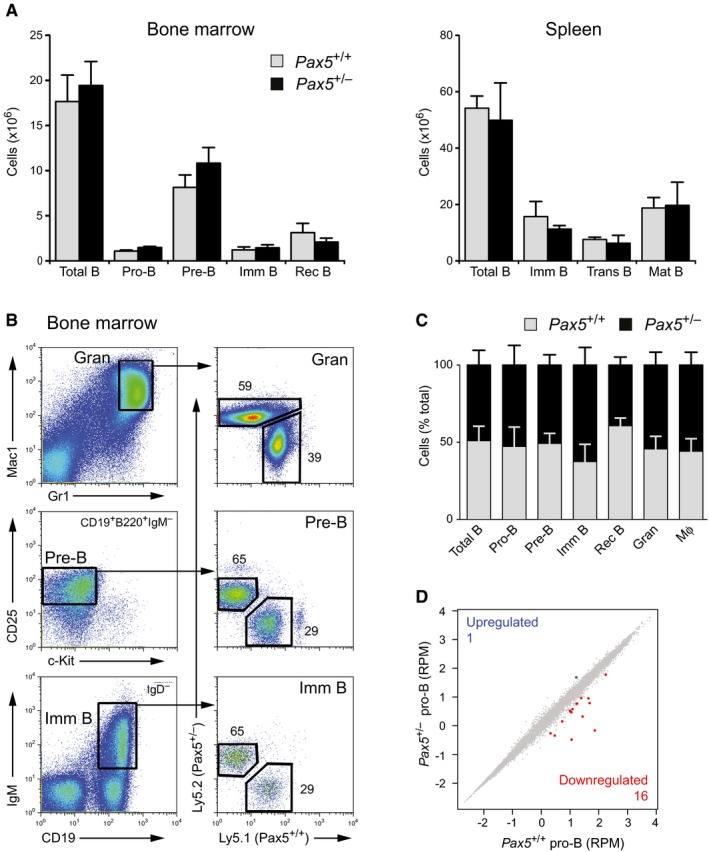
- Absolute cell numbers of the indicated cell types were determined by flow cytometric analysis of the bone marrow and spleen from 6‐week‐old Pax5 +/+ and Pax5 +/− mice (n = 5 per genotype) on the C57BL/6 background. Data are presented as average cell number with SEM. Imm, immature; Trans, transitional; Mat, mature; Rec, recirculating. See Materials and Methods for flow cytometric definition of the different cell types.
- Competitive bone marrow transplantation. Lethally irradiated Pax5 +/+ (Ly5.1) mice were reconstituted with a 1:1 mixture of bone marrow cells from Pax5 +/− (Ly5.2) and Pax5 +/+ (Ly5.1) mice. Granulocytes (Gran), pre‐B, and immature B cells in the bone marrow of a chimeric mouse were analyzed by flow cytometry 12 weeks after injection.
- The relative contribution of cells of the Pax5 +/− (Ly5.2) and Pax5 +/+ (Ly5.1) genotype to the development of the indicated cell types was determined by flow cytometric analysis of the bone marrow from four chimeric mice 12 weeks after injection. Data are represented as average percentage with SEM. The relative frequency of the Pax5 +/+ and Pax5 +/− B lymphocytes at the indicated developmental stages was not significantly different (P > 0.1), as demonstrated by two‐way analysis of variance (ANOVA). Mϕ, macrophage.
- Scatter plot of gene expression differences between ex vivo sorted Pax5 +/+ and Pax5 +/− pro‐B cells, based on two (Pax5 +/+) or three (Pax5 +/−) independent RNA‐seq experiments. The normalized expression data of individual genes (indicated by dots) were plotted as RPM values. Genes with an expression difference of > threefold, an adjusted P‐value of < 0.05 (DESeq2), and a TPM value of > 5 (in one of the two cell types) are colored in blue or red. RPM, reads per gene per million mapped sequence reads. TPM, transcripts per million (Wagner et al, 2012). As revealed by RNA‐seq analysis, the Pax5 mRNA was expressed in Pax5 +/− pro‐B cells at 58% of the mRNA level observed in Pax5 +/+ pro‐B cells.
Impaired B‐cell development in Pax5 Etv6/+ and Pax5 Foxp1/+ mice
We next established mouse models for studying the role of the PAX5‐ETV6 and PAX5‐FOXP1 fusion proteins in B‐ALL development. To recapitulate the corresponding human translocations as closely as possible, we inserted human cDNA sequences, starting in exon 4 and encoding the remaining PAX5‐ETV6 or PAX5‐FOXP1 protein, in frame into exon 4 of the mouse Pax5 locus to generate the Pax5 Etv6 and Pax5 Foxp1 knock‐in alleles (Fig 2A, and Appendix Fig S2A and B). We furthermore inserted a stop codon together with an IRES‐luciferase indicator gene into Pax5 exon 4 to create the Pax5 Prd allele expressing only the DNA‐binding paired domain (Prd) of Pax5 (Fig 2A and Appendix Fig S2C). Importantly, all three alleles were designed to express their Pax5 fusion or Prd protein under the control of the endogenous Pax5 promoter and B‐cell‐specific enhancer (Decker et al, 2009) similar to human PAX5 translocations. Immunoblot analysis of nuclear extracts with a Pax5 paired domain‐specific antibody indeed revealed that the Pax5 Etv6/+, Pax5 Foxp1/+, and Pax5 Prd/+ pro‐B cells expressed the Pax5‐Etv6, Pax5‐Foxp1, or Prd protein at the same level as wild‐type Pax5 (Fig 2B).
Figure 2. Impaired B‐cell development in Pax5 Etv6/+ and Pax5 Foxp1/+ mice.
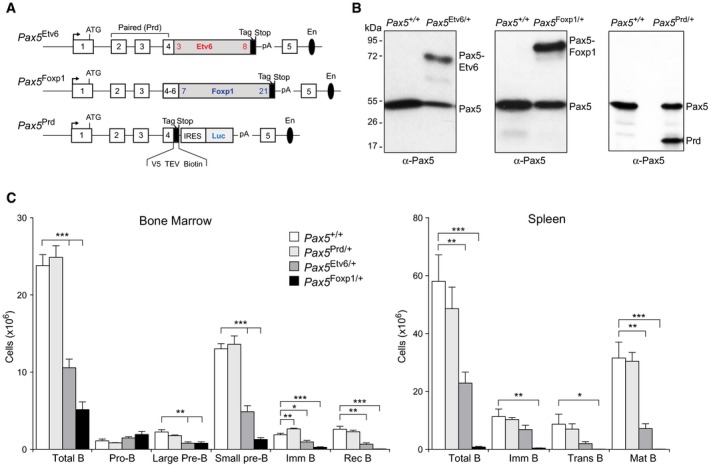
- Schematic diagram of the Pax5 Etv6, Pax5 Foxp1, and Pax5 Prd alleles. Human cDNA sequences, starting in exon 4 and encoding the remaining PAX5‐ETV6 or PAX5‐FOXP1 protein, were inserted in frame into exon 4 of the mouse Pax5 locus to generate the Pax5 Etv6 and Pax5 Foxp1 knock‐in alleles (Appendix Fig S2A and B). The first and last exons encoding the human ETV6 and FOXP1 cDNA are indicated. Notably, the human and mouse Pax5 protein sequences encoded from exon 1 to exon 6 contain only one amino acid substitution (human Ser13 to mouse Ile13), which is present upstream of the paired domain (first functional domain of Pax5) in the very N‐terminal sequence encoded by exon 1 (Adams et al, 1992). The Pax5 Prd allele was generated by insertion of a stop codon together with an IRES‐luciferase indicator gene into Pax5 exon 4 (Appendix Fig S2C). The C‐terminal tag sequence (in black) contains an epitope for the V5 antibody, two cleavage sites for the TEV protease, and a biotin acceptor sequence (Biotin). A black oval denotes the B‐cell‐specific enhancer (En) in intron 5 (Decker et al, 2009).
- Expression of Pax5 proteins in Pax5 +/+, Pax5 Etv6/+, Pax5 Foxp1/+, and Pax5 Prd/+ pro‐B cells. Nuclear extracts prepared from short‐term cultured pro‐B cells of the indicated genotypes were analyzed by immunoblotting with a Pax5 antibody recognizing the N‐terminal Prd domain. The size of marker proteins is indicated in kilodaltons (kDa) to the left. Weak bands represent potential degradation products.
- B‐cell development in Pax5 +/+, Pax5 Prd/+, Pax5 Etv6/+, and Pax5 Foxp1/+ mice. Absolute cell numbers were determined for the indicated cell types by flow cytometric analysis of the bone marrow and spleen from 6‐week‐old mice of the indicated genotypes (n = 5 per genotype). Data are represented as average cell number with SEM and were analyzed by the Student's t‐test; *P < 0.05, **P < 0.01, ***P < 0.001. For abbreviations, see Fig 1A.
Source data are available online for this figure.
Notably, B‐lymphopoiesis was normal in the bone marrow and spleen of Pax5 Prd/+ mice (Fig 2C) similar to Pax5 +/− mice (Fig 1). In marked contrast, B‐cell development was strongly impaired in Pax5 Etv6/+ and Pax5 Foxp1/+ mice (Fig 2C). Other hematopoietic cell types were, however, present in normal numbers in the bone marrow and thymus of Pax5 Etv6/+ and Pax5 Foxp1/+ mice (Appendix Fig S2G), which is consistent with the B‐cell‐specific expression of both knock‐in alleles under the control of the Pax5 locus (Appendix Fig S2A and B). Splenic B‐cell subsets were almost completely absent in Pax5 Foxp1/+ mice, while transitional and mature B cells were strongly diminished in the spleen of Pax5 Etv6/+ mice (Fig 2C). In the bone marrow, small pre‐B, immature, recirculating, and total B cells were strongly reduced in both knock‐in mice (Fig 2C). Moreover, the large pre‐B cells expressing the pre‐BCR were also decreased in Pax5 Etv6/+ and Pax5 Foxp1/+ mice, whereas pro‐B cells were moderately increased. Together, these data demonstrate that both Pax5 fusion proteins arrest B‐cell development at the transition from pro‐B to pre‐B cells. This B‐cell developmental defect is furthermore caused by the fusion partner rather than solely by the Pax5 paired domain.
Gene regulation by Pax5‐Etv6 and Pax5‐Foxp1 in pro‐B cells
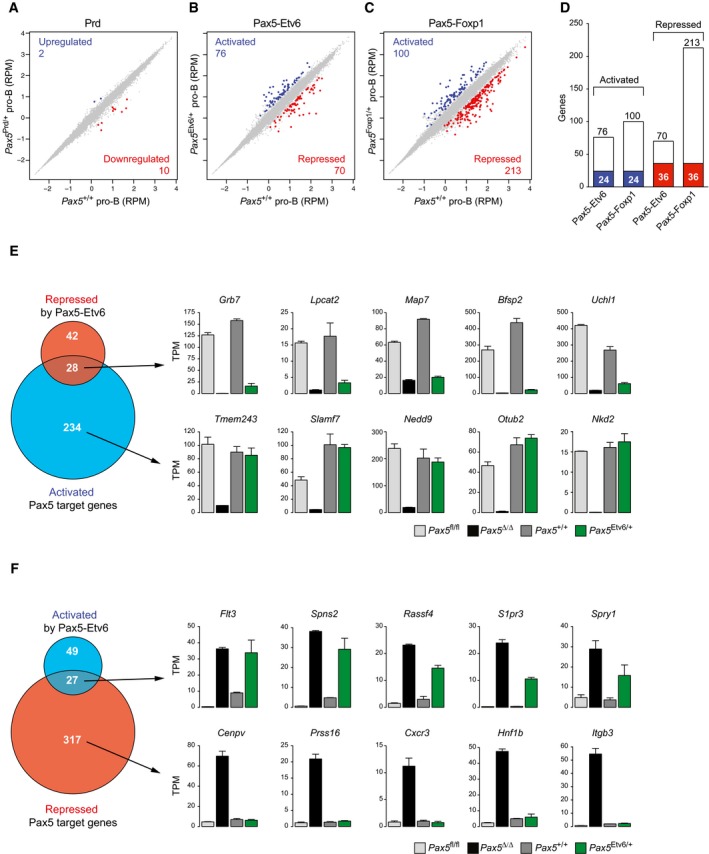
To gain insight into the function of Pax5‐Etv6 and Pax5‐Foxp1 in early B‐cell development, we compared sorted Pax5 +/+ pro‐B cells with Pax5 Prd/+, Pax5 Etv6/+, or Pax5 Foxp1/+ pro‐B cells (Appendix Fig S3A) by RNA‐seq. Differentially expressed genes were identified in all RNA‐seq comparisons, if they revealed an expression difference of > threefold, an adjusted P‐value of < 0.05 and a TPM value of > 5 in one of the two cell populations. Consistent with normal B‐cell development in Pax5 Prd/+ mice, only two genes were upregulated and 10 genes were downregulated in Pax5 Prd/+ pro‐B cells (Fig 3A and Appendix Fig S1B). Moreover, eight of these genes were similarly downregulated also in Pax5 +/− pro‐B cells (Appendix Fig S1A), indicating that the Pax5 Prd allele behaves like a Pax5 null allele. In contrast, the Pax5‐Etv6 protein activated 76 genes and repressed 70 genes in Pax5 Etv6/+ pro‐B cells (Fig 3B and Table EV1). Notably, Pax5‐Foxp1 activated a similar number of genes (100), but repressed three times more genes (213) compared to Pax5‐Etv6 (Fig 3C and Table EV1), which could explain the stronger B‐cell developmental defect observed in Pax5 Foxp1/+ mice (Fig 2C). The overlap of activated (24) or repressed (36) genes was relatively small for Pax5‐Etv6 and Pax5‐Foxp1 (Fig 3D), indicating that the two fusion proteins predominantly regulated different gene sets.
Figure 3. Gene regulation by Pax5‐Etv6 and Pax5‐Foxp1 in pro‐B cells.
-
A–CScatter plot of gene expression differences between ex vivo sorted Pax5 +/+ and Pax5 Prd/+ (A), Pax5 Etv6/+ (B), or Pax5 Foxp1/+ (C) pro‐B cells, based on four (Pax5 Etv6/+) or two (other genotypes) independent RNA‐seq experiments. The normalized expression data of individual genes (indicated by dots) were plotted as RPM values. Genes with an expression difference of > threefold, an adjusted P‐value of < 0.05 (DESeq2), and a TPM value of > 5 (in one of the two cell types) are colored in blue or red, corresponding to activation (upregulation) or repression (downregulation) by Prd, Pax5‐Etv6, or Pax5‐Foxp1, respectively.
-
DOverlap of Pax5‐Etv6‐ and Pax5‐Foxp1‐regulated genes in pro‐B cells. Commonly activated or repressed genes are indicated by blue and red bars, respectively.
-
EVenn diagram indicating the overlap between activated Pax5 target genes (Revilla‐i‐Domingo et al, 2012) and Pax5‐Etv6‐repressed genes in pro‐B cells. The expression of genes indicated to the right was determined by RNA‐seq of in vitro cultured Pax5 fl/fl (light gray) and Vav‐Cre Pax5 fl/fl (Pax5∆/∆, black) pro‐B cells as well as ex vivo sorted Pax5 +/+ (dark gray) and Pax5 Etv6/+ (green) pro‐B cells. Gene expression is shown as normalized expression value (TPM) with SEM, based on at least two independent RNA‐seq experiments for each cell type. Notably, six activated Pax5 target genes, which were apparently “repressed” by Pax5‐Etv6, showed decreased expression already in response to a twofold reduction in full‐length Pax5 expression in Pax5 +/− pro‐B cells (Appendix Fig S1A).
-
FVenn diagram indicating the overlap between repressed Pax5 target genes and Pax5‐Etv6‐activated genes in pro‐B cells, as described in (E).
Pax5 fusion proteins are thought to act as dominant‐negative proteins to interfere with the wild‐type function of Pax5 in B cells (Bousquet et al, 2007; Mullighan et al, 2007; Kurahashi et al, 2011; Kawamata et al, 2012). To test this hypothesis, we determined the overlap of the Pax5‐Etv6‐repressed genes with the previously identified activated Pax5 target genes in pro‐B cells (Revilla‐i‐Domingo et al, 2012). This analysis revealed that only 28 activated Pax5 target genes were repressed by Pax5‐Etv6 in pro‐B cells, as exemplified by the Bfsp2, Grb7, Lpcat2, Map7, and Uchl1 genes (Fig 3E). The vast majority (234) of the activated Pax5 target genes was, however, not affected by Pax5‐Etv6, as shown by the normal expression of Nedd9, Nkd2, Otub2, Slamf7, and Tmem243 in wild‐type and Pax5 Etv6/+ pro‐B cells (Fig 3E). Likewise, only 27 of the repressed Pax5 target genes (Revilla‐i‐Domingo et al, 2012) were activated by Pax5‐Etv6 in pro‐B cells, as indicated by Flt3, Rassf4, S1pr3, Spns2, and Spry1 (Fig 3F). By contrast, 317 repressed Pax5 target genes were not activated in Pax5 Etv6/+ pro‐B cells, as illustrated by Cenpv, Cxcr3, Hnf1b, Itgb3, and Prss16 (Fig 3F).
A similar situation was observed for Pax5‐Foxp1, which repressed 54 of all 262 activated Pax5 target genes and activated only 21 of all 344 repressed Pax5 target genes in Pax5 Foxp1/+ pro‐B cells (Appendix Fig S3C and D). Notably, 15 activated Pax5 target genes, exemplified by Grb7, Lpcat2, and Map7, were similarly repressed by Pax5‐Etv6 and Pax5‐Foxp1, whereas other activated Pax5 target genes were repressed only by Pax5‐Etv6 (Bfsp2, Uchl1) or Pax5‐Foxp1 (Otub2, Nkd2) in pro‐B cells (Fig 3E and Appendix Fig S3C). Likewise, 11 repressed Pax5 target genes were similarly activated by Pax5‐Etv6 and Pax5‐Foxp1, as shown for Flt3 and Spns2, whereas other repressed Pax5 target genes were only activated by Pax5‐Etv6 (Rassf4, S1pr3, Spry1) or Pax5‐Foxp1 (Cybb, Gpr97, Sema6d) in pro‐B cells (Fig 3F and Appendix Fig S3D). In summary, these data demonstrate that Pax5‐Etv6 and Pax5‐Foxp1 largely regulate different genes in pro‐B cells and do not generally function as dominant‐negative proteins to antagonize the normal function of Pax5.
Pax5‐Etv6 binds to active promoters and enhancers
To investigate the genome‐wide binding of the Pax5‐Etv6 and Prd proteins in pro‐B cells, we additionally inserted a C‐terminal biotin acceptor sequence at the last codon of these proteins in the respective knock‐in allele, thus allowing discrimination of the tagged protein from the co‐expressed full‐length Pax5 protein (Appendix Fig S2A–C). Notably, the Escherichia coli biotin ligase BirA efficiently biotinylated the Pax5‐Etv6 and Prd proteins in Pax5 Etv6/+ Rosa26 BirA/+ and Pax5 Prd/+ Rosa26 BirA/+ pro‐B cells, which facilitated streptavidin‐mediated pull‐down of the two proteins in contrast to the co‐expressed Pax5 protein (Appendix Fig S2E and F). The Pax5‐Foxp1 protein was, however, not biotinylated in Pax5 Foxp1/+ Rosa26 BirA/+ pro‐B cells (data not shown), possibly because the C‐terminal tag sequences may be located inside of a Pax5‐Foxp1 protein complex, thus preventing access for the ligase BirA. In vitro cultured Pax5 Etv6/+ Rosa26 BirA/+ and Pax5 Prd/+ Rosa26 BirA/+ pro‐B cells were analyzed by streptavidin‐mediated chromatin precipitation coupled with deep sequencing (Bio‐ChIP‐seq) (Revilla‐i‐Domingo et al, 2012). Peak calling with a stringent P‐value of < 10−10 identified 30,604 Pax5‐Etv6 peaks and 12,768 Prd peaks compared to the 24,947 Pax5 peaks that were previously identified by Bio‐ChIP‐seq of Pax5 Bio/Bio pro‐B cells (Revilla‐i‐Domingo et al, 2012). Multiple overlap analysis revealed distinct binding patterns for the different Pax5 proteins, as illustrated by a Venn diagram, heat maps, and density diagrams (Fig 4A and B) as well as by protein binding at individual target genes (Fig 4D). Whereas about half of all Pax5‐Etv6 peaks (16,537; sectors a, b) were also bound by Pax5, there was a relatively large fraction of unique Pax5‐Etv6 peaks (12,200; sector g) and unique Pax5 peaks (5,041; sector f). This binding pattern was confirmed by analysis of the 300 top‐ranked peaks with the de novo motif‐discovery program MEME‐ChIP (Machanick & Bailey, 2011), which identified only the Ets motif in the unique Pax5‐Etv6 peaks (sector g) and only the Pax5 motif in the unique Pax5 peaks (sector f) in contrast to the presence of both motifs in the common binding sites of Pax5‐Etv6 and Pax5 (sectors a, b) (data not shown). Notably, the Prd protein mainly bound to a subset of the Pax5 peaks (8,047; sectors a, d), indicating that Prd failed to compete with full‐length Pax5 for binding to a large class of Pax5‐binding sites (16,184; sectors b, f) in Pax5 Prd/+ pro‐B cells.
Figure 4. Genome‐wide Pax5‐Etv6 binding identifies regulated Pax5‐Etv6 target genes.
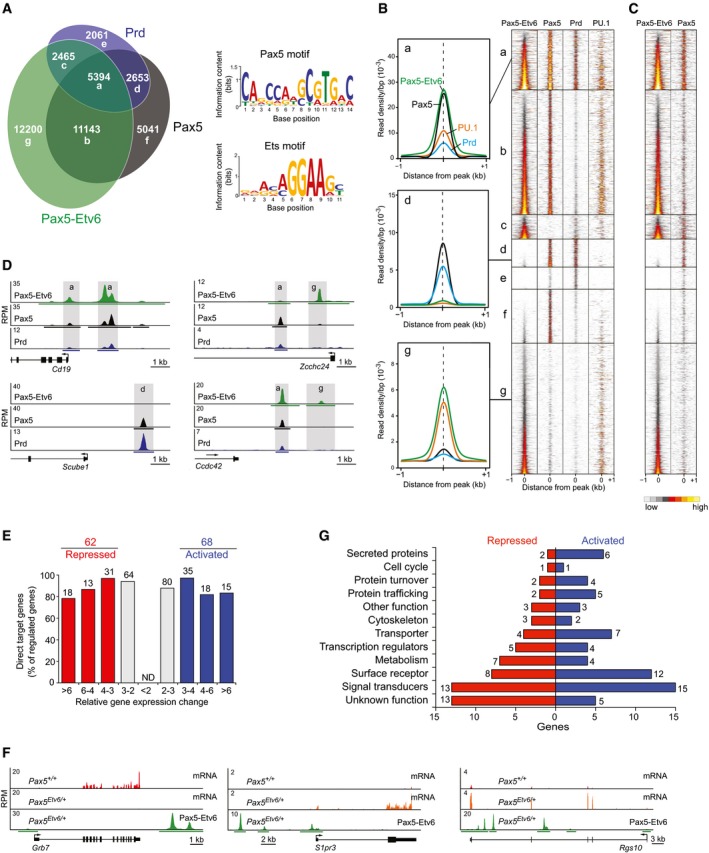
- Multiple overlap of Pax5‐Etv6, Pax5, and Prd binding in pro‐B cells. Peaks, which were identified by Bio‐ChIP‐seq of Pax5 Bio/Bio, Pax5 Etv6/+ Rosa26 BirA/+, and Pax5 Prd/+ Rosa26 BirA/+ pro‐B cells, were called by the MACS program with a P‐value of < 10−10, and their number is indicated in each sector of the Venn diagram. De novo motif‐discovery analysis of the common peaks (sector a) identified the indicated Ets and Pax5 motifs with E‐values (MEME‐ChIP) of 1.3 × 10−35 and 4.4 × 10−31, respectively.
- Density heat maps of all Pax5‐Etv6, Pax5, and Prd peaks. An additional heat map shows binding of the Ets protein PU.1 (Spi1), which was determined by Bio‐ChIP‐seq analysis of Spi1 Bio/Bio Rosa26 BirA/BirA pro‐B cells (Schwickert et al, 2014). The heat maps are shown for a region extending from −1 kb to +1 kb relative to the peak summit and were sorted according to the increasing density of Pax5‐Etv6 binding. To the left, the average binding densities of Pax5‐Etv6 (green), Pax5 (black), Prd (blue), and PU.1 (orange) are shown for the binding sites present in sectors a, d, and g, respectively.
- Binding of Pax5‐Etv6 and full‐length Pax5 in Pax5 Etv6/+ pro‐B cells. Binding of full‐length Pax5 was determined by ChIP‐seq with an antibody recognizing the central and C‐terminal Pax5 sequences. A density scale from low (gray) to high (yellow) is shown.
- Binding of Pax5‐Etv6, Pax5, and Prd at the indicated genes in pro‐B cells. Horizontal bars below the ChIP‐seq tracks indicate binding regions identified by MACS peak calling. The exon–intron structure and a scale bar in kilobases (kb) are shown.
- Identification of activated and repressed Pax5‐Etv6 target genes in pro‐B cells. The number and percentage of Pax5‐Etv6 target genes are shown for the indicated fold gene expression differences between experimental Pax5 Etv6/+ and control Pax5 +/+ pro‐B cells, as defined in Fig 3B. ND, not determined.
- Expression of the repressed (Grb7) and activated (S1pr3, Rgs10) Pax5‐Etv6 target genes in ex vivo sorted Pax5 +/+ and Pax5 Etv6/+ pro‐B cells, as shown by RNA‐seq analysis (first two tracks). Pax5‐Etv6 binding was determined in Pax5 Etv6/+ Rosa26 BirA/+ pro‐B cells by Bio‐ChIP‐seq (last track). For further description, see (D).
- Functional classification and quantification (numbers) of the proteins encoded by activated and repressed Pax5‐Etv6 target genes in pro‐B cells.
We next addressed the question whether the full‐length Pax5 protein, which is expressed from the wild‐type allele in Pax5 Etv6/+ pro‐B cells, is still able to interact with its binding sites in the presence of Pax5‐Etv6. For this purpose, we performed ChIP‐seq analysis of full‐length Pax5 in Pax5 Etv6/+ pro‐B cells by using an anti‐Pax5 antibody recognizing the central and C‐terminal Pax5 sequences that are absent in the Pax5‐Etv6 protein. As shown in Fig 4C, Pax5 could bind to the same sites (sectors a, b) as Pax5‐Etv6 either on the second allele in the same Pax5 Etv6/+ pro‐B cells or in separate pro‐B cells, indicating that full‐length Pax5, in contrast to the Prd protein, can efficiently compete with Pax5‐Etv6 for binding sites.
We subsequently analyzed the chromatin state at the Pax5‐Etv6, Pax5, and Prd peaks based on the chromatin landscape of pro‐B cells that we previously determined by profiling of active and repressive histone marks and mapping of DNase I hypersensitive (DHS) sites (Revilla‐i‐Domingo et al, 2012). As shown in Appendix Fig S4A, Pax5‐Etv6 primarily bound to sites of open chromatin (DHS) containing active histone modifications (H3K4me2, H3K9ac, H3K27ac; sectors a–c, g), whereas these active marks as well as the repressive H3K27me3 modification were largely absent at unique Pax5‐ and Prd‐binding sites (sectors d–f) in pro‐B cells (Appendix Fig S4A). Taking advantage of the inverse correlation of H3K4me1 and H3K4me3 abundance at promoters and enhancers (Heintzman et al, 2007), we could demonstrate that Pax5‐Etv6 predominantly bound to promoters (H3K4me3high, H3K4me1low) and enhancers (H3K4me3low, H3K4me1high) that were embedded in active chromatin (Appendix Fig S4A).
Pax5‐Etv6 contains three protein domains that contribute to the complex DNA‐binding behavior of this fusion protein. In addition to the N‐terminal paired domain, the C‐terminal Ets domain functions as a second DNA‐binding region (Cazzaniga et al, 2001), whereas the more N‐terminally located pointed domain of Etv6 mediates homodimerization of Pax5‐Etv6 or heterodimerization with the endogenously expressed Etv6 protein (Fortschegger et al, 2014). Consistent with this notion, the common binding sites of Pax5‐Etv6 and Pax5 (sectors a, b) contained the Ets‐binding motif of Etv6 as well as the paired domain‐binding motif of Pax5 (Fig 4A), both of which resemble their published consensus recognition sequence (Wei et al, 2010; Revilla‐i‐Domingo et al, 2012). In agreement with the presence of the Ets motif, the Ets protein PU.1 bound to about half of all Pax5‐Etv6 peaks in pro‐B cells, indicating that these peaks contain functional Ets‐binding sites (Fig 4B and Appendix Fig S4B). Moreover, the Pax5‐Etv6 peaks had a broader width than the Pax5 peaks (Appendix Fig S4C), further demonstrating that the Ets domain of the fusion protein contributes to DNA binding. We next investigated whether the recognition sequence of the paired or the Ets domain is in the center of the Pax5‐Etv6 peaks. For this, we plotted the position of maximal binding density for each Pax5‐Etv6 peak (sectors a, b) relative to the Pax5 motif, which was identified by scanning with the Pax5 consensus motif shown in Fig 4A. This analysis indicated that the maximal binding densities of all Pax5‐Etv6 peaks were centered at the Pax5 motif (Appendix Fig S4D). In contrast, if the plot of maximal binding density was centered at the Ets motif located closest to the Pax5 motif, the binding densities showed a dip in the center and peaked on both sides of the Ets motif due to the increased density at the adjacent Pax5‐binding site (Appendix Fig S4E). We conclude therefore that the paired domain binds to the center of the peaks, while the Ets domain contributes to DNA‐binding by interacting with adjacent sequences.
Function of proteins encoded by regulated Pax5‐Etv6 target genes
Peak‐to‐gene assignment of the 30,604 Pax5‐Etv6 peaks identified 13,665 genes, which were bound by Pax5‐Etv6 in pro‐B cells. By determining the overlap between these Pax5‐Etv6‐bound genes (Fig 4A and B) and the Pax5‐Etv6‐regulated genes (Fig 3B), we identified only a relative small number of 68 potentially directly activated and 62 potentially directly repressed Pax5‐Etv6 target genes that were regulated more than threefold by Pax5‐Etv6 in pro‐B cells (Fig 4E and Table EV2). The RNA expression and Pax5‐Etv6 binding are shown for S1pr3 and Rgs10 or Grb7 as representative activated or repressed target genes, respectively (Fig 4F). The regulated Pax5‐Etv6 target genes code for proteins with distinct functions (Fig 4G and Appendix Fig S4F). Notably, we found 14 activated and eight repressed Pax5‐Etv6 target genes, which code for secreted proteins, cell surface receptors, signal transducers, and cytoskeletal proteins involved in cell adhesion and migration (Appendix Fig S4G). Consistent with this finding, we could demonstrate by performing Transwell migration assays that Pax5 Etv6/+ and Pax5 Foxp1/+ pro‐B cells migrated less efficiently toward a source of CXCL12 (SDF‐1α) than wild‐type pro‐B cells (Appendix Fig S4H), although Cxcr4 (encoding the receptor for CXCL12) was similarly expressed in pro‐B cells of all three genotypes (data not shown). In summary, the identification of relatively few regulated target genes defined a role of Pax5‐Etv6 in controlling cell migration and possibly adhesion, which could contribute to the oncogenic function of this Pax5 fusion protein in leukemic cells.
Loss of the Cdkn2a/b tumor suppressor locus cooperates with Pax5‐Etv6 in B‐ALL development
The majority of all Pax5 Etv6/+ and Pax5 Foxp1/+ mice were still alive after 1 year, and the three mice of each strain that died during the 1‐year observation period failed to develop leukemia (Fig 5A and Appendix Fig S5A), indicating that the expression of Pax5‐Etv6 or Pax5‐Foxp1 alone is not sufficient to promote tumor formation. As constitutive STAT5 activation was shown to cooperate with Pax5 heterozygosity to induce B‐cell leukemia in the mouse (Heltemes‐Harris et al, 2011), we next investigated whether the expression of constitutively active STAT5 would accelerate leukemia formation in Pax5 Etv6/+ and Pax5 Foxp1/+ mice compared to Pax5 +/− mice. To this end, we used the EμSRα‐Stat5a(S711F) transgene (Joliot et al, 2006), which is expressed under the control of the Eμ enhancer and SV40 early promoter, contains the activating mutation S711F, and is subsequently referred to as constitutively active (ca) Stat5a (caStat5a). Pax5 Etv6/+ and Pax5 Foxp1/+ mice expressing the caStat5a transgene readily developed monoclonal B‐cell leukemias that expressed early B‐cell markers (IL‐7Rα, CD43, CD93) and were able to infiltrate different organs (Appendix Fig S5B–E). Surprisingly, however, the caStat5a Pax5 Etv6/+ and caStat5a Pax5 Foxp1/+ mice died of leukemia with similar kinetics as caStat5a Pax5 +/− mice (Appendix Fig S5A). These data strongly suggest that it is the inactivation of one Pax5 allele rather than the expression of the Pax5 fusion protein, which contributes to leukemogenesis in caStat5a Pax5 Etv6/+ and caStat5a Pax5 Foxp1/+ mice. In other words, caStat5a appears to be an inappropriate cooperating oncogene to reveal the tumorigenic role of Pax5‐Etv6 or Pax5‐Foxp1.
Figure 5. Leukemia development in Cdkn2ab +/− Pax5 Etv6/+ mice.
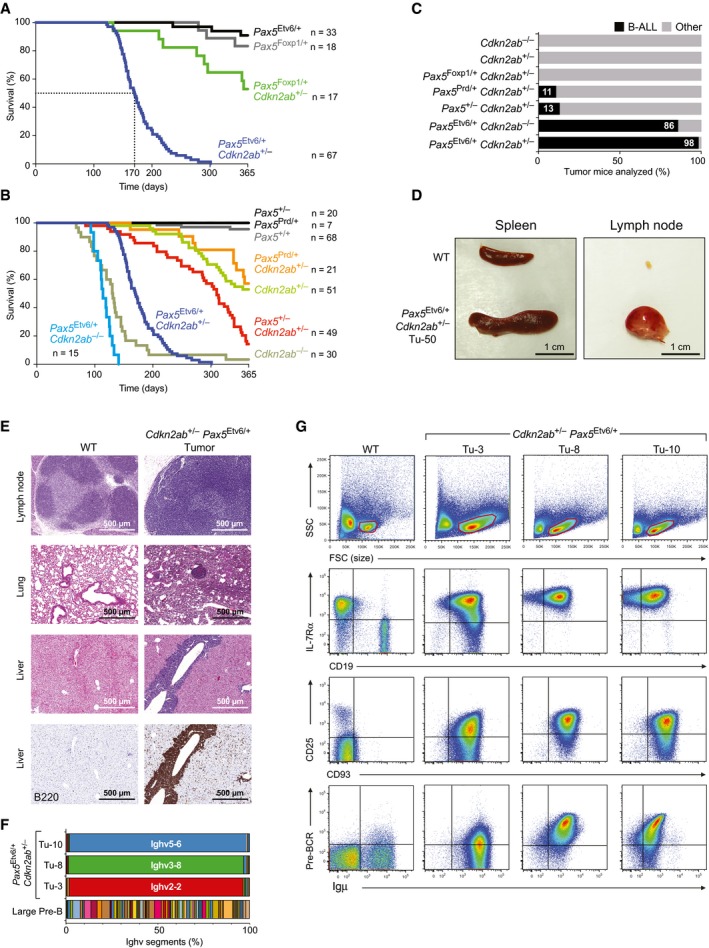
-
A, BKaplan–Meier survival analysis of mice of the indicated genotypes. The number (n) of mice analyzed is shown.
-
CPercentage of B‐cell leukemia (B‐ALL) in mice of the indicated genotypes at the time of analysis of the tumor mice. B‐ALL tumors were identified by the presence of enlarged lymph nodes and/or their characteristic cell surface protein expression that was determined by flow cytometric analysis, as described in (G). Other tumors refer to non‐B‐cell tumors, which were not further characterized.
-
DSize comparison of the spleen and lymph nodes from a wild‐type (WT) mouse and Cdkn2ab +/− Pax5 Etv6/+ tumor mouse (Tu‐50).
-
EEosin–hematoxylin‐stained sections of the lymph node, lung, and liver of a WT mouse and Cdkn2ab +/− Pax5 Etv6/+ tumor mice (Tu‐29 lymph node; Tu‐50 lung and liver). Immunohistochemical staining of B cells with an anti‐B220 antibody on adjacent liver sections from the wild‐type and tumor mice is shown in the bottom row. The infiltration pattern of all tumors is shown in Table EV3.
-
FMonoclonality of three Cdkn2ab +/− Pax5 Etv6/+ tumors. The expression of each VH gene across the Igh locus was determined as TPM value by RNA‐seq of three Cdkn2ab +/− Pax5 Etv6/+ tumors (Tu‐3, 8, 10) and is displayed as a percentage of the total expression of all VH genes. Ighv2‐2 = VHQ52.2.4; Ighv3‐8 = VH36‐60.8.74; Ighv5‐6 = VH7183.7.10.
-
GFlow cytometric analysis of lymph node cells from a WT mouse and three Cdkn2ab +/− Pax5 Etv6/+ tumor mice (Tu‐3, 8, 10). Cells within the forward and side scatter (FCS/SSC) gates (red) were analyzed.
In human B‐ALL, PAX5‐ETV6 is generated by the formation of a dic(9;12)(p13;p13) chromosome, which results in the loss of the reciprocal translocation product, thus eliminating one allele of the CDKN2A/B tumor suppressor locus (Cazzaniga et al, 2001; Strehl et al, 2003) (Appendix Fig S5F). Hence, heterozygous loss of this tumor suppressor locus could be the relevant mutation cooperating with Pax5‐Etv6 expression in leukemia induction. The Cdkn2a/b locus encodes three cell cycle inhibitory proteins: p15Ink4b (encoded by Cdkn2b) as well as p16Ink4a and p19Arf (both encoded by Cdkn2a) (Gil & Peters, 2006). We thus used the Cdkn2ab − allele, containing a deletion of all three open reading frames (Krimpenfort et al, 2007), to generate compound heterozygous Cdkn2ab +/− Pax5 Etv6/+, Cdkn2ab +/− Pax5 Foxp1/+, Cdkn2ab +/− Pax5 Prd/+, and Cdkn2ab +/− Pax5 +/− mice. Whereas 53% of the Cdkn2ab +/− Pax5 Foxp1/+ mice were still alive after 1 year, all Cdkn2ab +/− Pax5 Etv6/+ mice died with a median time of 170 days (Fig 5A). The survival curve of Cdkn2ab +/− Pax5 Foxp1/+ and Cdkn2ab +/− Pax5 Prd/+ mice was similar to that of the Cdkn2ab +/− mice, indicating that the delayed tumor development in these mice is caused by the loss of one Cdkn2ab allele rather than the mutant Pax5 allele (Fig 5A and B). Moreover, the survival of Cdkn2ab +/− Pax5 +/− mice was also significantly prolonged compared to that of Cdkn2ab +/− Pax5 Etv6/+ mice. Flow cytometric analysis of lymphoid organs from tumor mice revealed the presence of B‐cell leukemia in 98% of all Cdkn2ab +/− Pax5 Etv6/+ mice in contrast to 11% of the Cdkn2ab +/− Pax5 Prd/+ mice and 13% of the Cdkn2ab +/− Pax5 +/− mice (Fig 5C). B‐cell leukemia was, however, not detected in Cdkn2ab +/− Pax5 Foxp1/+ and Cdkn2ab +/− mice, indicating that heterozygous Cdkn2ab loss does not cooperate with Pax5‐Foxp1 expression in leukemia induction. Notably, tumor development in homozygous Cdkn2ab −/− Pax5 Etv6/+ mice was significantly accelerated compared to heterozygous Cdkn2ab +/− Pax5 Etv6/+ mice, but was equally fast as in Cdkn2ab −/− mice (Fig 5B). Importantly, however, the tumor spectrum of Cdkn2ab −/− and Cdkn2ab −/− Pax5 Etv6/+ mice was very different, as only the Cdkn2ab −/− Pax5 Etv6/+ mice (86%) developed B‐cell leukemia (Fig 5C), indicating that Pax5‐Etv6 expression was able to drive B‐ALL development in Cdkn2ab −/− mice. Together, these data demonstrate that the loss of Cdkn2ab is a relevant cooperating mutation to induce B‐cell leukemia in Pax5 Etv6/+ mice.
Characterization of the Cdkn2ab +/− Pax5 Etv6/+ tumor mice revealed that the spleen and lymph nodes were enlarged (Fig 5D) and that the tumor cells infiltrated many different organs (Fig 5E and Table EV3) and gave rise to overt leukemia within 4 weeks after transplantation in syngeneic mice (data not shown). The tumor cells isolated from the lymph node were furthermore of monoclonal origin, as they predominantly expressed only one VH gene of the Igh locus, as shown by RNA‐seq (Fig 5F). Flow cytometric analysis revealed large blastic tumor cells compared to the small quiescent B cells of wild‐type lymph nodes (Fig 5G). Abundant blast cells were also detected in the peripheral blood of Cdkn2ab +/− Pax5 Etv6/+ tumor mice (Appendix Fig S5G). With the exception of B220 expression, the cell surface phenotype of Cdkn2ab +/− Pax5 Etv6/+ tumors was very homogeneous, which is indicated by the percentage of all analyzed tumors expressing the following cell surface markers: CD19+ (100%), B220+ (52%), IL‐7Rα+ (100%), CD43+ (100%), CD93+ (100%), Igμ+ (100%), CD2+ (100%), CD25+ (93%), Flt3− (100%), c‐Kit− (96%), CD23− (100%), CD21− (90%), IgD− (96%) (Fig 5G; data not shown). Notably, the Igμ protein was often expressed as part of the pre‐BCR on the tumor cells (Fig 5G). Collectively, the observed cell surface phenotype strongly suggests that the tumor cells are arrested at the large pre‐BCR+ cell stage of early B‐cell development. We therefore conclude that the Cdkn2ab +/− Pax5 Etv6/+ tumors have the characteristic features of B‐ALL and thus appear to be a faithful model for human PAX5‐ETV6+ B‐ALL.
Molecular functions of the Pax5‐Etv6 oncoprotein in mouse B‐ALL formation
To investigate the transcriptional role of Pax5‐Etv6 in leukemia development, we determined the transcriptome of three Cdkn2ab +/− Pax5 Etv6/+ tumors by RNA‐seq. Notably, the wild‐type Cdkn2a and Cdkn2b alleles were expressed in one tumor (Tu‐8), giving rise to p16Ink4a and low p15Ink4b mRNA expression, respectively (Fig 6A). Unexpectedly, however, no sequence reads could be detected at exons 1 and 2 of Cdkn2a and Cdkn2b in two tumors (Tu‐3 and Tu‐10), indicating that the respective wild‐type sequences may have been deleted in these leukemias (Fig 6A). Analysis of tumor DNA by PCR genotyping indeed detected only the deleted Cdkn2ab − allele in Tu‐3 and Tu‐10 in contrast to the presence of both wild‐type and deleted Cdkn2ab alleles in Tu‐8 (Fig 6B). Loss of the wild‐type Cdkn2ab allele was furthermore tumor‐specific, as shown by PCR comparison of tail and tumor DNA from three different mice (Fig 6C). Moreover, the wild‐type Cdkn2ab allele was lost in seven (78%) of nine tumors analyzed (Fig 6B and C). Together, these data demonstrated a frequent loss of heterozygosity in Cdkn2ab +/− Pax5 Etv6/+ tumors, which revealed a strong selection pressure to delete the wild‐type Cdkn2a/b genes during leukemia development.
Figure 6. Identification of regulated Pax5‐Etv6 target genes in mouse B‐ALL .
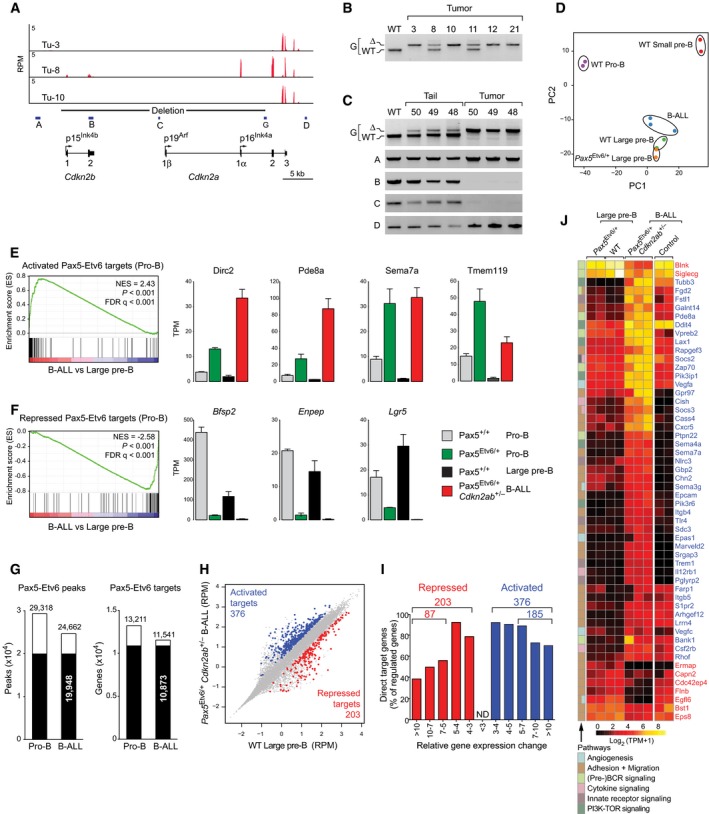
-
AExpression of Cdkn2a and Cdkn2b in three Cdkn2ab +/− Pax5 Etv6/+ tumors (Tu‐3, 8, 10), as revealed by RNA‐seq. The exon–intron structure of both genes is indicated together with the promoters giving rise to mRNA encoding p15Ink4b, p16Ink4a, and p19Arf. The Cre‐mediated deletion of the Cdkn2ab − allele is shown together with the positions of the PCR primer pairs A–D and the genotyping PCR primer pair G.
-
BGenotyping PCR analysis of DNA isolated from the indicated Cdkn2ab +/− Pax5 Etv6/+ tumors. The PCR fragments corresponding to the deleted (∆) and wild‐type (WT) Cdkn2ab alleles are indicated to the left of the gel image.
-
CPCR analysis of DNA isolated from the tail and tumor of the indicated Cdkn2ab +/− Pax5 Etv6/+ tumor mice by using the primer pairs indicated to the left of the gel image.
-
DPrincipal component analysis based on two or three RNA‐seq experiments for each of the indicated cell types and Cdkn2ab +/− Pax5 Etv6/+ B‐ALL tumors.
-
E, FGSEA analysis of the 68 activated (E) or 62 repressed (F) Pax5‐Etv6 target genes identified in pro‐B cells (Fig 4E), as compared to the ranked log2‐fold gene expression changes in Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs versus wild‐type large pre‐B cells (left). The expression of selected genes is shown for the four indicated cell types (right) and is presented as normalized expression value (TPM) with SEM, based on at least two independent RNA‐seq experiments for each cell type. NES, normalized enrichment score; FDR, false discovery rate.
-
GOverlap of Pax5‐Etv6 peaks and Pax5‐Etv6 target genes between Pax5 Etv6/+ Rosa26 BirA/+ pro‐B cells and Cdkn2ab +/− Pax5 Etv6/+ Rosa26 BirA/+ B‐ALL cells (Tu‐43), as determined by Bio‐ChIP‐seq. Peaks were called by MACS with a stringent P‐value of < 10−10.
-
HScatter plot of gene expression differences between Cdkn2ab +/− Pax5 Etv6/+ B‐ALL tumors and wild‐type large pre‐B cells, based on three (B‐ALL) and two (pre‐B) independent RNA‐seq experiments. The normalized expression data of individual genes (indicated by dots) were plotted as RPM values. Pax5‐Etv6‐bound genes with an expression difference of > threefold, an adjusted P‐value of < 0.05 (DESeq2), and a TPM value of > 5 (in one of the two cell types) are colored in blue or red, corresponding to activated or repressed Pax5‐Etv6 target genes, respectively.
-
IIdentification of activated and repressed Pax5‐Etv6 target genes in Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs. The number and percentage of Pax5‐Etv6 target genes are shown for the indicated fold gene expression differences between Cdkn2ab +/− Pax5 Etv6/+ tumors and wild‐type large pre‐B cells.
-
JHeat map displaying the differential expression of activated (blue) and repressed (red) Pax5‐Etv6 target genes in wild‐type large pre‐B cells (n = 2) and Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (n = 3), which were analyzed by hierarchical clustering. The 54 target genes shown were selected for an expression difference of > fivefold and for encoding a protein implicated in one of the indicated pathways. The expression of the indicated genes is also shown for Pax5 Etv6/+ large pre‐B cells (n = 2) and control B‐ALLs (n = 2) driven by Myc, NrasG12D, and loss of Trp53 (for details, see Appendix Supplementary Methods). The log2 [TPM+1] expression value of each gene is visualized according to the indicated scale. The pathway annotation is shown to the left.
Source data are available online for this figure.
As shown by principal component analysis of RNA‐seq data, the Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs were related most closely to wild‐type large pre‐B cells and only slightly more distantly to Pax5 Etv6/+ large pre‐B cells in marked contrast to pro‐B and small pre‐B cells (Fig 6D), which is consistent with the observed cell surface phenotype of these tumors (Fig 5G). For gene expression analyses, we therefore compared the Cdkn2ab +/− Pax5 Etv6/+ B‐ALL tumors with wild‐type large pre‐B cells. By gene set enrichment analysis (GSEA), we could demonstrate that the activated Pax5‐Etv6 target genes determined in pro‐B cells (Fig 4E) were significantly enriched as upregulated genes in Cdkn2ab +/− Pax5 Etv6/+ B‐ALL tumors (Fig 6E), while the repressed Pax5‐Etv6 target genes (Fig 4E) were significantly enriched as expressed genes in large pre‐B cells (Fig 6F). These GSEA data therefore demonstrate that many regulated Pax5‐Etv6 target genes identified in pro‐B cells were similarly regulated in Cdkn2ab +/− Pax5 Etv6/+ B‐ALL tumors.
To identify novel tumor‐specific Pax5‐Etv6 target genes, we determined the genome‐wide Pax5‐Etv6 binding pattern in Cdkn2ab +/− Pax5 Etv6/+ Rosa26 BirA/+ tumor cells by Bio‐ChIP‐seq, which identified 24,662 Pax5‐Etv6‐binding sites defining 11,541 Pax5‐Etv6 target genes (Fig 6G). Notably, the overlap of Pax5‐Etv6 binding sites and target genes was very high between the Pax5 Etv6/+ pro‐B cells and Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (Fig 6G). In support of this, the Pax5‐Etv6 binding pattern at individual genes was similar in both cell types (Appendix Fig S6A). Of all Pax5‐Etv6‐bound genes, only 376 genes were > threefold upregulated and 203 genes were > threefold downregulated in Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs compared to large pre‐B cells (Fig 6H and I). For functional annotation of these potentially directly regulated Pax5‐Etv6 target genes, we selected the genes with a > fivefold gene expression difference, which resulted in 185 activated and 87 repressed Pax5‐Etv6 target genes identified in Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (Fig 6I, Appendix Fig S6B and Table EV4). These regulated Pax5‐Etv6 target genes coded for proteins of different functions (Appendix Fig S6C). Interestingly, the two most prominent functional classes encoded by activated target genes consisted of 29 cell surface receptors and 43 signal transducers, while eight cell surface proteins and nine signal transducers were encoded by repressed target genes (Appendix Fig S6C and D). Hence, these data suggest that gene regulation by Pax5‐Etv6 likely contributes to altered cell signaling in Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs.
By categorizing the regulated target genes according to pathways, we identified activated (act) and repressed (rep) target genes coding for proteins implicated in the following processes and pathways; adhesion and migration (22 act, 6 rep), (pre‐)BCR signaling (5 act, 2 rep), PI3K‐TOR signaling (6 act), cytokine signaling (5 act), innate receptor signaling (5 act), and angiogenesis (4 act, 1 rep) (Fig 6J). Most of these pathway genes were not only strongly deregulated in Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs compared to large pre‐B cells, but also compared to an alternative murine B‐ALL model driven by Myc, NrasG12D, and loss of Trp53 (Fig 6J), which further supported the notion that these genes are directly regulated by Pax5‐Etv6 in B‐cell leukemia. Notably, these regulated target genes were similarly expressed in the non‐leukemic Pax5 Etv6/+ and wild‐type large pre‐B cells (Fig 6J), indicating that Pax5‐Etv6 regulated these genes only in the oncogenic environment of the Cdkn2ab +/− Pax5 Etv6/+ tumors. In conclusion, we have identified regulated Pax5‐Etv6 target genes with important functions in several signaling pathways that likely contribute to B‐ALL development in Cdkn2ab +/− Pax5 Etv6/+ mice.
Expression of Pax5‐Etv6‐regulated genes in human B‐ALLs
We next investigated whether the activated and repressed Pax5‐Etv6 target genes identified in mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs are similarly regulated in human PAX5‐ETV6+ B‐ALLs. To this end, we analyzed nine human B‐ALL samples by RNA‐seq. The presence of the PAX5‐ETV6 translocation in all nine B‐ALL samples was determined by DNA‐FISH analysis as well as by expression of the PAX5‐ETV6 fusion transcript (Appendix Fig S7A, data not shown). Although the ratio between the full‐length PAX5 and PAX5‐ETV6 fusion transcripts varied for individual B‐ALL samples, the average ratio of all nine human B‐ALLs was close to 1 (48.7%/51.3%; Appendix Fig S7B). A similar average ratio (46.6%/53.4%) was determined for three mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (Appendix Fig S7C), which further validated this mouse tumor as a model for the human disease. Analogous to the gene expression analysis of the mouse B‐ALLs, we compared the RNA‐seq data of the nine human PAX5‐ETV6+ B‐ALL samples with RNA‐seq data of CD34−CD10+CD19+ pre‐B cells, which were isolated from bone marrow samples of healthy donors. As shown by GSEA analysis, a large fraction of the activated Pax5‐Etv6 target genes identified in mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (Fig 6I) were significantly enriched as upregulated genes in the human PAX5‐ETV6+ B‐ALLs compared to human pre‐B cells (Fig 7A and B), whereas no significant enrichment was observed for repressed Pax5‐Etv6 target genes (data not shown).
Figure 7. Expression of Pax5‐Etv6‐regulated genes in human B‐ALLs.
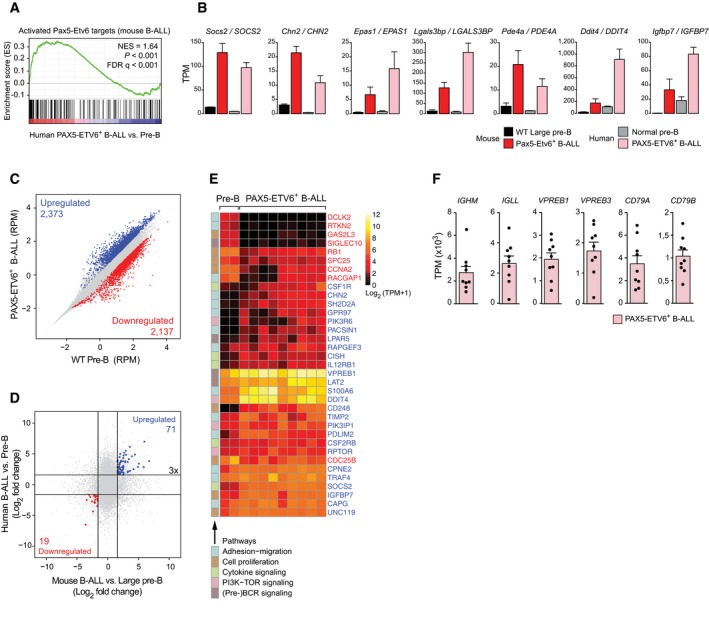
- GSEA analysis of the 185 (> fivefold) activated Pax5‐Etv6 target genes identified in mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (Fig 6I), as compared to the ranked log2‐fold gene expression changes in human PAX5‐ETV6+ B‐ALLs (n = 9) versus CD34−CD10+CD19+ pre‐B cells of healthy donors (n = 2). NES, normalized enrichment score; FDR, false discovery rate.
- The expression of selected genes of the core GSEA signature is shown for mouse Cdkn2ab +/− Pax5 Etv6/+ (Pax5‐Etv6+) B‐ALLs and wild‐type large pre‐B cells as well as for human PAX5‐ETV6+ B‐ALLs and normal pre‐B cells. The gene expression data are presented as normalized expression values (TPM) with SEM, based on two (WT large pre‐B and human normal pre‐B cells), three (Cdkn2ab +/‐ Pax5 Etv6/+ B‐ALL) and nine (human PAX5‐ETV6+ B‐ALL) independent RNA‐seq experiments.
- Scatter plot of gene expression differences between human PAX5‐ETV6+ B‐ALLs and normal human pre‐B cells. The normalized expression data of individual genes (indicated by dots) were plotted as RPM values. Genes with an expression difference of > threefold, an adjusted P‐value of < 0.05 (DESeq2), and a TPM value of > 5 (in one of the two cell types) are colored in blue or red, corresponding to gene upregulation (activation) or downregulation (repression) in the B‐ALL tumors, respectively.
- Expression of the 71 commonly activated (blue) and 19 commonly repressed (red) Pax5‐Etv6 target genes in mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs and human PAX5‐ETV6+ B‐ALLs. The log2‐fold expression change observed between the mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs and wild‐type large pre‐B cells (horizontal axis) as well as between human PAX5‐ETV6+ B‐ALLs and normal pre‐B cells (vertical axis) is plotted for each gene.
- Heat map displaying differentially expressed genes in normal human pre‐B cells (n = 2) and human PAX5‐ETV6+ B‐ALLs (n = 9), which were analyzed by hierarchical clustering. The indicated genes code for proteins implicated in the pathways shown to the left and were identified as activated (26, blue) or repressed (9, red) Pax5‐Etv6 target genes with an expression difference of > threefold in Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (Fig 6I). The log2 [TPM+1] expression value of each gene is visualized according to the indicated scale. SIGLEC10 (human) corresponds to Siglecg (mouse).
- Expression of the genes encoding pre‐BCR components in the nine human PAX5‐ETV6+ B‐ALLs. The expression value of each B‐ALL sample is shown as a dot. The data are presented as average expression value with SEM.
Notably, 2,373 genes were > threefold upregulated and 2,137 genes were > threefold downregulated in the human PAX5‐ETV6+ B‐ALL samples compared to human pre‐B cells (Fig 7C). The considerably higher (fivefold–sixfold) number of differentially expressed genes identified in human PAX5‐ETV6+ B‐ALLs compared to the mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs could have been explained by the fact that we used the quiescent small pre‐B cells as a reference cell type in the human comparison in contrast to the cycling large pre‐B cells in the mouse comparison. However, when we analyzed the expression differences between mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs and wild‐type small pre‐B cells, the number of differentially expressed genes was only 1.7‐fold increased (data not shown). Consequently, the larger number of differentially expressed genes in human PAX5‐ETV6+ B‐ALLs indicates that these tumors differ more substantially from pre‐B cells than the mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs. Of the 376 activated and 203 repressed target genes identified in mouse Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs (Fig 6I), 71 genes were also activated and 19 genes were repressed in human PAX5‐ETV6+ B‐ALLs (Fig 7D, Appendix Fig S7D and Table EV5). Among these genes, 26 activated and nine repressed genes code for proteins involved in pre‐BCR signaling (4), cytokine signaling (5), PI3K‐TOR signaling (4), cell proliferation (8), and adhesion–migration (14) (Fig 7E and Appendix Fig S7E). We next analyzed the expression of the different components of the pre‐BCR in all nine PAX5‐ETV6+ B‐ALLs. As shown in Fig 7F, the mRNAs coding for the extracellular components (IGHM [Igμ], IGLL [λ5], VPREB1, VPREB3) and the signal‐transducing chains (CD79A [Igα], CD79B [Igβ]) of the pre‐BCR were on average highly expressed (> 1,000 TPM) in these B‐ALLs, which therefore likely express the pre‐BCR on their surface. This conclusion was further supported by GSEA analysis, as genes, that are upregulated in human pre‐BCR+ B‐ALLs relative to pre‐BCR− B‐ALLs (Geng et al, 2015), were also significantly enriched as upregulated genes in human PAX5‐ETV6+ B‐ALLs compared to control PAX5‐JAK2+ B‐ALLs, while downregulated genes in pre‐BCR+ B‐ALLs were also downregulated in PAX5‐ETV6+ B‐ALLs (Appendix Fig S7F). In summary, the establishment of a faithful mouse B‐ALL model facilitated the identification of Pax5‐Etv6 target genes, some of which are similarly regulated in human PAX5‐ETV6+ B‐ALLs, are involved in pre‐BCR signaling and may thus contribute to leukemia development in both species.
Discussion
Due to its frequent association with loss‐of‐function mutations, PAX5 is known to function as a haploinsufficient tumor suppressor gene in human B‐ALL (Mullighan et al, 2007). Less is known about the smaller subset of PAX5 mutations that result in the expression of different PAX5 fusion proteins with novel transcriptional activities (Nebral et al, 2009; Coyaud et al, 2010). The function of two PAX5 fusion proteins, PAX5‐ETV6 and PAX5‐PML, has so far been studied by retroviral overexpression in mouse B‐cell progenitors, which revealed little insight into the gene regulation by these transcription factors (Fazio et al, 2008, 2013; Qiu et al, 2011; Cazzaniga et al, 2015). Moreover, analysis of primary human PAX5‐ETV6+ B‐ALLs can also provide only limited information about the role of PAX5‐ETV6 in B‐ALL development. To bypass these limitations, we have generated a physiological mouse model expressing the human PAX5‐ETV6 protein, which allowed us to gain detailed insight into the developmental function, genome‐wide binding pattern, regulated target gene profile, cooperating mutations, and oncogenic role of PAX5‐ETV6 in B‐ALL formation.
As Pax5 is an essential regulator of B‐cell development (Nutt et al, 1999; Cobaleda et al, 2007), it has been assumed that PAX5 heterozygosity contributes to B‐ALL formation in human patients (Mullighan et al, 2007) and mouse models (Heltemes‐Harris et al, 2011; Liu et al, 2014) by arresting B‐lymphopoiesis at the large pre‐B‐cell stage. Here, we have shown, however, that the loss of one Pax5 allele alone does not affect B‐cell development in heterozygous Pax5 +/− mice as well as in competitive bone marrow chimeras that usually reveal even minor developmental defects of mutant hematopoietic cells. It remains though a possibility that Pax5 heterozygosity contributes, together with other cooperating mutations, to the B‐cell developmental arrest in the oncogenic environment of B‐ALLs. In contrast, expression of the Pax5‐Etv6 and Pax5‐Foxp1 fusion proteins under the control of the Pax5 locus severely affected B‐cell development already at the pro‐B to pre‐B‐cell transition in Pax5 Etv6/+ and Pax5 Foxp1/+ mice. Importantly, the severity of the B‐cell developmental defect was determined by the fusion partner and not by the Pax5 paired domain polypeptide, whose expression on its own did not interfere with B‐lymphopoiesis. Gene expression analysis in pro‐B cells failed to identify a signature of Pax5‐Etv6‐ or Pax5‐Foxp1‐regulated genes that code for proteins with functions in pre‐BCR signaling. However, several regulated genes encoded proteins implicated in the control of cell migration and/or adhesion and, consistent with this finding, Pax5 Etv6/+ and Pax5 Foxp1/+ pro‐B cells showed diminished migration. Hence, deregulation of these genes by Pax5‐Etv6 or Pax5‐Foxp1 could explain, at least in part, the observed developmental phenotype, given the recent evidence that decreased migration combined with increased adhesion is associated with impaired transition from the pro‐B to the pre‐B‐cell stage (Joshi et al, 2014; Schwickert et al, 2014).
PAX5 fusion proteins are thought to function as competitive inhibitors by interfering with the transcriptional activity of the wild‐type PAX5 protein that is simultaneously expressed in the leukemic cells. This model is primarily based on transient transfection assays in non‐B cells, whereby increasing amounts of a PAX5 fusion protein prevent the activation of a Pax5‐dependent reporter gene by wild‐type PAX5 (Bousquet et al, 2007; Mullighan et al, 2007; Kurahashi et al, 2011; Kawamata et al, 2012). Interestingly, several fusion partner proteins contain an oligomerization domain (Kawamata et al, 2012; Fortschegger et al, 2014). Moreover, the oligomerization motif of the PAX5‐C20orf112 protein is known to strongly increase the DNA‐binding affinity of this fusion protein, which likely results from the interaction of multiple PAX5 paired domains with the same DNA‐binding region (Kawamata et al, 2012). Hence, PAX5 fusion proteins could act as dominant‐negative proteins by preventing wild‐type PAX5 from interacting with its genomic binding sites (Kawamata et al, 2012). Both, the PAX5‐ETV6 and PAX5‐FOXP1 proteins also function as dominant‐negative proteins in transient transfection assays (Mullighan et al, 2007) and contain, in addition to the PAX5 paired domain, a second DNA‐binding region (Ets [ETV6] or forkhead [FOXP1] domain) as well as a dimerization motif (pointed [ETV6] or coiled‐coil [FOXP1] domain) (Cazzaniga et al, 2001; Mullighan et al, 2007). However, contrary to the existing model, we have shown here that Pax5‐Etv6 and Pax5‐Foxp1 were able to repress only a small fraction of the activated Pax5 target genes in pro‐B cells, while they activated an equally small subset of the repressed Pax5 target genes. Interestingly, the two Pax5 fusion proteins predominantly regulated different genes, demonstrating that the partner protein determines the transcriptional activity of the Pax5 fusion protein. Moreover, both Pax5 and Pax5‐Etv6 interacted with a large set of common binding sites in the genome, thus demonstrating efficient competition of both proteins for the same binding sites in Pax5 Etv6/+ pro‐B cells. Detailed analysis of the binding pattern furthermore indicated that Pax5‐Etv6 simultaneously uses both the paired domain of Pax5 and the Ets domain of Etv6 for DNA sequence recognition. Hence, our data do not support the prevailing model of a common dominant‐negative mechanism for Pax5 fusion proteins, but rather indicate that different Pax5 fusion proteins have distinct transcriptional activities dictated by their partner proteins.
Tumor development did not occur in heterozygous Pax5 Etv6/+, Pax5 Foxp1/+, and Pax5 +/− mice, which revealed an additional requirement for cooperating mutations. The tumor suppression function of Pax5 was previously identified by the cooperation of Pax5 heterozygosity with constitutive activation of STAT5, JAK1, or JAK3, leading to B‐ALL development in Pax5 +/− mice (Heltemes‐Harris et al, 2011; Liu et al, 2014; Dang et al, 2015). Notably, tumor development was not further accelerated in caStat5a Pax5 Etv6/+ or caStat5a Pax5 Foxp1/+ mice, indicating that only the loss of one wild‐type Pax5 allele, but not the expression of the Pax5 fusion proteins, is essential for leukemia formation in this mouse model. Due to the non‐reciprocal nature of the PAX5‐ETV6 translocation, all PAX5‐ETV6+ B‐ALLs lack one allele of the CDKN2A/B tumor suppressor locus (Cazzaniga et al, 2001; Strehl et al, 2003), which encodes two cyclin‐dependent kinase inhibitors restricting G1/S phase progression as well as the Arf protein preventing Trp53 degradation (Gil & Peters, 2006). Consistent with this finding, heterozygous Cdkn2ab +/− Pax5 Etv6/+ mice developed, with a high penetrance of 98%, B‐ALL tumors that were arrested at the large pre‐B‐cell stage and infiltrated many different organs. The monoclonal nature of the Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs points to additional cooperating mutations that contribute to tumor development. One of these cooperating events was the observed loss of heterozygosity at the Cdkn2ab locus, as the wild‐type Cdkn2ab allele was absent in ~80% of all Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs. Importantly, the generation of B‐cell leukemia was significantly accelerated in homozygous Cdkn2ab −/− Pax5 Etv6/+ mice, thus indicating that loss of heterozygosity provides a strong selective advantage for B‐ALL development. In agreement with this observation, homozygous deletion of the CDKN2A/B locus is also detected in half of all human PAX5‐ETV6+ B‐ALL cases analyzed (Schinnerl et al, 2015). Hence, the Cdkn2ab +/− Pax5 Etv6/+ tumors recapitulate many of the salient features of human PAX5‐ETV6+ B‐ALL and thus constitute a faithful mouse model for this human disease.
Tumor development was strongly delayed and mainly driven by the Cdkn2ab heterozygosity in Cdkn2ab +/− Pax5 +/− and Cdkn2ab +/− Pax5 Prd/+ mice. Only 11–13% of these tumors had a B‐ALL phenotype, demonstrating that heterozygous loss of Cdkn2ab can only inefficiently cooperate with Pax5 heterozygosity in B‐ALL development. Surprisingly, Cdkn2ab +/− Pax5 Foxp1/+ mice also failed to generate B‐cell leukemia, indicating that Cdkn2ab heterozygosity is not sufficient to cooperate with Pax5‐Foxp1 in driving B‐ALL development. Although Pax5‐Foxp1 induces a more pronounced B‐cell developmental arrest than Pax5‐Etv6, it appears that the nature of the cooperating mutation is more decisive than the developmental arrest in driving B‐ALL development by Pax5 fusion proteins. Finally, tumor development occurred with a similarly short latency in Cdkn2ab −/− mice and Cdkn2ab −/− Pax5 Etv6/+ mice, although the tumor spectrum is known to be very broad in Cdkn2ab −/− mice (Krimpenfort et al, 2007) in contrast to the predominant development of B‐ALLs in Cdkn2ab −/− Pax5 Etv6/+ mice. Hence, the expression of Pax5‐Etv6 can efficiently drive tumor development in the B‐cell lineage of Cdkn2ab −/− mice, which unequivocally identifies Pax5‐Etv6 as a potent B‐cell‐specific oncoprotein.
The large pre‐B cell was identified as the likely cell of origin for the development of Cdkn2ab +/− Pax5 Etv6/+ B‐ALLs based on global gene expression analysis and cell surface phenotyping. Regulated Pax5‐Etv6 target genes in B‐ALL were identified by RNA‐seq comparison of Cdkn2ab +/− Pax5 Etv6/+ tumors and large pre‐B cells combined with genome‐wide Pax5‐Etv6 binding. One large functional class of regulated target genes codes for proteins implicated in cell migration and/or adhesion, which include chemokine and cell adhesion receptors (activated: Cxcr5, Sema7a, Epcam, Gpr97, Lrrn4, Itgb4, Itgb5, S1pr2; repressed: Erma, Bst1) as well as intracellular signal transducers involved in controlling rearrangements of the actin cytoskeleton (activated: Fdg2, Cass4, Srgap3, Arhgef12, Marveld2, Gbp2, Rhof, Farp1, Chn2, Rapgef3; repressed: Cdc42ep4, Eps8). It is therefore conceivable that the Pax5‐Etv6‐mediated regulation of these genes contributes to the observed tissue infiltration of Cdkn2ab +/− Pax5 Etv6/+ tumor cells. Three highly activated Pax5‐Etv6 target genes, Cish, Socs2, and Socs3, code for negative regulators of the JAK‐STAT signaling pathway (Alexander & Hilton, 2004) and may thus actively suppress cytokine signaling in Cdkn2ab +/− Pax5 Etv6/+ B‐ALL cells. Pax5‐Etv6‐mediated gene regulation furthermore strengthens pre‐BCR signaling, as three activated genes code for the surrogate light chain Vpreb2, the receptor‐proximal Zap70 kinase, and the signaling adaptor Bank1, whereas two repressed genes code for the inhibitory co‐receptor Siglecg (Hoffmann et al, 2007) and the central adaptor Blnk (Slp65). Importantly, Blnk has been identified as a tumor suppressor in B‐ALL, as its deficiency prevents calcium signaling and further differentiation to small pre‐B cells, while promoting cell proliferation and survival through the PI3K‐AKT pathway of pre‐BCR signaling (Jumaa et al, 2003). Hence, the strong repression of Blnk by Pax5‐Etv6 likely contributes to B‐ALL development in Cdkn2ab +/− Pax5 Etv6/+ mice.
Human PAX5‐ETV6+ B‐ALLs also appear to express the pre‐BCR together with a gene expression signature that is characteristic of pre‐BCR+ B‐ALLs (Geng et al, 2015). Hence, PAX5‐ETV6+ B‐ALLs may belong to the recently identified class of pre‐BCR+ B‐ALLs (Geng et al, 2015). Notably, the tumor suppressor gene BLNK was repressed in four of nine human PAX5‐ETV6+ B‐ALLs (data not shown). In addition, one‐sixth of the regulated Pax5‐Etv6 target genes identified in mouse Cdkn2ab +/− Pax5 Etv6/+ tumors were similarly activated or repressed in human PAX5‐ETV6+ B‐ALLs, indicating that some of these genes may contribute to leukemia development in both species. In summary, the establishment of a faithful mouse model for a specific subset of human B‐ALLs has provided novel insight into the molecular mechanisms, by which the oncoprotein PAX5‐ETV6 contributes to leukemogenesis. Hence, our data demonstrate for the first time that PAX5 can function not only as a haploinsufficient tumor suppressor, but also as a potent oncoprotein as part of PAX5 fusion proteins in the same tumor entity, B‐ALL.
Materials and Methods
Detailed methods can be found in the Appendix Supplementary Methods available online.
Mice
The following mice were maintained on the C57BL/6 genetic background: Pax5 +/− (Urbánek et al, 1994), Cdkn2ab +/− (Krimpenfort et al, 2007), Rosa26 BirA/BirA (Driegen et al, 2005), and transgenic EμSRα‐Stat5a(S711F) mouse strain (Joliot et al, 2006). All animal experiments were carried out according to valid project licenses, which were approved and regularly controlled by the Austrian Veterinary Authorities.
Generation of Pax5 Etv6/+, Pax5 Foxp1/+, and Pax5 Prd/+ mice
All three alleles were generated by homologous recombination in ES cells using targeting vectors that were cloned as shown in Appendix Fig S2A–C and described in detail in the corresponding figure legend.
Flow cytometric definition of mouse hematopoietic cell types
The different hematopoietic cell types were identified by flow cytometry as follows: pro‐B (CD19+B220+c‐Kit+CD2−[or CD25−]IgM−IgD−), large pre‐B cells (CD19+B220+CD2+c‐Kit−IgM−IgD−FSChi), small pre‐B (CD19+B220+CD2+[or CD25+]c‐Kit−IgM−IgD−FSClo), immature B (CD19+B220+IgMhiIgDlo), transitional B (CD19+B220+IgMhiIgDhi), mature B (CD19+B220+IgMlo IgDhi), recirculating B (CD19+B220+IgMlo IgDhi), total B cells (CD19+B220+), granulocytes (Gr1hiMac1hi), and macrophages (Mac1+M‐CSFR+).
Transplantations
Lymph node cells from tumor mice (0.1–1 × 106 donor cells per mouse) were injected into sublethally irradiated C57BL/6 mice (4.5–6 Gy). Tumor development was monitored, and mice were sacrificed when losing more than 10% of body weight or showing signs of terminal illness.
Patient samples and human cells
Normal human pre‐B cells were sorted as CD34−CD19+CD10+ cells from the bone marrow of healthy donors. Informed consent for all patient samples was obtained from all subjects.
Bio‐ChIP‐sequencing
Pro‐B cells isolated from the bone marrow of Rosa26 BirA/+ Pax5 Etv6/+ or Rosa26 BirA/+ Pax5 Prd/+ mice were cultured for 4–5 days on OP9 cells in the presence of IL‐7. These pro‐B cells or Cdkn2ab +/− Pax5 Etv6/+ Rosa26 BirA/+ B‐ALL cells were used for Bio‐ChIP‐seq analysis (Table EV6) as described (Schwickert et al, 2014).
RNA‐sequencing
RNA from ex vivo sorted B cells was isolated with the RNeasy Plus Mini Kit (Qiagen). mRNA was obtained by two rounds of poly(A) selection and used for library preparation and Illumina deep sequencing (Table EV6) as described (Minnich et al, 2016).
Bioinformatic analysis of RNA‐ and ChIP‐seq data
The bioinformatic analysis of RNA‐ and ChIP‐seq data (Table EV6) was performed as described in detail (Minnich et al, 2016).
De novo motif discovery
For de novo motif discovery, we used the MEME‐ChIP suite (version 4.9.1) (Machanick & Bailey, 2011) to predict the most significant motifs present in the 300 base pairs centered at the peak summit of the top 300 sequences, as sorted by the fold enrichment score of the MACS program.
Gene set enrichment analysis
Gene set enrichment analysis was performed using the GSEA software from the Broad Institute (Subramanian et al, 2005).
Accession numbers
The mouse RNA‐seq and ChIP‐seq data are available at the Gene Expression Omnibus (GEO) repository under the accession numbers GSE84987. The human RNA‐seq data are available upon request.
Author contributions
LS performed most experiments; SJ performed the pro‐B‐cell migration assays and in vitro culture experiments of mouse B‐ALL cells; AA and BW generated the Pax5 Etv6 and Pax5 Foxp1 alleles; MF and MJ performed the bioinformatic analysis of all RNA‐seq and ChIP‐seq data, respectively; MR and JZ provided RNA‐seq data of mouse control B‐ALLs; SS, MLdB, and MS provided human PAX5‐ETV6+ B‐ALL samples; CGM provided RNA‐seq data of human pre‐B cells; LS and MB planned the project, designed the experiments, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 6
Acknowledgements
We thank A. Berns (Netherlands Cancer Institute, Amsterdam) for providing Cdkn2ab +/− mice, J. Ghysdael (Curie Institute, Paris) for the transgenic EμSRα‐Stat5a(S711F) mouse strain, I. Bilic for chromatin ChIP‐seq data, C. Theussl and J. Wojciechowski for blastocyst injection, K. Aumayr and her colleagues for flow cytometric sorting, A. Sommer and his colleagues (Vienna Biocenter Core Facilities GmbH) for Illumina sequencing, and A. N. Kavirayani (Vienna Biocenter Core Facilities GmbH) for histopathological tumor analysis. This research was supported by Boehringer Ingelheim, an ERC Advanced Grant (291740‐LymphoControl) from the European Community's Seventh Framework Program, the Austrian Science Fund (P‐21604‐B13), and the Austrian Industrial Research Promotion Agency (Headquarter Grant FFG‐834223).
The EMBO Journal (2017) 36: 718–735
See also: A Alsadeq & H Jumaa (March 2017)
References
- Adams B, Dörfler P, Aguzzi A, Kozmik Z, Urbánek P, Maurer‐Fogy I, Busslinger M (1992) Pax‐5 encodes the transcription factor BSAP and is expressed in B lymphocytes, the developing CNS, and adult testis. Genes Dev 6: 1589–1607 [DOI] [PubMed] [Google Scholar]
- Alexander WS, Hilton DJ (2004) The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol 22: 503–529 [DOI] [PubMed] [Google Scholar]
- Bousquet M, Broccardo C, Quelen C, Meggetto F, Kuhlein E, Delsol G, Dastugue N, Brousset P (2007) A novel PAX5‐ELN fusion protein identified in B‐cell acute lymphoblastic leukemia acts as a dominant negative on the wild‐type PAX5. Blood 109: 3417–3423 [DOI] [PubMed] [Google Scholar]
- Cazzaniga G, Daniotti M, Tosi S, Giudici G, Aloisi A, Pogliani E, Kearney L, Biondi A (2001) The paired box domain gene PAX5 is fused to ETV6/TEL in an acute lymphoblastic leukemia case. Cancer Res 61: 4666–4670 [PubMed] [Google Scholar]
- Cazzaniga V, Bugarin C, Bardini M, Giordan M, te Kronnie G, Basso G, Biondi A, Fazio G, Cazzaniga G (2015) LCK over‐expression drives STAT5 oncogenic signaling in PAX5 translocated BCP‐ALL patients. Oncotarget 6: 1569–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, Busslinger M (2007) Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 449: 473–477 [DOI] [PubMed] [Google Scholar]
- Coyaud E, Struski S, Prade N, Familiades J, Eichner R, Quelen C, Bousquet M, Mugneret F, Talmant P, Pages M‐P, Lefebvre C, Penther D, Lippert E, Nadal N, Taviaux S, Poppe B, Luquet I, Baranger L, Eclache V, Radford I et al (2010) Wide diversity of PAX5 alterations in B‐ALL: a Groupe Francophone de Cytogénétique Hématologique study. Blood 115: 3089–3097 [DOI] [PubMed] [Google Scholar]
- Dang J, Wei L, de Ridder J, Su X, Rust AG, Roberts KG, Payne‐Turner D, Cheng J, Ma J, Qu C, Wu G, Song G, Huether RG, Schulman B, Janke L, Zhang J, Downing JR, van der Weyden L, Adams DJ, Mullighan CG (2015) PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood 125: 3609–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T, Pasca di Magliano M, McManus S, Sun Q, Bonifer C, Tagoh H, Busslinger M (2009) Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity 30: 508–520 [DOI] [PubMed] [Google Scholar]
- Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M (2006) Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity 24: 269–281 [DOI] [PubMed] [Google Scholar]
- Driegen S, Ferreira R, van Zon A, Strouboulis J, Jaegle M, Grosveld F, Philipsen S, Meijer D (2005) A generic tool for biotinylation of tagged proteins in transgenic mice. Transgenic Res 14: 477–482 [DOI] [PubMed] [Google Scholar]
- Fazio G, Palmi C, Rolink A, Biondi A, Cazzaniga G (2008) PAX5/TEL acts as a transcriptional repressor causing down‐modulation of CD19, enhances migration to CXCL12, and confers survival advantage in pre‐BI cells. Cancer Res 68: 181–189 [DOI] [PubMed] [Google Scholar]
- Fazio G, Cazzaniga V, Palmi C, Galbiati M, Giordan M, te Kronnie G, Rolink A, Biondi A, Cazzaniga G (2013) PAX5/ETV6 alters the gene expression profile of precursor B cells with opposite dominant effect on endogenous PAX5. Leukemia 27: 992–995 [DOI] [PubMed] [Google Scholar]
- Fortschegger K, Anderl S, Denk D, Strehl S (2014) Functional heterogeneity of PAX5 chimeras reveals insight for leukemia development. Mol Cancer Res 12: 595–606 [DOI] [PubMed] [Google Scholar]
- Geng H, Hurtz C, Lenz KB, Chen Z, Baumjohann D, Thompson S, Goloviznina NA, Chen WY, Huan J, LaTocha D, Ballabio E, Xiao G, Lee JW, Deucher A, Qi Z, Park E, Huang C, Nahar R, Kweon SM, Shojaee S et al (2015) Self‐enforcing feedback activation between BCL6 and pre‐B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell 27: 409–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Peters G (2006) Regulation of the INK4b‐ARF‐INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 7: 667–677 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Heltemes‐Harris LM, Willette MJ, Ramsey LB, Qiu YH, Neeley ES, Zhang N, Thomas DA, Koeuth T, Baechler EC, Kornblau SM, Farrar MA (2011) Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J Exp Med 208: 1135–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Kerr S, Jellusova J, Zhang J, Weisel F, Wellmann U, Winkler TH, Kneitz B, Crocker PR, Nitschke L (2007) Siglec‐G is a B1 cell‐inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol 8: 695–704 [DOI] [PubMed] [Google Scholar]
- Horcher M, Souabni A, Busslinger M (2001) Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity 14: 779–790 [DOI] [PubMed] [Google Scholar]
- Joliot V, Cormier F, Medyouf H, Alcalde H, Ghysdael J (2006) Constitutive STAT5 activation specifically cooperates with the loss of p53 function in B‐cell lymphomagenesis. Oncogene 25: 4573–4584 [DOI] [PubMed] [Google Scholar]
- Joshi I, Yoshida T, Jena N, Qi X, Zhang J, Van Etten RA, Georgopoulos K (2014) Loss of Ikaros DNA‐binding function confers integrin‐dependent survival on pre‐B cells and progression to acute lymphoblastic leukemia. Nat Immunol 15: 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumaa H, Bossaller L, Portugal K, Storch B, Lotz M, Flemming A, Schrappe M, Postila V, Riikonen P, Pelkonen J, Niemeyer CM, Reth M (2003) Deficiency of the adaptor SLP‐65 in pre‐B‐cell acute lymphoblastic leukaemia. Nature 423: 452–456 [DOI] [PubMed] [Google Scholar]
- Kawamata N, Pennella MA, Woo JL, Berk AJ, Koeffler HP (2012) Dominant‐negative mechanism of leukemogenic PAX5 fusions. Oncogene 31: 966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimpenfort P, Ijpenberg A, Song J‐Y, van der Valk M, Nawijn M, Zevenhoven J, Berns A (2007) p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 448: 943–946 [DOI] [PubMed] [Google Scholar]
- Kurahashi S, Hayakawa F, Miyata Y, Yasuda T, Minami Y, Tsuzuki S, Abe A, Naoe T (2011) PAX5‐PML acts as a dual dominant‐negative form of both PAX5 and PML. Oncogene 30: 1822–1830 [DOI] [PubMed] [Google Scholar]
- Liu GJ, Cimmino L, Jude JG, Hu Y, Witkowski MT, McKenzie MD, Kartal‐Kaess M, Best SA, Tuohey L, Liao Y, Shi W, Mullighan CG, Farrar MA, Nutt SL, Smyth GK, Zuber J, Dickins RA (2014) Pax5 loss imposes a reversible differentiation block in B‐progenitor acute lymphoblastic leukemia. Genes Dev 28: 1337–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, Bailey TL (2011) MEME‐ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedovic J, Ebert A, Tagoh H, Busslinger M (2011) Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol 111: 179–206 [DOI] [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bönelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, Busslinger M (2016) Multifunctional role of the transcription factor Blimp‐1 in coordinating plasma cell differentiation. Nat Immunol 17: 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan‐Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui C‐H, Relling MV, Evans WE, Shurtleff SA, Downing JR (2007) Genome‐wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446: 758–764 [DOI] [PubMed] [Google Scholar]
- Nebral K, Denk D, Attarbaschi A, König M, Mann G, Haas OA, Strehl S (2009) Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia 23: 134–143 [DOI] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M (1999) Commitment to the B‐lymphoid lineage depends on the transcription factor Pax5. Nature 401: 556–562 [DOI] [PubMed] [Google Scholar]
- Qiu JJ, Chu H, Lu X, Jiang X, Dong S (2011) The reduced and altered activities of PAX5 are linked to the protein‐protein interaction motif (coiled‐coil domain) of the PAX5‐PML fusion protein in t (9;15)‐associated acute lymphocytic leukemia. Oncogene 30: 967–977 [DOI] [PubMed] [Google Scholar]
- Revilla‐i‐Domingo R, Bilic I, Vilagos B, Tagoh H, Ebert A, Tamir IM, Smeenk L, Trupke J, Sommer A, Jaritz M, Busslinger M (2012) The B‐cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J 31: 3130–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, Busslinger M (2007) Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity 27: 49–63 [DOI] [PubMed] [Google Scholar]
- Schinnerl D, Fortschegger K, Kauer M, Marchante JRM, Kofler R, Den Boer ML, Strehl S (2015) The role of the Janus‐faced transcription factor PAX5‐JAK2 in acute lymphoblastic leukemia. Blood 125: 1282–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickert TA, Tagoh H, Gültekin S, Dakic A, Axelsson E, Minnich M, Ebert A, Werner B, Roth M, Cimmino L, Dickins RA, Zuber J, Jaritz M, Busslinger M (2014) Stage‐specific control of early B cell development by the transcription factor Ikaros. Nat Immunol 15: 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehl S, König M, Dworzak MN, Kalwak K, Haas OA (2003) PAX5/ETV6 fusion defines cytogenetic entity dic(9;12)(p13;p13). Leukemia 17: 1121–1123 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbánek P, Wang Z‐Q, Fetka I, Wagner EF, Busslinger M (1994) Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell 79: 901–912 [DOI] [PubMed] [Google Scholar]
- Wagner GP, Kin K, Lynch VJ (2012) Measurement of mRNA abundance using RNA‐seq data: RPKM measure is inconsistent among samples. Theory Biosci 131: 281–285 [DOI] [PubMed] [Google Scholar]
- Wei G‐H, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, Yan J, Talukder S, Turunen M, Taipale M, Stunnenberg HG, Ukkonen E, Hughes TR, Bulyk ML, Taipale J (2010) Genome‐wide analysis of ETS‐family DNA‐binding in vitro and in vivo . EMBO J 29: 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 6