

Abstract
Mitochondria exert important control over plasma membrane (PM) Orai1 channels mediating store‐operated Ca2+ entry (SOCE). Although the sensing of endoplasmic reticulum (ER) Ca2+ stores by STIM proteins and coupling to Orai1 channels is well understood, how mitochondria communicate with Orai1 channels to regulate SOCE activation remains elusive. Here, we reveal that SOCE is accompanied by a rise in cytosolic Na+ that is critical in activating the mitochondrial Na+/Ca2+ exchanger (NCLX) causing enhanced mitochondrial Na+ uptake and Ca2+ efflux. Omission of extracellular Na+ prevents the cytosolic Na+ rise, inhibits NCLX activity, and impairs SOCE and Orai1 channel current. We show further that SOCE activates a mitochondrial redox transient which is dependent on NCLX and is required for preventing Orai1 inactivation through oxidation of a critical cysteine (Cys195) in the third transmembrane helix of Orai1. We show that mitochondrial targeting of catalase is sufficient to rescue redox transients, SOCE, and Orai1 currents in NCLX‐deficient cells. Our findings identify a hitherto unknown NCLX‐mediated pathway that coordinates Na+ and Ca2+ signals to effect mitochondrial redox control over SOCE.
Keywords: CRAC channel, mitochondrial redox, NCLX, SOCE, sodium signaling
Subject Categories: Membrane & Intracellular Transport, Metabolism
Introduction
The store‐operated Ca2+ entry (SOCE) pathway mediating the highly Ca2+ selective Ca2+ release‐activated Ca2+ (CRAC) current is the major Ca2+ influx route in non‐excitable cells (Putney, 1986; Potier & Trebak, 2008). The pathway is activated by interaction between the plasma membrane Ca2+ entry channel, Orai1 (Feske et al, 2006; Vig et al, 2006; Zhang et al, 2006), and the endoplasmic reticulum (ER) Ca2+ sensor protein, STIM1 (Liou et al, 2005; Zhang et al, 2005). Numerous studies have revealed that mitochondria are also key regulators of SOCE activity (Hoth et al, 1997; Deak et al, 2014). However, the exact mechanisms by which mitochondria facilitate SOCE are poorly understood. With the steep mitochondrial membrane potential as its driving force, Ca2+ permeates into the mitochondrial matrix via the recently identified mitochondrial Ca2+ uniporter (MCU; Baughman et al, 2011; De Stefani et al, 2011; Patron et al, 2013). Ca2+ is extruded from the mitochondria by an electrogenic mitochondrial 3Na+/1Ca2+ exchanger (NCLX; Palty et al, 2010) which is powered by the same mitochondrial membrane potential. This constant cycling of Ca2+ in and out of mitochondria functions as a dynamic and important signaling link between cytosolic Ca2+ signals and metabolic activity maintained by the regulation of key Krebs cycle enzymes as well as the F1/F0‐ATPase pump (Szabadkai & Duchen, 2008). The NCLX is a member of superfamily of Na+/Ca2+ exchangers and shares with them the hallmark of α1/α2 repeats that form the catalytic cation transport domain. It belongs to a distinct branch of this family termed CAX and is the single representative of this group in mammalian cells (Lytton, 2007; He & O'Halloran, 2014). It also has a unique selectivity to monovalent cations being capable of transporting either Li+ or Na+ in exchange for Ca2+, while other members of the family are inert to Li+ (Palty et al, 2010). Because the mitochondrial Ca2+ efflux rate is 2‐3 orders of magnitude slower than the rate of mitochondrial Ca2+ influx, the efflux represents a rate limiting step controlling the duration and magnitude of mitochondrial Ca2+ transients (Rudolf et al, 2004). In addition, by virtue of transporting at least 3 Na+ per 1 Ca2+, NCLX is the major mitochondrial Na+ influx pathway (Baysal et al, 1994; Jung et al, 1995; Nita et al, 2014a). In fact, the affinity of the NCLX for Na+ is ~10 mM and is therefore highly tuned to minor changes in cytosolic Na+ (Nita et al, 2014a).
Studies utilizing metabolic inhibitors and NCLX blockers suggest that mitochondria exert crucial regulation of CRAC channel activity (Malli et al, 2003; Ardon et al, 2009; Feldman et al, 2010). In immune cells, mitochondria are closely associated with the ER‐PM junctions in which CRAC channels function (Hoth et al, 1997; Rizzuto et al, 1998, 2012; Alvarez et al, 1999; Berridge et al, 2003; Singaravelu et al, 2011). Because of their spatial proximity and Ca2+ shuttling capacity, it was proposed that mitochondria buffer Ca2+ at the vicinity of CRAC channels thus eliminating the well‐established Ca2+‐dependent inactivation of the CRAC pathway (Parekh, 2008; Demaurex et al, 2009). However, in other cell types, for example, endothelial cells, mitochondria are more remotely located and are therefore less likely to directly buffer Ca2+ in the vicinity of CRAC channels (Demaurex et al, 2009; Giacomello et al, 2010; Naghdi et al, 2010; Nunes & Demaurex, 2014). Yet, remarkably, even in these cell types, the mitochondria can still impinge on CRAC activity through an as yet unidentified mechanism.
Interestingly, SOCE is also closely associated with the regulation of cytosolic Na+ and many studies have demonstrated that Na+ influx, mediated by non‐selective transient receptor potential canonical (TRPC) channel members, occurs in response to receptor‐mediated activation of CRAC channels (Lemos et al, 2007; Baryshnikov et al, 2009; Lee et al, 2010). Thus, Na+ signaling events are closely associated with SOCE and communication with mitochondria as recently reported (Reyes et al, 2013); however, the regulatory basis of this interaction remains poorly understood. SOCE is also strongly regulated by redox signals. These control several key steps including the interaction between STIM1 and Orai1 (Hawkins et al, 2010), and Ca2+ permeation by the Orai1 channel through modification of a redox‐sensitive cysteine located on the Orai1 channel (Bogeski et al, 2010). Despite the fact that mitochondria are the major effectors of cellular redox, it is still not understood how mitochondrial redox impacts SOCE activity and mitochondrial Ca2+ signaling.
In the current study, we have sought to understand the cross talk between NCLX and SOCE. We reveal that expression of NCLX is required for SOCE and CRAC channel regulation through a mitochondrial proximity‐independent pathway. We show that such regulation does not involve the elimination of Ca2+‐dependent inactivation of CRAC channels through mitochondrial buffering of Ca2+. We found instead that integrated Ca2+ and Na+ signals act synergistically to promote mitochondrial Ca2+ shuttling that modulates mitochondrial redox status, thereby controlling SOCE activity.
Results
SOCE and CRAC currents are regulated by mitochondrial NCLX
To determine the role of the mitochondrial Na+/Ca2+ exchanger, NCLX, we reduced its expression in HEK293T cells using previously characterized siNCLX (Palty et al, 2010). Consistent with previous results, HEK293T cells transfected with siNCLX displayed over twofold reduction in expression of NCLX protein compared to siControl cells with no significant change in the expression of STIM1 and Orai1 (Fig 1A). To determine the effect of NCLX knockdown on SOCE, we measured Ca2+ influx in Fura‐2‐loaded cells after depleting ER Ca2+ stores with Ca2+‐free Ringer containing ATP (100 μM) and thapsigargin (TG; 1 μM) followed by superfusion with Ca2+‐containing solutions (1.8 mM) while monitoring the rate of Ca2+ influx. Silencing of NCLX expression led to a 2.4‐fold reduction in Ca2+ influx rate (*P < 0.05) compared to cells transfected with siControl (1.2E‐02 versus 5E‐03; Fig 1B). As an additional strategy, we have employed the same experimental paradigm in cells expressing the plasma membrane‐targeted Ca2+ probe, GCamp5 (Akerboom et al, 2012) to determine the localized change in Ca2+ concentration at the vicinity of the plasma membrane. This localized Ca2+ influx signal in siNCLX‐transfected cells was also strongly (10‐fold) reduced compared to Ca2+ signals observed in siControl‐transfected cells (1.16E‐03 versus 1.1E‐04; *P < 0.05; Fig 1C). However, NCLX knockdown had no effect on STIM1 and Orai1 protein expression determined by Western blotting (Fig 1A). To directly establish whether NCLX has a role in controlling SOCE thereby ruling out indirect effects of cellular Ca2+ pumping or changes in membrane potential, we measured store depletion‐activated CRAC currents by whole‐cell electrophysiological recordings (Fig 1D–F). CRAC currents were recorded in Ca2+‐containing bath solutions and further amplified by exchanging bath solutions with divalent‐free (DVF). Such replacement immediately after break‐in (first DVF exchange) led to the appearance of a relatively small linear Na+ current, reflecting cell membrane or seal leak due to the absence of divalent cations (Fig 1D). During subsequent DVF exchanges, Ca2+ store depletion had taken place and the DVF inward current increased steadily while the outward current remain unchanged. In such experiments, the leak subtracted store depletion‐activated CRAC current recorded in DVF bath solution, displayed inward rectification typical of CRAC currents measured in Ca2+‐containing bath solutions. These CRAC Na+ currents also showed the typical rapid depotentiation in DVF bath solutions (Fig 1D). The current–voltage (I–V) relationships revealed inward rectification with a highly positive reversal potential and complete block by low concentrations of lanthanides (5 μM Gd3+; Fig 1D), consistent with the properties of CRAC currents recorded in many other cells. Just as NCLX knockdown reduced the store‐operated Ca2+ entry response, it also led to a twofold–threefold inhibition of both CRAC‐mediated Ca2+ and Na+ currents (Fig 1E and F). To determine whether the NCLX‐dependent changes in SOCE and CRAC are mediated by a change in STIM1‐Orai1 interactions, we monitored co‐localization of STIM1‐YFP and Orai1‐CFP. We determined that puncta formation after store depletion (Fig 2A and B) as well as enhanced STIM1‐Orai1 FRET (Fig 2C) was not affected by NCLX knockdown. Importantly, the time course of STIM1/Orai1 FRET interaction upon store depletion is indistinguishable between siControl and siNCLX cells (Fig EV1 and Movies EV1 and EV2). In contrast to the inhibitory effects that NCLX knockdown had on Ca2+ signals and CRAC current, the knockdown of MCU did not inhibit SOCE and CRAC currents in HEK293T cells (Appendix Fig S1).
Figure 1. NCLX controls SOCE activity.
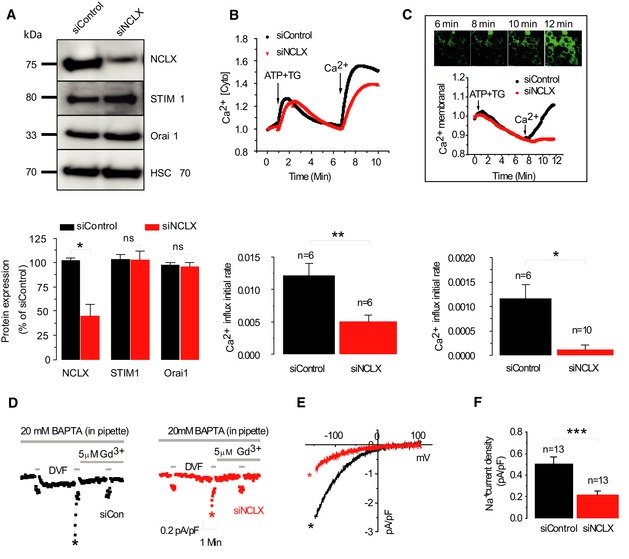
- Upper panel: Western blot was performed with 100 μg of protein samples obtained from HEK293T cells transfected with either siControl or siNCLX. Lower panel: Densitometry analysis of immunoblots of siControl‐transfected cells (n = 3) versus siNCLX‐transfected cells (n = 3) representing normalized expression levels (% of control) of NCLX, STIM1 and Orai1.
- Upper panel: HEK293T cells were transfected with either scrambled siRNA construct (siControl, black) or siNCLX (red) and loaded with Fura‐2. SOCE was triggered by ATP (100 μM) and thapsigargin (TG, 1 μM), and Fura‐2 fluorescence was monitored as described in Materials and Methods. Lowe panel: Averaged rates of Ca2+ rise in siNCLX‐silenced cells from several independent recordings (n = 6) versus siControl‐transfected cells (n = 6).
- Upper panel: Fluorescence of the membrane calcium sensor, GCamp5, targeted to the plasma membrane following treatment as described above using confocal microscopy. The scale bar represents 10 μm. Middle panel: Traces of membrane‐localized Ca2+ responses were measured in HEK293T cells co‐transfected with either the siNCLX (red) or siControl (black) and GCamp5‐expressing plasmid. The lower panel shows the averaged rates of membrane‐localized Ca2+ rise either in siControl cells (n = 6) or siNCLX cells (n = 10).
- Electrophysiological recordings were performed on the same batch of transfected cells used for Western blot and shown in panel (A). Representative time courses of whole‐cell CRAC currents activated by dialysis of 20 mM BAPTA through the patch pipette and taken at −100 mV from cells transfected either with siControl (black trace) or siNCLX (red trace).
- Representative I–V relationships are taken from traces in (D) where indicated by color‐coded asterisks.
- Statistical analysis on Na+ CRAC currents measured at −100 mV is shown.
Figure 2. NCLX knockdown has no effect on Orai1 and STIM1 interaction following store depletion.
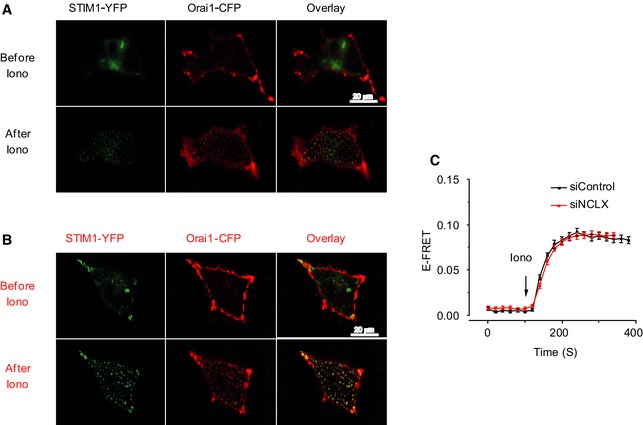
-
A, BHEK293T cells stably expressing Orai1‐CFP and STIM1‐YFP were transfected with either control siRNA (A) or NCLX siRNA (B) and incubated for 72 h. E‐FRET experiments were then performed. The scale bar represents 20 μm.
-
CSilencing of NCLX by siNCLX (red) has no effect on Orai1 and STIM1 colocalization after store depletion induced by applying 2.5 μM ionomycin to the bath solution, compared to siControl (black; means ± SEM).
Figure EV1. NCLX knockdown has no effect on Orai1/STIM1 interactions measured with FRET.
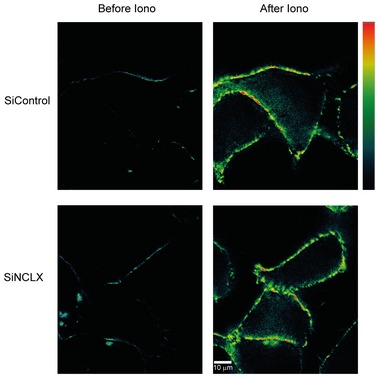
HEK293T cells expressing Orai1‐CFP and STIM1‐YFP were transfected with either control siRNA or NCLX siRNA and effect on Orai1/STIM1 corrected FRET after store depletion with 2.5 μM ionomycin (Iono) were compared. Bar scale: warmer colors indicate high FRET. The scale bar represents 10 μm.
Cultured primary aortic vascular smooth muscle cells (VSMCs) display robust SOCE as previously characterized in detail (Potier et al, 2009). We sought to determine whether similar cross talk between SOCE and mitochondrial NCLX exists in these cells. Silencing of NCLX expression led to a fourfold reduction in store‐operated Ca2+ influx rate measured with Fura‐2 (*P < 0.05) compared to cells transfected with siControl (2.1E‐03 versus 5.2E‐04; Fig 3A). The electrophysiological recordings of CRAC currents in VSMCs also showed twofold–threefold reduction in CRAC current density upon NCLX knockdown (Fig 3B and C). Together, these results indicate that mitochondrial NCLX is required for optimal activation of SOCE and CRAC currents.
Figure 3. Effect of NCLX knockdown on SOCE in primary vascular smooth muscle cells (VSMCs).
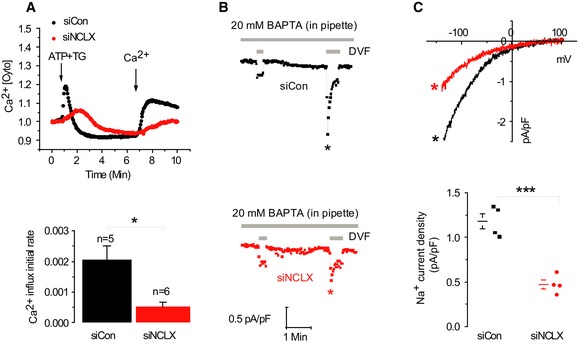
-
AUpper panel: Fluorescence traces of cytosolic Ca2+ responses in VSMC cells. Lower panel: Averaged rates of cytosolic Ca2+ influx in siControl cells (n = 5) and siNCLX cells (n = 6).
-
B, CElectrophysiological CRAC recordings in cells as in (A) using the same protocol used in Fig 2. Statistical analysis on Na+ CRAC currents measured at −100 mV is shown in (C).
Mitochondrial control of store‐operated Ca2+ signaling is known to occur as a result of buffering the Ca2+ entering through CRAC channels by virtue of the close proximity of mitochondria to the channels (Glitsch et al, 2002). This mode of regulation is revealed when CRAC channel activation is measured in the presence of low Ca2+ buffer in the patch pipette (e.g. 0.1 mM EGTA). In contrast, other studies suggested that an elusive proximity‐independent mechanism is mediating mitochondrial regulation of CRAC channels (Giacomello et al, 2010; Pizzo et al, 2012). To address this, we determined whether NCLX regulation of CRAC current is dependent on Ca2+ buffering by analyzing CRAC current under varying cytosolic Ca2+ buffering conditions. Although the inhibitory effect of NCLX knockdown on CRAC channels persisted when CRAC channels were activated by a high concentration of a fast Ca2+ buffer (20 mM BAPTA; Figs 1 and 3), we considered that NCLX might be more intimately coupled to CRAC channels and therefore higher buffering capacity would be needed to recapitulate this close interaction. As shown in Fig 4A and B, the inhibitory effect of NCLX knockdown was essentially maintained whether 20 mM or 50 mM BAPTA was used indicating that NCLX is unlikely to be acting only through buffering cytosolic Ca2+ to relieve the Ca2+‐mediated negative feedback on CRAC channels. Thus, our results indicate that the regulation of SOCE by mitochondrial NCLX is at least partially mediated by a mechanism distinct from Ca2+ buffering in the vicinity of the mouth of CRAC channels.
Figure 4. NCLX does not control CRAC channels through buffering of cytosolic Ca2+ or mitochondrial energization.
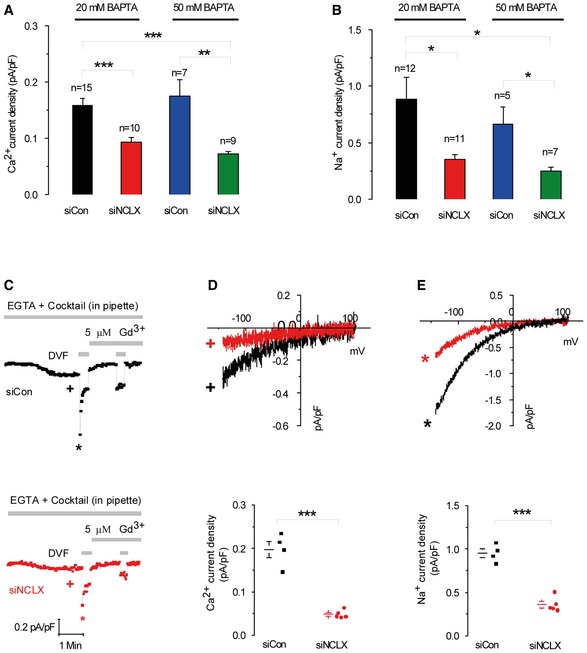
-
A, BAnalysis of Ca2+ (A) and Na+ (B) CRAC currents measured at −100 mV in HEK293T cells transfected with either siControl or siNCLX using the indicated concentrations of BAPTA (20 mM versus 50 mM) in the pipette solution to activate CRAC and buffer cytosolic Ca2+.
-
C–EElectrophysiological CRAC recordings were performed on cells where store depletion was induced by the use of a pipette solution containing low buffering capacity (0.1 mM EGTA) with IP3 (30 μM) and a cocktail to energize the mitochondria whose composition is listed in the Results section. Representative time courses of whole‐cell current development at −100 mV (D, E) and measured in Ca2+‐containing and DVF solutions, respectively, by comparison with siControl. At the end of recordings, 5 μM Gd3+ was used to inhibit CRAC currents. Representative I–V relationships of Ca2+ (D) and Na+ (E) CRAC in cells transfected with siControl or siNCLX are taken from traces in (C) where indicated by color‐coded asterisks. Statistical analysis on Ca2+ and Na+ CRAC currents measured at −100 mV (D, E).
A similar inhibitory effect of NCLX knockdown was observed in cells dialyzed with a mitochondrial‐energizing solution (see Materials and Methods). Thus, when the mitochondrial‐energizing cocktail was used, CRAC currents were 2.8‐fold bigger than those recorded with conventional pipette solution (Fig EV2). Importantly, and consistently with the results described in Fig 4A and B, we found that even under conditions of energized mitochondria (and hence enhanced mitochondrial Ca2+ buffering), NCLX knockdown still inhibits CRAC current (Fig 4C–E), further supporting our conclusion that NCLX acts through a pathway independent of mitochondrial Ca2+ buffering.
Figure EV2. CRAC channels are enhanced under physiological conditions of energized mitochondria.
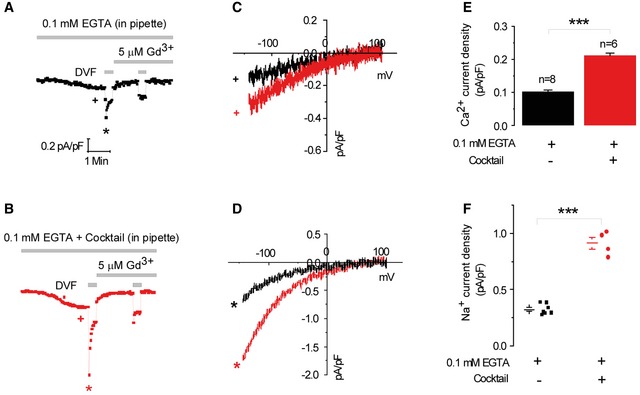
-
A, BElectrophysiological CRAC recordings were performed on HEK293T cells where store depletion was induced by the use of a pipette solution containing 0.1 mM EGTA with IP3 with (B) and without (A) inclusion of a cocktail to energize the mitochondria. At the end of recordings, 5 μM Gd3+ was used to inhibit CRAC currents.
-
C, DRepresentative I–V relationships of Ca2+ (C) and Na+ (D) CRAC in are taken from traces in (A) and (B) where indicated by color‐coded asterisks.
-
E, FStatistical analysis on Ca2+ and Na+ CRAC currents measured at −100 mV.
Sodium transport by NCLX controls Ca2+ influx via SOCE
One important distinction between NCLX and MCU is that the latter is solely a Ca2+ transporter while the former mediates exchange of three Na+ per Ca2+ ion and therefore exerts dual control of Ca2+ and Na+ transport (Pitts, 1979; Reeves & Hale, 1984). We asked whether Na+ plays a role in regulating SOCE by comparing the SOCE responses in cells superfused with bath solutions containing either physiological concentrations of Na+ (140 mM) or an equal concentration of NMDG+, a large monovalent cation that is not transported. Notably, replacing extracellular Na+ with NMDG+ led to a threefold decrease in store‐operated Ca2+ influx rate (Fig 5A) [5E‐03 versus 1.53E‐02; ***P < 1E‐03]. However, when Na+ ions were replaced with Li+, a substrate ion for NCLX but not of plasma membrane Na+/Ca2+ exchanger (NCX; Palty et al, 2004), the store‐dependent Ca2+ influx was rescued and was similar to that of the Na+‐containing Ringer solution (1.13E‐02 versus 1.49E‐02, respectively; Fig 5B, lower panel). Furthermore, silencing of NCLX expression combined with replacing Na+ ions in the Ringer's solutions with NMDG+ led to eightfold reduction in Ca2+ influx [1.49E‐02 and 1.8E‐03, respectively; ***P < 1E‐03 (Fig 5B)]. To determine whether intracellular Na+ rise is required for SOCE activation, cytosolic Na+ rise was triggered by blocking the Na+/K+ ATPase by preincubation with ouabain (100 μM; Fig 5C) in the presence of Na+ to load the cytosol with Na+. As expected, application of ouabain in the presence of extracellular Na+ led to an increase in cytosolic Na+ concentrations (Appendix Fig S2). We then compared the rates of store‐dependent Ca2+ influx in cells initially pretreated with ouabain in the presence of Na+ and then superfused with NMDG+ (without Na+) versus untreated cells superfused with either NMDG+ or Na+. Ouabain pretreatment in cells subsequently superfused with NMDG+‐containing bath solution had a similar effect of supporting SOCE to that of extracellular Na+ (Fig 5C, upper panel). Thus, preloading the cytosol with Na+ is sufficient for restoring store‐dependent Ca2+ influx rate in NMDG+‐containing bath solutions to similar values as in Na+‐containing bath solutions (1.78E‐02 and 1.41E‐02, respectively; Fig 5C, lower panel), but not in cells superfused with NMDG+ without preincubation with ouabain (8.5E‐03, **P < 0.01). This set of experiments indicates that the cytosolic rise in Na+ is required for full activation of SOCE.
Figure 5. Extracellular Na+ signaling controls Ca2+ influx through SOCE and CRAC channels.
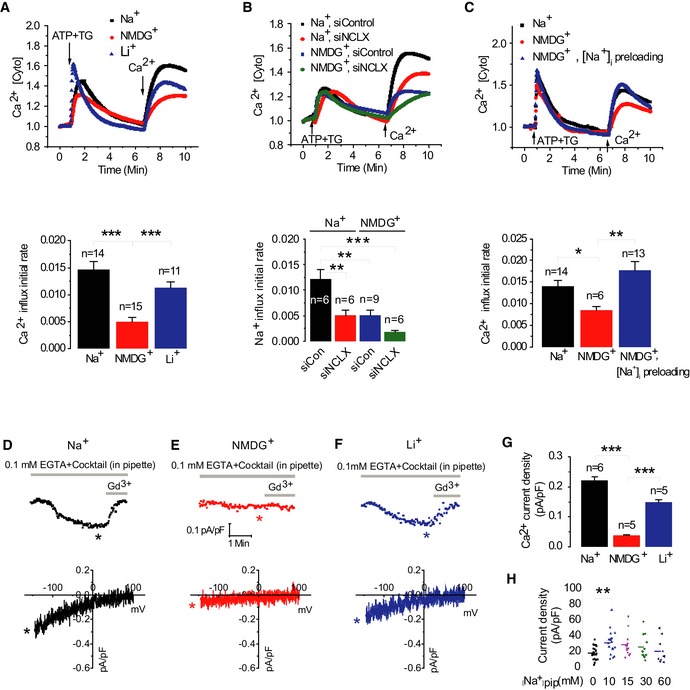
-
A, BUpper panel: Cytosolic Ca2+, Ca2+ [Cyto], responses monitored in HEK293T cells after store depletion by ATP and TG as in Fig 1B and upon extracellular Na+ ions replacement by either NMDG+ or Li+ ions. Lower panel: Averaged rates of cytosolic Ca2+ influx. The combined effects of NMDG+ together with silencing of NCLX on cytosolic Ca2+ influxes via SOCE are presented in (B).
-
CUpper panel: After being loaded with Fura‐2, intracellular Na+, [Na+]i was preloaded by preincubation for 20 min with ouabain (100 μM) in the presence of Na+, following loading of intracellular Na+ SOCE protocol was applied as in Fig 1 using NMDG+‐containing Ringer. Lower panel: Averaged Ca2+ influx rates in NMDG+ [Na+]i preloaded cells (n = 6), Na+ control cells (n = 14), and cells perfused with NMDG+ alone (n = 13).
-
D–HRepresentative Ca2+ CRAC current recordings and corresponding color‐matched I–V relationships measured in HEK293T cells with different monovalent cation‐based bath solution is shown in (D) for Na+, in (E) for NMDG+, and in (F) for Li+; 20 mM Ca2+ was included in all bath solutions. Statistical analysis on Ca2+ CRAC currents measured at −100 mV in all three conditions is shown in (G). The effects of cytosolic Na+ concentration on Ca2+ CRAC currents. (H) The whole‐cell patch clamp recordings were performed in HEK293T cells transiently expressing eYFP‐STIM1 and CFP‐Orai1. Scatter plots represent Ca2+ CRAC currents activated by dialysis through the patch pipette of a solution containing 20 mM BAPTA and the different concentration of Na+. Currents were measured at −100 mV and shown as current density pA/pF.
We then asked whether a rise in cytosolic monovalent cations will have a similar effect on CRAC currents in HEK293T cells. Bath solutions containing Ca2+ (20 mM) in the presence of the indicated cations (Na+, NMDG+ or Li+) were used to measure Ca2+ CRAC currents. The pipette solution contained 0.1 mM EGTA, IP3 to deplete stores, together with the mitochondrial‐energizing cocktail described above. As shown in Fig 5D–G, the presence of either Na+ or the NCLX substrate Li+ in the bath solution led to development of inwardly rectifying Ca2+ CRAC currents that were blocked by 5 μM Gd3+. However, replacing Na+ in the bath solution with NMDG+ inhibited Ca2+ CRAC current activation in response to store depletion by IP3. A scatter blot representation of these data showing individual recordings with mean ± SE is shown in Fig EV3.
Figure EV3. Substitution of Na+ by NMDG+ inhibits CRAC currents in HEK293T and RBL cells.
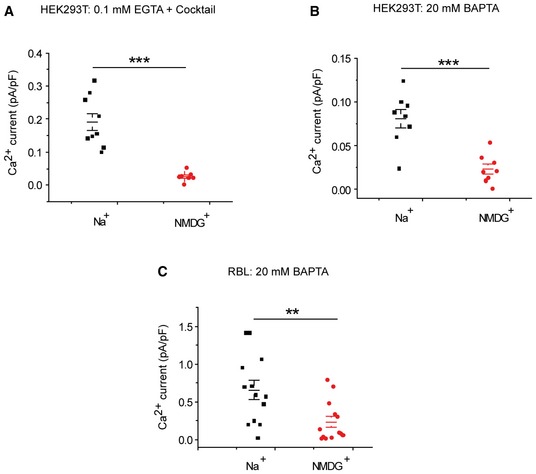
-
A–CScatter plots representing Ca2+ CRAC currents activated either by dialysis through the patch pipette of a solution containing either 0.1 mM EGTA+IP3 (A) or 20 mM BAPTA (B) in HEK293T cells, or in RBL cells activated by dialysis of a solution containing 20 mM BAPTA (C). P‐values (A‐C) indicate the results of an unpaired student's t‐test.
We obtained similar results when CRAC currents were activated in either HEK293T cells (Fig EV3A and B) or rat basophilic leukemia cells (RBL; Fig EV3C). Recordings in HEK293T cells were performed under two conditions: either with store depletion achieved by inclusion of IP3 and mitochondrial‐energizing cocktail together with 0.1 mM EGTA in the patch pipette (Fig EV3A) or with 20 mM BAPTA in the patch pipette (Fig EV3B) in the presence of either extracellular Na+ or NMDG+. Although CRAC current was consistently inhibited by extracellular NMDG+ under all conditions, extracellular NMDG+‐mediated inhibition of CRAC current was more pronounced when CRAC currents were activated by 0.1 mM EGTA together with 30 μM IP3 (Fig EV3A and B). Note that the slightly smaller potency of Li+ versus Na+ in stimulating CRAC and SOCE is consistent with the lower exchange rate of Li+ versus Na+ mediated by NCLX (Carafoli et al, 1974). This corresponds to the functional fingerprint of NCLX and reveals that NCLX is the integrating component between Na+ signaling and SOCE (Palty et al, 2004).
To determine the role of intracellular Na+ in mediating the action of CRAC current, we performed another set of experiments in which HEK293T cells were co‐transfected with STIM1 and Orai1 to generate large CRAC currents. Using these cells, whole‐cell patch clamp recordings were performed after store depletion induced by dialysis with 20 mM BAPTA through the patch pipette together with bath solution containing 135 mM Na+ and 20 mM Ca2+ in the presence of varying concentrations of Na+ in the patch pipette (Fig 5H). HEK293T cells co‐expressing STIM1 and Orai1 exhibited enhanced CRAC currents when the pipette solution contained 10 mM or higher Na+ concentrations (Fig 5H), providing additional support for the role of Na+ in optimal CRAC current activation.
Additional support for the cross talk between Na+ and Ca2+ ions on CRAC channel function was derived from direct monitoring of Na+ influx, applying the SOCE protocol to cells loaded with the fluorescent Na+ dye Asanta Natrium (Fig 6A and B). Depletion of Ca2+ stores triggered a 4.5‐fold rise in Na+ influx rate compared to control cells with intact Ca2+ stores [2.60E‐04 and 5.87E‐05, respectively, **P < 0.01] (Fig 6A, lower panel). This finding is consistent with previous studies documenting store‐dependent Na+ influx (Poburko et al, 2009). Na+ influx was observed only in the presence of extracellular Na+ indicating that it reflects Na+ permeation into cells and not release form putative intracellular Na+ stores (Fig 6B, upper panel) [2E‐04 versus ‐3E‐05, **P < 0.01] (Fig 6B, lower panel). We further asked whether the cytosolic Na+ rise is propagating into the mitochondria by monitoring changes in mitochondrial Na+ in cells preloaded with the mitochondrial Na+ reporter, CoroNa red (Jayaraman et al, 2001a,b). As shown in Fig 6C, SOCE activation was linked to a strong mitochondrial Na+ influx, consistent with the major role of NCLX in mediating mitochondrial Na+ uptake (Palty et al, 2010; Nita et al, 2014a). Mitochondrial Na+ uptake was however reduced in cells superfused with NMDG+ Ringer compared to cells superfused with Na+‐containing Ringer, (2.3E‐04 and 5E‐04, respectively, *P < 0.05; Fig 6C), indicating that mitochondrial Na+ influx is preceded by Na+ influx into the cell across the plasma membrane. Previous studies demonstrated that NCLX is tuned to sense small changes in cytosolic Na+ and is therefore strongly activated by a rise in cytosolic Na+ triggered, for example, by voltage gated Na+ channels in pancreatic β cells (Nita et al, 2014a). We therefore asked whether Na+ influx is activating SOCE by triggering activation of NCLX. We monitored mitochondrial Ca2+ extrusion via the mitochondrial NCLX (Palty et al, 2010) by performing the SOCE protocol as described in Fig 1 while monitoring mitochondrial Ca2+ in the absence or presence of extracellular Na+. The presence of Na+ ions in the Ringer solution strongly accelerated mitochondrial Ca2+ efflux (Fig 6D, upper panel). In contrast, mitochondrial Ca2+ efflux rate was reduced by 2.5‐fold in the presence of NMDG+ compared to cells that were superfused with Na+‐containing Ringer (2.26E‐04 and 5.7E‐04, respectively, **P < 0.01, Fig 6D, lower panel).
Figure 6. Depletion of intracellular Ca2+ stores triggers cytosolic Na+ influx which drives NCLX‐associated mitochondrial Na+ influx and Ca2+ efflux.
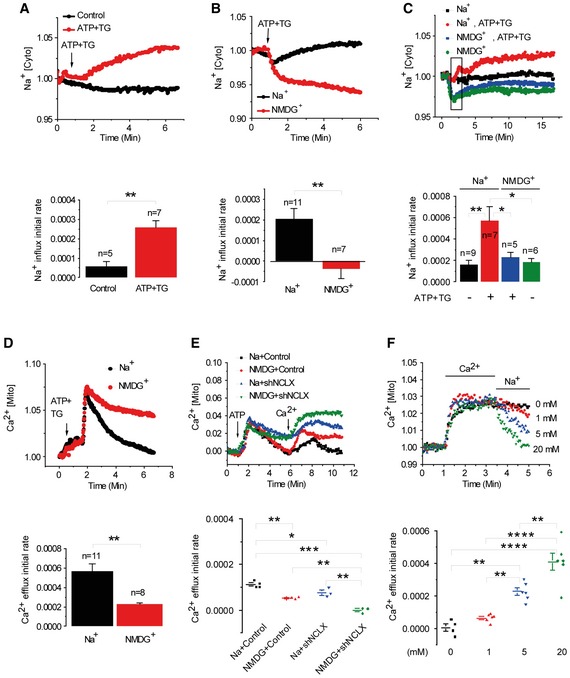
- Upper panel: Fluorescence traces of cytosolic Na+ responses, Na+ [Cyto], in ATP‐depleted cells (n = 7) versus control non‐depleted HEK293T cells (n = 5) loaded with Asanta Natrium. Na+ influx averaged rates are shown in the lower panel.
- Upper panel: Fluorescence traces of cytosolic Na+. Lower panel: Averaged rates of cytosolic Na+ influx in cells superfused with either Na+‐ (n = 11) or NMDG+ (n = 7)‐containing Ringer's solution.
- Upper panel: Traces of mitochondrial Na+ transport in cells preloaded with the mitochondrial Na+ dye, CoroNa red in HEK293T cells. Lower panel: Mitochondrial Na+ influx rates in the presence (n = 7) or absence (n = 5) of Na+.
- Upper panel: Traces of mitochondrial Ca2+, Ca2+ [Mito], recorded by monitoring RP‐mt fluorescence following Ca2+‐store depletion in HEK293T cells by ATP and TG as in Fig 1B in the presence of Na+ or NMDG+. Lower panel: Mitochondrial Ca2+ efflux in cells in the presence of NMDG+ (n = 8) compared to Na+ (n = 11).
- Upper panel: Traces of Ca2+ [Mito] of cell superfused with ATP alone in Ca2+‐free Ringer followed by superfusion with Ca2+ Ringer with or without Na+ in shControl cells versus shNCLX cells. Lower panel: Rates of traces shown in the upper panel in the presence (n = 4) or absence (n = 3) of Na+ ions in cells transfected with shControl (n = 3) or shNCLX (n = 3).
- Upper panel: Cytosolic Na+‐dose dependence of Ca2+ [Mito] efflux. Lower panel: Mitochondrial Ca2+ efflux rates at the indicated concentrations of Na+ [mM] [0 (n = 4); 1, 5 and 20 (n = 6)].
Thus far, we determined that the mitochondrial Ca2+ efflux triggered by robust store depletion using ATP+TG is Na+ dependent. Next, we sought to determine whether this is also the case when SOCE is activated using more physiological means (i.e. with purinergic ATP stimulation alone; Fig 6E, upper panel). As expected, absence of Na+ led to twofold lower ATP‐activated mitochondrial Ca2+ efflux than the control (with Na+; 1.13E‐04 and 5.13E‐05, respectively, **P < 0.01, Fig 6E, lower panel) and the combination of using NMDG+ Ringer together with silencing NCLX resulted in the lowest Ca2+ efflux rate (−1.02E‐06). Silencing of NCLX alone shows 3.6‐fold decrease in the Ca2+ efflux rate versus control (7.85E‐04 and 2.18E‐04, respectively; Fig EV4). As shown in Fig 6F, mitochondrial Ca2+ efflux following mitochondrial Ca2+ entry is strictly dependent on the presence of Na+, consistent with our previous studies (Nita et al, 2014a). Na+ dose dependence analysis of mitochondrial Ca2+ efflux revealed an apparent Km of 9.7 mM in HEK293T cells (Fig 6F), in agreement with values previously reported for cardiac myocytes (Cai et al, 2016) and consistent with Na+ physiological concentrations, which are typically within 4–16 mM range (Bers et al, 2003). Altogether our results indicate that Ca2+ influx through the SOCE pathway requires a cytosolic Na+ rise to propagate within mitochondria through the exchange activity of NCLX which activates mitochondrial Ca2+ efflux. The results further suggest that this Na+ is required for activating mitochondrial Ca2+ shuttling by NCLX hence ensuring optimal activation of SOCE and CRAC currents.
Figure EV4. Knockdown of NCLX expression inhibits mitochondrial Ca2+ efflux and Na+ uptake.
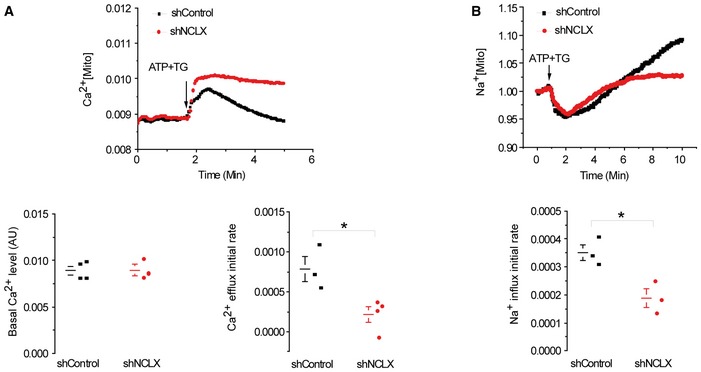
- Upper panel: Mitochondrial Ca2+ was monitored in cells expressing the mitochondrial Ca2+ sensor RP‐mt. Mitochondrial Ca2+ transient was evoked by application of ATP and TG, added when indicated. Lower right panel: Rates of mitochondrial Ca2+ efflux derived from the upper panel. Lower left panel: The basal Ca2+ level (arbitrary units, AU) of shControl (n = 3) versus shNCLX (n = 4).
- Upper panel: Mitochondrial Na+ uptake in HEK293T cells preloaded with the mitochondrial Na+ sensor, CoroNa red. Na+ signals were evoked by ATP and TG added when indicated. Lower panel: Rates of mitochondrial Na+ influx derived from the upper panel.
SOCE and CRAC current are regulated by mitochondrial redox
Mitochondrial redox state is elicited by mitochondrial Ca2+ transients and in turn regulates SOCE. We reasoned that the NCLX knockdown effect on SOCE is mediated by mitochondrial redox (De Marchi et al, 2014). To determine whether NCLX knockdown affects mitochondrial redox state, we applied the SOCE protocol as described in Fig 1 in cells expressing the redox sensor roGFP1 targeted to the mitochondrial matrix. Store depletion by ATP and thapsigargin (TG) in the absence of external Ca2+ followed by addition of Ca2+ to the bath solution, triggered transient changes in the mitochondrial redox state, temporarily reducing mitochondrial free oxygen radical levels in the mitochondrial matrix (Fig 7A). In contrast, knockdown of NCLX expression was followed by a twofold inhibition of this redox change (Fig 7A). Consistent with the role of NCLX in controlling the mitochondrial redox state, the knockdown of NCLX was followed by a fivefold lower NADH/NAD+ ratio, monitored by intrinsic fluorescence (2.4E‐04 and 1.2E‐03, respectively, *P < 0.05; Fig 7B, lower panel). These results indicate that the redox transients maintained by NCLX limit free radical bursts and thereby prevent oxidant‐mediated inhibition of SOCE/CRAC.
Figure 7. Mitochondrial catalase (m‐catalase) rescues SOCE, CRAC, and mitochondrial redox responses after NCLX knockdown.
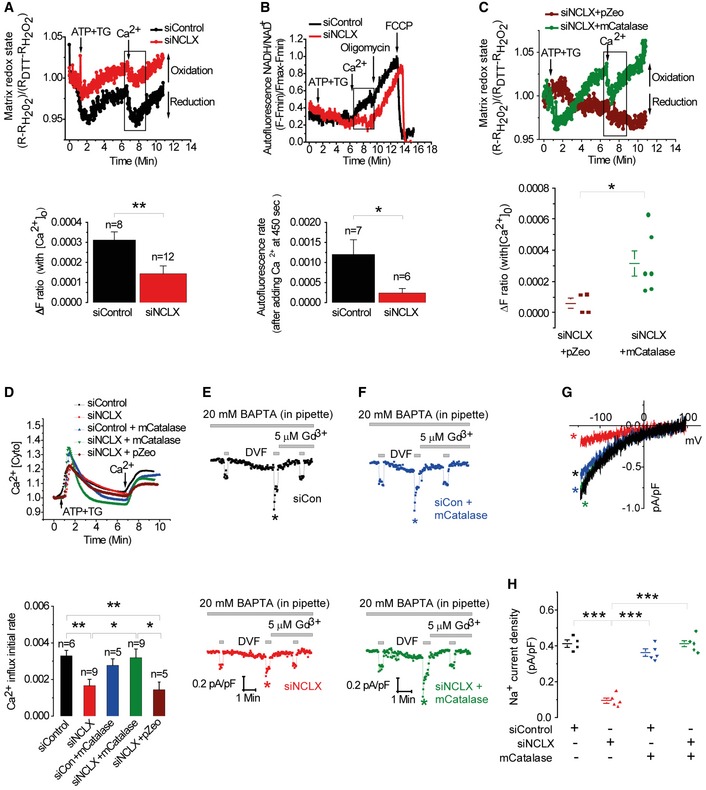
-
AUpper panel: Traces of mitochondrial roGFP1 fluorescence in HEK293T cells co‐transfected with either siControl or siNCLX and Ca2+‐ store depleted as described in Fig 1B. Changes in roGFP1 410/480 ratio fluorescence were determined using minimal and maximal values obtained by application of 100 mM DTT and 10 mM H2O2, respectively, as described in Materials and Methods. Lower panel: ΔF ratio of redox change after Ca2+ restoration to the external milieu in siControl cells (n = 8) compared to siNCLX cells (n = 12).
-
BUpper panel: Effect of NCLX on redox state determined by monitoring NAD(P)H intrinsic fluorescence in HEK293T cells, transfected with either siNCLX or siControl, and treated as described in Fig 1B. Oligomycin or FCCP were used for calibration and added where indicated. Lower panel: Averaged autofluorescence rates after adding Ringer's solution containing Ca2+.
-
CUpper panel: Effect of mitochondrial catalase (pZeoSV2 + mCat) expression. Lower panel: Rates of redox change in siNCLX + pZeoSV2 + (pZeo) empty vector transfected cells (n = 4) compared to siNCLX cells transfected with m‐catalase (n = 6).
-
DUpper panel: Fluorescence traces of cytosolic Ca2+ responses in HEK293T cells co‐transfected with either siControl or siNCLX together with pZeo or m‐catalase. Lower panel: Averaged rates of Ca2+ influx in cells transfected with siNCLX (n = 9) or cells co‐transfected with siNCLX and either m‐catalase (n = 9) or pZeo (n = 5) compared to cells transfected with siControl (n = 6) or co‐transfected with siControl and m‐catalase (n = 5).
-
E–HCRAC electrophysiological recordings in HEK293T cells activated by dialysis of 20 mM BAPTA through the patch pipette under the same conditions of transfection used in (B). I–V relationships are shown in (G) and statistical analysis on Na+ CRAC currents measured at −100 mV is shown in (H). m‐Catalase is routinely co‐transfected with a plasmid encoding eGFP for identification of transfected cells.
Hence, the SOCE/CRAC activity suppressed by NCLX knockdown could be rescued by mitochondrial targeted expression of an H2O2‐metabolizing enzyme, mitochondrial catalase (m‐catalase). We determined whether m‐catalase can restore the mitochondrial redox response in NCLX‐silenced cells. Note that the m‐catalase expression fully rescued the SOCE‐dependent increase in reduced state of the mitochondrial matrix (Fig 7C). We then compared SOCE rate in siControl versus siNCLX cells with or without m‐catalase expression. Remarkably, overexpression of m‐catalase in NCLX knockdown cells was sufficient to recover SOCE activity and Ca2+ influx rate (Fig 7D). Further support for the link between Na+ and NCLX in controlling SOCE by redox is demonstrated by our finding that expression of m‐catalase rescued SOCE even in the absence of extracellular Na+ (Fig EV5).
Figure EV5. Na+ has no effect on SOCE in the presence of m‐catalase.
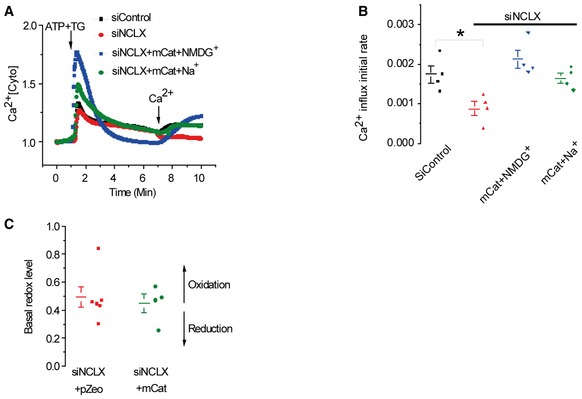
-
AHEK293T cells were transfected with either siControl or siNCLX with or without m‐catalase. Cytosolic Ca2+ responses were monitored in HEK293T cells after store depletion by ATP and TG as in Fig 1B in the presence or absence of Na+ ions.
-
B, CAveraged rates (means ± SEM) of Ca2+ influx in either siControl cells (n = 4), siNCLX (n = 4), siNCLX+m‐catalase with Ringer containing Na+ (n = 4), or NMDG+ (n = 4) are shown in (B). P‐values indicate the results of a one‐way ANOVA test followed by Tukey post hoc analysis. *P < 0.05. The basal redox levels of siNCLX+pZeo cells (n = 6) versus siNCLX+m‐catalase (n = 4) are shown in (C).
We then asked whether the rescue of the mitochondrial redox transient by m‐catalase could restore CRAC currents suppressed by knockdown of NCLX expression. Consistent with the above SOCE results (Fig 7C and D), we observed that knockdown of NCLX expression was followed by a strong inhibition of CRAC current (Fig 7E–H). Remarkably, targeting catalase to the mitochondria was sufficient to rescue CRAC currents in cells in which NCLX was knocked down (Fig 7F and H). We compared the basal redox state of siNCLX cells vs siNCLX cells expressing m‐catalase and found that siNCLX cells are slightly more oxidized, although this difference was not significant (Fig EV5C). Together, our results strongly support a novel paradigm whereby NCLX control of the mitochondrial redox state participates in mitochondrial communication with SOCE.
Since our results (Fig 2) indicate that the NCLX effect on SOCE is not mediated via disruption of STIM1 and Orai1 interactions, we reasoned that the mitochondria redox response evoked by NCLX is targeting a redox‐sensitive residue on Orai1. Orai1 possesses three cysteine residues potentially modified by oxidants, Cys126, Cys143, and Cys195. One of the Orai1 cysteine residues, Cys195, has recently emerged as a redox responsive element within this channel (Bogeski et al, 2010). We therefore focused on Cys195 within Orai1 and compared the wild‐type SOCE response (Fig 8A) to the SOCE response of either Orai1 or Orai1 C195S coexpressed with STIM1 in either siControl‐ or siNCLX‐transfected HEK293T cells (Fig 8B and C). Remarkably, while the silencing of NCLX was followed by reduction in native as well overexpressed Orai1‐dependent Ca2+ influx, mutating Cys195 to serine (C195S) fully rescued Orai1‐dependent SOCE in NCLX knockdown cells (Fig 8C). To establish the role of this residue in mediating the NCLX‐dependent redox effect on Orai1, we analyzed Orai1‐mediated CRAC currents in HEK293T cells expressing STIM1 with either Orai1 or the Orai1‐C195S mutant (Fig 8D–I) while depleting internal stores and strongly buffering cytosolic Ca2+ with 20 mM BAPTA, as defined in Fig 1. Consistent with data obtained with SOCE measurements using Fura‐2, the inhibitory effect of NCLX knockdown was also observed on CRAC currents in cells over‐expressing STIM1 and Orai1. In contrast, mutating the redox‐sensitive cysteine 195 to serine (C195S) fully rescued Orai1‐mediated CRAC currents following the knockdown of NCLX. Mutation of all three cysteines Cys126, Cys143, and Cys195 in Orai1 to serine also fully rescued Orai1‐mediated CRAC currents following the knockdown of NCLX (Fig 8J–L). CRAC currents mediated by this triple Orai1 mutant were significantly bigger than those mediated by either Orai1 or Orai1‐C195S (Orai1; 17.62, C195S‐Orai1; 16.52, C126S/C143S/C195S‐Orai1; 51.59). Taken together, our results indicate that the mitochondrial redox transients controlled by NCLX prevent CRAC channel inhibition mediated by oxidation of cysteine 195 on Orai1.
Figure 8. C195S mutation in Orai1 prevents the inhibitory effect of SOCE and CRAC current caused by NCLX knockdown.
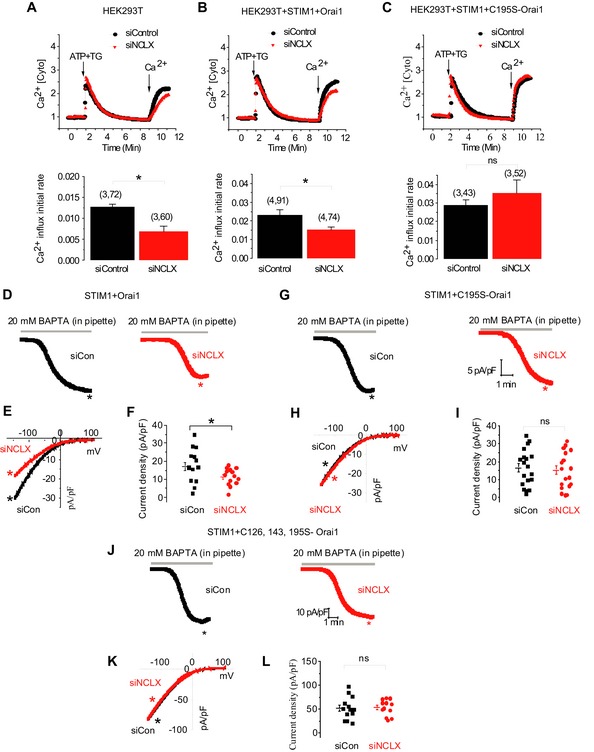
-
A–CHEK293T cells were transfected with either control siRNA (black) or NCLX siRNA (red) and incubated for 72 h (A). Cells were then transfected again with siRNA along with either plasmids, STIM1 and Orai1 (B), or plasmids, STIM1 and C195S‐Orai1 (C), and incubated for another 24 h. The numbers in bar graphs (i.e., (x, y)) represent the total number of independent recording (x) and the total number of cells from all recordings (y). SOCE was triggered as described in Fig 1B, and Fura‐2 fluorescence was monitored as described in Materials and Methods. Averaged rates of Ca2+ influx were shown in the lower panels.
-
D–LRepresentative time course traces (D and G) of Orai1‐ and Orai1‐C195S‐mediated CRAC currents activated by dialysis through the patch pipette of a solution containing 20 mM BAPTA and recorded in a bath solution containing 20 mM Ca2+. I–V curves are shown in (E and H) which are taken from traces as indicated by color‐coded asterisks. Scatter blots (F and I) depict maximal CRAC currents values taken at −100 mV and represented as current densities (pA/pF). Similar CRAC current recordings from HEK293T cells expressing STIM1 and C126S/C143S/C195S‐Orai1 triple mutant are shown in (J–L).
Discussion
Although a role for mitochondrial Ca2+ signaling in regulating SOCE is well established, the mode of communication between these two domains is not understood. The recent identification of the mitochondrial Ca2+ transporters enables more selective molecular tools to specifically control their activity and study their role in controlling SOCE. In this study, we have focused on NCLX whose rate of mitochondrial Ca2+ removal is two orders of magnitude slower than the mitochondrial Ca2+ influx. Thus, NCLX is the rate limiting component in mitochondrial Ca2+ shuttling. NCLX is also a Na+ transporter that mediates the exchange of 3 Na+ per 1 Ca2+ making it the major mitochondrial Na+ uptake pathway (Baysal et al, 1994; Jung et al, 1995). Na+ transport was previously linked to SOCE. Thus, it was proposed that Na+ influx occurs through concomitant activation of non‐selective TRPC cation channels in response to pharmacological or receptor‐mediated store depletion (Parekh & Putney, 2005). However, the role of cytosolic Na+ in regulating the highly Ca2+ selective Orai1 channel is still poorly understood. In this study, we sought to address the following questions. Does NCLX control SOCE‐dependent Ca2+ signaling? And, if so, what is the mechanism of NCLX‐mediated regulation of SOCE? Our results indicate that NCLX is required for SOCE and support a redox effect of NCLX on CRAC channels that is independent of mitochondrial Ca2+ buffering.
An important mode of cross talk between CRAC channels and mitochondria is based on the ability of mitochondria to control Ca2+ concentrations at the vicinity of the CRAC channel thereby decreasing their Ca2+‐dependent inactivation. Such interaction between the mitochondria and CRAC channels is particularly prominent in immune cells (Rizzuto et al, 1998, 2012; Alvarez et al, 1999; Berridge et al, 2003; Singaravelu et al, 2011), in which inhibition of MCU and, more recently, molecular knockdown of MCU have demonstrated that SOCE is inhibited by the block of mitochondrial Ca2+ uptake in immune cells (Gilabert et al, 2001; Ma & Beaven, 2011). In other cell types, however, the proximity of the mitochondria to CRAC channels is less pronounced (Naghdi et al, 2010). Studies employing organellar Ca2+ reporters targeted, for example, to the outer mitochondrial membrane and the plasma membrane suggest that mitochondria are not mediating strong Ca2+ changes at the vicinity of the CRAC channel (Giacomello et al, 2010). Furthermore, inhibition of mitochondrial efflux by the NCLX inhibitor CGP‐37157b enhances mitochondrial Ca2+ uptake rather than decreasing it and yet leads to similar inhibition of SOCE. These findings indicate that in addition to the Ca2+‐dependent inactivation, there is an additional missing link that participates in the cross talk between the mitochondria and SOCE. The first question that we have addressed is whether a Ca2+‐dependent inactivation or a distinct regulatory mode is solely responsible for the cross talk between NCLX and SOCE. If Ca2+‐dependent inactivation is the predominant mode at play, then buffering of cytosolic Ca2+ should rescue CRAC currents when NCLX is silenced. Our results indicate that Ca2+ buffering is not involved in NCLX‐mediated control of SOCE. While it may be argued that chelation by EGTA is not sufficiently strong or rapid, we have used the stronger and faster Ca2+ chelator BAPTA at high concentrations (up to 50 mM) and failed to rescue CRAC currents upon NCLX knockdown.
Our results further indicate that store‐associated activation of Na+ influx across the plasma membrane is essential for NCLX function thereby maintaining SOCE and CRAC currents (Fig 9) based on our following findings. First, consistent with previous results (Rosker et al, 2004; Eder et al, 2007; Lemos et al, 2007; Demaurex et al, 2009), we find that store depletion is followed by cytosolic Na+ influx which is then transported by NCLX to the mitochondria. Second, we find that the SOCE‐associated cytosolic Na+ influx is required to activate NCLX‐dependent mitochondrial Ca2+ extrusion. The requirement for cytosolic Na+ rise for activation of NCLX is consistent with the low affinity of this exchanger for Na+ such that under resting cytosolic Na+ levels, NCLX will be inactive while a small increase in cytosolic Na+ will activate a large mitochondrial Ca2+ efflux by NCLX. The rise of cytosolic Na+ in response to store depletion can occur, for example, by the well described Ca2+‐dependent activation of non‐selective TRPC cation channels (Lemos et al, 2007; Lee et al, 2010). Thus, NCLX can be physiologically considered as a Na+‐activated Ca2+ exchanger. Consistent with this hypothesis, we also found that extracellular Na+ is required to fully activate CRAC channels. Thus, Na+‐dependent activation of NCLX is required for full activation of SOCE. Perhaps most remarkably, Li+, the substrate cation of NCLX but not of plasma membrane NCX members, can effectively replace Na+ in supporting SOCE and CRAC currents. Notably, SOCE and CRAC channel activity monitored in the presence of Li+ is slightly lower than that of Na+ thus bearing the exact monovalent cation finger print of NCLX selectivity (Carafoli et al, 1974; Palty et al, 2004). The latter finding also provides strong evidence for a distinct role of NCLX in the regulation of CRAC currents because plasma membrane NCX members are inert to Li+. However, it is important to note that the reverse mode of the plasma membrane NCX triggered by a SOCE‐associated Na+ rise (e.g. through TRPC6) is also contributing to cytosolic Ca2+ rise (Borin et al, 1993; Lemos et al, 2007, 2007). Unlike NCLX, the NCX effect on cytosolic Ca2+ is not mediated through direct action on CRAC conductance. Therefore, our current and previous studies (Borin et al, 1993; Lemos et al, 2007; Nita et al, 2014a,b) indicate that NCLX and NCX concertedly amplify SOCE‐dependent cytosolic Ca2+ signals but through different mechanisms. For NCX, the SOCE‐dependent Na+ rise triggers its reverse mode leading to increased cytosolic Ca2+ while for NCLX it triggers forward activation that can lead to maintenance of CRAC channel activity (Fig 9). How does the powering of NCLX by cytosolic Na+ culminate in the activation of SOCE and CRAC currents? Mitochondria are not only the metabolic engine and a major hub for Ca2+ signaling, they function as major regulators of cellular redox potential (Szabadkai & Duchen, 2008; Nunes & Demaurex, 2014) that link between activation of NCLX to SOCE and CRAC currents.
Figure 9. Na+ and Ca2+ signals are regulated by NCLX and mediate mitochondrial redox control of SOCE.
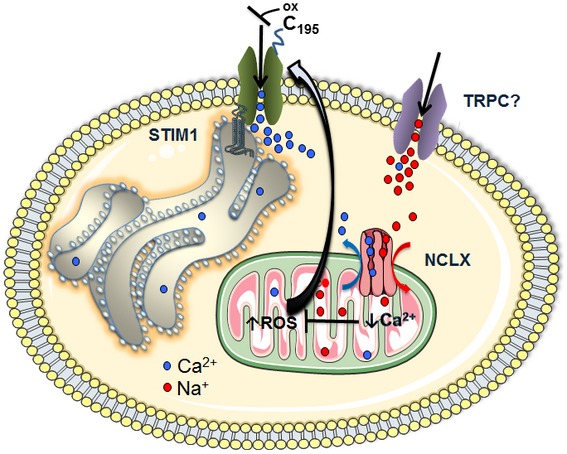
A schematic illustration of the Na+ and Ca2+ signaling networks involving NCLX and SOCE cross talk. Ca2+ depletion of intracellular stores activates store‐operated Ca2+ entry (SOCE) pathway mediated through the highly Ca2+ selective, Ca2+ release‐activated Ca2+ (CRAC) currents. SOCE activation involves interaction upon store depletion of the endoplasmic reticulum (ER) Ca2+ STIM1 with the plasma membrane channel protein Orai1. Upon store depletion, rise of cytosolic Na+ also occurs, apparently, reflecting the activation of non‐selective transient potential channel canonical (TRPC) channel isoforms. Cytosolic Na+ is required for powering NCLX to activate mitochondrial Ca2+ efflux, thus ensuring proper mitochondrial Ca2+ shuttling. This tight regulation by NCLX of mitochondrial matrix Ca2+ homeostasis prevents the excessive Ca2+ rise in mitochondrial matrix that would otherwise lead to increases in mitochondrial reactive oxygen species (ROS) and subsequent inhibition of SOCE via oxidation of a reactive cysteine at position C195 on an extracellular loop of Orai1.
Our results indicate that NCLX regulation of SOCE persists following the knockdown of MCU. Although the reason for this observation is not entirely clear, it is important to note that mitochondrial Ca2+ transients initiated by MCU reflect only ~5% of the total mitochondrial Ca2+. The rest, ~95% of the mitochondrial Ca2+ pool, is an insoluble/soluble pool consisting mainly of the phosphate Ca2+ salt. The solubility of the latter pool is strongly pH dependent and therefore any change with matrix pH; for example, metabolic activity will trigger a matrix‐free Ca2+ change and thereby a change in mitochondrial redox. Affinity and sensitivity of the mitochondrial Ca2 sensitive dyes are, however, less than optimal (Pizzo et al, 2012). Therefore, these changes are often left unnoticed. Consistent with this hypothesis, our pervious and current results show that the apparent duration of mitochondrial Na+ fluxes (which in contrast to Ca2+ is unbuffered) is much longer than mitochondrial Ca2+ transients (Palty et al, 2010; Nita et al, 2014a), indicating that NCLX is pumping out Ca2+ for much more extensive periods than the “apparent” mitochondrial Ca2+ transients. Finally, in contrast to MCU, NCLX is dually linked to Ca2+ and Na+ signaling which we show here is critical for redox regulation and the SOCE response.
Regulation by redox has recently emerged as a critical mode of SOCE regulation (Bogeski et al, 2010; Hawkins et al, 2010; Nunes & Demaurex, 2014).
There are several studies which show regulation of m‐catalase (Naziroglu, 2012; Littlejohns et al, 2014). However, the H2O2 clearing rate of catalase is mainly dependent on the levels of expression of catalase (Rodriguez et al, 2000). Therefore, over‐expressed catalase targeted to the mitochondria (as described here) is expected to enhance H2O2 detoxification in the mitochondria generated by abnormal mitochondrial Ca2+ rise.
Our previous results indicate that expression and activity of NCLX are critical in triggering a mitochondrial redox signal (De Marchi et al, 2014). Our current results demonstrating that catalase rescues the mitochondrial redox response and CRAC/SOCE in NCLX‐deficient cells, suggest that diffusible H2O2 is the link between NCLX and SOCE (Fig 9). While buffering of cytosolic Ca2+ failed to rescue SOCE and CRAC currents in NCLX knockdown cells, expression of mitochondrial catalase leads to a full recue of SOCE in NCLX knockdown cells. It may be argued that this effect is indirect and associated, for example, with ER Ca2+ release. However, our electrophysiological analysis in cells expressing the mitochondrial catalase also show rescue of CRAC currents. Redox potential is a key regulator of SOCE that can act on multiple targets in this pathway. The oligomerization of STIM1 and its interaction with Orai1 are redox‐sensitive. However, our results indicate that the oligomerization of STIM1 and its interaction with Orai1 (as well as their expression) are not affected by knockdown of NCLX, arguing against such a scenario. Instead, our results suggest that the target of the mitochondrial redox response is an Orai1 cysteine residue (Cys195). Oxidation of the latter by H2O2, leads to CRAC channel inhibition (Bogeski et al, 2010). Furthermore, our finding showing that substitution of cysteine 195 in Orai1 with serine (C195S) is sufficient to rescue SOCE and CRAC currents indicate that redox‐dependent regulation of SOCE by mitochondria is mediated through cysteine 195 of Orai1 (Fig 9).
In conclusion, the results of this study identify a long sought and novel mode of interaction between the mitochondria and SOCE. This pathway integrates Na+ and Ca2+ signals that converge on the mitochondrial NCLX Na+/Ca2+ exchanger, to modulate cellular redox which controls SOCE and CRAC currents, mediated by a redox‐sensitive site on Orai1 (Fig 9).
Materials and Methods
Cell culture and transfection
HEK293T cells (human embryonic kidney cell line) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1% penicillin/streptomycin, 2 mM l‐glutamine. Transfection of HEK293T cells was performed using the CaPO4 precipitation protocol in cultures of 30–50% confluence, as previously described (Palty et al, 2004). Vascular smooth muscle cells (VSMCs) were cultured on glass coverslips and transfected with DharmaFECT 1 (Dharmacon, T‐2001). SiRNA NCLX or siControl was diluted in DharmaFECT siRNA transfection reagent, incubated ~20 min at room temperature and then added in the antibiotics‐free media. The efficiency of transfection was assessed by visualizing Dharmacon siGLO Red (Dharmacon, D‐001630‐02‐05) transfection particles according to the protocol provided by manufacturer. The transfection efficiency, for siNCLX delivery as determined by siGlo fluorescent marker was high, ~90%. VSMCs were isolated as previously described (Potier et al, 2009) and maintained in culture 45% DMEM and 45% Ham's F12 with 10% FBS supplemented with l‐glutamine) at 37°C, 5% CO2, and 100% humidity, passaged and used within 1–5 passages. Rat basophilic leukemia (RBL‐2H3) mast cells were obtained from ATCC, cultured in EMEM with 2 mmol/l l‐glutamine and 10% FBS and maintained in a 37°C, 5% CO2 humidified incubator as described previously (Abdullaev et al, 2008).
Plasmid and siRNA preparation
Double‐stranded siRNAs used to silence NCLX expression were obtained from Applied Biosystems as previously described (Palty et al, 2010). The human NCLX shRNA plasmid was obtained from Sigma (Mission TRC shRNA Target Set TRCN‐5045). The shMCU and shControl plasmids were a gift from Fabiana Perocchi (TRCN0000133861; Baughman et al, 2011). The membrane‐targeted GCAMP5 was obtained from Addgene [pN1 Lck‐GCaMP5G (Plasmid #34924)]. The RP‐mt plasmid was provided by Atsushi Miyawaki (Wako, Japan; Nagai et al, 2001). The roGFP1 was kindly gifted by Roger Y. Tsien (UCSD; Dooley et al, 2004). The pZeoSV2+ mCat and pZeoSV2+ empty vector were provided by J. Andres Melendez (Albany, USA; McCarthy et al, 2013). The plasmid of eYFP‐STIM1 was kindly gifted by Tobias Meyer; the plasmid of CFP‐Orai1 was provided by James W. Putney; and the plasmids of C195S‐Orai1‐eGFP and C126S/C143S/C195S‐Orai1‐eGFP were constructed by Mohamed Trebak's laboratory. All those tagged or mutant of STIM1 and Orai1 plasmids are used for patch clamp experiments.
Immunoblot analysis
Immunoblot was performed with 100 μg of protein samples obtained from HEK293T cells transfected with either siControl or siNCLX. Detection for NCLX, STIM1, and Orai1 proteins was performed 96 h post‐transfection using specific antibodies; HSC 70 loading control is also shown. Anti‐NCLX polyclonal antibody was generated in our laboratory as previously described (Palty et al, 2010) while anti‐STIM1 monoclonal antibody was from BD Biosciences, anti‐Orai1 polyclonal antibody was from Alomone Labs, and anti‐HSC 70 was from Santa Cruz Biotechnology.
Fluorescent Ca2+ and Na+ imaging
The imaging system consisted of an Axiovert 100 inverted microscope (Zeiss, Oberaue, Germany), Polychrome V monochromator (Till Photonics, Planegg, Germany) and a Sensi‐Cam cooled charge‐coupled device (PCO, Kelheim, Germany). Fluorescence images were acquired with Imaging WorkBench 6.0 software (Axon Instruments, Foster City, CA, USA). Ca2+ imaging was performed in HEK293T and VSMCs cells that were attached onto coverslips and superfused with Ringer's solution containing (in mM): 126 NaCl, 5.4 KCl, 0.8 MgCl2, 20 HEPES, 1.8 CaCl2, and 15 glucose; pH was adjusted to 7.4 with NaOH or NMDG+ in Na+‐free Ringer's solutions (Palty et al, 2010). For cytosolic Ca2+ measurements, HEK293T and VSMCs cells were loaded with Fura‐2 AM (2 μM) for 25 min at room temperature or 4 μM for 45 min at 37°C, respectively (Bisaillon et al, 2010). Cell were excited with 340/380 nm wavelength light and imaged using a 510‐nm long‐pass filter, as described previously (Jonkers & Henquin, 2001). For measurements of plasma membrane‐localized Ca2+ signals, HEK293T cells were transfected with 1 μg of GCamp5 which is targeted solely to the plasma membrane. The membrane‐targeted Gcamp5 was excited at 485 nm, and fluorescence was monitored at 510 nm emission (Akerboom et al, 2012). Mitochondrial Ca2+ measurements were performed in HEK293T cells expressing ratiometric mitochondrial pericam (mitopericam), which is targeted solely to the inner membrane of mitochondria. The mitochondrial pericam fluorescence in HEK293T cells was acquired at 430 nm excitation and 550 nm emission, as described previously (Nagai et al, 2001). Cytosolic Na+ levels were recorded in Asanta Natrium Green‐2 (Teflabs, Jackson Springs, NC, USA)‐loaded HEK293T cells excited with 485 nm and imaged at 510‐nm long‐pass filter. It is well known that CoroNa Red is localized to the mitochondria (Yang et al, 2004; Baron et al, 2005; see in Appendix Fig S3). Therefore, mitochondrial Na+ signals were monitored in cells loaded with CoroNa Red (Molecular Probes) at excitation of 568 nm and emission at 590 nm, respectively (Lemos et al, 2007). Note that at 50 s, Na+ fluorescence falls a bit. This change is apparent in the presence or absence of Na+ and therefore a fluorescence artifact due to addition of solution.
For all single‐cell imaging experiments, traces of averaged responses were analyzed and plotted using Origin Labs software. The fluorescent Na+ or Ca2+ signals were normalized once again to the averaged baseline signal (F/F 0 or R/R 0), obtained at the beginning of the measurements. The influx and efflux rates were derived from a linear fit of the fluorescence change during 30 s (Palty et al, 2010; Nita et al, 2012). Averaged rates of the fluorescent Na+ or Ca2+ responses, over n (indicated in the figure legends) experiments, are presented in the bar graphs and analyzed using Origin software.
Mitochondrial redox state measurements
Ratiometric measurements of the mitochondrial redox state were performed on using the same instrument as described above using the mitochondrial targeted, genetically encoded sensor roGFP1. Cells were exited at 410 and 480 nm and emission was collected at 535 nm. Images were acquired every 2 s. The 410/480 fluorescence ratios were normalized by obtaining the maximum and minimum ratios (obtained after addition of 1 mM H2O2 and of 10 mM DTT, respectively; De Marchi et al, 2014). The fluorescence signals of roGFP1 were normalized to the averaged baseline signal (F/F 0 or R/R 0), obtained at the beginning of the measurements.
Measurements of NADH fluorescence
NAD(P)H intrinsic fluorescence in HEK293T cells was fluorescently monitored (360 nm excitation and 440 nm emission) in an inverted microscope (see Fluorescent Ca2+ and Na+ imaging), as previously reported (Nita et al, 2012) and calibrated by superfusing the cell with a Ringer's solution containing 2 μM FCCP, protonophore carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (Ascent Laboratories, Asc‐081) and 1 μM oligomycin (Merck Millipore, MBS495455) at the end of the experiments.
Confocal microscopy
HEK293T cells were grown onto six‐well chamber slides (Nunc, Rochester, NY, U.S.A.) and transfected with membrane‐targeted GCamp5 (0.2 μg of DNA/well). Forty‐eight hours following transfection, samples were examined by fluorescence confocal microscopy (LSM510 system, Carl Zeiss, Jena, Germany) using 480 nm (Ca2+ insensitive) excitation laser and 505‐nm long‐pass emission filter as previously described (Feldman et al, 2010). Briefly, baseline picture was captured followed by incubation with 2 μM of thapsigargin (TG) for 3 min. Then, cells were perfused by Ringer's solution containing 5 mM of CaCl2 before starting recordings.
Föster resonance energy transfer measurements
These measurements were done as described previously (Navarro‐Borelly et al, 2008; Wang et al, 2014; Zhou et al, 2015; Cai et al, 2016). To determine resonance energy transfer (FRET) signals between Orai1‐CFP and STIM1‐YFP, we used the Leica DMI 6000B fluorescence microscope equipped with CFP (438Ex/483Em), YFP (500Ex/542Em), FRET (438Ex/542Em) filters. Fluorescence intensities captured with the CFP, YFP, and FRET filters were collected at room temperature using a 40× oil objective (N.A.1.35; Leica) and processed using Slidebook 6.0 software (Intelligent Imaging Innovations). Three‐channel corrected FRET was calculated using the following formula: FC = IDA− Fd/Dd * IDD− Fa/Da * IAA, where IDD, IAA, and IDA represent the background‐subtracted CFP, YFP, and FRET images, respectively. And FC represents the corrected energy transfer, Fd/Dd represents measured bleed‐through of CFP through the FRET filter (0.457), and Fa/Da is measured bleed‐through of YFP through the FRET filter (0.19). Although the CFP signal was constant in Orai1‐CFP stable cells, there was some variation in transiently expressed STIM1‐YFP levels. To minimize the variation, only cells with similar YFP intensity were analyzed. Also, the above FRETc values were normalized against acceptor fluorescence (FYFP) to generate normalized FRET signals, which compensated for these variations in YFP expression. Thus, normalized FRET (NFRET) = FRETc/IAA. Average FRET measurements shown are typical of at least three independent analyses ± SEM. Same data pool was also calculated for EFRET with following formula (Zal & Gascoigne, 2004): Eapp = Fc / (FC + G*IDD), where the FC = IDA− Fd/Dd * IDD − Fa/Da * IAA. G is the instrument‐specific constant and was determined as described (Zal & Gascoigne, 2004; Navarro‐Borelly et al, 2008). G value is 1.9 ± 0.1 (n = 32 cells). Only cells with similar YFP/CFP ratio were used for EFRET analysis.
Patch clamp electrophysiology
Conventional whole‐cell patch clamp recordings were carried out using an Axopatch 200B and Digidata 1440A (Molecular Devices, LLC, Sunnyvale, CA) as previously published (Gonzalez‐Cobos et al, 2013; Zhang et al, 2013, 2014). Pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Inc., Sarasota, FL) with a P‐97 flaming/brown micropipette puller (Sutter Instrument Company, Novato, CA) and polished with DMF1000 (World Precision Instruments). Resistances of filled glass pipettes were 2–4 MΩ. Series resistances were < 10 MΩ. The liquid‐junction potential offset was around 5 mV and was corrected. Only cells with tight seals (> 13 GΩ) were selected for break‐in. Cells were maintained at a 0 mV holding potential during experiments and subjected to voltage ramps from −140 to +100 mV lasting 250 ms every 2 s. All experiments were performed at room temperature (20–25°C). Clampfit 10.1 software was used for data analysis. The solutions employed for patch clamp recordings are as follows.
Bath solution
115 mM Na+‐methanesulfonate, 10 mM CsCl, 1.2 mM MgSO4, 10 mM HEPES, 20 mM CaCl2, and 10 mM glucose (pH was adjusted to 7.4 with NaOH). For Li+ based bath, we replaced Na+‐methanesulfonate with Li+‐methanesulfonate; for N‐methyl‐D‐glutamine (NMDG+)‐based bath, we replaced Na+‐methanesulfonate with NMDG+.
Pipette solution (20 mM BAPTA)
115 mM Cs‐methanesulfonate, 20 mM Cs‐1,2‐bis‐(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid (Cs‐BAPTA), 8 mM MgCl2, and 10 mM HEPES (pH adjusted to 7.2 with CsOH). For high buffer, we replaced 20 mM Cs‐BAPTA with 50 mM in pipette solution.
Pipette solution (0.1 mM EGTA)
135 mM Cs+‐methanesulfonate, 8 mM NaCl, 1.0 mM MgCl2, 0.1 mM EGTA, and 10 mM HEPES, 0.03 mM IP3 (pH adjusted to 7.2 with CsOH). For cocktail, we added 2 mM pyruvic acid, 2 mM malic acid, 1 mM NaH2PO4, 0.5 mM cAMP, 2 mM ATP, 0.5 mM GTP to the pipette solution.
To energize the mitochondria a pipette solution containing a cocktail (2 mM pyruvic acid, 2 mM malic acid, 1 mM NaH2PO4, 0.5 mM cAMP, 0.5 mM GTP, 0.5 mM MgCl2) was used. Note, that the mitochondria also led to enhanced CRAC current only when activated in the presence of weak to moderate concentrations of EGTA (< 0.6 mM EGTA; Gilabert & Parekh, 2000; Parekh, 2008), presumably through enhanced mitochondrial Ca2+ buffering and reduced Ca2+‐dependent negative feedback on CRAC channels. When indicated, we performed CRAC recordings using a pipette solution containing 0.1 mM of the chelator EGTA combined with IP3.
Divalent‐free (DVF) bath solution
155 mM Na‐methanesulfonate, 10 mM HEDTA, 1 mM EDTA, and 10 mM HEPES (pH 7.4, adjusted with NaOH).
Statistical analysis
The results of the experiments are presented as the means ± SEM of ≥ 4 experiments (n), using 5–30 cells in each condition. Statistical significance for all experiments was determined using either unpaired two‐tailed Student's t‐test or one‐way ANOVA test followed by Tukey post hoc analysis. Values of P < 0.05 were considered significant.
Author contributions
IS, MT, TBKN, XZ, and DLG wrote the manuscript; TBKN, XZ, AE, SR, YZ, RKM, and MG performed the experiments; JAS, NH, MH, DLG, MT, and IS provided experimental oversight. IS, MT, TBKN, XZ, and AE designed the experiments; IS, MT, TBKN, XZ, MH, and DLG interpreted the data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1A
Acknowledgements
This study was funded by the DIP and ISF grants to IS and MH and by grants R21AG050072, R01HL097111 and R01HL123364 from the NIH and by the American Heart Association grant 14GRNT18880008 and Qatar National Research Fund (QNRF) grant # NPRP8‐110‐3‐021 to M.T. We wish to thank Dr. J. Andres Melendez (SUNY Albany) for the generous gift of the m‐catalase construct and Dr. Iulia Nita for helping with the NAD(P)H experiments.
The EMBO Journal (2017) 36: 797–815
Contributor Information
Mohamed Trebak, Email: mtrebak@psu.edu.
Israel Sekler, Email: sekler@bgu.ac.il.
References
- Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M (2008) Stim1 and Orai1 mediate CRAC currents and store‐operated calcium entry important for endothelial cell proliferation. Circ Res 103: 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S et al (2012) Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci 32: 13819–13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez J, Montero M, Garcia‐Sancho J (1999) Subcellular Ca(2+) dynamics. News Physiol Sci 14: 161–168 [PubMed] [Google Scholar]
- Ardon F, Rodriguez‐Miranda E, Beltran C, Hernandez‐Cruz A, Darszon A (2009) Mitochondrial inhibitors activate influx of external Ca(2+) in sea urchin sperm. Biochim Biophys Acta 1787: 15–24 [DOI] [PubMed] [Google Scholar]
- Baron S, Caplanusi A, van de Ven M, Radu M, Despa S, Lambrichts I, Ameloot M, Steels P, Smets I (2005) Role of mitochondrial Na+ concentration, measured by CoroNa red, in the protection of metabolically inhibited MDCK cells. J Am Soc Nephrol 16: 3490–3497 [DOI] [PubMed] [Google Scholar]
- Baryshnikov SG, Pulina MV, Zulian A, Linde CI, Golovina VA (2009) Orai1, a critical component of store‐operated Ca2+ entry, is functionally associated with Na+/Ca2+ exchanger and plasma membrane Ca2+ pump in proliferating human arterial myocytes. Am J Physiol Cell Physiol 297: C1103–C1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal K, Jung DW, Gunter KK, Gunter TE, Brierley GP (1994) Na(+)‐dependent Ca2+ efflux mechanism of heart mitochondria is not a passive Ca2+/2Na+ exchanger. Am J Physiol 266: C800–C808 [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517–529 [DOI] [PubMed] [Google Scholar]
- Bers DM, Barry WH, Despa S (2003) Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res 57: 897–912 [DOI] [PubMed] [Google Scholar]
- Bisaillon JM, Motiani RK, Gonzalez‐Cobos JC, Potier M, Halligan KE, Alzawahra WF, Barroso M, Singer HA, Jourd'heuil D, Trebak M (2010) Essential role for STIM1/Orai1‐mediated calcium influx in PDGF‐induced smooth muscle migration. Am J Physiol Cell Physiol 298: C993–C1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogeski I, Kummerow C, Al‐Ansary D, Schwarz EC, Koehler R, Kozai D, Takahashi N, Peinelt C, Griesemer D, Bozem M, Mori Y, Hoth M, Niemeyer BA (2010) Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal 3: ra24 [DOI] [PubMed] [Google Scholar]
- Borin ML, Goldman WF, Blaustein MP (1993) Intracellular free Na+ in resting and activated A7r5 vascular smooth muscle cells. Am J Physiol 264: C1513–C1524 [DOI] [PubMed] [Google Scholar]
- Cai X, Zhou Y, Nwokonko RM, Loktionova NA, Wang X, Xin P, Trebak M, Wang Y, Gill DL (2016) The orai1 store‐operated calcium channel functions as a hexamer. J Biol Chem 291: 25764–25775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C (1974) The release of calcium from heart mitochondria by sodium. J Mol Cell Cardiol 6: 361–371 [DOI] [PubMed] [Google Scholar]
- De Marchi U, Santo‐Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N (2014) NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+‐induced NAD(P)H production and modulating matrix redox state. J Biol Chem 289: 20377–20385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck‐Weiermair M, Hallstrom S, Graier WF, Malli R (2014) IP3‐mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J Cell Sci 127: 2944–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaurex N, Poburko D, Frieden M (2009) Regulation of plasma membrane calcium fluxes by mitochondria. Biochim Biophys Acta 1787: 1383–1394 [DOI] [PubMed] [Google Scholar]
- Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY (2004) Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem 279: 22284–22293 [DOI] [PubMed] [Google Scholar]
- Eder P, Probst D, Rosker C, Poteser M, Wolinski H, Kohlwein SD, Romanin C, Groschner K (2007) Phospholipase C‐dependent control of cardiac calcium homeostasis involves a TRPC3‐NCX1 signaling complex. Cardiovasc Res 73: 111–119 [DOI] [PubMed] [Google Scholar]
- Feldman B, Fedida‐Metula S, Nita J, Sekler I, Fishman D (2010) Coupling of mitochondria to store‐operated Ca(2+)‐signaling sustains constitutive activation of protein kinase B/Akt and augments survival of malignant melanoma cells. Cell Calcium 47: 525–537 [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441: 179–185 [DOI] [PubMed] [Google Scholar]
- Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T (2010) Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store‐operated Ca2+ channels. Mol Cell 38: 280–290 [DOI] [PubMed] [Google Scholar]
- Gilabert JA, Parekh AB (2000) Respiring mitochondria determine the pattern of activation and inactivation of the store‐operated Ca(2+) current I(CRAC). EMBO J 19: 6401–6407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilabert JA, Bakowski D, Parekh AB (2001) Energized mitochondria increase the dynamic range over which inositol 1,4,5‐trisphosphate activates store‐operated calcium influx. EMBO J 20: 2672–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch MD, Bakowski D, Parekh AB (2002) Store‐operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J 21: 6744–6754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Cobos JC, Zhang X, Zhang W, Ruhle B, Motiani RK, Schindl R, Muik M, Spinelli AM, Bisaillon JM, Shinde AV, Fahrner M, Singer HA, Matrougui K, Barroso M, Romanin C, Trebak M (2013) Store‐independent Orai1/3 channels activated by intracrine leukotriene C4: role in neointimal hyperplasia. Circ Res 112: 1013–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, Kurosaki T, Joseph SK, Gill DL, Madesh M (2010) S‐glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol 190: 391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, O'Halloran DM (2014) Analysis of the Na+/Ca2+ exchanger gene family within the phylum Nematoda. PLoS ONE 9: e112841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS (1997) Mitochondrial regulation of store‐operated calcium signaling in T lymphocytes. J Cell Biol 137: 633–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Joo NS, Reitz B, Wine JJ, Verkman AS (2001a) Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc Natl Acad Sci USA 98: 8119–8123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Song Y, Vetrivel L, Shankar L, Verkman AS (2001b) Noninvasive in vivo fluorescence measurement of airway‐surface liquid depth, salt concentration, and pH. J Clin Invest 107: 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers FC, Henquin JC (2001) Measurements of cytoplasmic Ca2+ in islet cell clusters show that glucose rapidly recruits beta‐cells and gradually increases the individual cell response. Diabetes 50: 540–550 [DOI] [PubMed] [Google Scholar]
- Jung DW, Baysal K, Brierley GP (1995) The sodium‐calcium antiport of heart mitochondria is not electroneutral. J Biol Chem 270: 672–678 [DOI] [PubMed] [Google Scholar]
- Lee KP, Yuan JP, So I, Worley PF, Muallem S (2010) STIM1‐dependent and STIM1‐independent function of transient receptor potential canonical (TRPC) channels tunes their store‐operated mode. J Biol Chem 285: 38666–38673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos VS, Poburko D, Liao CH, Cole WC, van Breemen C (2007) Na+ entry via TRPC6 causes Ca2+ entry via NCX reversal in ATP stimulated smooth muscle cells. Biochem Biophys Res Commun 352: 130–134 [DOI] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+‐store‐depletion‐triggered Ca2+ influx. Curr Biol 15: 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlejohns B, Pasdois P, Duggan S, Bond AR, Heesom K, Jackson CL, Angelini GD, Halestrap AP, Suleiman MS (2014) Hearts from mice fed a non‐obesogenic high‐fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS ONE 9: e100579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J (2007) Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J 406: 365–382 [DOI] [PubMed] [Google Scholar]
- Ma HT, Beaven MA (2011) Regulators of Ca(2+) signaling in mast cells: potential targets for treatment of mast cell‐related diseases? Adv Exp Med Biol 716: 62–90 [DOI] [PubMed] [Google Scholar]
- Malli R, Frieden M, Osibow K, Zoratti C, Mayer M, Demaurex N, Graier WF (2003) Sustained Ca2+ transfer across mitochondria is essential for mitochondrial Ca2+ buffering, sore‐operated Ca2+ entry, and Ca2+ store refilling. J Biol Chem 278: 44769–44779 [DOI] [PubMed] [Google Scholar]
- McCarthy DA, Ranganathan A, Subbaram S, Flaherty NL, Patel N, Trebak M, Hempel N, Melendez JA (2013) Redox‐control of the alarmin, Interleukin‐1alpha. Redox Biol 1: 218–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A (2001) Circularly permuted green fluorescent proteins engineered to sense Ca2+ . Proc Natl Acad Sci USA 98: 3197–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghdi S, Waldeck‐Weiermair M, Fertschai I, Poteser M, Graier WF, Malli R (2010) Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1‐Orai1‐dependent store‐operated Ca2+ entry. J Cell Sci 123: 2553–2564 [DOI] [PubMed] [Google Scholar]
- Navarro‐Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M (2008) STIM1‐Orai1 interactions and Orai1 conformational changes revealed by live‐cell FRET microscopy. J Physiol 586: 5383–5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naziroglu M (2012) Molecular role of catalase on oxidative stress‐induced Ca(2+) signaling and TRP cation channel activation in nervous system. J Recept Signal Transduct Res 32: 134–141 [DOI] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Fishman D, Ozeri E, Rutter GA, Sensi SL, Khananshvili D, Lewis EC, Sekler I (2012) The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS ONE 7: e46649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Kantor C, Rutter GA, Lewis EC, Sekler I (2014a) Pancreatic beta‐cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J 28: 3301–3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Lewis EC, Sekler I (2014b) A crosstalk between Na channels, Na/K pump and mitochondrial Na transporters controls glucose‐dependent cytosolic and mitochondrial Na signals. Cell Calcium 57: 69–75 [DOI] [PubMed] [Google Scholar]
- Nunes P, Demaurex N (2014) Redox regulation of store‐operated Ca2+ entry. Antioxid Redox Signal 21: 915–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, Sekler I (2004) Lithium‐calcium exchange is mediated by a distinct potassium‐independent sodium‐calcium exchanger. J Biol Chem 279: 25234–25240 [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan‐Barmatz V, Herrmann S, Khananshvili D, Sekler I (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 107: 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Putney JW Jr (2005) Store‐operated calcium channels. Physiol Rev 85: 757–810 [DOI] [PubMed] [Google Scholar]
- Parekh AB (2008) Mitochondrial regulation of store‐operated CRAC channels. Cell Calcium 44: 6–13 [DOI] [PubMed] [Google Scholar]
- Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, Wright L, Pallafacchina G, Terrin A, Mammucari C, Rizzuto R (2013) The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. J Biol Chem 288: 10750–10758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts BJ (1979) Stoichiometry of sodium‐calcium exchange in cardiac sarcolemmal vesicles. Coupling to the sodium pump. J Biol Chem 254: 6232–6235 [PubMed] [Google Scholar]
- Pizzo P, Drago I, Filadi R, Pozzan T (2012) Mitochondrial Ca(2)(+) homeostasis: mechanism, role, and tissue specificities. Pflugers Archiv 464: 3–17 [DOI] [PubMed] [Google Scholar]
- Poburko D, Liao CH, van Breemen C, Demaurex N (2009) Mitochondrial regulation of sarcoplasmic reticulum Ca2+ content in vascular smooth muscle cells. Circ Res 104: 104–112 [DOI] [PubMed] [Google Scholar]
- Potier M, Trebak M (2008) New developments in the signaling mechanisms of the store‐operated calcium entry pathway. Pflugers Archiv 457: 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M (2009) Evidence for STIM1‐ and Orai1‐dependent store‐operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J 23: 2425–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW Jr (1986) A model for receptor‐regulated calcium entry. Cell Calcium 7: 1–12 [DOI] [PubMed] [Google Scholar]
- Reeves JP, Hale CC (1984) The stoichiometry of the cardiac sodium‐calcium exchange system. J Biol Chem 259: 7733–7739 [PubMed] [Google Scholar]
- Reyes RC, Verkhratsky A, Parpura V (2013) TRPC1‐mediated Ca2+ and Na+ signalling in astroglia: differential filtering of extracellular cations. Cell Calcium 54: 120–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13: 566–578 [DOI] [PubMed] [Google Scholar]
- Rodriguez AM, Carrico PM, Mazurkiewicz JE, Melendez JA (2000) Mitochondrial or cytosolic catalase reverses the MnSOD‐dependent inhibition of proliferation by enhancing respiratory chain activity, net ATP production, and decreasing the steady state levels of H(2)O(2). Free Radic Biol Med 29: 801–813 [DOI] [PubMed] [Google Scholar]
- Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K (2004) Ca(2+) signaling by TRPC3 involves Na(+) entry and local coupling to the Na(+)/Ca(2+) exchanger. J Biol Chem 279: 13696–13704 [DOI] [PubMed] [Google Scholar]
- Rudolf R, Mongillo M, Magalhaes PJ, Pozzan T (2004) In vivo monitoring of Ca(2+) uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol 166: 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaravelu K, Nelson C, Bakowski D, de Brito OM, Ng SW, Di Capite J, Powell T, Scorrano L, Parekh AB (2011) Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J Biol Chem 286: 12189–12201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Duchen MR (2008) Mitochondria: the hub of cellular Ca2+ signaling. Physiology 23: 84–94 [DOI] [PubMed] [Google Scholar]
- Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R (2006) CRACM1 multimers form the ion‐selective pore of the CRAC channel. Curr Biol 16: 2073–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, Gill DL (2014) Distinct Orai‐coupling domains in STIM1 and STIM2 define the Orai‐activating site. Nat Commun 5: 3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KT, Pan SF, Chien CL, Hsu SM, Tseng YZ, Wang SM, Wu ML (2004) Mitochondrial Na+ overload is caused by oxidative stress and leads to activation of the caspase 3‐ dependent apoptotic machinery. FASEB J 18: 1442–1444 [DOI] [PubMed] [Google Scholar]
- Zal T, Gascoigne NR (2004) Photobleaching‐corrected FRET efficiency imaging of live cells. Biophys J 86: 3923–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437: 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD (2006) Genome‐wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release‐activated Ca(2+) channel activity. Proc Natl Acad Sci USA 103: 9357–9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gonzalez‐Cobos JC, Schindl R, Muik M, Ruhle B, Motiani RK, Bisaillon JM, Zhang W, Fahrner M, Barroso M, Matrougui K, Romanin C, Trebak M (2013) Mechanisms of STIM1 activation of store‐independent leukotriene C4‐regulated Ca2+ channels. Mol Cell Biol 33: 3715–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Clemens RA, Liu F, Hu Y, Baba Y, Theodore P, Kurosaki T, Lowell CA (2014) STIM1 calcium sensor is required for activation of the phagocyte oxidase during inflammation and host defense. Blood 123: 2238–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang X, Wang X, Loktionova NA, Cai X, Nwokonko RM, Vrana E, Wang Y, Rothberg BS, Gill DL (2015) STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat Commun 6: 8395 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1A