

Abstract
In innate immune responses, induction of type‐I interferons (IFNs) prevents virus spreading while viral replication is delayed by protein synthesis inhibition. We asked how cells perform these apparently contradictory activities. Using single fibroblast monitoring by flow cytometry and mathematical modeling, we demonstrate that type‐I IFN production is linked to cell's ability to enter dsRNA‐activated PKR‐dependent translational arrest and then overcome this inhibition by decreasing eIF2α phosphorylation through phosphatase 1c cofactor GADD34 (Ppp1r15a) expression. GADD34 expression, shown here to be dependent on the IRF3 transcription factor, is responsible for a biochemical cycle permitting pulse of IFN synthesis to occur in cells undergoing protein synthesis inhibition. Translation arrest is further demonstrated to be key for anti‐viral response by acting synergistically with MAVS activation to amplify TBK1 signaling and IFN‐β mRNA transcription, while GADD34‐dependent protein synthesis recovery contributes to the heterogeneous expression of IFN observed in dsRNA‐activated cells.
Keywords: cGAMP, integrated stress response, puromycin, RIG‐I‐like receptors, stress granules
Subject Categories: Immunology
Introduction
Many viruses generate double‐stranded (ds)RNA replication intermediates within infected host cells. These viral determinants elicit innate immune responses and subsequent type‐I IFN and pro‐inflammatory cytokine production through the triggering of nucleic acid (NA)‐sensing pathways (Kawai & Akira, 2006). Several families of molecules are known to detect dsRNA or its synthetic mimic polyriboinosinic:polyribocytidylic acid (poly(I:C)), including endosomal TLR3 and the cytosolic RNA helicases RIG‐I and MDA5 (RLRs) (Pichlmair & Reis e Sousa, 2007; Kawai & Akira, 2010; Goubau et al, 2013). Signaling through RIG‐I or MDA5 requires the mitochondrial anti‐viral signaling protein (MAVS), which predominantly localizes to the mitochondrial outer membrane or on peroxisomes (Kawai et al, 2005; Belgnaoui et al, 2011). The MAVS signaling pathway results in the activating phosphorylation of several kinases and their downstream targets, including the transcription factors IRF3 and NF‐κB. IRFs are activated by TANK‐binding kinase 1 (TBK1) and by IKKε, whereas NF‐κB is activated by IKKα and IKKβ (Li et al, 2011). Activation of RLR/MAVS signaling in infected cells results in the production of high levels of pro‐inflammatory cytokines and type‐I IFNs. These orchestrate anti‐viral protection in neighboring tissues and stimulate innate and adaptive immunity against invading pathogens. Surprisingly, type‐I IFN production is detected in only a minority (15–40%) of infected cells (Zawatzky et al, 1985). The relatively small proportion of responding cells was attributed to cellular variability in the expression levels or activities of key innate immunity components, leading to the stochastic expression of the IFN‐β gene (Zhao et al, 2012). Stochastic production is thought to limit type‐I IFN secretion levels and protect the host organism from its inherent toxicity, while maintaining anti‐viral effects (Zhao et al, 2012). Different models, mostly based on gene transcription analyses, have been proposed to explain this phenomenon (Zhao et al, 2012; Patil et al, 2015; Zhang et al, 2015). They involve mostly different expression levels of innate sensors in individual cells and the existence of transcriptional feedback loops linked to the paracrine activity of secreted IFN‐β (Hwang et al, 2013). Despite being informative, these models are not completely satisfying, since they do not take into consideration the protein synthesis inhibition triggered by dsRNA or virus detection, and initiated upon phosphorylation of translation initiation factor eIF2α by protein kinase RNA‐activated (PKR or EIF2AK2), which is likely to impact on IFN production and the translation of type‐I IFN‐stimulated genes (ISGs) mRNAs, such as RLRs or PKR itself (Williams, 2001).
At least two apparently antagonist programs are initiated in infected cells upon cytosolic dsRNA detection. One, triggered by RIG‐I‐like receptors (RLRs), is dedicated to innate transcriptional responses and cytokine production that promotes systemic immunity. The second program is PKR dependent and prevents viral replication by blocking mRNA translation in individual cells, promoting stress granules (SGs) formation and apoptosis (Reineke et al, 2012). This PKR‐dependent program is likely inhibitory for the RLR‐triggered program, suggesting that a complex integration process of protein synthesis inhibition with innate sensing is necessary for cells to respond efficiently to viruses or dsRNA (Claudio et al, 2013). Here, we describe how MAVS‐ and IRF3‐dependent expression of the GADD34 phosphatase‐1 (PP1) cofactor resolves this antagonistic situation and how, together with PKR‐dependent translation inhibition, contributes to the amplification of IRF3 activation and the apparent stochasticity of type‐I IFN‐β production in response to dsRNA or vesicular stomatitis virus (VSV) infection. The coordination of these different biochemical pathways leads over time to cycles of protein synthesis inhibition and activation leading to IFN production in selected individual cells. Mathematical modeling supports these observations and suggests that integration of the different pathways described here is sufficient to orchestrate a stochastic production of type‐I IFN within a population of cells responding to dsRNA.
Results
The transcriptional response of GADD34ΔC/ΔC MEFs to poly(I:C)
We previously showed that GADD34 activity is necessary for IFN‐β and IL‐6 production upon PKR‐dependent translation inhibition in MEFs stimulated with poly(I:C) or infected with Chikungunya virus (ChikV) (Clavarino et al, 2012). To confirm that GADD34‐driven PP1 phosphatase activity is the only critical function required to allow cytokine production, we expressed the herpes simplex viral effector ICP34.5, which increases eIF2α dephosphorylation (Mohr & Sonenberg, 2012) in dsRNA‐stimulated GADD34ΔC/ΔC MEFs. Ectopic GADD34 or ICP34.5 expression rescued IFN‐β production (Fig 1A), confirming that eIF2α dephosphorylation is required to sustain cytokine production upon dsRNA‐dependent protein synthesis inhibition. GADD34 induction is therefore a primary event in the establishment of the cell response to dsRNA. To evaluate globally the role of GADD34 on this response, a comparative microarray‐based mRNA transcription analysis of WT and GADD34ΔC/ΔC MEFs, lipofected with poly(I:C), was performed (Fig 1B). A total of 648 differently upregulated genes (DEGs) in WT and 354 in GADD34ΔC/ΔC MEFs were found after exposure to poly(I:C) (Fig 1B and Appendix Table S1). Among them, 257 were upregulated in both WT and GADD34ΔC/ΔC MEFs, 391 were only upregulated in WT and 97 only in GADD34ΔC/ΔC MEFs. Ingenuity pathway analysis indicated that most DEGs were found to be significantly associated with canonical pathways participating in anti‐viral transcription programs (Fig 1C). Among the commonly upregulated genes were the IRF3‐dependent genes Ifnb1, Ifna4, and Il6, together with Irf7 and other type‐I IFN‐stimulated genes (ISGs) including Oasl1, Isg15, Ifit3, and Ifih1. Additional IPA “Upstream Regulators” searches confirmed that the principal transcription regulators controlling the response to poly(I:C) in both cell types were IRF3, IFN‐β, IFNAR, or STAT1 (Fig 1D). Identification of these regulators demonstrates that at least two different transcriptional activation programs co‐exist upon dsRNA sensing. These are governed by direct stimulation of the RLR/MAVS/IRF3 pathway and by autocrine activation of the interferon receptor (IFNβ/IFNAR/STAT1), among other cytokine receptors. The majority of DEGs were unique to WT MEFs. Many of these DEGs were associated with antigen presentation and anti‐viral activities (Fig 1C and Appendix Table S1). This strongly contrasted with the lack of any significant enrichment in known pathways for the 97 genes uniquely upregulated in GADD34ΔC/ΔC MEFs (Appendix Table S1). This result confirmed the existence of an altered transcription response to dsRNA in GADD34ΔC/ΔC MEFs, but also revealed that GADD34 seems to have little function in controlling transcription factor activation directly, but rather acts through its protein synthesis regulatory role on cytokines and proteins (including transcription factors) induced by dsRNA. We were surprised by the induction of many ISGs in GADD34ΔC/ΔC MEFs, since these cells are severely impaired in their ability to produce type‐I IFN (Fig 1A) and should therefore display minimal paracrine triggering of IFNAR and subsequent ISG transcription (Clavarino et al, 2012). A subset of ISGs are known to be induced directly by IRFs (Grandvaux et al, 2002; Daffis et al, 2007), including those coding for Ifit1, Ifit3, Cxcl10, Isg15, Oasl1, Rsad2, Nfkbiz, Ccrl2 (Lazear et al, 2013). These were all found to be induced in GADD34ΔC/ΔC MEFs, confirming their likely independence from IFNAR; however, genes previously described as strictly IFN‐β dependent, like Oas1, Oas2, Ifna2, Ddx58, Gbp5, Mx2, or Isg20 (Lazear et al, 2013), were also expressed in these cells despite their incapacity to produce cytokines and transcription factors like IRF7. ISG induction in GADD34ΔC/ΔC MEFs might either reflect cell specificity and a broader role of IRFs in their transcription than anticipated, or it might be the result of the production of small amounts of IFN‐β (Fig 1A), sufficient to trigger tonic IFNAR signaling, but insufficient to induce a full level response. In fact, many of the genes co‐expressed by WT and GADD34ΔC/ΔC have been found to be downregulated in immune cells deficient for IFNAR at steady state and potentially exposed to low‐chronic‐dose IFN (Mostafavi et al, 2016). The lack of induction of emblematic type‐I IFN‐induced genes coding for MHC I, IFNα1, PKR, or RNASEL in GADD34ΔC/ΔC cells further suggests the existence of ISG subgroups displaying different induction thresholds or requirement for transcription factor combinations that are revealed by the profound reduction in cytokine production and protein synthesis associated with GADD34 deficiency.
Figure 1. Comparative transcriptional analysis of WT and GADD34ΔC/ΔC MEFs responding to poly(I:C).
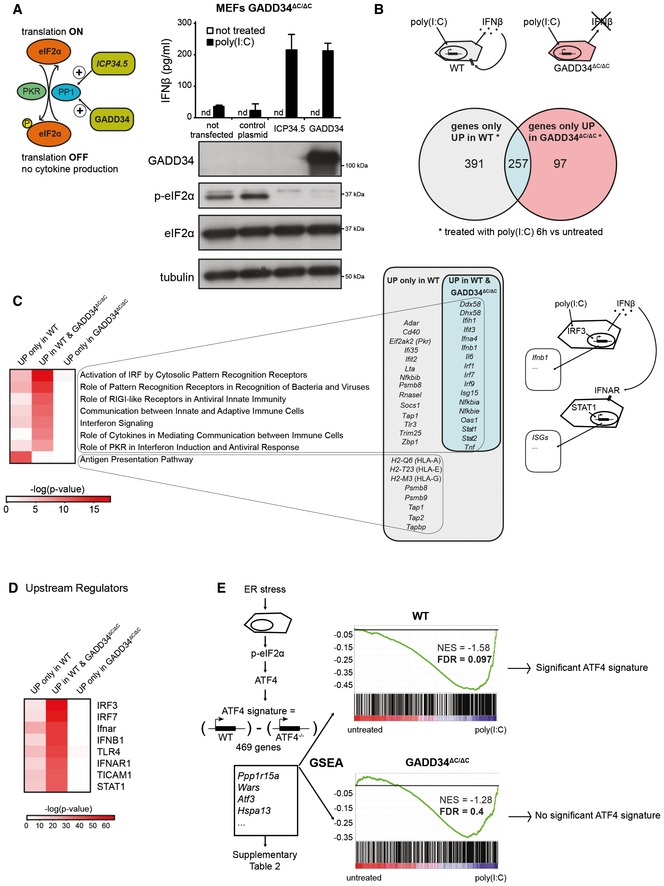
- Left: Graphical abstract of GADD34 and herpes simplex viral protein ICP34.5 effects on translation initiation. ICP34.5 mimics PP1 cofactor GADD34 activity and contributes to dephosphorylation of eIF2α. As a result, the translation inhibition mediated by eIF2α kinases such as PKR is relieved. Right: GADD34ΔC/ΔC MEFs were transfected with the indicated plasmid constructs 24 h before HMW poly(I:C) delivery. After 6 h of treatment, culture supernatants were collected and IFN‐β production was measured by ELISA (mean ± SD of three independent experiments). Representative detection of GADD34, eIF2α, and p‐eIF2α by immunoblot in the lysate of the same group of cells. Tubulin is used as a loading control. “nd” stands for “not detected”.
- Top: Schematic representation of WT cells treated with poly(I:C) and producing IFN‐β. This cytokine triggers specific signaling via IFNAR receptor in an autocrine or paracrine manner, whereas GADD34ΔC/ΔC MEFs do not produce IFN‐β. Bottom: Venn diagram representation of statistically upregulated genes in WT and GADD34ΔC/ΔC MEFs after 6 h of HMW poly(I:C) treatment. The complete sets of genes are detailed in Appendix Table S1.
- Heat maps of selected pathways found enriched by ingenuity pathway analysis (IPA) for the DEGs depicted in Venn diagrams. Selected genes belonging at least to one of the IPA pathways are shown and grouped according to their expression specificity. Right: Schematic representation of poly(I:C)‐treated cells showing IRF3‐dependent induction of genes, like IFN‐β, and the concomitant induction in neighboring cells of IFN‐stimulated genes (ISGs) after IFNAR stimulation.
- Heat‐map representation of selected putative upstream regulators found enriched by IPA for the different DEGs sets.
- Gene set enrichment analyses (GSEAs) of a defined ATF4 gene expression regulated signature (Appendix Table S2), on the pairwise comparisons of WT MEFs untreated vs poly(I:C) (left panel) and GADD34ΔC/ΔC MEFs untreated vs poly(I:C) (right panel). The more the ATF4 regulated signature gene set is differentially expressed between conditions, the more the bar code is shifted to the corresponding extremity. NES: normalized enrichment score; FDR: false discovery rate.
GADD34 activity is required for ATF4‐dependent transcription in response to poly(I:C)
PKR triggering by dsRNA and subsequent eIF2α phosphorylation should trigger the ATF4/CHOP pathway (Han et al, 2013) and promote GADD34 expression as part of the negative feedback loop returning protein synthesis to normal levels. This process is commonly observed during the integrated stress response (ISR) driven by PERK activation in the endoplasmic reticulum (ER) (Novoa et al, 2001; Ron & Walter, 2007; Clavarino et al, 2012), during which ATF4 synthesis is necessary for GADD34 transcription (Ron & Walter, 2007; Han et al, 2013). We took advantage of available genomic data (GSE49598) comparing mRNA expression profiles of WT vs ATF4−/− MEFs treated with the ER stress‐inducer tunicamycin to define the core of an ATF4‐dependent gene expression signature (Appendix Table S2). Gene set enrichment analysis (GSEA) (Subramanian et al, 2005), which allows statistical assessment of changes in the expression of a predefined set of genes in different conditions, indicated that ATF4‐dependent transcription was significantly increased (FDR < 0.1) in WT cells stimulated with poly(I:C) compared with non‐treated cells (Fig 1E); however, this was not the case in activated GADD34ΔC/ΔC MEFs (FDR 0.4) (Fig 1E). Surprisingly, increased p‐eIF2α levels did not augment ATF4 translation nor associated transcriptional signature in these cells, and importantly, GADD34 mRNA remained strongly induced by poly(I:C) (Appendix Fig S1A; Clavarino et al, 2012). As anticipated, ATF4 was detected in nuclear extracts of WT MEFs exposed to dsRNA or ER stress‐triggering drug thapsigargin (thapsi), but was, however, absent from GADD34ΔC/ΔC MEF extracts, confirming biochemically the results of the GSEA (Appendix Fig S1B). GADD34 expression can therefore be induced in the absence of ATF4 synthesis, thus contrasting with what is normally observed during the ISR (Claudio et al, 2013; Fig 2A).
Figure 2. GADD34 is part of the primary innate immune response to dsRNA.
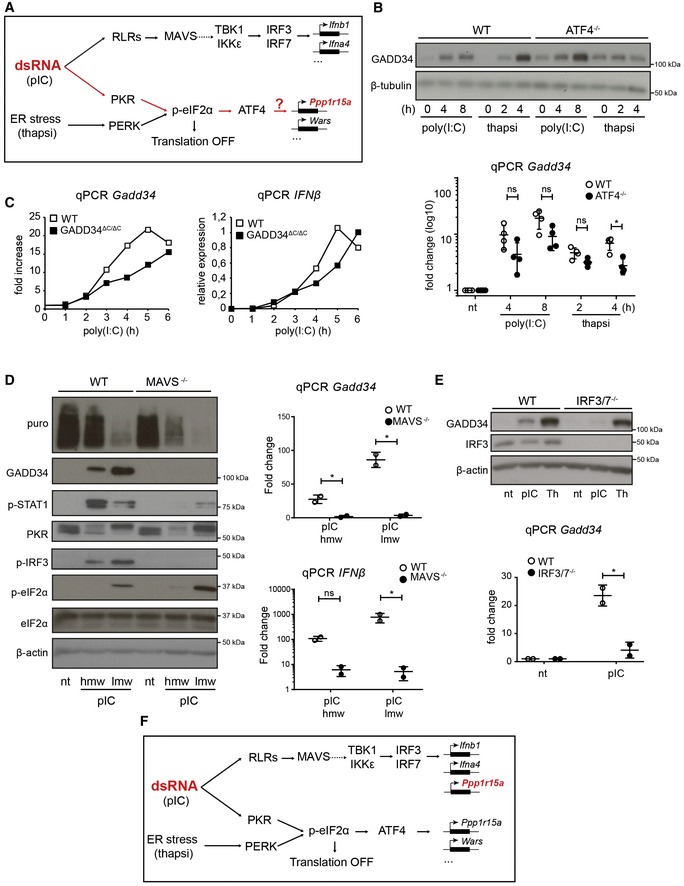
- Schematic representation of known signaling pathways involved during dsRNA response and the UPR. Two distinct pathways are triggered in the cytosol of infected cells: RIG‐I‐like receptors (RLRs) detect dsRNA and trigger IRF3 activation via MAVS, while PKR stimulation leads to protein translation inhibition and ATF4‐dependent gene transcription (e.g., GADD34), as observed after PERK activation during thapsigargin‐induced ER stress.
- WT and ATF4−/− MEFs were treated with HMW poly(I:C) for 4 h and 8 h, or with thapsigargin (thapsi) for 2 h and 4 h. Expression of GADD34 was analyzed by immunoblot and by qPCR (mean ± SD of five independent experiments). Tubulin is shown as a loading control for immunoblot.
- WT and GADD34ΔC/ΔC MEFs were stimulated with LMW poly(I:C). GADD34 and IFNB mRNA expression was monitored by qPCR for 6 h after dsRNA delivery.
- WT and MAVS−/− MEFs were analyzed by immunoblot (left) and by qPCR (right panels) after stimulation with poly(I:C) (pIC). Protein synthesis was determined using puromycin labeling followed by immunoblot with the anti‐puromycin mAb. GADD34, p‐STAT1, PKR, p‐IRF3, eIF2α, and p‐eIF2α levels were monitored by immunoblot. Actin is shown as a loading control. Fold increase compared to non‐treated cells in normalized mRNA levels. Each point represents result of one independent experiment.
- GADD34 expression was determined by immunoblot and by qPCR in WT and IRF3/7−/− MEFs after HMW poly(I:C) or thapsigargin (Th) treatment. qPCR are the mean ± SD of three independent experiments (“nt” stands for “not treated”).
- Schematic representation of predicted signaling pathways involved during dsRNA response and the UPR, according to the results shown in (B–D). GADD34 induction belongs to the primary transcriptional response consecutive to dsRNA sensing and is dependent on IRF3/IRF7 transcription factors, together with IFN‐β.
IRF3 drives GADD34 expression
Intrigued by the possibility that GADD34 transcription could be ATF4‐independent, we monitored the expression of GADD34 in WT and ATF4−/− MEFs after poly(I:C) delivery (Fig 2A and B). GADD34 mRNA transcription was clearly augmented after 4–8 h of poly(I:C) stimulation in both cell types, and only appeared significantly reduced in ATF4−/− MEFs stimulated with thapsigargin for 4 h (Fig 2B). At the protein level, moderate expression of GADD34 was detected at steady state in ATF4−/− cells, probably mirroring their adaptation to in vitro culture conditions. However, despite ATF4 inactivation, GADD34 was strongly induced in response to poly(I:C) contrasting with thapsigargin treatment during which additional expression of the PP1 cofactor was not observed. ATF4 is therefore not strictly required for GADD34 transcription but could still eventually contribute to the magnitude of the response to dsRNA. GADD34 transcription in WT and GADD34ΔC/ΔC MEFs was found to match the induction IFNB mRNA both in kinetics and intensity, suggesting that these two genes are co‐regulated (Fig 2C). This prompted us to examine GADD34 expression in cells inactivated for critical upstream regulators of type‐I IFN transcription, including MAVS (Kawai et al, 2005; Seth et al, 2005) and different IRFs (Honda & Taniguchi, 2006).
Using MAVS‐deficient MEFs, we observed that both IFN‐β and GADD34 expression were profoundly altered after dsRNA delivery, along with IRF3 and STAT1 phosphorylation (Fig 2D). MAVS−/− MEFs displayed normal levels of PKR activation, however, followed by accentuated eIF2α phosphorylation and translation inhibition, reflecting the absence of GADD34 expression (Fig 2D). MAVS activation normally results in IRF3 dimerization and translocation into the nucleus, where it drives type‐I IFN transcription (Fig 2A) (Honda & Taniguchi, 2006; Goubau et al, 2013; Ourthiague et al, 2015). We further investigated GADD34 induction in IRF3/IRF7 double‐KO (IRF3/7−/−) MEFs and found that similar to MAVS‐deficient cells, GADD34 mRNA transcription and synthesis remained undetectable upon poly(I:C) delivery, while thapsigargin treatment induced a comparable response in control and IRF3/7−/− MEFs (Fig 2E). IRF3/7−/− cells stimulated with dsRNA also expressed normally ATF4 (Appendix Fig S1C), suggesting that IRF3 and maybe IRF7 are the major transcription factors controlling GADD34 expression after detection of cytosolic dsRNA by RLRs (Fig 2F). Treatment of these cells with recombinant IFN‐β increased RIG‐I levels, but did not rescue GADD34 expression, further demonstrating its transcriptional dependence on IRFs and not on IFNAR‐associated STAT1/STAT2‐dependent transcription (Appendix Fig S1D).
Protein synthesis inhibition prevents the induction of negative regulators of TBK1 and potentiates TBK1 and IRF3 activation
GADD34 transcription and protein expression are strongly altered in the absence of PKR or eIF2α phosphorylation (Clavarino et al, 2012). Given the implication of IRFs in GADD34 induction, levels of p‐IRF3 and GADD34 expression were investigated in both WT and PKR−/− MEFs exposed to poly(I:C) (Fig 3A and B). In WT MEFs, GADD34 was upregulated concomitantly with IRF3 phosphorylation, while in the absence of PKR, GADD34 synthesis was abolished and a strong reduction in IRF3 phosphorylation was observed. In coordination with diminished levels of p‐IRF3, both IFN‐β and GADD34 mRNA transcription levels were decreased in PKR−/− MEFs (Fig 3B), suggesting a potentiation role of the eIF2α kinase on MAVS signaling and/or IFNB mRNA stabilization (Schulz et al, 2010). Despite lower mRNA levels however, IFN‐β protein production was augmented in the absence of PKR (Fig EV1A), confirming that the general translation arrest normally caused by dsRNA detection can be detrimental for cytokine production and that IFN‐β is not regulated by specific regulatory sequences favoring its translation upon eIF2α phosphorylation (Starck et al, 2016).
Figure 3. Translation inhibition amplifies the transcriptional response to dsRNA.
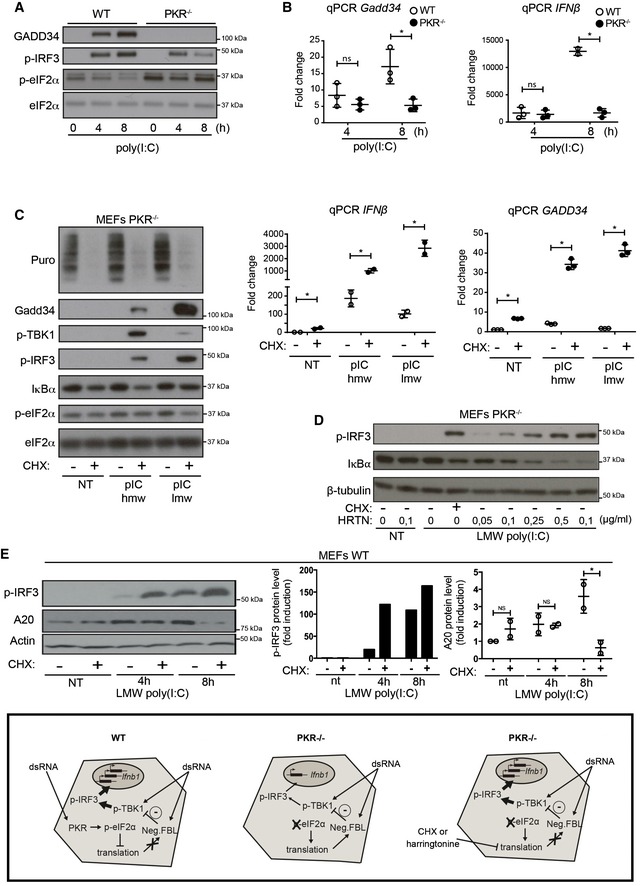
-
A, BWT and PKR−/− MEFs were stimulated with HMW poly(I:C) for 4 h and 8 h. (A) GADD34, p‐IRF3, and p‐eIF2α/eIF2α were detected by immunoblot. (B) GADD34 and IFNB mRNA expression were determined by qPCR in WT and PKR−/− MEFs. Results are the mean ± SD of three independent experiments.
-
CPKR−/− MEFs were treated or not with 5 μg/ml of cycloheximide (CHX), together with poly(I:C) for 8 h. Protein synthesis was monitored using puromycin labeling followed by immunoblot with an anti‐puromycin mAb. Expression of the indicated proteins was determined by immunoblot (left). GADD34 and IFNB mRNA levels were quantified by qPCR (right). Data are representative of three independent experiments. Results are the mean ± SD of three independent experiments.
-
DPKR−/− MEFs were treated or not with the indicated concentration of harringtonine, before monitoring p‐IRF3 levels and IκBα expression by immunoblot after 8 h of dsRNA exposure. Non‐treated (nt, control without poly(I:C)) and CHX treatment were used as negative and positive references. Tubulin is shown as a loading control.
-
EWT MEFs were treated with LMW poly(I:C) for the indicated time before monitoring p‐IRF3 levels and A20 level by immunoblot (left panel). P‐IRF3 (middle) and A20 protein levels (right) were quantified by ImageJ quantification. Graphs represent data normalized to non‐treated samples, n = 2. A schematic representation of the experimental design and corresponding results is boxed at the bottom of the figure. Results are the mean ± SD of three independent experiments.
Figure EV1. IFNAR1‐independent potentiation of nucleic acid sensors by translation inhibition.
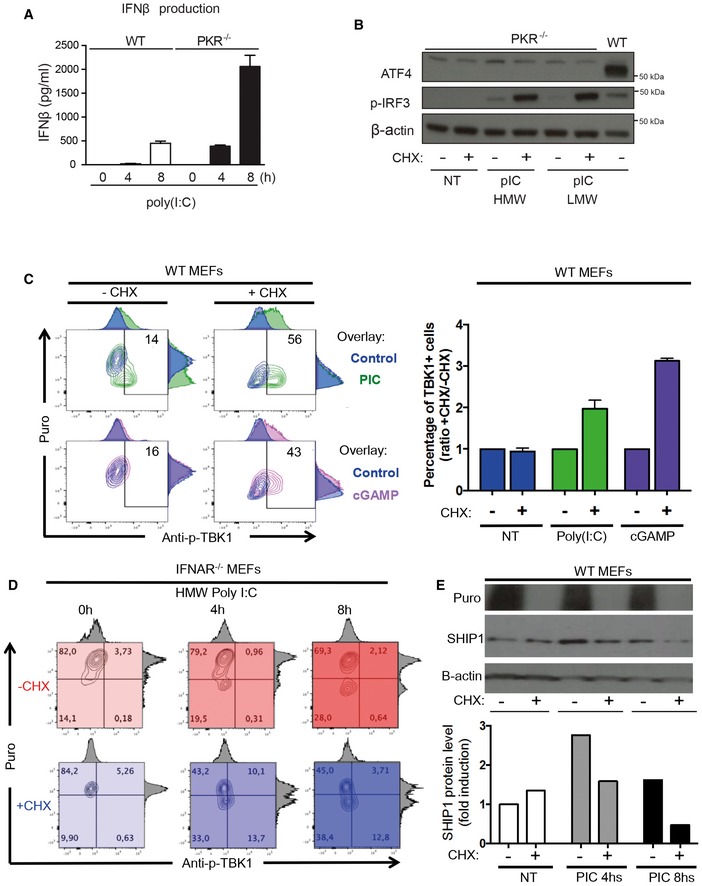
- IFN‐β production was measured by ELISA in cell culture supernatants of WT and PKR−/− MEFs that were stimulated with HMW poly(I:C) for 4 and 8 h. Results are the mean ± SD of three independent experiments.
- PKR−/− MEFs were left untreated (NT) or were treated with HMW or LMW poly(I:C) for 8 h in the presence or not of 5 μg/ml of cycloheximide (CHX). ATF4 and phospho‐IRF3 levels in nuclear extracts were detected by immunoblot. β‐actin was used as equal loading control. WT MEFs were treated with LMW poly(I:C) for 4 h and the corresponding nuclear extract was used as a positive control for ATF4 detection.
- WT MEFs were lipofected with poly(I:C) (green) or cGAMP (pink) for the indicated time in the presence or absence of CHX. Cells were pulsed with puromycin prior to collection, staining with anti‐puromycin and anti‐pTBK1 and flow cytometry analysis. Quantification is presented as histograms on the right (n ≥ 2). Bars are mean ± SD.
- IFNAR1−/− MEFs were lipofected with poly(I:C) for the indicated time in the presence (shades of blue) or absence (shades of red) of CHX. Cells were pulsed with puromycin prior to collection, staining with anti‐puromycin and anti‐pTBK1, and flow cytometry analysis.
- IFNAR1−/− MEFs were lipofected with poly(I:C) for the indicated time in the presence or absence of CHX. Cells were pulsed with puromycin prior to collection and immunoblot detection of puromycin and SHIP‐1. β‐actin was used as equal loading control. Quantification of the blot is presented below.
PKR activity has been associated with several signaling cascades including enhanced NF‐κB activity (McAllister et al, 2012) and activation of the NLRP3 inflammasome (Lu et al, 2012). However, the importance of PKR‐dependent eIF2α phosphorylation and translation inhibition has not been evaluated in the context of IRF3 phosphorylation. Artificial inhibition of protein synthesis using a low dose of cycloheximide (CHX) or harringtonine in PKR−/− MEFs enabled us to dissect the role of translation arrest independently of other potential signaling functions attributed to PKR. In the absence of PKR, treatment with CHX together with poly(I:C) delivery had a strong potentiating effect on both p‐TBK1 and p‐IRF3, causing a subsequent increase in IFNB and GADD34 mRNA transcription (Fig 3C). Synthesis of GADD34 independently of PKR expression and eIF2α phosphorylation occurred in cells undergoing translation inhibition, confirming that protein synthesis reduction itself is capable of augmenting IRF3 signaling and associated GADD34 expression. Despite abundant GADD34 synthesis, ATF4 induction was not observed in nuclear extracts of PKR−/− MEFs treated with poly(I:C) and CHX (Fig EV1B), again demonstrating the independence of GADD34 transcription from ATF4 expression. Harringtonine effectively blocks translation initiation by inhibiting elongation during the first rounds of peptide bond formation (Robert et al, 2009) and therefore mimics the impact of p‐eIF2α and translation inhibition on IRF3 signaling (Fig 3D). IRF3 phosphorylation was strongly increased in a dose‐dependent manner following harringtonine treatment, with higher doses of the drug causing a quasi‐complete loss of I‐κBα, demonstrating both its efficiency and the potential impact of translation inhibition on negative regulators of IRF3 and NF‐κB activation (Deng et al, 2004; McAllister et al, 2012).
We further tested whether protein synthesis inhibition reinforces TBK1 and IRF3 activation in a different NA‐sensing context than poly(I:C) and PKR activation by artificially activating stimulator of interferon genes (STING) with the cyclic dinucleotide cGAMP (Ishikawa et al, 2009). cGAMP, a dinucleotide normally produced by the DNA‐binding protein cGAS (cGAMP synthase), has no documented effect on protein synthesis and triggers strong type‐I IFN production in exposed cells (Gao et al, 2013). To refine our analysis, we optimized the previously described puromycylation procedure “SUNSET” to quantify protein synthesis by intracellular flow cytometry (Schmidt et al, 2009) and follow the consequences of both poly(I:C) and cGAMP detection on TBK1 phosphorylation in individual MEFs over time. Cytometry revealed that, upon poly(I:C) exposure, the cells showing the strongest protein synthesis inhibition also displayed higher levels of p‐TBK1 levels, which were further potentiated by CHX treatment (Fig EV1C). As expected from the absence of protein synthesis inhibition, the initial levels of p‐TBK1 induction in cGAMP‐stimulated cells were lower than after poly(I:C) treatment (Fig EV1C). Artificial protein synthesis inhibition (CHX), however, strongly synergized with the dinucleotide and augmented TBK1 phosphorylation equivalently to what was observed with poly(I:C) (Fig EV1C), thus confirming the importance of protein synthesis regulation for the fine‐tuning of the TBK1/IRF3 signaling pathway in response to NA. Interestingly, when the situation was examined in poly(I:C)‐treated IFNAR1−/− MEFs, translation inhibition was not efficiently implemented and TBK1 phosphorylation remained minimal in these cells (Fig EV1E). As observed for IRF3/IRF7−/− cells (Fig EV1D), chronic and induced IFNAR stimulation is therefore presumably required to allow RLRs and PKR expression to reach threshold levels capable of detecting dsRNA and trigger both TBK1 and PKR (Balachandran et al, 2004). Interestingly, the presence of CHX compensated partially IFNAR deficiency by again increasing TBK1 phosphorylation (Fig EV1E), which was now well detected by intracellular flow and appeared proportional to the cumulated levels of protein synthesis inhibition (poly(I:C) + CHX).
These results suggest that translation sensitive factors involved in TBK1 activation are likely to be IFN independent. As already shown for IκBα and the NF‐κB pathway (Fig 3D), translation inhibition could also impact inhibitors of the TBK1 signaling pathway. We therefore turned our attention toward A20 (also known as TNFAIP3), a potent anti‐inflammatory molecule that inhibits multiple intracellular signaling cascades and is induced by NF‐κB‐dependent signals (Ma & Malynn, 2012). A20 is a de‐ubiquitinating enzyme (DUB) that restricts the duration and intensity of NF‐κB signaling, but has also been shown to interact with TBK1 and IKKε to suppress IRF3 activation in a negative feedback loop (Saitoh et al, 2005). A20 expression was measured in MEFs after poly(I:C) stimulation in the presence or absence of CHX (Fig 3E). Immunoblots showed that A20 accumulation induced by poly(I:C) was prevented by CHX treatment concomitantly with increased p‐IRF3 levels (Fig 3E, right and center panels). This was also the case for the SHIP‐1 phosphatase, which is also involved in p‐TBK1 negative regulation (Gabhann et al, 2010), and whose induction by poly(I:C) was inhibited by CHX treatment (Fig EV1C). Protein synthesis inhibition impairs therefore the expression of several negative feedback regulatory molecules, like IκBα, SHIP‐1, and A20, and contributes to mount a commensurate response to the microbial threat by potentiating TBK1/IRF3 signaling.
Protein synthesis inhibition reinforces innate anti‐viral responses
We next evaluated the importance of protein synthesis inhibition and GADD34 expression in response to viral infection. We chose to monitor vesicular stomatitis virus (VSV) infection given it sensitivity to type‐I IFN and PKR activity (Balachandran et al, 2000). Upon infection with a VSV strain encoding GFP as an additional transcription unit (Obuchi et al, 2003), levels of protein synthesis and viral GFP were monitored by flow cytometry in WT and GADD34ΔC/ΔC MEFs (Fig 4). Analysis of VSV‐inoculated MEFs evidenced the presence after 8 h of two main populations of infected cells, one presenting strong translation inhibition (Puro−GFP+/−, P2 dark gray and P3 light green quadrants) and a second that was clearly infected but did not undergo protein synthesis arrest (Puro+GFP+, P4 dark green quadrant) (Fig 4A). After 12 h, all remaining live cells had their protein synthesis inhibited (P2 and P3), with little sign of recovery. The WT subpopulation arrested in translation contained approximately 65% of the total cells, with a minority displaying low GFP levels (P3), suggesting that upon VSV infection, protein synthesis inhibition takes place rapidly and prevents the accumulation of high GFP levels in infected cells, GADD34 being mostly synthetized between 8 and 12 h of infection (Fig 4B). As expected from the lack of IFN‐β release in the culture media (Fig 4B), infected GADD34ΔC/ΔC MEFs, still displaying normal protein synthesis activity after 8 h (P4), expressed higher levels of viral GFP than their WT counterparts (P1 + P4). GADD34ΔC/ΔC MEFs were more susceptible to infection overall with most cells being GFP positive after 8 h (Fig 4A and B), and interestingly most translation‐arrested cells (P3) expressed slightly higher GFP levels than equivalent WT cells (Fig 4A), suggesting that in the absence of IFN‐β, reduced PKR levels (Figs 1C and EV1D) could slow down translation arrest in GADD34ΔC/ΔC MEFs, allowing some early viral GFP expression to occur before complete inhibition of protein synthesis and IFN‐β induction. Uncontrolled VSV infection in PKR−/− and IFNAR1−/− MEFs, indicated by a massive accumulation of viral GFP in these cells (P4, Fig 4B), confirmed that PKR activity and type‐I IFN signaling are absolutely necessary to mediate translation inhibition and control VSV replication. We attempted to revert these phenotypes by treating infected cells with a low dose of CHX to both reinforce TBK1 phosphorylation and limit viral replication. Translation inhibition was extremely efficient at reducing GFP‐VSV replication in susceptible MEFs (Fig 4C), while, like for poly(I:C) detection, TBK1 phosphorylation was strongly enhanced by CHX treatment of these infected cells (Fig 4D).
Figure 4. VSV infection is controlled by GADD34 expression and translation inhibition.
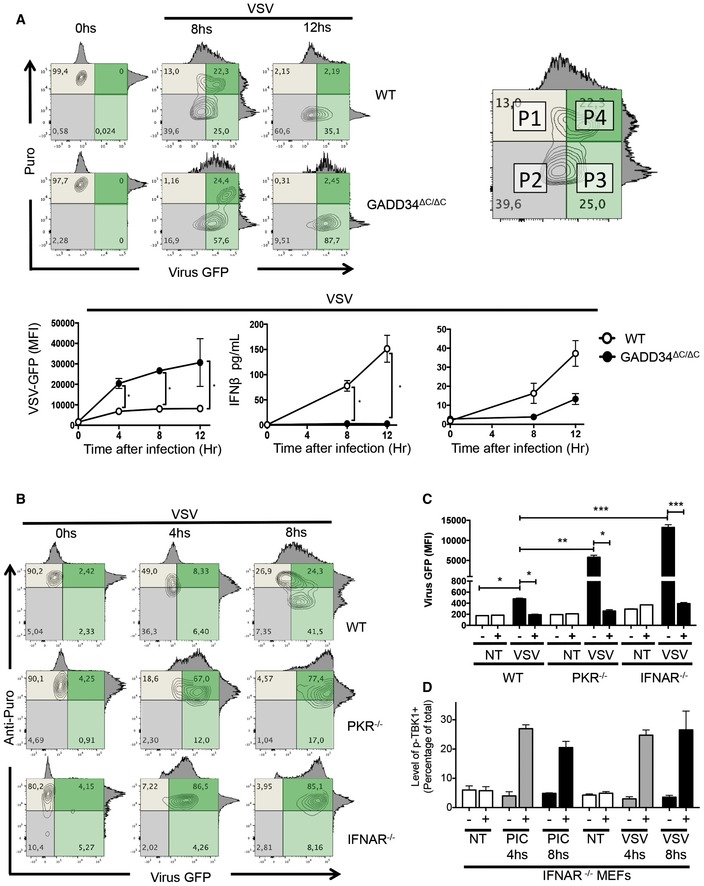
- WT and Gadd34ΔC/ΔC MEFs were inoculated with VSV‐GFP for the indicated time points. As depicted in the contour plots, the level of total translation (puromycin incorporation, Puro y‐axis, gray) and viral GFP expression (x‐axis, green)) were measured using anti‐puromycin staining and flow cytometry. An example of how flow quadrants (P1–P4) are labeled is shown (right). The kinetics of VSV‐GFP expression (GFP MFI) in WT and Gadd34ΔC/ΔC MEFs is shown (lower left panel). Induction of GADD34 measured by FACS and concentration of IFN‐β in the supernatant of VSV‐infected WT, and Gadd34ΔC/ΔC MEFs are shown in the lower panel.
- WT, PKR−/−, and IFNAR1−/− MEFs were inoculated with VSV‐GFP (green) for 4–8 h prior to being subjected to puromycin incorporation and anti‐puromycin staining (gray) for flow cytometry analysis.
- WT, PKR−/−, and IFNAR1−/− MEFs were inoculated with VSV‐GFP in the presence or absence of 5 μg/ml of cycloheximide (CHX) and viral GFP levels were measured by flow cytometry after 8 h of treatment.
- IFNAR1−/− MEFs were inoculated with VSV‐GFP or lipofected with poly(I:C) for the indicated time in the presence or absence of CHX. Cells were collected and stained with anti‐pTBK1 and quantified by flow cytometry.
Translation inhibition appears therefore to prevent viral replication not only by limiting viral protein synthesis, but also through a reinforcement of innate signaling pathways leading to enhanced IRF3 phosphorylation and type‐I IFN mRNA induction. Interestingly, in IFNAR1−/− MEFs treated with poly(I:C) or VSV, the levels of p‐TBK1 were strongly potentiated by CHX treatment, confirming that this effect is IFN independent. Thus, in normal circumstances, the intensity of the anti‐viral response triggered by dsRNA sensing will be linked to the kinetics of PKR activation and GADD34 expression, which through their antagonist actions on p‐eIF2α and translation initiation will balance the magnitude of IRF3‐dependent transcriptional responses together with the intensity of protein synthesis arrest occurring differently in each infected cell.
GADD34 is required for stress granules disassembly
To further address the molecular details of eIF2α phosphorylation and translation arrest in the response to dsRNA, we examined the formation of stress granules (SGs) with respect to GADD34 activity. SGs form in response to a wide range of cellular stresses and contain mRNAs and components of the translational 48S pre‐initiation complex, as well as other RNA‐binding proteins such as TIA‐1 or G3BP1, which also interacts with PKR and restricts viral infection (Kedersha et al, 2000, 2005; Reineke et al, 2015). Given the reciprocal interference of SG formation with protein synthesis, WT and GADD34ΔC/ΔC MEFs were treated with poly(I:C), which gives a more homogenous response than viral infection, prior to confocal microscopy analysis to visualize concomitantly G3BP1 localization in SG, IFN‐β production, protein synthesis using puromycin incorporation, and IFNB mRNA localization by FISH (Figs 5A and EV2A). IFN‐β expression, always associated with puromycin incorporation, was only observed in a small number of WT MEFs, suggesting that active protein synthesis and stochastic IFN production could be linked. Most WT and GADD34ΔC/ΔC MEFs arrested for translation displayed large G3BP1‐positive SGs that contained IFNB mRNAs (Figs 5A and EV2A), further explaining the absence of cytokine production in these cells despite active mRNA transcription, and associating SG formation with the storage of IFNB mRNA upon translation inhibition. To obtain reliable measurements of the capacity of both cell types to form SGs, G3BP1‐positive granules were counted using mosaic microscopy and software‐assisted quantification. We also evaluated whether SG induction in these cells depended uniquely on the dominant negative role of p‐eIF2α on guanine exchange factor (GEF) eIF2B using the pharmacological ISR inhibitor (ISRIB) that prevents inhibition of eIF2B and promotes translation initiation and stress granule assembly despite eIF2α phosphorylation (Sekine et al, 2015; Sidrauski et al, 2015; Fig 5B). A large majority of GADD34ΔC/ΔC cells rapidly displayed SGs when treated with thapsigargin (Fig 5C, upper panel), with ISRIB having a profound inhibitory effect on SG induction in these cells, thus confirming its capacity to prevent protein synthesis inhibition during ER stress (Sidrauski et al, 2015). Interestingly, while thapsigargin induced SGs in a relatively small fraction of WT cells (< 15%), SG induction by poly(I:C) was far more efficient (50%) and even enhanced in GADD34‐deficient cells after 1 h (Fig 5C, lower panel). Importantly in both cell types, ISRIB had no impact on SG formation after 1 h of poly(I:C) treatment (Fig 5C, lower panel), suggesting that the conditions of SG formation or maintenance between thapsigargin and poly(I:C) induction are critically different. When SGs were monitored over longer periods (Fig 5D), a progressive decrease in SG‐positive WT MEFs occurred between 3 h and 6 h after poly(I:C) treatment, strongly contrasting with GADD34‐deficient MEFs, which kept forming SGs (Fig 5D). Taken together, these results suggest that GADD34 expression promotes SG disappearance and facilitates the release of IFNB mRNAs to allow their productive translation. Interestingly, the role of GADD34 in this context appears to be independent of its effect on the interaction between p‐eIF2α and eIF2B, given that ISRIB treatment is unable to compensate for GADD34 deficiency and thereof associated lack of eIF2α dephosphorylation. These observations are further supported by the fact that ISRIB treatment does not rescue IFN‐β production (Fig 5E), nor protein synthesis in poly(I:C)‐treated GADD34ΔC/ΔC MEFs (Fig EV2B and C). This insensitivity to ISRIB suggests further that GADD34 expression is required for another function than strictly relieving the dominant negative inhibitory activity of p‐eIF2α over eIF2B to fully rescue protein synthesis and allow cytokine production in response to dsRNA.
Figure 5. Stress granules formed during response to poly(I:C) contain IFNB mRNAs and are resistant to ISRIB.
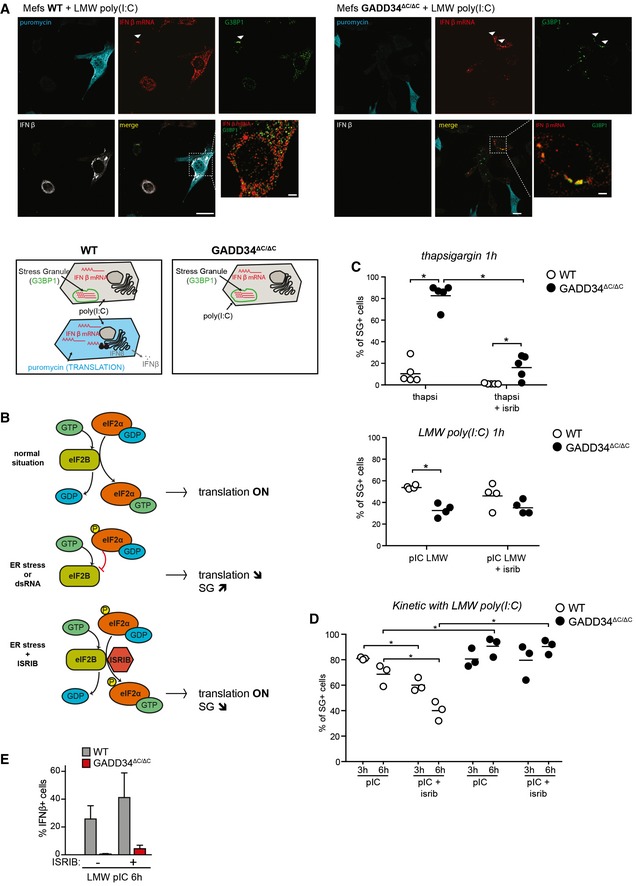
- Fluorescence in situ hybridization (FISH) staining for IFNB mRNA, combined with immunofluorescence confocal microscopy to detect protein translation (puromycin), stress granules (G3BP1), and IFN‐β protein in WT and GADD34ΔC/ΔC MEFs stimulated with LMW poly(I:C) for 6 h and labeled with puromycin for 10 min. Scale bars = 10 μm. Examples of colocalization between IFNB mRNA and G3BP1 in stress granules are indicated by arrowheads. Schematic representation of the different situations observed in both cell lines is shown below.
- Graphical abstract of the mechanism of action of ISR inhibitor (ISRIB). The small molecule ISRIB is able to prevent the dominant negative effects of p‐eIF2α on the guanine exchange factor (GEF) activity of eIF2B in cells undergoing ER stress. ISRIB facilitates therefore translation initiation even in the presence of large amounts of p‐eIF2α and is able to reverse the effects of a stress response such as translation decrease or stress granule formation.
- Percentages of SG‐containing cells within the total cell population were determined using software‐assisted quantification (see Appendix Supplementary Materials and Methods for details) from immunofluorescence mosaic images of WT and GADD34ΔC/ΔC MEFs stimulated with thapsigargin (left panel) or LMW poly(I:C) (right panel) for 1 h. When indicated, ISRIB was added to the cells at the time of stimulation. Each plot corresponds to one replicate out of five (left) or four (right) independent experiments. Means are represented with bars. The total number of counted cells in each replicate was comprised between 500 and 1,500. P‐values were calculated using a Student's t‐test, *P < 0.05.
- Same as (C) except that MEFs were stimulated with LMW poly(I:C) for 3 h or 6 h.
- Percentile of IFN‐β‐producing cells determined by flow cytometry. The results are the mean ± SD of four independent experiments. ISRIB has little impact on the protein synthesis inhibition triggered by dsRNA stimulation of PKR.
Figure EV2. Level of total translation, IFN production and IFN mRNA in WT and KO upon dsRNA stimulation.
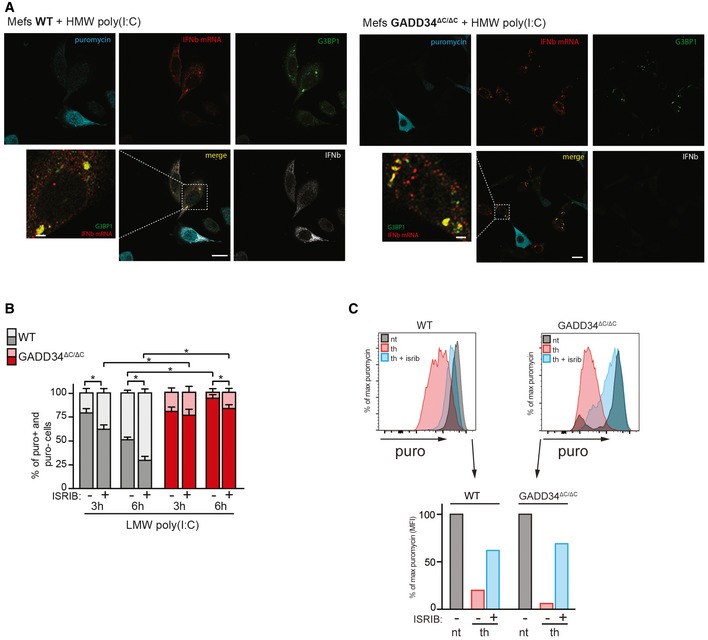
- Fluorescence in situ hybridization (FISH) for IFNB mRNA, combined with immunofluorescence staining for protein synthesis (with an anti‐puromycin mAb), for stress granules (with anti‐G3BP1 Ab) and for IFN‐β protein in WT and GADD34ΔC/ΔC MEFs stimulated with HMW poly(I:C) for 6 h and labeled with puromycin for 10 min. Scale bars = 10 μm.
- WT and GADD34ΔC/ΔC MEFs were stimulated with LMW poly(I:C) for indicated times in the presence of ISRIB and puromycin for protein synthesis measurement by FACS. Percentiles of translating (puro+, light color) and non‐translating cells (puro−, dark color) were determined by flow cytometry from fluorescence intensity of individual cells and are represented as cumulative bars for WT (gray) and GADD34ΔC/ΔC MEFs (red). P‐values were calculated using a Student's t‐test, *P < 0.05. Mean ± SD.
- Similar experiments were conducted with thapsigargin and ISRIB. In UPR conditions, ISRIB rescued protein synthesis efficiently, confirming the efficacy of the drug on this pathway. Although ISRIB abrogates the impact of eIF2α phosphorylation on translation initiation during ER stress, it does not efficiently restore protein synthesis and cytokines production in cells responding to dsRNA.
Cells exposed to dsRNA undergo protein synthesis inhibition prior to producing type‐I IFN
Given the importance of dsRNA detection kinetics to allow protein synthesis inhibition and type‐I IFN transcription, single‐cell analysis by intracellular flow cytometry was again performed to follow the quantitative impact of translation arrest and GADD34 expression over time on IFN‐β production (Fig 6). When WT and GADD34ΔC/ΔC MEFs were lipofected with poly(I:C) for 3 h and 6 h, several populations of cells could be distinguished based on their levels of puromycin incorporation and ability to produce GADD34, p‐eIF2α, and/or IFN‐β (Figs 6 and EV3). With time, like for VSV infection, dsRNA delivery induced translation inhibition in a large proportion (up to 80%), but not all individual cells (Fig 6A), this despite a rapid uptake of poly(I:C) in most cells (Fig EV3A). The proportion of these two populations was similar in WT and in GADD34‐deficient cells 3 h after poly(I:C) delivery (Fig 6A, B and D), but after 6 h, significant differences in the proportion of translating (P1 + P4) and non‐translating (P2 + P3) cells could be detected. A large majority of GADD34ΔC/ΔC cells were arrested in translation (> 90%), whereas the equivalent population of puromycin‐negative cells (P2 + P3) in WT MEFs was reduced from 79 to 51% between 3 h and 6 h, in a manner correlated with GADD34 induction (P3) and its capacity to restore protein synthesis (P4). GADD34 expression was monitored using an antibody raised against the N‐terminal part of the protein, enabling detection of both complete GADD34 and truncated GADD34ΔC. After 3 h of poly(I:C) delivery in WT MEFs, a majority of GADD34‐expressing cells had not restored translation (Fig 6B, P3), while after 6 h, protein synthesis recovery was observed in 15.5% of the cells (P4), representing half of the GADD34‐positive population (P3 + P4). Translation recovery never occurred in GADD34ΔC/ΔC cells (Fig 6B and D), as indicated by the accumulation of cells with the highest level of p‐eIF2α in the puromycin‐negative cell quadrant (P3, Fig EV3B). Interestingly, after 6 h of activation, 6% of WT cells displayed both active protein synthesis and moderate eIF2α phosphorylation, while the population displaying normal translation levels nearly doubled (P1, 34%) (Fig EV3B), confirming that a majority of MEFs exposed to poly(I:C) first undergo PKR‐dependent inhibition of protein synthesis, prior to expressing GADD34 (P3, Fig 6B), which in turn promotes eIF2α dephosphorylation and progressively restores translation in about 15–30% of these cells (P4, Figs 6B and D, and EV5B).
Figure 6. Protein synthesis cycling in response to dsRNA is revealed by intracellular flow cytometry.
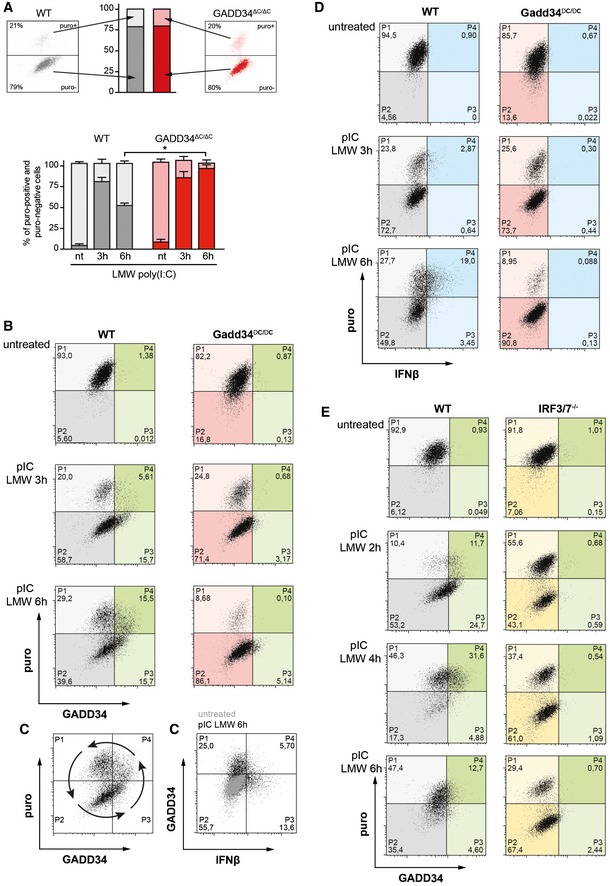
- Two‐dimensional plots of fluorescence intensity of individual WT (left panel, grey) and GADD34ΔC/ΔC MEFs (right panel, red) stimulated with poly(I:C) for 3 h prior to staining for puromycin (y‐axis) and GADD34 (x‐axis). The percentiles of translating (puro+, light color) and non‐translating cells (puro−, dark color) were determined from the plots of fluorescence intensity of individual cells and are represented as cumulative bars for WT (grey) and GADD34ΔC/ΔC MEFs (red). Mean ± SD of four independent experiments is shown for not treated 0 h (nt), and 3 and 6 h poly(I:C)‐treated MEFs. P‐values were calculated using a Student's t‐test, *P < 0.05. Translation recovery at 6 h is around 50% of WT cells and hardly occurs in GADD34ΔC/ΔC cells.
- Two‐dimensional plots of fluorescence intensity of individual WT (left panels, gray and green) and GADD34ΔC/ΔC MEFs (right panels, red and green) stimulated with poly(I:C) for 0, 3, 6 h prior to staining for puromycin (y‐axis) and GADD34 (x‐axis). Translating cells are detected in the two upper quadrants (P1 and P4), cells in which protein synthesis is inhibited are in the two lower quadrants (P2 and P3), and cells expressing GADD34 are shifted in the two right quadrants (P3 and P4, green). The progression over time of the dsRNA‐stimulated cells through the different quadrants suggests a cycle and potentially, an oscillation of protein synthesis and IFN‐β production.
- Same as (B) with GADD34 staining on the y‐axis and IFN‐β on the x‐axis. Cells that produce IFN‐β rarely express the GADD34 protein, despite a common transcriptional regulation. This indicates that full translation recovery is necessary for IFN‐β production, a situation that is likely promoting a rapid GADD34 loss.
- Same experiment as in (B) but cells expressing IFN‐β are in the two right quadrants (blue). IFN‐β production only occurs in the cells that are active in protein synthesis.
- Two‐dimensional plots of fluorescence intensity of WT (left panels) and IRF3/IRF7−/− MEFs (right panels) stimulated with poly(I:C) for 0, 2, 4, and 6 h prior to staining for puromycin (y‐axis) and GADD34 (x‐axis). IRFs are necessary for GADD34 expression and their inactivation renders cell incapable of restoring protein synthesis after dsRNA‐induced inhibition. Interestingly, in this set of control MEFs, a rapid cycle of translation arrest, restoration, and again arrest can be observed during the 6 h of the experiment, matching the synthesis of GADD34.
Figure EV3. Levels of intracellular poly(I:C) uptake and eIF2α phosphorylation in WT and GADD34ΔC/ΔC MEFs.
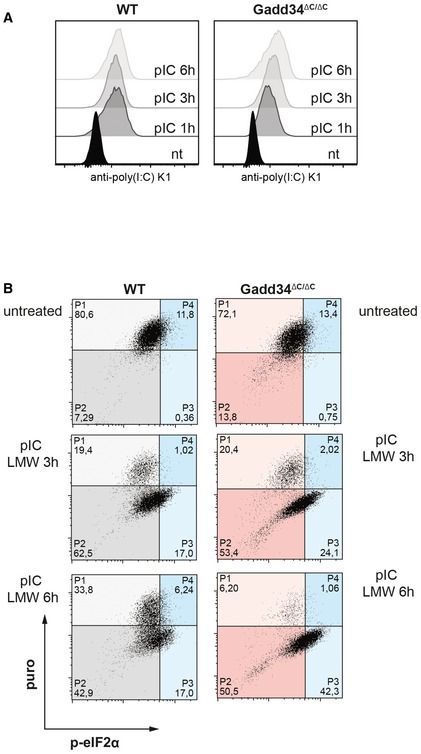
- WT and GADD34ΔC/ΔC MEFs were treated with poly(I:C) for 1, 3, and 6 h before fixation and permeabilization. Poly(I:C) was detected with specific K1 mAb using intracellular flow cytometry.
- Two‐dimensional plots of fluorescence intensity of individual WT (grey and blue) and GADD34ΔC/ΔC MEFs (red and blue) stained for puromycin (y‐axis) and p‐eIF2α (x‐axis). Percentage of cells in each quadrant is indicated. Cells expressing p‐eIF2α are shifted in the two right quadrants (P3 and P4, blue).
Figure EV5. Mathematical modeling of protein synthesis oscillation upon increased GADD34 half‐life and reduced parameters variability.
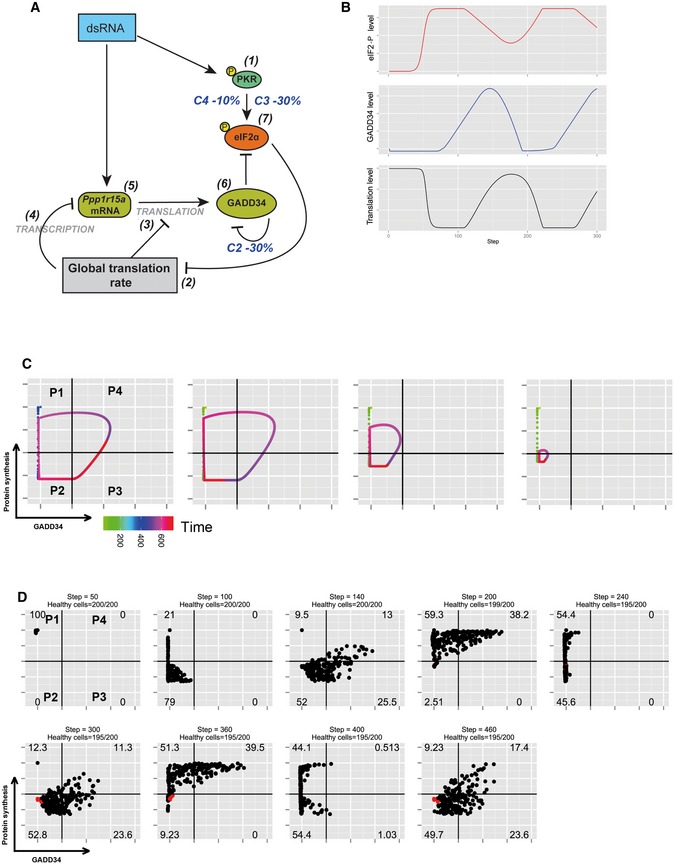
- Schematic representation of the biochemical relationships and interdependences used to establish the mathematical model described in the Materials and Methods section. For this simulation, parameter C3 of equation (7) and parameter C2 of equation (6) were decreased by 30% and parameter C4 of equation (7) by 10%, while random variability was kept at 10%.
- Graphic representation over time of model‐based levels of p‐eIF2α (red), protein synthesis (black), and GADD34 activity (blue), demonstrating the more rapid oscillatory nature of protein synthesis with these modified parameters.
- Representative single‐cell simulation showing the progression across quadrants (P1–P4) representing protein synthesis intensity and GADD34 levels over time (rainbow color gradient). Cells undergo more rapid and compact cycles of protein synthesis inhibition and rescue.
- Grouped simulations of 200 cells over 450 time steps. By introducing 10% of variability in most parameters with fixed delays, virtual cell behavior mimics closely the experimental situation observed in Fig 5D. Few dead cells are represented in red and subtracted from the indicated cell percentiles in each quadrant.
Only a fraction of WT cells displaying active protein synthesis was found to produce IFN‐β (P4, Fig 6D), while GADD34ΔC/ΔC MEFs were incapable of producing IFN‐β (Fig 6D). Most cells producing IFN‐β expressed little GADD34 (P4, Fig 6C), suggesting that its rapid degradation by the proteasome (half‐life < 15 min, Fig EV4; Brush & Shenolikar, 2008), together with efficient inhibition of its synthesis upon eIF2α dephosphorylation, strongly decreased its expression at the peak of translation restoration and IFN‐β production. These results also confirm that IFN‐β and cytokine production only occurs in cells expressing sufficient GADD34 to efficiently promote eIF2α dephosphorylation and re‐establish active translation.
Figure EV4. GADD34 half‐life upon poly(I:C) treatment.
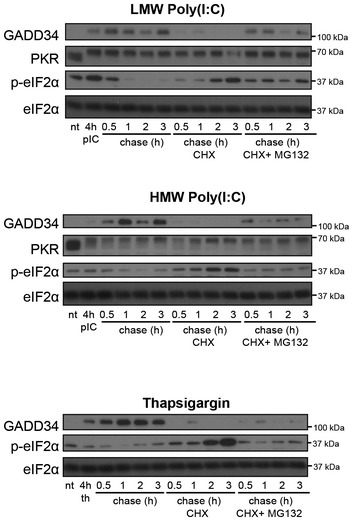
WT MEFs were treated with HMW poly(I:C), LMW poly(I:C), or thapsigargin (th) for 4 h, prior to a chase of 0.5, 1, 2, and 3 h with fresh medium. When indicated, cycloheximide (CHX) +/− MG132 were added to the medium during the chase. GADD34, PKR, p‐eIF2α, and total eIF2α were detected by immunoblot with specific antibodies.
IRF3/IRF7−/− MEFs do not induce GADD34 upon dsRNA detection and should therefore be unable to overcome PKR‐dependent eIF2α phosphorylation, displaying a protein synthesis inhibition similar to GADD34ΔC/ΔC MEFs (Fig 6E). As anticipated, unstimulated WT and IRF3/IRF7−/− MEFs displayed similar level of protein synthesis, which was rapidly downregulated upon dsRNA delivery (P2, Fig 5E). Although a higher proportion of non‐responder cells was found in the IRF3/IRF7−/− cells (P1), translation arrest occurred in more than 60% of the cells (P2) without any signs of GADD34 expression nor protein synthesis recovery (P3 and P4). Thus, despite a slightly increased threshold of dsRNA sensing in these IRF‐deficient cells (Balachandran et al, 2004), as observed in IFNAR1−/− MEFs, PKR levels were still sufficient to induce protein synthesis inhibition in response to poly(I:C). However, much as in GADD34ΔC/ΔC cells, IRF3/IRF7−/− MEFs were incapable of mediating eIF2α dephosphorylation, rendering translation inhibition poorly reversible. Interestingly, protein synthesis in the matching control MEFs was very efficiently arrested after 2 h of stimulation (P2), immediately followed by GADD34 expression (P3) and protein synthesis recovery (P4 + P1) within 4 h. However, within 2 additional hours, a novel cycle of translation arrest seemed to be initiated, since the puromycin‐negative cell population (P2) was doubled (17–35%) and GADD34 expression was strongly decreased at this time of activation (P3 + P4).
Altogether, these results indicate that translation arrest is a key step in the response to dsRNA, since it amplifies IRF3 signaling and augments IFN‐β transcription, prior to GADD34 synthesis and restoration of protein and cytokine synthesis. We have thus elucidated a direct link between transcription regulation by IRFs and the capacity of cells to translate proteins upon dsRNA and viral detection. This finding has many implications for the global understanding of innate responses to pathogens and IRF‐dependent transcription. As these events are dynamic over time and linked both to transcription and translation regulation, dsRNA‐stimulated cells engage in what looks like cycles of consecutive translation arrest and recovery (Fig 6B). Stochastic expression of IFN‐β could therefore reflect the cycling levels of active protein synthesis in cells responding to dsRNA asynchronously and representing around 20–30% of the total cell population at a given time point in our experimental system.
Mathematic modeling is consistent with protein synthesis cycling in individual cells exposed to dsRNA
Given the observation that protein synthesis inhibition and rescue after GADD34 expression seems to cycle in cells responding to dsRNA (Fig 6B), we used mathematical modeling to explore whether the integration of the different unraveled biochemical pathways could adopt an oscillating behavior, which when extended to a larger cell population could account for our experimental observations. We thus constructed a mathematical model (see Materials and Methods) that describes the evolution of eIF2α and protein synthesis regulatory parameters for a single cell. This discrete time model is based on the scheme depicted in Fig 7A, in which the main relationships involved were the PKR activation by dsRNA and subsequent eIF2α phosphorylation leading to translation arrest, followed by the induction of the negative regulatory feedback loop‐driven by short‐lived GADD34. These experimentally characterized interactions were put in equation taking into account their dependences and time or delay of activity. The formulation models each interaction (arrows, Fig 7A), associated with a parameter adjusting the dependency of the downstream vertex on the upstream one. These dependencies are mainly linear except for PKR activation by dsRNA and protein synthesis inhibition by p‐eIF2α, which both depend on sigmoidal laws (inverted for p‐eIF2α), and thus describe an activation/saturation behavior. These choices are supported by our observation that PKR activation (phosphorylation) remains constant over the time frame of the experiments (Figs 2D and EV4). By default, each vertex‐associated variable level at a time step t depends on both its own level (negatively in the case of GADD34, modeling its degradation rate) and those of its upstream regulators at the previous time step t−1. We also considered a delayed effect, (i) for GADD34 levels in relation to protein synthesis given their strict mutual dependence (e.g., GADD34(t) depends on p(t−d1), d1 > 1), and (ii) for eIF2α dephosphorylation in response to GADD34 expression (d2)). We did not take into consideration SGs' impact, since their formation and destruction are linked to eIF2α phosphorylation and GADD34 expression, and thus obey to the same equations.
Figure 7. Mathematical modeling supports the existence of protein synthesis waves of cells exposed to dsRNA.
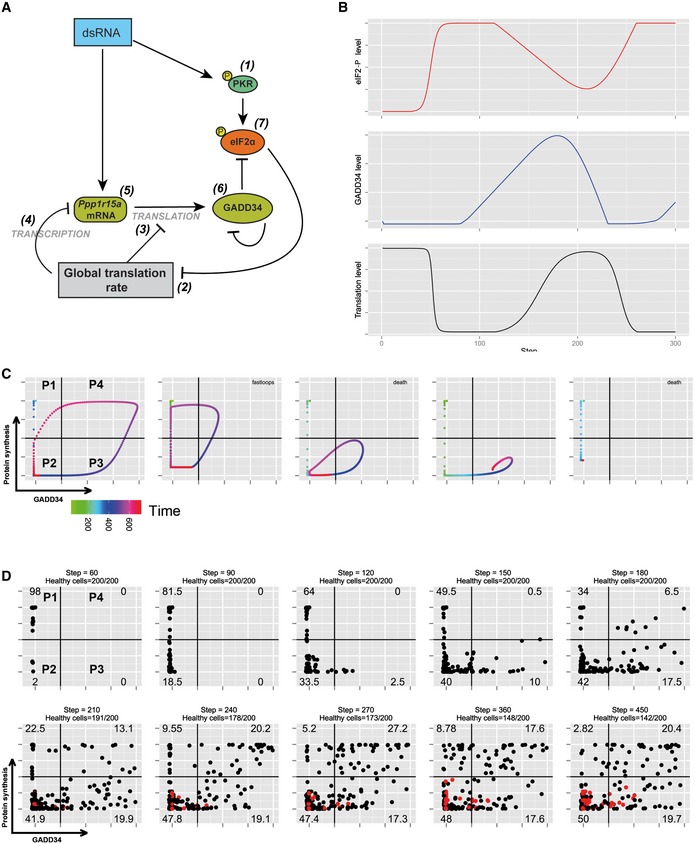
- Schematic representation of the biochemical relationships and interdependences used to establish the mathematical model described in the Materials and Methods section.
- Graphic representation over time of model‐based levels of p‐eIF2α (red), protein synthesis (black), and GADD34 activity (blue).
- Representative single‐cell simulation showing the progression across quadrants (P1–P4) representing protein synthesis intensity and GADD34 levels over time (rainbow color gradient). Cells can undergo several cycles of protein synthesis inhibition and rescue (fastloops), while some cells are unable to rescue protein synthesis and are likely to die (death).
- Grouped simulations of 200 cells over 450 time steps. By introducing 30% of variability in most parameters with fixed delays, virtual cell behavior mimics closely the experimental situation observed in Fig 5B. Dead cells are represented in red and subtracted from the indicated cell percentiles in each quadrant.
The relative heterogeneity in the experimental estimations of the parameters, as well as their orders of magnitude that clearly depend on the measurement strategies, prevents this model from being quantitatively predictive. Nevertheless, these estimations gave us access, with good confidence, to the type (shape) of dependencies that define the natural dynamic behavior of the system. Simulation of eIF2 and GADD34 and global translation‐level dynamics for realistic parameter values demonstrate that oscillations can be intrinsically present in such model (Fig 7B). Moreover, the model contains two network motifs, (i) an incoherent type‐1 feed‐forward loop (IFFL), and (ii) a repressilator, both of them described in the literature as generating oscillatory dynamics (see Materials and Methods). Given the relatively broad error margin in the measurement of our parameters, and considering the existing heterogeneity among individual cells, we introduced up to 30% of random variability in most parameters values with fixed time delays. Predicted dynamics in the GADD34 translation‐level plan for different randomly perturbed values of parameters (initial condition in the P1 quadrant, rainbow gradient) is shown for chosen representative virtual cells (Fig 7C). Many of the simulations (cells) show an oscillatory behavior, with at least two passages though protein synthesis inhibition and generally one rescue (P1 through P4, fast loops) in the chosen simulation time. The speed of transition from one protein synthesis status to the other was irregular in each individual cell; however, even with levels of variability as high as 30%, the cyclic nature of protein synthesis inhibition remained a characteristic of most behaviors. Several cells did not display cyclic behavior and adopted convergent dynamics toward a fix point. In reality, such cell behavior could correspond to death or apoptosis and was therefore indicated as such in the different simulations, if they remained for 150 periods in the same quadrant. We performed simulations of larger virtual populations by integrating the behavior of 200 randomly perturbed single cells over 450 time steps and gradually removing “dead” cells (red) from the counts (Fig 7D). The progression of the virtual cell populations across the four quadrants representing the intensity of protein synthesis and GADD34 levels over time was strikingly consistent with what was observed experimentally by FACS in the WT MEFs (Fig 6B). Starting from 100% of virtual cells active in protein synthesis in P1, this population was rapidly arrested in translation (P2) after the initiation of the simulation. This proportion fell to 34% after 180 time steps, while GADD34 expression first and protein synthesis second increased to reach, respectively, 17.5% (P3) and 6.5% (P4), proportions similar to what is observed experimentally after 3 h of poly(I:C) treatment (Fig 6B). From this time step on, cells displaying both active protein synthesis and GADD34 expression increased (P4), with a step 210 mimicking the experimental situation at 6 h, although cells active in translation without GADD34 expression (P1) had a relative lower representation in the model. From single‐cell tracts (Fig 7C), we observed that passage from P4 toward P2 across P1 was faster than other transitions, thus explaining the lower proportion of cells found in P1 at step 210. From this step on, the virtual cells population left its state of transition and entered an asymptotic regimen in which most cells moved from one quadrant to the other, except for an immobile fraction, which was considered as dying (red dots). These simulations clearly suggest that protein synthesis can adopt an oscillatory behavior in cells exposed to dsRNA and that the molecular machinery and interdependency relationships described in Fig 6A are sufficient to explain the stochastic behavior of protein synthesis and IFN‐β production observed experimentally.
We tested the robustness of the model by adjusting several parameters to reproduce the experimental situation observed in Fig 6E, during which WT cells behave more homogeneously and transit from one stage to the other more rapidly than the WT MEFs used in Fig 6B. We anticipated that by reducing random variability (± 10%) while increasing GADD34 stability (C2, −30% degradation) and decreasing PKR activity (C3, −30%), virtual cells could tend toward a similar population dynamics (Fig EV5). Indeed, GADD34 stability is likely to represent a central adjustment parameter across cell types, since it depends on ubiquitination and proteasome activity that are adapted in a cell‐specific manner to different growth conditions and environmental cues. Starting from 100% of cells active in translation (P1), these new set of parameters influenced the progression of the virtual population that showed more rapid and compact undulations of protein synthesis (Fig EV5B and C), with single‐cell behaviors capable of going through two consecutive cycles of protein synthesis and rescue (Fig EV5C). Upon population simulation (Fig EV5D), the cell distribution observed at step 140 or 300 were equivalent to what is observed after 2 h of poly(I:C) stimulation. Steps 200 or 360 displayed a cell distribution similar to the experimental situation at 4 h, while steps 240 and 400 could be considered equivalent to 6 h of poly(I:C) exposure, although the proportions of cells residing in P3 and P4 appeared reduced compared with the experimental conditions. The period required to achieve these cycles is very regular and suggests that 1 virtual time step is equivalent to 2 min of experimental time. Our different simulation attempts also indicate that the delays (d1 and d2) between the global translation inhibition, eiF2α phosphorylation status, and GADD34 synthesis or its loss, all have a strong influence on the general progression of the virtual cells across the quadrants and require minimal variation to reflect our experimental results. Thus, the number of protein synthesis cycles occurring after dsRNA detection is predicted to be highly variable and dependent on these delays, as well as the half‐life of GADD34 that defines the capacity of individual cells to rapidly re‐establish protein synthesis, while avoiding cell death.
Discussion
We describe here how two apparently opposite signaling programs, mediated by the kinase PKR on the one hand and RLR/MAVS IFN triggering pathway on the other, synergize, and provide a molecular frame to explain some stochastic aspects of type‐I IFN production. These signaling cascades, both elicited by dsRNA detection, are tightly coordinated, allowing efficient protein synthesis inhibition preventing viral replication, followed by abundant cytokine production in the same cells. We propose that GADD34, the inducible cofactor of PP1c, is the key molecule involved in resolving this antagonistic situation. The role of ATF4 and GADD34 in eIF2α dephosphorylation to restore protein synthesis after ER stress has been extensively studied (Novoa et al, 2001). We show, however, that the GADD34 transcriptional induction by poly(I:C) strictly requires the transcription factors IRF3 and possibly IRF7, but not ATF4, which remains a hallmark of the UPR. This dependency on IRFs indicates that GADD34 belongs to a group of genes induced during the primary innate immune response to dsRNA, together with key anti‐viral mediators like IFN‐β and IFN‐α (Ourthiague et al, 2015), and as suggested by ChIP‐seq analysis of genome‐wide occupancy of IRF3 and p65/RELA, performed on a human B lymphocytic cell line infected with Sendai virus (Freaney et al, 2013). In addition to the lack of ATF4‐associated transcriptional signatures in GADD34ΔC/ΔC MEFs exposed to poly(I:C), our analysis reveals that ISGs can be ranked in at least two subsets displaying different transcriptional requirements. For instance, the MHC I‐associated genes, Pkr and several members of the tripartite motif‐containing protein (Trim) family (Trim 6, 12c, 14, 25, 26, 30a, 30b, and 56) (Jefferies et al, 2011) are only upregulated in WT MEFs, suggesting that these genes display critical differences in timing and threshold of activation by type‐I IFN. The profoundly reduced capacity of GADD34ΔC/ΔC MEFs to produce cytokines therefore reveals different modes of ISG induction, potentially linked to the translation and activation of different combinations of transcription factors (e.g., ATF4 or IRF7) to achieve optimal and coordinated responses.
Using PKR−/− MEFs, we showed that downregulation of global translation initiation following dsRNA detection contributes to the amplification of IRF3 phosphorylation and thus GADD34 and IFNB expression. The most likely explanation for low GADD34 mRNA induction in PKR−/− MEFs in response to poly(I:C) would therefore not be the absence of ATF4 induction directly, as previously proposed (Clavarino et al, 2012), but would rather be linked to the lack of protein synthesis inhibition and a consecutive decrease in IRF3 activation in these cells. PKR‐dependent activation of IRF3 has been observed during mutant vaccinia virus infection of HeLa cells (Zhang & Samuel, 2008), and PKR, GCN2, or PERK have been proposed to promote innate responses against viral infection by enhancing IFN production through MAVS signaling and augmented NF‐κB activation (Jiang et al, 2003; McAllister et al, 2012). Higher NF‐κB activation is promoted by the destabilization of its inhibitory chaperone IκBα and importantly by its lack of neosynthesis upon translation inhibition (Jiang et al, 2003). This situation can be extrapolated to other regulatory proteins, like the deubiquitinase A20, which was shown to regulate TBK1 and IRF3 activation. We confirmed that a decreased rate of protein synthesis in PKR−/− MEFs treated with poly(I:C) strongly enhances IRF3 phosphorylation, while decreasing both IκBα, A20, and SHIP‐1 levels. This also implies that these regulators are submitted to a tight control of their homeostasis through rapid proteasome‐mediated degradation. The concomitant implication of several of these negative regulators does not exclude the existence of additional pathways regulating the MAVS cascade, such as the activating dephosphorylation of RIG‐I and MDA5 by PP1 in human cells (Wies et al, 2013; Mesman et al, 2014). Protein synthesis inhibition functions therefore as an amplification signal, synergizing with innate signaling to favor primary anti‐viral genes transcription and GADD34 synthesis. Translation inhibition should be considered as a “danger” signal potentiating innate responses commensurate with the pathogenic threat (Argüello et al, 2015) and recently suggested for legionella infection (Pierre & Gatti, 2013). Cells use SG formation to adapt to different stresses and therefore are likely to adopt specific features according to the nature of the insults encountered. SGs, in addition to their important role in controlling IFNB mRNA stability, can serve as signaling hubs for PKR and RLRs (Reineke & Lloyd, 2015) and could thus be important for translation inhibition‐associated signaling. Their breakdown is also directly influenced by IRF3 translocation and GADD34 expression, thus allowing IFN production in coordination with a regulation of innate signaling and potentially its termination.
Measuring translation intensity using intracellular flow cytometry was essential to dissect kinetics of anti‐viral responses at the cellular level. Even though lipofected poly(I:C) enters cells rapidly and homogeneously, PKR does not trigger translation arrest uniformly. This heterogeneity in the early response to dsRNA could depend on PKR levels, as well on the physiological status of the cells, including cell cycle stage or expression of transcription factors like IRF3. Apart from this small number of non‐responder cells, GADD34 induction was mostly observed in cells arrested in translation and was absolutely required to recover normal levels of protein synthesis and produce IFN‐β. IRF3‐dependent expression of GADD34 is therefore a prerequisite for cytokine production by dsRNA‐activated MEFs, while the intensity its proteasome‐dependent degradation will influence the duration and intensity of IFN‐β expression both transcriptionally and translationally.
Mathematical modeling further established that protein synthesis cycling is compatible with our experimental results. The robustness of the model indicates that the different biochemical loops included in the modeling (Fig 7A) are sufficient to obtain a pattern of protein synthesis regulation equivalent over time to the one observed in individual cells (Fig 6). This model can be simplified by removing the transcriptional regulation of the Ppp1r15a gene and considering the production of GADD34 as a protein only (Appendix Supplementary Materials and Methods). We feel, however, that to respect biological principles, the transcriptional regulatory step had to be included in the presented model. This model points toward a dynamic dsRNA‐dependent oscillation of translation, with a periodicity directly linked to GADD34 degradation rate and intensity of PKR signaling (Dalet et al, 2015). In support of this hypothesis, the majority of cells that restored their protein synthesis and are thus capable of producing IFN‐β, no longer expressed GADD34. Translation oscillation would also be consistent with the dynamic behavior of SG formation following infection with various RNA viruses (Ruggieri et al, 2012). At least one cycle could be observed over 6 h of poly(I:C) exposure (Fig 6E), but many factors linked to cell metabolism and differentiation are likely to influence these events within a cell population. Translation oscillation may resolve the apparent dilemma resulting from concomitant translation inhibition and IFN‐β secretion in the same dsRNA‐exposed or infected cell (Fig 7). Cell‐to‐cell variability in the expression levels of some limiting factors of the RLR signaling pathway, among them type‐I IFN‐inducible RLRs and IRF7, has been evoked as the primary reason for stochastic IFN production in fibroblasts (Zhao et al, 2012). However, given the dependency of GADD34 expression on RLR signaling through MAVS and IRFs, our results support a direct contribution of protein synthesis inhibition and GADD34 expression to the stochasticity of IFN‐β production.
Materials and Methods
Cells, reagents, and plasmid transfection
Matched wild‐type (129 SvEv) and PKR−/− MEFs (Yang et al, 1995: #98) were a gift from Caetano Reis e Sousa (Cancer Research UK, London); matched wild‐type (129 SvEv), ATF4−/−, GADD34ΔC/ΔC MEFs were a gift from David Ron (Cambridge Institute for Medical Research, UK). Matched wild‐type and IRF3/7−/− MEFs were a gift from Michael Diamond (Washington University, Saint Louis). Matched wild‐type (C57/Bl6) and IPS‐1−/− MEFs were a gift from Matthew Albert (Pasteur Institute, Paris). Matched wild‐type (C57/Bl6) and IFNAR1−/− MEFs were generated from knockout mice. MEFs were cultured in DMEM, 5% FBS (Sigma) and 50 μM 2‐mercaptoethanol, except IPS‐1−/− MEFs and their matched WT, which were cultured with 15% FBS, 2 mM glutamine, MEM non‐essential and essential amino acids. MEFs were treated for the indicated time with 100 MOI VSV‐GFP (Whelan et al, 2000), or lipofected with 2.5 μg/ml of HMW poly(I:C) or 2 μg/ml of LMW poly(I:C) (InvivoGen) in combination with Lipofectamine 2000 (Invitrogen). Thapsigargin (SIGMA) and ISRIB (gift from Carmela Sidrausky and Peter Walter, UCSF, San Francisco) were used at 500 nM and 750 nM, respectively. Brefeldin A (BFA) and cycloheximide (CHX) (both from SIGMA) were used at 10 μg/ml and 5 μg/ml, respectively. The plasmid GADD34 in pCMV6‐XL5 mammalian expression was from OriGene (Rockville). The plasmid coding for herpes simplex virus ICP34.5 was a gift from Ian Mohr (New York University, NY, USA). As control plasmid, we used a pmax‐GFP vector (Amaxa).
RNA extraction
For qPCR, RNAs were isolated from cells using the RNeasy miniprep kit (QIAGEN) combined with a DNA digestion step (RNase‐free DNase set, QIAGEN). For microarray samples, cells were lysed in a Nonidet P‐40‐containing buffer (0.5%) before a short centrifugation. Proteins from supernatants were denatured with 1% SDS, 10 mM EDTA and 200 μg/ml proteinase K at 37°C for 30 min. RNA was extracted with phenol:chloroform pH 4.5 (Ambion), precipitated with isopropanol (v/v) at −20°C for 30 min, and washed with 70% v/v ethanol. RNAs were further purified with RNeasy miniprep kit (QIAGEN) combined with a DNA digestion step (RNase‐free DNase set, QIAGEN). RNA integrity was measured by capillary electrophoresis using the Agilent RNA 6000 Pico Chip kit (Agilent Technologies) in an Agilent 2100 Bioanalyzer, according to the manufacturer's instructions. A RIN (RNA integrity number) score > 7 was required for samples to be analyzed by microarray.
Microarray data
The microarray data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE77777.
Quantitative PCR
After RNA extraction and purification, cDNA was synthesized using the Superscript II reverse transcriptase (Invitrogen) and random hexamer primers (Thermo Scientific). Quantitative PCR amplification was carried out using complete SYBR Green PCR master mix (Applied Biosystems) and 200 nM of each specific primer; 5 μl of diluted cDNA template was added to 15 μl of PCR mix, and the amplification was tracked via SYBR Green incorporation by an Applied Biosystems thermal cycler. cDNA concentration in each sample was normalized to GAPDH expression. The primers used for gene amplification were the following:
GADD34 (S 5′‐GACCCCTCCAACTCTCCTTC‐3′, AS 5′‐CTTCCTCAGCCTCAGCATTC‐3′);
IFN‐β (S 5′‐CCCTATGGAGATGACGGAGA‐3′, AS 5′‐ACCCAGTGCTGGAGAAATTG‐3′);
GAPDH (S 5′‐TGGAGAAACCTGCCAAGTATG‐3′, AS 5′‐GTTGAAGTCGCAGGAGACAAC‐3′).
Flow cytometry
Cells were fixed and permeabilized with cytofix/cytoperm buffer (BD Biosciences) for 15 min at 4°C and washed with perm/wash buffer (BD Biosciences) before antibody staining at 4°C. The antibodies used to detect GADD34 and p‐eIF2α[Ser52] were identical to the ones used for immunoblotting. The secondary antibody used was conjugated to R‐PE (Invitrogen). Intracellular IFN‐β was detected with a FITC‐conjugated monoclonal antibody (RMMB‐1, PBL InterferonSource). Addition of brefeldin A (10 μg/ml) was necessary to detect IFN‐β after HMW poly(I:C) treatment. Data were acquired on a Canto II (BD) and analyzed using FlowJo (Tree Star). For phospho‐TBK1, phosflow cytofix and phosflow perm/wash were used (BD) to fix and permeabilize cells as indicated by manufacturers. Phospho‐TBK1 (Ser172) was detected using a PE (directly)‐conjugated rabbit mAb (D52C2).
Translation intensity measurement
Puromycin labeling for measuring the intensity of translation was performed as previously described (Schmidt et al, 2009). Puromycin (Sigma, min 98% TLC, cell culture tested, P8833, diluted in PBS) was added at 2.5 μg/ml in the culture medium, and the cells were incubated for 10 min at 37°C and 5% CO2. Cells were washed with PBS prior to cell lysis and immunoblotting with the anti‐puromycin 12D10 antibody (Merck Millipore). For immunofluorescence, after PBS wash, cells were fixed with 3% PFA for 15 min at room temperature before permeabilization and antibody staining. For flow cytometry, cells were fixed and permeabilized with cytofix/cytoperm buffer (BD Biosciences) and stained with the same antibody diluted in perm/wash buffer (BD Biosciences) and with a secondary antibody coupled to Alexa Fluor 647.
Immunoblotting
Cells were lysed in CytoBuster protein extraction reagent (Novagen), supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific). Protein quantification was performed using the BCA Protein Assay (Pierce). 20–50 μg of soluble material was loaded on 8% or 10% SDS‐PAGE before immunoblotting and chemiluminescence detection (SuperSignal West Pico Chemiluminescent Substrate, Pierce). Nuclear extraction was performed using the Nuclear Extract kit (Active Motif). Rabbit polyclonal antibody recognizing ATF4 (CREB‐2, C‐20) and mouse monoclonal antibodies recognizing PKR (B‐10) and CHOP (GADD153, B‐3) were from Santa Cruz Biotechnology. GADD34, IκBα, p‐eIF2α[Ser52], and β‐tubulin rabbit polyclonal antibodies were purchased from Proteintech, Cell Signaling Technologies, Invitrogen, and Abcam, respectively. Rabbit monoclonal antibodies anti‐p‐STAT1[Tyr701] (56D8), p‐TBK1[Ser72] (D52C2), p‐IRF3[Ser396] (4D4G), IRF3 (D83B9), eIF2α (D7D3), A20(D13H3), and SHIP1(D1163) were from Cell Signaling Technologies. Mouse monoclonal antibodies for β‐actin and HDAC‐1 (10E2) were purchased from Sigma and Cell Signaling Technologies. Secondary antibodies were from Jackson ImmunoResearch Laboratories.
Immunofluorescence combined with fluorescent mRNA in situ hybridization (FISH)
Cells on coverslips were fixed with 3.7% formaldehyde for 15 min, then permeabilized in 70% EtOH overnight and blocked with 5% bovine serum albumin (Sigma) containing ribonucleoside vanadyl complex (2 mM). Cells were stained for puromycin as described above, and G3BP1 with a rabbit polyclonal antibody from Millipore. Corresponding fluorophore‐conjugated secondary antibodies from Invitrogen were used. RNase inhibitor (Rnasin, Promega) was added to the staining buffer. For FISH, cells were then washed with 10% formamide in 2× SSC. Fluorescent probes (Quasar 570) against IFN‐β mRNA (Stellaris, Biosearch Technologies) were diluted in hybridization buffer containing dextran sulfate 10 mg/ml and 10% formamide in 2× SSC, according to the manufacturer's instructions. Probes were incubated with cells for 4 h at 37°C. After two washes of 30 min at 37°C, coverslips were mounted with Prolong Gold containing DAPI (Invitrogen), and confocal microscopy images were acquired with a LSM580 (Carl Zeiss) using a 63× objective and accompanying imaging software.
Stress granules detection and scoring
Stress granules detection was performed by mosaic image acquisition of 49 planes with an Axio Observer.Z1 (Carl Zeiss) equipped with the software Zen 2 and a ORCA flash 4 camera. SGs were detected using an antibody recognizing G3BP1 (Millipore) and secondary antibody conjugated to Alexa 488. Nuclei were stained with DAPI. The fluorescence of the two channels was captured through the objective Plan Apochromat 40×/0.95 and the fluorescence filters DAPI (BP 445/50) and GFP (BP525/50). The images were then processed individually with a homemade pipeline using the software “Cell profiler 2.1.1” (Carpenter et al, 2006; CellProfiler: image analysis software for identifying and quantifying cell phenotypes, www.cellprofiler.org). The pipeline uses the nucleus DAPI staining as a primary object to perform cell segmentation and identify and count the granules by cell.
Mathematical model
To build the single‐cell model, we have established a set of equations describing the relationships shown in Fig 6A. Every relationship shown in this figure has been designed as an equation to account for its effect on the global model. The construction of these equations has requested the establishment of several assumptions about the shape of the relationship that could bind variables. Some of these assumptions are supported by experimental quantitative measurements that were conducted to observe them specifically (data not shown). Others are from choice of representation of the biological behavior (activation/saturation effect, linear relationship, etc.). Note that our goal was not to implement a biologically accurate model but to show that a certain type of behavior extended to a cell population can allow obtaining phenomena like those observed experimentally. In this, the choices imposed on equations, although several are supported by quantitative measures, remain completely arbitrary. We present below the equations in detail:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
where t is the step number, representing the discretized time, Ds is the level of double‐stranded RNA, P is the level of phosphorylated PKR, E is the level of phosphorylated eIF2, Gr is the level of GADD34 RNA, and Gp is the level of GADD34 protein. Tl is the level of global translation (ratio between 0 and 1), Tlg is the level of translation of GADD34 RNA (ratio between 0 and 1), Tcg is the level of transcription of GADD34 gene (ratio between 0 and 1), S1 is the sigmoïd function S1(x) = 1/(1 + exp(ax + c)), and S2 is the decreasing sigmoid function S2(x) = 1−1/(1 + exp(a′x + c′)). C1 is the parameter representing the impact of the presence of dsRNA on the level of produced GADD34 RNA. The chosen default value for this parameter is 1.4. C2 is the parameter representing the GADD34 protein degradation along the time. The default value for this parameter is 1 and modified to 0.7 in Fig EV5. C3 is the parameter representing the impact of the presence of phosphorylated PKR on the phosphorylation of eIF2. The default value for this parameter is 10 and modified to 7 in Fig EV5. C4 is the parameter representing the impact of the presence of GADD34 protein on dephosphorylation of phosphorylated eIF2. The default value for this parameter is 2.5 and modified to 2.25 in Fig EV5. d1 is the time delay between the evolution of global translation and the evolution of GADD34 transcription and translation. The default value for this parameter is 30. d2 is the time delay between the evolution of GADD34 and the dephosphorylation of p‐eIF2. The default value for this parameter is 30. The values of the parameters used to define the two sigmoid functions (a, a′, c, and c′ in functions S1 and S2) were defined in order to adapt the sigmoids to the variables ranges of values.
-
(1)
This equation describes the level of phosphorylated/activated PKR in the presence of double‐stranded RNA (dsRNA). The level of dsRNA has been chosen constant along the time and its relationship with phosphorylated PKR is described as an activation/saturation model (sigmoid function S1).
-
(2)
This equation describes the level of global translation in the presence of phosphorylated eIF2. Here again, an activation/saturation model has been chosen to describe this relationship (inverted sigmoid function S2).
-
(3, 4)
These equations describe, respectively, the level of translation of GADD34 RNA and the level of transcription of GADD34 gene. Both are linked in an inverted linear relationship with the global level of translation, implying that when the global translation is lowered by the presence of phosphorylated eIF2, the level of GADD34 RNA and protein may increase. Note the delay in response (d1) used to take into account duration of biological processes.
-
(5)
This equation describes the level of GADD34 RNA as an accumulation depending on a production level coming from the transcription of the GADD34 gene and a consumption level coming from the translation of the RNA into protein.
-
(6)
This equation describes the level of GADD34 protein as an accumulation depending on a production level coming from translation of the GADD34 RNA into protein and a consumption due to the constant degradation of the GADD34 protein.
-
(7)
This equation describes the level of phosphorylated eIF2 as the equilibrium between a production level coming from the presence of phosphorylated PKR (inducing phosphorylation of eIF2) and a consumption coming from the phosphatase activity of GADD34 (inducing a dephosphorylation of p‐eIF2).
These equations have several parameters (C1‐C4, d1, d2) whose value, for some, could be approximated by the results of some experiments (data not shown) but for the most part have been chosen so that the resulting dynamics can realize an oscillating behavior. Note that the values of these parameters remain relatively robust since we can observe that disruption of the order of 30% of their initial value continue to allow the existence of oscillatory behavior in the vast majority of cases with just variations in the periods and amplitudes of the oscillations. The model is therefore not strictly subject to the chosen values, and sets of relatively variable parameter values can realize the same type of oscillations.
In fact, we can analyze the network of the model in order to look at network motifs like described in Milo et al (2002). It appears that the model network contains two main motifs: (i) An incoherent type‐1 feed‐forward loop (IFFL) (Mangan et al, 2006) described by two signal pathways starting from the same origin and arriving at the same target but acting in opposite directions (one pathway activates the target and the other represses it). In our case, this IFFL can be found from dsRNA to Elf2α‐p where the pathway through Ppp1r15a RNA and GADD34 repress the production of Elf2a‐p and the pathway through PKR‐p activates it. (ii) A repressilator, first described in Elowitz and Leibler (2000), by a ring of three genes each expressing a protein that represses the next gene in the loop. In our case, the repressilator is composed of Elf2α‐p inhibiting global translation, which inhibits translation of GADD34, itself inhibiting Elf2α‐p.
Each of these motifs is known to generate oscillatory dynamics, and we may then expect that our model network generates such kind of dynamics. This is what is observed using the range of parameters described above.
For simulations of a population of virtual cells, we chose to run the simulations on 200 cells. To take into account the biological heterogeneity that may exist between the cells, we apply a random perturbation of the parameters C1 to C4 described above in the order of 30% of their initial value. In addition, we considered that the cells could not begin their evolution at the same time. We therefore allocated a random delay from each cell chosen uniformly 1 and 200 time steps. Thus, each cell is given a set of randomly chosen parameters, and the corresponding equations were integrated along time for the evolution of cells. A panel of variable trajectories is then produced, and we can observe the behavior at the population level. In these simulations, it appears some behaviors that are no longer cyclical but convergent, that is, the variables converge to finite fixed values, which, depending on the situation, can be likened to cell death or anergy. Despite the strong perturbations applied to the model, we never observed divergent behaviors (i.e., variables tending to infinity over time).
Statistical analysis
Data showing multiple experiments are displayed as individual dots with mean ± SD. Holm–Sidak tests were used to determine P‐values.
Other technical details are available from the Appendix Supplementary Materials and Methods.
Author contributions
AD, RJA, AC, EG, and PP designed research and analyzed data. AD, RJA, AC, VC, AM, JP, and MR performed research. LS and SJ designed and ran mathematical simulations. T‐PVM, MD, and TW performed bioinformatics analysis. TW and MAS performed microarray analysis. MF designed image analysis software. LS, SJ, AD, RJA, EG, and PP wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
A.D. and the laboratory received financial support from the Innate Immunocytes in Health and Disease (I2HD) Collaborative Project between CIML, AVIESAN, and SANOFI. TPVM was funded by the European Research Council under the European Community's Seventh Framework Programme (FP7/2007–2013 Grant Agreement number 281225 to MD for the SystemsDendritic project). AC received a MNERT and ARC PhD fellowship. The laboratory is “Equipe de la Fondation de la Recherche Médicale” (FRM) sponsored by the grant DEQ20140329536. The project was supported by Agence Nationale de la Recherche (ANR) grants “ANR 10‐BLAN‐1236”, “ANR 10‐BLAN‐1308”, “ANR‐12‐BSV2‐0025‐01”, “ANR‐12‐ISV3‐0002”. This work has also been carried out with the support of A*MIDEX‐AAP‐INME‐1401‐140512 “CSI”, “INFORM Labex ANR‐11‐LABX‐0054”, “DCBIOL Labex ANR‐11‐LABEX‐0043” with grants from ANR‐10‐IDEX‐0001‐02 PSL* and A*MIDEX‐ANR‐11‐IDEX‐0001‐02 funded by the “Investissements d'Avenir” French government program managed by the ANR. The research was also supported by the Ilídio Pinho Foundation and FCT through the Institute for Biomedicine—iBiMED contract UID/BIM/04501/2013 and PTDC/IMI‐IMU/3615/2014. We acknowledge financial support from n° ANR‐10‐INBS‐04‐01 France Bio Imaging and the ImagImm CIML imaging core facility. We acknowledge Drs. Lawrence and Bebien (CIML), as well as Drs. Cazaret and Grelier (Lyon) for providing GFP–vesicular stomatitis virus.
The EMBO Journal (2017) 36: 761–782
Contributor Information
Evelina Gatti, Email: gatti@ciml.univ-mrs.fr.
Philippe Pierre, Email: pierre@ciml.univ-mrs.fr.
References
- Argüello RJ, Rodriguez Rodrigues C, Gatti E, Pierre P (2015) Protein synthesis regulation, a pillar of strength for innate immunity? Curr Opin Immunol 32: 28–35 [DOI] [PubMed] [Google Scholar]
- Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN (2000) Essential role for the dsRNA‐dependent protein kinase PKR in innate immunity to viral infection. Immunity 13: 129–141 [DOI] [PubMed] [Google Scholar]
- Balachandran S, Thomas E, Barber GN (2004) A FADD‐dependent innate immune mechanism in mammalian cells. Nature 432: 401–405 [DOI] [PubMed] [Google Scholar]
- Belgnaoui SM, Paz S, Hiscott J (2011) Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol 23: 564–572 [DOI] [PubMed] [Google Scholar]
- Brush MH, Shenolikar S (2008) Control of cellular GADD34 levels by the 26S proteasome. Mol Cell Biol 28: 6989–7000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio N, Dalet A, Gatti E, Pierre P (2013) Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J 32: 1214–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavarino G, Claudio N, Couderc T, Dalet A, Judith D, Camosseto V, Schmidt EK, Wenger T, Lecuit M, Gatti E, Pierre P (2012) Induction of GADD34 is necessary for dsRNA‐dependent interferon‐beta production and participates in the control of Chikungunya virus infection. PLoS Pathog 8: e1002708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Keller BC, Gale M Jr, Diamond MS (2007) Cell‐specific IRF‐3 responses protect against West Nile virus infection by interferon‐dependent and ‐independent mechanisms. PLoS Pathog 3: e106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalet A, Gatti E, Pierre P (2015) Integration of PKR‐dependent translation inhibition with innate immunity is required for a coordinated anti‐viral response. FEBS Lett 589: 1539–1545 [DOI] [PubMed] [Google Scholar]
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D (2004) Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 24: 10161–10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Leibler S (2000) A synthetic oscillatory network of transcriptional regulators. Nature 403: 335–338 [DOI] [PubMed] [Google Scholar]
- Freaney JE, Kim R, Mandhana R, Horvath CM (2013) Extensive cooperation of immune master regulators IRF3 and NFkappaB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep 4: 959–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabhann JN, Higgs R, Brennan K, Thomas W, Damen JE, Ben Larbi N, Krystal G, Jefferies CA (2010) Absence of SHIP‐1 results in constitutive phosphorylation of tank‐binding kinase 1 and enhanced TLR3‐dependent IFN‐beta production. J Immunol 184: 2314–2320 [DOI] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ (2013) Cyclic GMP‐AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341: 903–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, Deddouche S, Reis e Sousa C (2013) Cytosolic sensing of viruses. Immunity 38: 855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J (2002) Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon‐stimulated genes. J Virol 76: 5532–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ (2013) ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Taniguchi T (2006) IRFs: master regulators of signalling by Toll‐like receptors and cytosolic pattern‐recognition receptors. Nat Rev Immunol 6: 644–658 [DOI] [PubMed] [Google Scholar]
- Hwang SY, Hur KY, Kim JR, Cho KH, Kim SH, Yoo JY (2013) Biphasic RLR‐IFN‐beta response controls the balance between antiviral immunity and cell damage. J Immunol 190: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA‐mediated, type I interferon‐dependent innate immunity. Nature 461: 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies C, Wynne C, Higgs R (2011) Antiviral TRIMs: friend or foe in autoimmune and autoinflammatory disease? Nat Rev Immunol 11: 617–625 [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC (2003) Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF‐kappaB in response to diverse cellular stresses. Mol Cell Biol 23: 5651–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) MAVS, an adaptor triggering RIG‐I and Mda5‐mediated type I interferon induction. Nat Immunol 6: 981–988 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S (2006) Innate immune recognition of viral infection. Nat Immunol 7: 131–137 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S (2010) The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 11: 373–384 [DOI] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P (2000) Dynamic shuttling of TIA‐1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 151: 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke‐Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169: 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich‐Zugich J, Moses AV, Gale M Jr, Fruh K, Diamond MS (2013) IRF‐3, IRF‐5, and IRF‐7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wang L, Berman M, Kong YY, Dorf ME (2011) Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35: 426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes‐Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson‐Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488: 670–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Malynn BA (2012) A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 12: 774–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, Itzkovitz S, Zaslaver A, Alon U (2006) The incoherent feed‐forward loop accelerates the response‐time of the gal system of Escherichia coli . J Mol Biol 356: 1073–1081 [DOI] [PubMed] [Google Scholar]
- McAllister CS, Taghavi N, Samuel CE (2012) Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF‐2alpha‐mediated translational control. J Biol Chem 287: 36384–36392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesman AW, Zijlstra‐Willems EM, Kaptein TM, de Swart RL, Davis ME, Ludlow M, Duprex WP, Gack MU, Gringhuis SI, Geijtenbeek TB (2014) Measles virus suppresses RIG‐I‐like receptor activation in dendritic cells via DC‐SIGN‐mediated inhibition of PP1 phosphatases. Cell Host Microbe 16: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R, Shen‐Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U (2002) Network motifs: simple building blocks of complex networks. Science 298: 824–827 [DOI] [PubMed] [Google Scholar]
- Mohr I, Sonenberg N (2012) Host translation at the nexus of infection and immunity. Cell Host Microbe 12: 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Yoshida H, Moodley D, LeBoite H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang SY, Feng T, Lee M, Casanova JL, Clark JD, Hegen M, Telliez JB, Hacohen N, De Jager PL, Regev A, Mathis D, Benoist C (2016) Parsing the interferon transcriptional network and its disease associations. Cell 164: 564–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34‐mediated dephosphorylation of eIF2alpha. J Cell Biol 153: 1011–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obuchi M, Fernandez M, Barber GN (2003) Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol 77: 8843–8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ourthiague DR, Birnbaum H, Ortenlof N, Vargas JD, Wollman R, Hoffmann A (2015) Limited specificity of IRF3 and ISGF3 in the transcriptional innate‐immune response to double‐stranded RNA. J Leukoc Biol 98: 119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Fribourg M, Ge Y, Batish M, Tyagi S, Hayot F, Sealfon SC (2015) Single‐cell analysis shows that paracrine signaling by first responder cells shapes the interferon‐beta response to viral infection. Sci Signal 8: ra16 [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Reis e Sousa C (2007) Innate recognition of viruses. Immunity 27: 370–383 [DOI] [PubMed] [Google Scholar]
- Pierre P, Gatti E (2013) Loss of translation: a stealth weapon against pathogens? Nat Immunol 14: 1203–1205 [DOI] [PubMed] [Google Scholar]
- Reineke LC, Dougherty JD, Pierre P, Lloyd RE (2012) Large G3BP‐induced granules trigger eIF2alpha phosphorylation. Mol Biol Cell 23: 3499–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke LC, Kedersha N, Langereis MA, van Kuppeveld FJ, Lloyd RE (2015) Stress granules regulate double‐stranded RNA‐dependent protein kinase activation through a complex containing G3BP1 and Caprin1. MBio 6: e02486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke LC, Lloyd RE (2015) The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 89: 2575–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert F, Carrier M, Rawe S, Chen S, Lowe S, Pelletier J (2009) Altering chemosensitivity by modulating translation elongation. PLoS One 4: e5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529 [DOI] [PubMed] [Google Scholar]
- Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, Bankhead P, Hiet MS, Kallis S, Alvisi G, Samuel CE, Lohmann V, Kaderali L, Rohr K, Frese M, Stoecklin G, Bartenschlager R (2012) Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12: 71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Yamamoto M, Miyagishi M, Taira K, Nakanishi M, Fujita T, Akira S, Yamamoto N, Yamaoka S (2005) A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol 174: 1507–1512 [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6: 275–277 [DOI] [PubMed] [Google Scholar]
- Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, Takeuchi O, Akira S, Kaufman RJ, Reis e Sousa C (2010) Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7: 354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine Y, Zyryanova A, Crespillo‐Casado A, Fischer PM, Harding HP, Ron D (2015) Stress responses. Mutations in a translation initiation factor identify the target of a memory‐enhancing compound. Science 348: 1027–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 122: 669–682 [DOI] [PubMed] [Google Scholar]
- Sidrauski C, McGeachy AM, Ingolia NT, Walter P (2015) The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 4: e05033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, Martins‐Green M, Shastri N, Walter P (2016) Translation from the 5' untranslated region shapes the integrated stress response. Science 351: aad3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Barr JN, Wertz GW (2000) Identification of a minimal size requirement for termination of vesicular stomatitis virus mRNA: implications for the mechanism of transcription. J Virol 74: 8268–8276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, Gack MU (2013) Dephosphorylation of the RNA sensors RIG‐I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38: 437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR (2001) Signal integration via PKR. Sci STKE 2001: re2 [DOI] [PubMed] [Google Scholar]
- Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M, Weissmann C (1995) Deficient signaling in mice devoid of double stranded RNA‐dependent protein kinase. EMBO J 14: 6095–6106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawatzky R, De Maeyer E, De Maeyer‐Guignard J (1985) Identification of individual interferon‐producing cells by in situ hybridization. Proc Natl Acad Sci USA 82: 1136–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Samuel CE (2008) Induction of protein kinase PKR‐dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter MAVS signaling. J Biol Chem 283: 34580–34587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Tian T, Zou X (2015) Negative feedback contributes to the stochastic expression of the interferon‐beta gene in virus‐triggered type I interferon signaling pathways. Math Biosci 265: 12–27 [DOI] [PubMed] [Google Scholar]
- Zhao M, Zhang J, Phatnani H, Scheu S, Maniatis T (2012) Stochastic expression of the interferon‐beta gene. PLoS Biol 10: e1001249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2
Source Data for Figure 3