

Abstract
DNA-encoded synthesis is rekindling interest in combinatorial compound libraries for drug discovery and in technology for automated and quantitative library screening. Here, we disclose a microfluidic circuit that enables functional screens of DNA-encoded compound beads. The device carries out library bead distribution into picoliter-scale assay reagent droplets, photochemical cleavage of compound from the bead, assay incubation, laser-induced fluorescence-based assay detection, and fluorescence-activated droplet sorting to isolate hits. DNA-encoded compound beads (10-μm diameter) displaying a photocleavable positive control inhibitor pepstatin A were mixed (1920 beads, 729 encoding sequences) with negative control beads (58 000 beads, 1728 encoding sequences) and screened for cathepsin D inhibition using a biochemical enzyme activity assay. The circuit sorted 1518 hit droplets for collection following 18 min incubation over a 240 min analysis. Visual inspection of a subset of droplets (1188 droplets) yielded a 24% false discovery rate (1166 pepstatin A beads; 366 negative control beads). Using template barcoding strategies, it was possible to count hit collection beads (1863) using next-generation sequencing data. Bead-specific barcodes enabled replicate counting, and the false discovery rate was reduced to 2.6% by only considering hit-encoding sequences that were observed on >2 beads. This work represents a complete distributable small molecule discovery platform, from microfluidic miniaturized automation to ultrahigh-throughput hit deconvolution by sequencing.
Keywords: DNA-encoded synthesis, combinatorial compound libraries, miniaturized automation
Introduction
Combinatorial chemistry is currently experiencing a renaissance, with DNA-templated1,2 and -encoded3,4 synthesis technologies squarely at the epicenter of this movement. DNA-encoded libraries (DELs) store complex small-molecule structural information in an associated DNA sequence,5 analogous to display-type technologies that associate a peptide with its encoding nucleic acid.6−8 DELs, however, bridge a significant chemotype divide by moving library content away from the space of naturally occurring biopolymers found in display libraries and into the more drug-like space of high-throughput screening (HTS) compound collections.4,9,10 The plunging costs of both DNA synthesis and next-generation sequencing (NGS) have positioned DEL technology as a promising companion to industrial HTS platforms11 because it offers access to larger compound collections and lead identification is more economical and expeditious.
Unlike conventional HTS in which compound library members are directly assayed for the desired function, leads from DELs are generated from selections, using binding as a surrogate for all function (e.g., inhibition, agonism). The DEL is incubated with the target, the unbound fraction is washed away, and the bound fraction is eluted, amplified, and sequenced.4 Ligands have been discovered in this fashion for several targets, including sirtuins,12 InhA,13 BCATm,14 and neurokinin-3 receptor.15 Analysis of the sequencing data from a DEL selection can predict relative binding affinities between hits,16 but compounds are ultimately resynthesized and evaluated individually. One can bias the hit pool by eluting target-bound library members using a known functional competitor ligand, but such ligands may not be available for every target. Beyond this measure, evaluating individual DEL members for function other than binding in the primary selection would require a single-molecule process, such as the first selections of catalytic RNA.17 In vitro compartmentalization,18 another single-molecule approach, has recently found use in single-molecule DEL binding assays.19
Combinatorial libraries prepared via solid-phase synthesis20−22 fundamentally differ from DELs in that individual, microscopic beads each display many copies of a single library member. This one-bead-one-compound (OBOC)22 format presents the opportunity to screen library members directly for function by liberating them from the bead surface into discrete volumes for analysis. For example, bead libraries have been spread on culture plates to discover antibacterial compounds,23 distributed into microplates to generate HTS compound library stock solutions,24 arrayed in various microfabricated ultralow-volume well arrays,25,26 and even nebulized into Petri dishes.27 Many of these approaches, however, lack automated strategies to address the bottlenecks associated with screening and structure elucidation, which are currently manual and serial.
Solutions to these OBOC handling and screening automation challenges are now within reach after a decade of component-level technology development in the field of microfluidics. The discovery of segmented flow or “droplet” microfluidics28,29 and its utility in high-throughput reaction monitoring30,31 led to a flood of components for droplet sorting,32,33 splitting,34 incubation,35 synchronization,36,37 and sampling38−40 among many others. Facile droplet-scale component integration has paved the way to advanced circuit architectures for highly sophisticated, multistage biochemical reaction assembly, processing, and analysis.41−45 In a previous study, we disclosed an integrated microfluidic processor for light-induced and -graduated high-throughput screening after bead release (hνSABR), wherein droplet-scale assay initiation with photochemical compound dosing control, incubation, and high-sensitivity detection occur in flow.46 In this study, droplet sorting was fully integrated into the hvSABR processor to perform a functional screen of a model DNA-encoded combinatorial compound bead library. Hit deconvolution by NGS provides a powerful, quantitative means of rejecting noise resulting from the Poisson process of bead encapsulation in droplets.
Results and Discussion
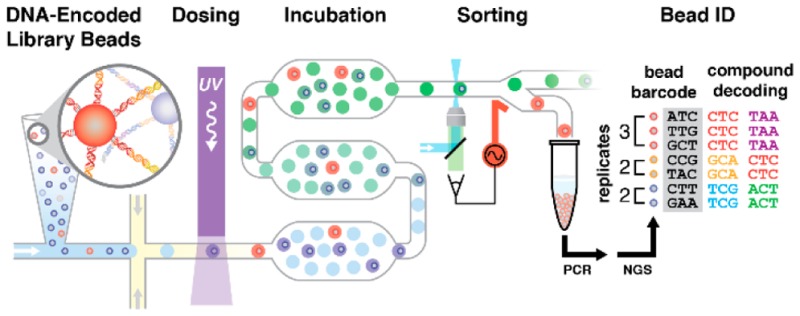
An integrated microfluidic circuit (Figure 1A) combines assay reagents, encapsulates model library beads into droplets, photochemically cleaves compound from the bead, incubates the dosed droplets, and sorts droplets based on their individual fluorescence intensity. Model library beads are suspended in a capped pipet tip, or “suspension hopper,”40 filled with bead hopper buffer (BHB) that was supplemented with sucrose (13% w/v) to tune density-driven sedimentation, and thereby bead introduction rate. Two aqueous inputs allow for mixing of assay reagent streams immediately prior to droplet generation. A mixture of assay probe and internal standard (R-phycoerythrin) in BHB is introduced via AQ1, which sweeps library beads from LIB toward the flow-focusing junction for encapsulation. Cathepsin D (CatD) in protease activity assay buffer is introduced via AQ2. Following library bead encapsulation in activity assay-filled droplets, an integrated waveguide precisely irradiates (λ = 365 nm) the droplet flow to induce photochemical cleavage of compound from the bead into the droplet volume. UV LED power was calibrated46 to maintain consistent UV intensity and thereby compound dosing (1–3 μM) between experiments. Droplets dosed with compound are then incubated (18 min) within a Frenz-type delay line (20 cm long, 1 mm wide, 133 μm deep).35 At the end of the delay line, droplets are focused back into single file. Additional spacing oil from OIL2 and guiding oil from OIL3 ensure adequate droplet separation and positioning before detection and sorting. Droplets enter the sorting junction (orange box),33 the confocal laser-induced fluorescence (LIF) detectors measure droplet fluorescence (2000 Hz), and the software makes a sort decision (Figure 1B). By default, all droplets flow through the sorting junction to OUT. If a droplet is determined to contain an active compound, a salt water electrode (4 M NaCl, orange)47 delivers a high-voltage AC pulse to the sorting junction, generating an electric field that deflects the desired droplet toward HIT output (Figure 1C).32
Figure 1.
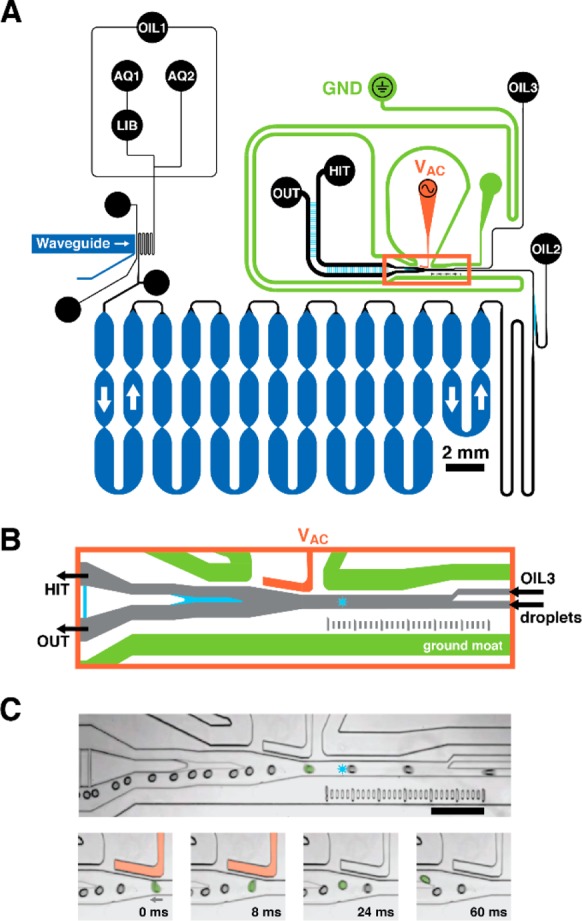
Microfluidic circuit schematic. (A) Oil, aqueous phase assay components 1 and 2, and model DNA-encoded library beads enter the device at inputs OIL1, AQ1 and AQ2, and LIB, respectively. OIL1 flow encapsulates library beads in droplets of assay reagent at the flow-focusing junction, where OIL1 meets the combined AQ1 and AQ2 streams. Droplets flow through a serpentine channel, where an integrated waveguide irradiates droplets with UV, photochemically liberating compound from the bead into the droplet volume. Droplets then flow into a deep, 20 cm-long channel for incubation before auxiliary oil inputs (OIL2, OIL3) separate and guide droplets at the sorting junction (orange box). Channel depths are color coded: shallow 32 μm (cyan), standard 57 μm (black, orange, green), and deep 133 μm (dark blue). (B) By default, droplets flow to the primary output (OUT) unless the droplet LIF profile (detection laser spot shown as a blue star) defines the droplet as a hit. When the system detects a hit-containing droplet, it energizes a salt water electrode (VAC, orange) that dielectrophoretically deflects the hit-containing droplet to the collection output (HIT). The gapped sort divider (cyan) facilitates droplet deflection while minimizing droplet splitting. A salt water ground moat (green) shields the incubation circuit. (C) Micrographs show a selected droplet (green, false color) deflecting into the HIT output channel in response to an 8-ms sorting voltage pulse (500 Vpp, 10 kHz). Neighboring droplets do not deflect and continue to OUT. Scale = 500 μm.
Model libraries containing two types of DNA-encoded library beads (10-μm diameter,Figure 2A) were prepared to assess droplet sorting performance and to investigate postsort sequence-based bead counting. A photocleavable linker is appended with either a positive control inhibitor (Glu-pepstatin A bead, 1) or negative control (N-acetyl Glu bead, 2). DNA-encoding sequences are installed modularly by split-and-pool enzymatic cohesive-end ligation onto headpiece DNA (HDNA)4 functionalized resin, which validated as supporting ligation and qPCR amplification of 52000 DNA molecules/bead.48 Split-and-pool combinatorial oligonucleotide ligations using 10 different encoding modules in encoding positions 1–4 (Supporting InformationT1–T2) afforded 104 distinct bead-specific barcodes (BSB) onto both 1 and 2 beads.49 Compound encoding region (ER) modules in encoding positions 5–10 were similarly installed. Bead set 1 was encoded using 3 different encoding modules at positions 5, 7, and 9, and 3 different modules at positions 6, 8, and 10, yielding (3 × 3)3 = 729 distinct ER sequences. Bead set 2 was encoded using 3 different encoding modules at positions 5, 7, and 9, and 4 different modules at positions 6, 8, and 10, yielding (3 × 4)3 = 1728 distinct ER sequences. The modules used to encode bead sets 1 and 2 were distinct from one another; there were no duplicated sequences between the 729 possible ER sequences of bead set 1 and the 1728 ER sequences of bead set 2. Combining the ER sequences for each bead set with the 104 BSB sequences employed, the 10-position encoding strategy produced a total of 7.29 × 106 and 1.73 × 107 unique sequences for bead sets 1 and 2, respectively. Ligation of a reverse primer module (Figure 2A, inset), displaying a position-specific overhang, randomized 8-nt unique molecular identifier (UMI, green, inset), and reverse primer binding site, completed the DNA encoding tag. Five bead lots (100 beads each) of encoded bead sets 1 and 2 were analyzed by qPCR and average encoding tags/bead were measured (1, 4.3 ± 0.8 × 104 tags/bead; 2, 2.5 ± 0.2 × 104 tags/bead). Positive control resin 1 was labeled with 5(6)-carboxytetramethylrhodamine (TMR) fluorophore (orange), allowing rapid visual differentiation of 1 from 2 in a mixture (Figure 2B).
Figure 2.
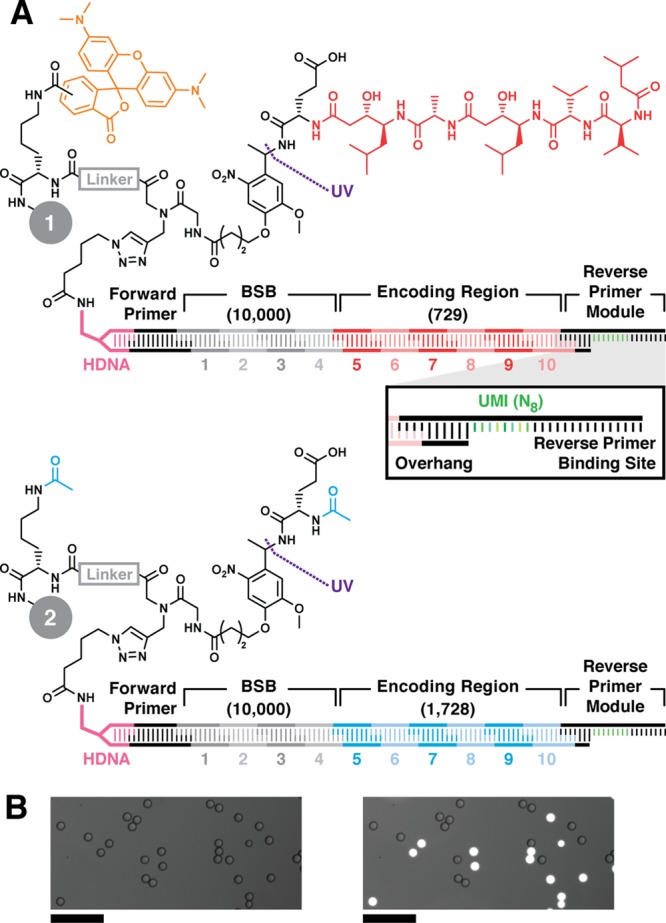
Model DNA-encoded library bead structures. Lysine, linker (gray), photocleavable linker, and Glu were sequentially coupled to 10-μm-diameter TentaGel resin. (A) Positive control inhibitor beads 1 display pepstatin A (red) coupled to Glu. The linker is labeled with 5(6)-carboxy TMR fluorophore (orange). Negative control beads 2 were prepared by acetylating the Glu α amine and linker amine (cyan). Bead sets were substoichiometrically functionalized with azido DNA headpiece (HDNA) via CuAAC. The DNA encoding sequence was installed by split-and-pool combinatorial enzymatic ligation. The BSB region contained 10 unique sequence modules at each of 4 positions (1—4, 104 possible BSBs). The encoding regions (ER, 5—10) contained either 729 (Glu-pepstatin A positive control beads, 1) or 1728 (N-acetyl-Glu negative control beads, 2) possible sequences. The DNA sequence terminates with ligation of a reverse primer module containing the reverse PCR primer binding site flanking an internal unique molecular identifier (UMI, green, inset). The UMI is a random 8-mer (65 536 possible sequences). (B) Micrographs of a model library containing positive and negative control beads 1 and 2 visualized in brightfield (left) and brightfield overlay with TMR fluorescence emission (λex = 550 nm; λem = 570 nm; right) illustrate facile differentiation between the two bead types. Scale = 100 μm.
DNA-encoded solid-phase synthesis (DESPS) of combinatorial libraries introduces numerous advantages, from synthesis scaling to analysis throughput. The sensitivity of PCR permits synthesis and encoding tag detection on miniaturized resin (10-μm diameter, 0.4 pmol/bead) and NGS massively parallelizes structure elucidation of hit-bead collections. Redundant libraries, in which multiple beads display the same compound library member, are particularly valuable for screening. Despite sacrificing screening throughput, redundancy adds statistical confidence to hit prediction. True positive hits tend to reproduce the desired screening outcome on multiple beads (replicates); false positive hits tend to be observed only once.50,51 The limited set of 729 encoding sequences for bead set 1 ensured that any given hit collected from screening a model library (∼1500 total 1) would likely share encoding region sequence with another hit bead. Differentiating replicates requires the BSBs, since the ER sequences would be identical otherwise.49 Downstream quantitative sequence counting requires the incorporation of UMIs.
The integrated circuit was first used to investigate droplet sorting performance in a hvSABR screen of a model library. Model library (∼100 000 beads, 1.5% bead set 1) was introduced via suspension hopper into droplets containing CatD, fluorogenic peptide probe, and internal standard. Droplets were irradiated, incubated, and detected using confocal LIF (Figure 3A). Each droplet’s LIF profile was compared to a predetermined threshold. CatD was uninhibited in both unoccupied droplets and droplets containing a negative control bead 2, efficiently digesting the fluorogenic probe and increasing droplet fluorescence in the 520 nm channel. These droplets flow by default to waste. CatD was inhibited in droplets containing at least one positive control hit bead 1. As a result, fluorogenic probe remained undigested and droplet fluorescence remained low. If the droplet fluorescence was below the inhibition threshold, the software triggered the generation of an electric field, which deflected the inhibitor bead-containing droplet for collection. Real-time data smoothing eliminated bead strike-related spikes in fluorescence intensity that would potentially compromise hit identification (Figure 3B). Histogram analysis of assay performance (Figure 3C) revealed two distinct droplet populations. CatD inhibition (positive, red) was observed in 1081 droplets over the 240 min experiment duration. Uninhibited droplet populations (negative, blue hues) were plotted as two 3 min time slices that represented the upper and lower extremes. Uninhibited droplet probe fluorescence values oscillated (period = 20 min, amplitude = 787 counts, Supporting Information S1) as did the internal standard signal (phycoerythrin, λem = 570 nm). Pairing the entire population of positive control droplet fluorescence values with either of the two negative control droplet populations to calculate assay quality score, Z′,52 gave Z′ = 0.75 and 0.63. A separate negative control experiment was conducted wherein model library beads were introduced into droplets, incubated, and detected without UV photochemical compound cleavage. Droplet fluorescence intensities under the inhibition threshold (1000 counts) were not detected under these conditions (Supporting Information S2).
Figure 3.
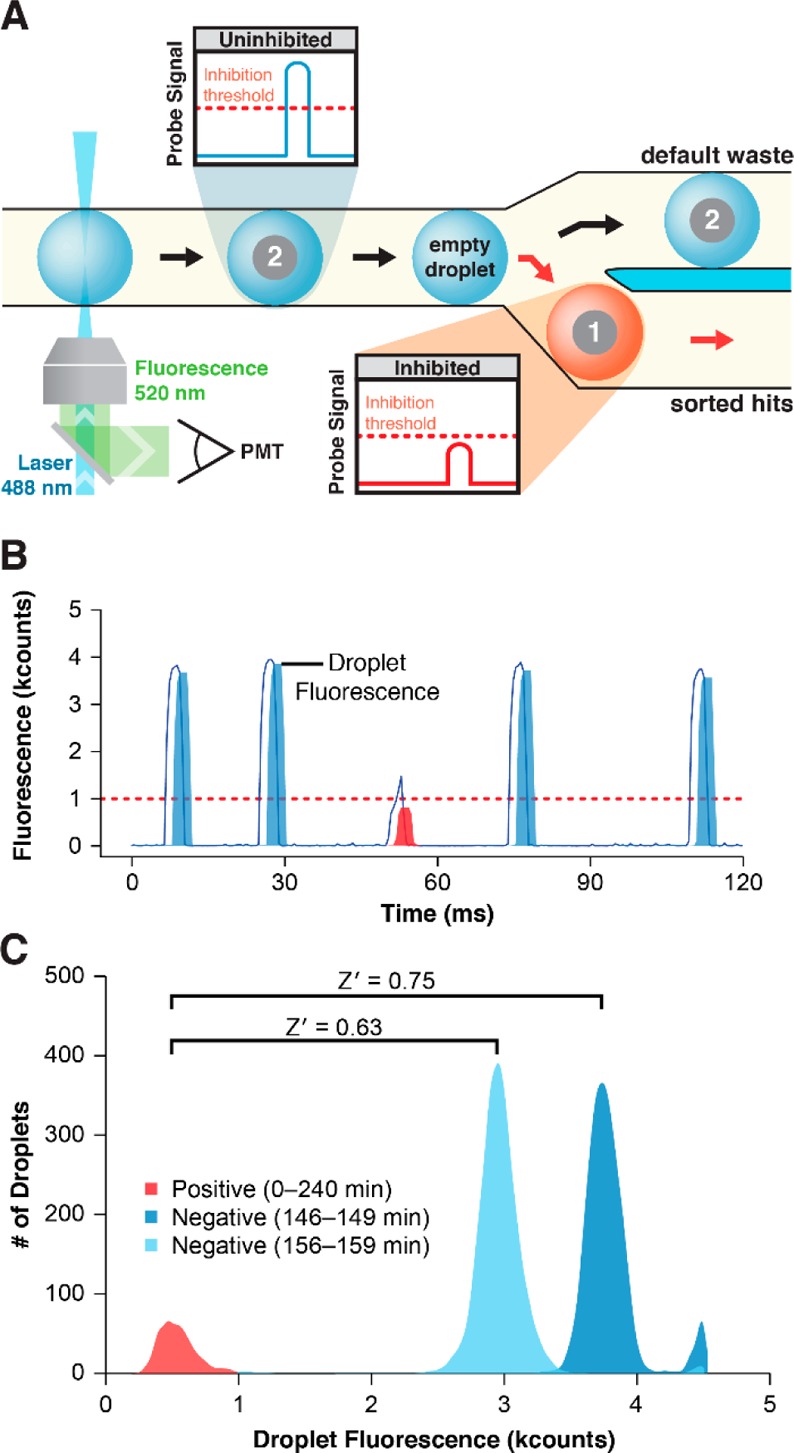
Droplet-based CatD activity assay. (A) Following incubation, assay droplets approach the sorting junction and traverse the LIF detection point (λex = 488 nm, blue cone). A PMT detects the droplet fluorescence emission (λem = 520 nm) to generate a droplet intensity profile. Droplets containing negative control beads 2 or no bead (empty droplet) exhibited high fluorescence intensity (blue droplet, blue trace). Droplets containing positive control inhibitor beads 1 exhibited low fluorescence intensity due to CatD inhibition (red droplet, red trace). If the profile maximum fell below a set threshold, the droplet was sorted and collected in the HIT output. (B) Raw droplet fluorescence emission signal (blue line) is plotted with real-time median-smoothing (filled). If droplet fluorescence was below the threshold (red dotted line), the droplet was a hit (red fill). (C) Droplet fluorescence maxima are plotted as a histogram to display assay performance. The hit droplet population for the entire run is plotted (0—240 min, 1081 droplets, red) and compared to 2 representative 3 min sections of negative droplet populations (146—149 min, 6161 droplets, dark blue; 156—159 min, 6408 droplets, light blue).
Robust assay performance was easily attainable (Z′ > 0.5) in <20 min incubation time due to the sensitivity of LIF detection and the large number of droplets sampled,46,53 however the experiment illuminated systematic error. The regular oscillation in droplet fluorescence may stem from fluctuations in the low AQ1 and AQ2 flow rates (AQ1, 0.3 μL/min; AQ2, 0.2 μL/min). Fluorescence emission oscillation in the internal standard, which is only present in AQ1, further corroborates this hypothesis. We are currently investigating the cause of this oscillatory behavior and methods to minimize its impact.
The model library screen also demonstrated that photochemical cleavage is required in order to observe enzyme inhibition in droplets. Pepstatin A is an extremely potent (IC50 < 1 nM)54 inhibitor of CatD activity and, despite this fact, library screening under conditions of no UV irradiation yielded no detectable hit beads. One might expect that all of the target would bind the bead surface driven by target-inhibitor interaction. After all, on-bead binding-based screens of OBOC libraries rely precisely on this concept. However, given reasonable affinity of the probe for target, the probe is likely competing for target-bead binding, continually driving the equilibrium toward solution-phase reaction.
Droplets with fluorescence below the inhibition threshold were sorted and collected. Droplet sorting performance was visually analyzed by overlaying the bright field and probe fluorescence images (Figure 4A, FAM), and by overlaying the positive control bead 1 fluorescence (TMR). Of the total 975 droplets in the plane of focus, 871 contained at least one 1 bead, while all 896 bead-occupied droplets contained 1247 1 + 2 beads. Bead set 1 occupancy was 871 droplets containing 890 beads. Co-encapsulation frequencies for droplets containing 1 and 2 were counted, plotted, and compared to a model Poisson distribution (Figure 4B). The total population of bead set 2 (357) included those coencapsulated with 1, (i.e., “passenger” beads; 252 droplets, 332 passenger beads), and those encapsulated without 1 (16 droplets, 25 false-positive beads). Within collected droplets, droplet occupancy agreed closely with a model Poisson distribution (droplet occupancy occupancy: λdrop = 0.68 beads/droplet). Deviation from Poisson (gray) increased at higher occupancy 5-bead events. Image analysis of droplet fluorescence indicated two high-intensity droplets. Medium-intensity droplets (n = 29) either contained no beads (n = 26) or contained only a negative control bead 2 (3 droplets).
Figure 4.
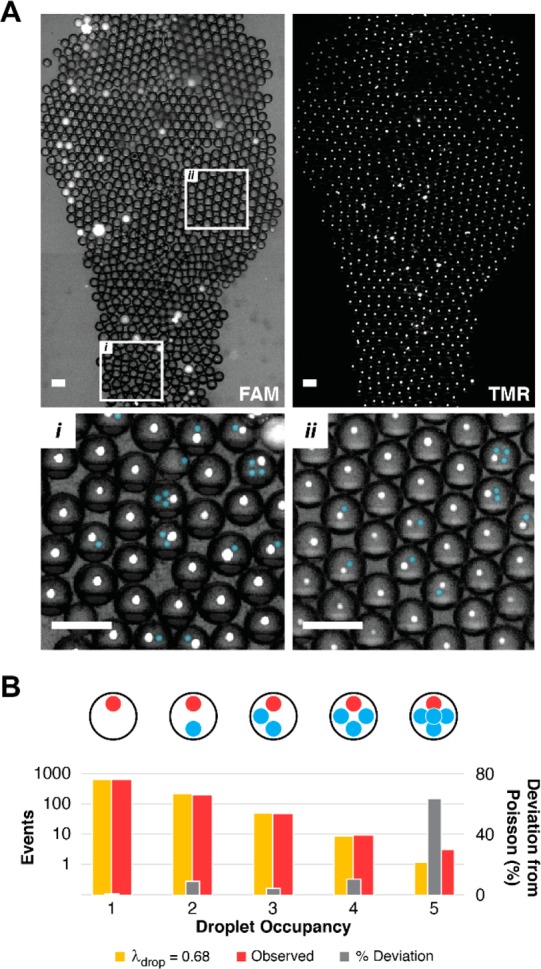
Sorted hit droplet collection. (A) Droplets exhibiting CatD inhibition were sorted, collected, and visually inspected. A brightfield/probe fluorescence overlay (FAM, top left) and positive control bead fluorescence (TMR, top right) confirmed the identity of positive control inhibitor beads 1. Magnified regions (i, ii) contain an overlay of brightfield and both fluorescence channels. Positive control beads 1 (white) are coencapsulated with negative control beads 2 (blue false color). (B) The overlay images were used to measure hit droplet occupancy (n = 975 droplets). For droplets containing at least one positive control inhibitor bead 1, the observed droplet occupancy frequency (red) was plotted against the occupancy frequency predicted by the Poisson distribution function (λdrop = 0.68, yellow). Scale = 100 μm.
System performance was evaluated by visual inspection to quantitate sources of negative control beads 2 in the hit bead collection. High-intensity droplets (n = 2) were likely collected as a result of mis-sorting. Medium-intensity droplets (n = 29), suggesting weak inhibition, were observed in the absence of a positive control bead. These droplets may have been contaminated by a bead-fragment that contributed a low dose of the highly potent pepstatin A. All other droplets in the FAM composite image showed pronounced inhibition, even droplets that were apparently empty, indicating that the system maintained a low error rate in sorting (0.2% sort failure). According to the overall sorting data set, the predominant source of 2 within the hit collection stemmed from Poisson-limited coencapsulation. Either increasing droplet generation frequency or slowing bead introduction (by increasing BHB solution density) rate is likely to reduce this source of 2 in the hit collection. Furthermore, we hypothesized that screening redundant libraries (>3 average replicates) and prioritizing replicate hit beads might form an additional mechanism for rejecting these otherwise randomly selected false positive hits.
To test this hypothesis, a hvSABR screen for DNA sequencing analysis was performed using another model library (60 000 beads, 3.2% bead set 1) and droplets were collected as described. The system performed 1518 droplet sorts, visual inspection of a subset of the sorted droplets (n = 1188) revealed 1532 total beads comprised of 1 (1166, 76%) and 2 (366, 24%), in agreement with the output of the previous screen (1: 890, 71%; 2: 357, 29%). The exact total bead count was unknown, therefore calibration standards containing positive control bead 1 lots (30, 100, 300, 1000, 3000 beads) were prepared by FACS in triplicate, and analyzed by qPCR (Supporting Information S3).
NGS analysis of the counted calibration bead set and the hit collection returned a set of DNA sequences with the number of reads and a list of UMIs for each sequence. Each encoding sequence’s UMI list was distance filtered (see Supporting Information) prior to counting. Sequences were rank-ordered by UMI counts (bead index) and plotted (Figure 5). For each lot size, the UMI counts descended to baseline as the bead index approached ∼85% of the expected aliquot size. A clear inflection point is observed near the expected number of beads for each lot sample (gray lines). The inflection point was evaluated by selecting the bead index with a maximum %ΔUMIw, a normalized derivative analysis of the rank-ordered UMI counts. The bead index cutoff values (25, 84, 270, 861, 2681) for the FACS-counted standard sets were consistent between standards (83%, 84%, 90%, 86%, 89%) and in agreement with the Poisson-limited sort yield specification for the instrument (>80%). The %ΔUMIw analysis of the hit bead collection indicated a bead index cutoff (1863) in agreement with the extrapolated estimate from visual inspection (1957).
Figure 5.
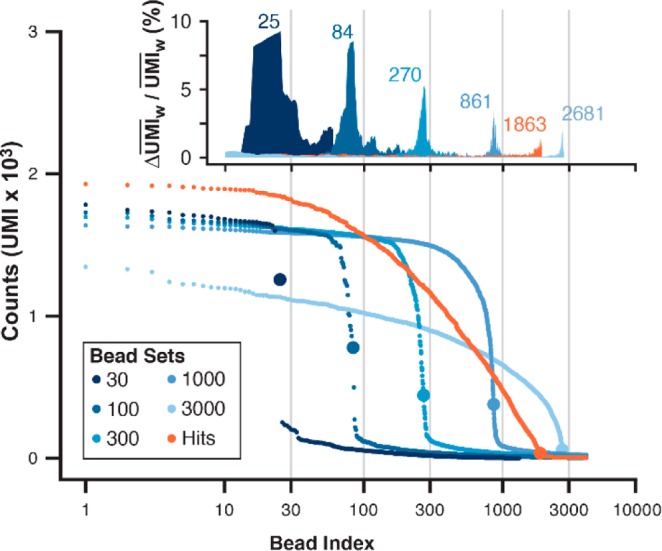
Hit bead collection quantitation by sequence analysis. Standards of positive control pepstatin A beads 1 were prepared by FACS in known lot sizes (30, 100, 300, 1000, 3000; blue hues). The DNA encoding tags of standard lots and the hit bead collection were each amplified in bulk and sequenced. Sequencing coverage for the hit bead collection and 3000-bead lot were 6-fold lower than other standard lots (30, 100, 300, 1000). Reads were aligned to a degenerate reference sequence, edited for single-base errors, aggregated by sequence, and counted by the N8 UMI after enforcing a Hamming distance of >1 per UMI. (A) Unique encoding sequences were rank ordered and plotted by UMI counts (bead index). Each plot contained an inflection (large data point) as the bead index approached the known lot size. (B) A normal weighted average UMI value (UMIw) was generated for each bead index, and the inflection point for each curve was approximated at the bead index yielding a maximum ΔUMIw/UMIw (% change in UMI; inset). The inflection x coordinate is the bead index cutoff (25, 84, 270, 861, 1863, 2681).
The bead index cutoff empirically defines the point at which sequencing reads no longer correspond to bead-derived encoding DNA. Sequencing reads from these “imposter” amplicons constitute a real interference in decoding the hit bead sequence pool as they align to the encoding reference sequence and contain otherwise legitimate ER sequences. The hvSABR output posed a challenge for bead counting because droplets encapsulate multiple beads based on the Poisson distribution,40 thus the droplet sort event count underestimates the actual number of beads collected. Analysis of the calibration bead lots demonstrated that the bead count can be inferred from the NGS data. By expanding the language to 10 encoding positions (1010 encoding depth) and augmenting the tag structure with both combinatorial BSBs and UMIs for molecule counting,4,55,56 it was possible to extract bead-derived structure-encoding hits from the NGS data for both control bead sets and hit bead collections from a model library screen. However, the heterogeneous effects of diverse library synthesis reaction conditions on encoding tag integrity may complicate the above approach,57 necessitating more sophisticated high-speed droplet imaging58 in order to obtain a direct bead count in situ.
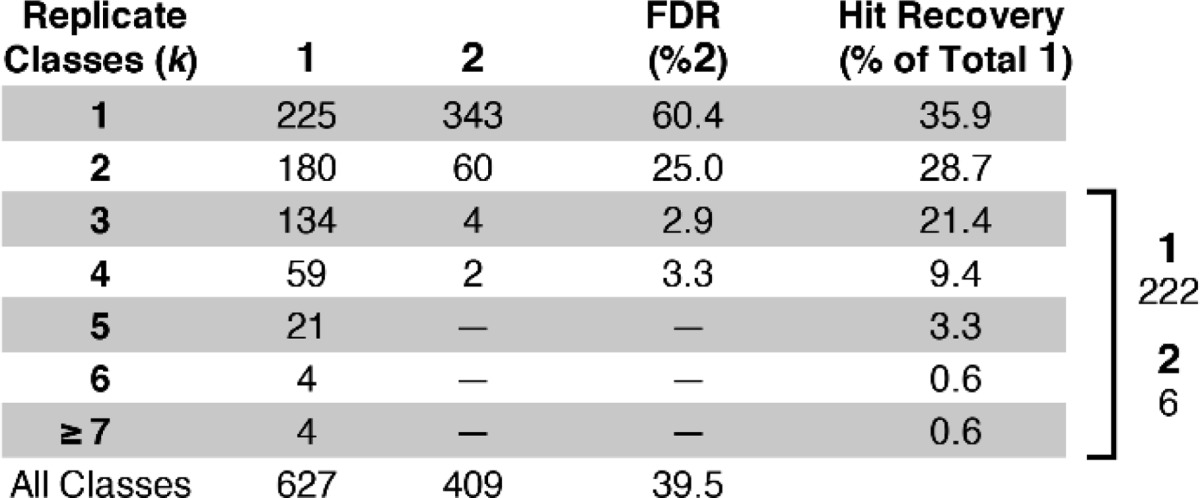
NGS analysis of the hit bead collection was consistent with the visual inspection and contained numerous replicate hit sequences. Using the bead index cutoff determined by %ΔUMIw inflection analysis (1863), bead sequences were decoded as either 1 or 2 based only on ER sequence, which represented unique compounds. Unique compounds (ER sequence) were then binned into replicate classes (k, Table 1) based on the number of BSBs associated with each ER sequence. The total bead counts of 1 (1380, 74%) and 2 (483, 26%) matched the visual analysis (76% and 24%, respectively). The replicate analysis experimentally demonstrated that Poisson-limited false positives in the hit collection can be readily rejected as randomly selected compounds. Significantly, large redundancy is not necessary to achieve this result. Our model library emulated a 60 000-bead screen at 3.2% hit rate (1920 1 beads) encoded such that hits were only present on average at 2.5-fold redundancy (729 ER sequences), meanwhile the negative control bead population (58 000 2 beads) was oversampled at 33-fold redundancy (1728 ER sequences). We observed a total of 627 of 729 possible 1 ER sequences, and 409 of 1728 possible 2 ER sequences. Partitioning these into replicate classes clearly differentiated authentic positives (1) and false positives (2). Each replicate class is summarized by a false discovery rate (FDR) and the hit recovery as a fraction of unique 1 contained within the class. Even in the limit of this modest hit redundancy and pessimistic oversampling of negative controls, when we only consider replicate classes k > 2, we isolate primarily 1 (222 unique ER) and few 2 (6 unique ER) giving a low FDR (2.6%).
Table 1. Hit Bead Collection by Replicate Class.
The validated microfluidic hvSABR lead identification architecture is now ready for analysis of several assays that have already been adapted to droplet scale. Example applications include biochemical assays of HIV protease activity40 or histone deacetylase activity,59 two recent enzyme targets that were explored in bids to move functional screening off the bead surface. There is also significant potential in exploring other challenging target classes, such as protein–protein interactions60,61 or revisiting the miniaturization of phenotypic assays.25,27 As assay scope expands, it will also likely become advantageous to adapt alternative detection strategies, such as fluorescence polarization,62 which could result in a new approach to screening combinatorial libraries directly for solution-phase ligand binding.
In conclusion, we used a model DNA-encoded compound library to demonstrate that hvSABR-based lead identification can economically identify functional library members with high confidence. The screen consumed 120 μL of mixed assay volume and surveyed up to 100 000 library beads (0.05 mg library beads) over 4 h of automated operation, demonstrating that the platform can interrogate a whole library using little material and without the need for robotic automation. Adding UMI and BSB features to the DNA encoding language represented minimal modifications to the DESPS protocol, but enabled powerful noise rejection at both the sequencing and hit structure identification stages. The hvSABR circuit thus provides distributable and economical library screening automation that matches the efficiency of DNA-encoded solid-phase combinatorial library synthesis and structure elucidation.
Experimental Procedures
Materials Sources
All reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. 5-Azidopentanoic acid, N-hydroxysuccinimide (NHS), 1,3-bis[tris(hydroxymethyl)methylamino]propane (Bis-Tris), tris(hydroxymethyl)aminomethane (Tris), 2-(N-morpholino)ethanesulfonic acid hydrate (MES), pepstatin A, N,N′-diisopropylcarbodiimide (DIC), ethyl 2-cyano-2-(hydroxyimino)acetate (Oxyma), 1-hydroxy-7-azabenzotriazole (HOAt), N,N′-diisopropylethylamine (DIEA), 2,4,6-trimethylpyridine (TMP), propargylamine, triisopropylsilane (TIPS), α-cyano-4-hydroxycinnamic acid (HCCA), bromoacetic acid (BAA), trifluoroacetic acid (TFA), acetic anhydride, mineral oil, triethylammonium acetate (TEAA, Life Technologies, Carlsbad, CA), M-280 streptavidin-coated magnetic resin (Life Technologies), biotin N-hydroxysulfosuccinimidyl ester (biotin-sNHS, Pierce Biotechnologies, Rockford, IL), Taq DNA polymerase (Taq, New England Biolabs, Ipswich, MA), 2′-deoxyribonucleotide triphosphate (dNTP, set of dATP, dTTP, dGTP, dCTP, Promega Corp., Milwaukee, WI), N-α-Fmoc-Lys(Mtt)–OH (AnaSpec, Inc., Fremont, CA), N-α-Fmoc-Arg(Pbf)–OH (AnaSpec), N-α-Fmoc-Gly-OH (AnaSpec), Fmoc-Glu(OtBu)–OH (AnaSpec), 5-(6)-carboxytetramethylrhodamine (TMR, AnaSpec), R-phycoerythrin (AnaSpec), HiLyte Fluor 488/QXL520-based FRET peptide substrate (AnaSpec), bovine serum albumin (BSA, Roche Diagnostics, Indianapolis, IN), 4-{4-[1-(9-fluorenylmethyloxycarbonylamino)ethyl]-2-methoxy-5-nitrophenoxy}butanoic acid (Fmoc-Photolinker-OH, Advanced ChemTech, Louisville, KY), DMF-A-6CS (Shin-Etsu, Akron, OH), KF-6038 (Shin-Etsu), polydimethylsiloxane (PDMS, Dow Corning, Midland, MI), trimethylsiloxy-terminated PDMS (200 cSt, Gelest Inc., Morrisville, PA), and Brilliant Violet 510 Streptavidin (BV-510, BioLegend, San Diego, CA) were used as provided.
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) was recrystallized three times in t-BuOH/H2O (1:1).63 Solvents used in solid-phase synthesis were dried over molecular sieves (3 Å, 3.2 mm pellets). All solid-phase synthesis was conducted in a UV-free environment.
Buffers and Oils
Cathepsin D protease activity assay buffer (100 mM NaOAc, 1 M NaCl, 1 mM EDTA, 2% DMSO, 1% BSA, 0.1% Tween 80, pH 4.7), MES buffer (100 mM MES, 1 M NaCl, 1 mM EDTA, 2% DMSO, 1% BSA, pH 6.5), bead hopper buffer (BHB, 1 mM sodium phosphate, 10 mM NaCl, 2% DMSO, 13% sucrose, 1% BSA, 0.1% Tween 80, pH 7.0), bind and wash buffer (BWB, 1 mM EDTA, 2 M NaCl, 10 mM Tris, pH 7.5), bind and wash buffer with Tween (BWBT, 1 mM EDTA, 2 M NaCl, 10 mM Tris, 0.1% Tween 20, pH 7.5), 10× Bis-Tris propane ligation buffer (BTPLB, 500 mM NaCl, 100 mM MgCl2, 10 mM ATP, 0.2% Tween 20, 100 mM Bis-Tris propane, pH 7.6), Bis-Tris propane wash buffer (BTPWB, 50 mM NaCl, 0.04% Tween 20, 10 mM Bis-Tris propane, pH 7.6), Bis-Tris propane breaking buffer (BTPBB 100 mM NaCl, 10 mM EDTA, 1% SDS, 1% Tween 20, 10 mM Bis-Tris propane, pH 7.6), click reaction buffer (CRB, 50% DMSO, 30 mM TEAA, 0.04% Tween 20, pH 7.5), 10X PCR buffer (2 mM each dNTP, 25 mM MgCl2, 500 mM KCl, 100 mM Tris, pH 8.3), 1X GC-PCR buffer (1× PCR buffer, 8% DMSO, 1 M betaine), and denaturing polyacrylamide gel electrophoresis loading buffer (GLB, 6 M urea, 0.5 mg/mL bromophenol blue, 12% w/v Ficoll 400, 1× TBE buffer, pH 8.5) were prepared in DI H2O. Oil for microfluidic droplet formation (4:20:76, w/w/w, KF-6038, DMF-A-6CS, mineral oil) was prepared gravimetrically and mixed with gentle rotation (14 h, 8 rpm) prior to use.
Dual-Scale Photolabile Linker Synthesis
Two aliquots of dual-scale resin were prepared. Each dual-scale aliquot contained TentaGel M NH2 resin (10 μm, 0.23 mmol/g, 30 mg, Rapp-Polymere, Tübingen, Germany) and TentaGel MB Rink amide resin (160 μm, 0.41 mmol/g, 5 mg, Rapp-Polymere). Mixed-scale resin was swelled in DMF (1 h, RT) and transferred into fritted spin-columns (Mobicol Classic, large filter, 10-μm pore size, MoBiTec GmbH, Goettingen, Germany). Linker construction proceeded via iterative cycles of manual solid-phase peptide synthesis. Each cycle included: (1) Fmoc deprotection (20% piperidine in DMF, 2 × 450 μL, 5 min first aliquot, 15 min second aliquot); (2) N-α-Fmoc-amino acid (36 μmol, 450 μL DMF) activation with COMU/DIEA (36 μmol/72 μmol), and incubation (2 min, RT); (3) N-α-Fmoc-amino acid coupling to resin by transferring activated acid (450 μL) to resin and incubating with rotation (15 min, RT, 8 rpm). Each amino acid coupling was repeated once.
After each deprotection and coupling step, reactants were expelled and the resin washed (DMF, 3 × 400 μL; DCM, 1 × 400 μL; DMF, 1 × 400 μL). N-α-Fmoc-Lys(Mtt)-OH, N-α-Fmoc-Gly-OH, N-α-Fmoc-Arg(Pbf)-OH, and N-α-Fmoc-Gly-OH were coupled sequentially. The pendant Fmoc-protected amine was deprotected and the resin washed (see above). The deprotected N-terminus was acylated by preparing a solution of BAA (112 μmol) and DIC (112 μmol) in DMF (450 μL) and incubating (2 min, RT), transferring the activated bromoacid (450 μL) to the resin, and incubating (15 min, 40 °C). The bromoacetylated resin was washed, propargylamine solution (1 M in DMF, 450 μL) was transferred to resin, and the resin was incubated with rotation (3 h, 50 °C, 8 rpm). The resin was washed, then N-α-Fmoc-Gly-OH was coupled as other amino acids above. The pendant Fmoc-protected amine was deprotected and the resin washed. Fmoc-photolinker-OH (36 μmol, 450 μL DMF) was activated with DIC/Oxyma/TMP (56.2 μmol/36 μmol/56.2 μmol) in DMF (450 μL), incubated (2 min, RT), then coupled to resin by transferring the activated acid (450 μL) to resin and incubating the resin with rotation (3 h, 37 °C, 8 rpm). Photolinker coupling reaction was repeated once, then the resin was washed (see above). Acetic anhydride (386 μmol) and TMP (386 μmol) were combined in DMF (450 μL), the solution added to resin, and the resin incubated (20 min, RT, 8 rpm). The pendant Fmoc-protected amine was deprotected and the resin washed. Fmoc-Glu(OtBu)-OH was coupled as above, and the resin was washed.
Dual-Scale Photolabile Glu-Pepstatin-A Resin Synthesis
An aliquot of dual-scale photolabile linker resin (35 mg) was deprotected (20% piperidine in DMF, 2 × 450 μL, 5 min first aliquot, 15 min second aliquot) and the resin washed (DMF, 3 × 400 μL; DCM, 1 × 400 μL; DMF, 1 × 400 μL). Pepstatin A (11.3 μmol) was activated with DIC/Oxyma/TMP (72 μmol/18 μmol/18 μmol) in DMF (450 μL), incubated (4 min, 60 °C) then coupled to resin by transferring the activated acid (450 μL) to resin and incubating with rotation (2 h, 60 °C, 8 rpm). Resin was washed (DMF, 3 × 400 μL; DCM, 3 × 400 μL), combined with Mtt deprotection cocktail (TFA/TIPS/DCM, 1:2:97, 400 μL) incubated with rotation (30 min, RT, 8 rpm), and washed (DCM, 2 × 450 μL). Resin was combined with a fresh aliquot of Mtt deprotection cocktail, incubated with rotation (30 min, RT, 8 rpm), and washed (DCM, 2 × 450 μL; DMF, 1 × 450 μL; 1% DIEA in DMF, 1 × 450 μL; DMF, 2 × 450 μL). 5(6)-TMR (39 μmol) was dissolved in DMSO (62 μL), combined with TMP (72 μmol), and COMU (36 μmol) in DMF (388 μL), and incubated (2 min, RT). The resin was combined with the activated 5(6)-TMR solution and incubated with rotation (120 min, 50 °C, 8 rpm). The resin was washed until washes were colorless and stored in DMF (4 °C).
Dual-Scale Photolabile N-Acetyl-Glu Resin Synthesis
An aliquot of dual-scale photolabile linker resin (35 mg) was deprotected (20% piperidine in DMF, 2 × 450 μL, 5 min first aliquot, 15 min second aliquot), washed (DMF, 3 × 400 μL; DCM, 3 × 400 μL), combined with Mtt deprotection cocktail (400 μL), incubated with rotation (30 min, RT, 8 rpm), and washed (DCM, 2 × 450 μL). Resin was combined with a fresh aliquot of Mtt deprotection cocktail, incubated with rotation (30 min, RT, 8 rpm), and washed (DCM, 2 × 450 μL; DMF, 1 × 450 μL; 1% DIEA in DMF, 1 × 450 μL; DMF, 2 × 450 μL). The resin was combined with acetic anhydride (386 μmol) and TMP (386 μmol) in DMF (450 μL), and incubated with rotation (20 min, RT, 8 rpm), washed (DMF, 3 × 400 μL; DCM, 1 × 400 μL; DMF, 1 × 400 μL), and stored in DMF (4 °C).
Photolabile Glu-pepstatin and N-Acetyl-Glu Resin Compound Characterization and Screening Bead Deprotection
Both resin samples were washed (DMF, 2 × 450 μL), sonicated (2 min, RT, B3510DTH, Branson Ultrasonics, Danbury, CT), and filtered (CellTrics 150-μm mesh, Sysmex-Partec, Lincolnshire, IL). The 160-μm resin particle samples (1 mg) were collected into fritted spin columns (Mobicol Classic), washed (DCM, 4 × 400 μL), and dried in vacuo. The dried 160-μm resin samples were combined with cleavage cocktail (90% TFA, 5% DCM, 5% TIPS, 450 uL), and incubated with rotation (1 h, RT, 8 rpm). Cleavage product was expelled into a tube, evaporated to dryness, resuspended (20% DMSO, 0.1% TFA in H2O, 100 μL), and analyzed using reversed-phase HPLC (X-Bridge BEH C18 column, 4.6 mm × 100 mm, 130 Å, 5 μm, Waters Corp., Milford, MA) with gradient elution (mobile phase A: ACN; mobile phase B: 0.1% TFA in H2O; 5%—75% A, 30 min) and absorbance detection (λ = 330 nm). Product fractions were collected, an aliquot (1 μL) was spotted to a MALDI-TOF MS target plate, dried, covered with HCCA matrix solution (1.5 mg/mL HCCA, dissolved in a 2:1 solution of aqueous 0.1% TFA:ACN) dried, and mass analyzed via MALDI-TOF MS (Microflex, Bruker Daltonics Inc., Billerica, MA). Major peaks in the HPLC chromatogram corresponded to photolabile N-acetyl-Glu (retention time = 9 min; theoretical [M + H]+ = 1019.5; observed [M + H]+ = 1019.7) and photolabile Glu-pepstatin TMR-labeled linker (retention time = 17 min; theoretical [M + H]+ = 2058; observed [M + H]+ = 2057). The 10-μm resin samples were collected during filtration into separate fritted spin columns (Mobicol Classic), washed (DCM, 3 × 400 μL), and dried in vacuo. The dried 10-μm resin samples were combined with deprotection cocktail (90% TFA, 5% DCM, 5% TIPS, 400 uL) and incubated (30 min, RT). The deprotection cocktail was drained, fresh cocktail was added (90% TFA, 5% DCM, 5% TIPS, 400 uL), and resin was incubated (30 min, RT). Deprotection cocktail was drained and the resin was washed (DCM, 3 × 400 μL; 1% DIEA in DMF, 2 × 400 μL; DMF, 2 × 400 μL; 1% DIEA in DMF, 1 × 400 μL; DMF, 2 × 400 μL), then incubated in DMF (overnight, RT).
Oligonucleotides
Oligonucleotides (Integrated DNA Technologies, Inc. Coralville, IA) were purchased as desalted lyophilate and used without further purification unless otherwise specified. Oligonucleotide ligation substrates were 5′-phosphorylated (/5Phos/). Amino-modified headpiece DNA (NH2-HDNA, /5Phos/GAGTCA/iSp9//iUniAmM//iSp9/TGACTCCC) was HPLC purified at the manufacturer and used without further purification. Oligonucleotides are indicated by “ ≈ ” followed by a 4-digit numeric identifier and “[+]” or “[−]” strand designation. The first digit groups the oligonucleotides by set: set 0 contains PCR primer sequences and sets 1 and 2 contain ER sequences. The second digit denotes position in the ER. The third and fourth digits index the different coding sequences in each set. Oligonucleotide paired (OP) stock solutions of complementary oligonucleotides (60 μM [+], 60 μM [−], 50 mM NaCl, 1 mM Bis-Tris, pH 7.6) were heated (5 min, 60 °C) and cooled to ambient (5 min, RT) before each use. These reagents are indicated with a “[±]” double-stranded designation. Table T1 is a concise look-up table for generating all oligonucleotide sequences. For example, ≈1302[+] is from set 1, and built by concatenating overhang X3XX[+] “/5Phos/GTT” with encoding sequence 1 × 02[+] “ACGGAGCA” to yield the sequence “/5Phos/GTTACGGAGCA.” The complement, ≈1302[−] is also from set 1, and built by concatenating overhang X3XX[−] “/5Phos/TAG” with encoding sequence 1 × 02[−] “TGCTCCGT” to yield the sequence “/5Phos/TAGTGCTCCGT.” Combining ≈1302[−] and ≈1302[+] and thermally processing as above yields the double-stranded coding module ≈1302[±], a position 3 OP stock solution (OP3) of set 1 parent sequence 02 (1 × 02). All sequences are written in the 5′ to 3′ direction. The new 10-position language positions are indicated in hexidecimal (0, 1, 2, ..., D, E, F), with “B” as the last position used for the 10-position language (Supporting Information T1).
Azido HDNA Synthesis, Purification, and Characterization
NH2–HDNA (300 nmol) was dissolved in phosphate buffer (200 mM, pH 8.0, 240 μL). 5-azidopentanoic acid NHS ester was prepared by dissolving NHS (9.6 μmoles), EDC (9.6 μmoles), and 5-azidopentanoic acid (7.2 μmoles) in DMF (20 μL) and incubating (30 min, 60 °C). The NH2–HDNA acylation reaction was assembled by sparging (N2, 1 min) the phosphate-buffered NH2–HDNA, adding 5-azidopentanoic acid NHS ester solution (22 μL), and incubating (2 h, RT). A fresh solution of 5-azidopentanoic acid NHS ester was prepared as described above, added to the acylation reaction, and the reaction incubated (1 h, RT). The reaction was quenched (1 M Tris, pH 7.6, 100 μL) and incubated (5 min, 60 °C). Azido-HDNA (N3–HDNA) product was precipitated twice in ethanol. The pellet was dried under N2, resuspended (20 mM TEAA, pH 8.0, 100 μL), and purified at semipreparative scale using reversed-phase HPLC (X-Bridge BEH C18 column, 10 mm × 150 mm, 130 Å, 5 μm, Waters Corp.) with gradient elution (mobile phase A H2O, 20 mM TEAA, pH 8; mobile phase B ACN; 5—12% B, 24 min). A product fraction aliquot (1 μL) was spotted to a MALDI-TOF MS target plate, dried, covered with matrix solution (18 mg/mL THAP, 7 mg/mL ammonium citrate dibasic in 1:1 ACN:H2O), dried and mass analyzed via MALDI-TOF MS (Microflex, Bruker Daltonics Inc.). Product-containing fractions (theoretical [M + H]+ = 5064; observed [M + H]+ = 5058) were pooled and evaporated to dryness.
HDNA Functionalization onto Photolabile Compound Resin
Photolabile Glu-pepstatin and photolabile N-acetyl-Glu resin (10 mg 10 μm, 0.5 mg 160 μm resin; 2.2 μmol) were each aliquoted, washed (CRB, 3 × 200 μL), combined with CRB (1 mL) and incubated (1 h, 40 °C). CuSO4 (3.1 μmol), TBTA (5.4 nmol), and ascorbic acid (15.6 μmol) were dissolved (66% v/v DMSO in H2O, 49.2 μL). N3–HDNA (11 nmol) and ascorbic acid (32 nmol) were dissolved in TEAA buffer (200 mM, pH 7.5, 16 μL). Bifunctional resin was washed (CRB, 1 mL), resuspended in CRB (1.3 mL), combined with Cu(II) solution (49 μL), mixed, and incubated with rotation (5 min, 40 °C, 15 rpm). The resin was centrifuged (30 s, 1000 rcf), combined with N3–HDNA solution (16 μL, 0.004 eq. to bead sites), vortexed immediately, and incubated with rotation (6 h, 40 °C, 15 rpm). Resin was centrifuged (2 min, 1,000 rcf), the supernatant removed, and the resin washed (BTPBB, 3 × 1 mL) and incubated with rotation (12 h, RT, 15 rpm). Resin was washed (BTPWB, 3 × 1 mL) then transferred into fritted spin-columns, washed (DI H2O, 3 × 1 mL; DMF, 3 × 1 mL), and stored in DMF (−20 °C).
Photolabile Glu-pepstatin and N-Acetyl-Glu Resin HDNA Coupling Validation
Wells of filtration microtiter plates (0.45 μm Hydrophobic PTFE MultiScreen Solvinert Filtre Plate, Merck Millipore Ltd., Cork, Ireland) were each wetted (DCM, 100 μL) then aliquots of HDNA-functionalized photolabile Glu-pepstatin, HDNA-functionalized photolabile N-acetyl-Glu resin (0.1 mg), and biotin-HDNA magnetic control resin (50 μg, prepared as previously described)48 were transferred into separate clean wells and washed (BTPWB, 6 × 150 μL, BTPLB; 1 × 100 μL). Reagents for the first enzymatic oligonucleotide ligation reaction, consisting of ≈0001[±] (1.4 nmol), ≈1109[±] (1.4 nmol), ≈2210[±] (1.4 nmol), ≈1301[±] (1.4 nmol), ≈2402[±] (1.4 nmol), ≈1503[±] (1.4 nmol), and T4 DNA ligase (4500 U) were combined (BTPLB, 450 μL) and aliquoted to HDNA-functionalized photolabile Glu-pepstatin resin (150 μL), HDNA functionalized photolabile N-acetyl-Glu resin (150 μL), and biotin-HDNA magnetic resin (150 μL). Plate wells were sealed with foil adhesive, and resin samples were incubated with shaking (3 h, RT, 600 rpm), then washed (BTPBB, 3 × 150 μL; BTPLB, 1 × 100 μL). Reagents for the second ligation reaction, consisting of ≈2604[±] (1.4 nmol), ≈1705[±] (1.4 nmol), ≈2806[±] (1.4 nmol), ≈1907[±] (1.4 nmol), ≈2A08[ ± ] (1.4 nmol), ≈0B01[ ± ] (1.4 nmol), and T4 DNA ligase (4500 U) were combined (BTPLB, 450 μL) and aliquoted to HDNA-functionalized photolabile Glu-pepstatin resin (150 μL), HDNA-functionalized photolabile N-acetyl-Glu resin (150 μL), and biotin-HDNA magnetic resin (150 μL). Plate wells were sealed and resin samples were incubated with shaking (3 h, RT, 600 rpm), then washed (BTPBB, 3 × 150 μL). qPCR mixture contained Taq (0.05 U/μL), oligonucleotide primers 5′-GCCGCCCAGTCCTGCTCGCTTCGCTAC-3′ and 5′-GTGGCACAACAACTGGCGGGCAAAC-3′ (0.3 μM each), and SYBR Green (0.2×, Life Technologies) in GC-PCR buffer (1×). Resin particles (HDNA functionalized photolabile Glu-pepstatin, HDNA functionalized photolabile N-acetyl-Glu) in BTPWB (100 beads/μL, 1 μL) were added to separate amplification reaction wells (20 μL, 10 replicates each). Each resin supernatant (1 μL) was added to respective negative control reaction wells (20 μL, 2 replicates). Biotin-HDNA magnetic resin (2000 beads/μL, 1 μL) was added to a positive control amplification reaction well (20 μL, 10 replicates). Template standards (100 amol, 10 amol, 1 amol, 100 zmol, 10 zmol, 1 zmol, 100 ymol, and 10 ymol in BTPWB) were added to separate reaction wells (20 μL). The reaction plate was thermally cycled (96 °C, 10 s; [95 °C, 8 s; 72 °C, 24 s] × 32 cycles; 72 °C, 120 s; C1000 Touch Thermal Cycler, Bio-Rad, Hercules, CA) with fluorescence monitoring (CFX-96 Real-Time System, Bio-Rad). Samples were quantitated (CFX Manager, version 3.1, Bio-Rad) using single-threshold Cq determination mode (300 RFU). Supernatant background was subtracted from respective single-particle measurements. Background-subtracted replicates were averaged and %RSD calculated.
FACS-Based Bead Lot Preparation and Sequencing
DNA-encoded photolabile Glu-pepstatin beads (1, 100 000 beads) were aliquoted (PBS, 1 mL) and sorted (BD FACS Jazz, BD Biosciences, San Jose, CA) into 96-well skirted PCR plate in replicates for each bead lot size (3 × 3000 beads, 3 × 1000, 3 × 300, 3 × 100, 3 × 30, 3 × 10, 3 × 3, 34 × 1 bead). Forward and side scatter were used to define a gate for the single-bead population. PBS was added to bead-occupied wells and template standard wells to equalize sorting-buffer volume (10 μL total). qPCR mixture contained Taq (0.05 U/μL), oligonucleotide primers 5′-GCCGCCCAGTCCTGCTCGCTTCGCTAC-3′ and 5′-GTGGCACAACAACTGGCGGGCAAAC-3′ (0.3 μM each), and SYBR Green (0.2×, Life Technologies) in GC-PCR buffer (1×). Template standards (100 amol, 10 amol, 1 amol, 100 zmol, 10 zmol, 1 zmol, 100 ymol, and 10 ymol in BTPWB) were added to separate reaction wells (30 μL). The reaction plate was thermally cycled (96 °C, 10 s; [95 °C, 8 s; 72 °C, 24 s] × 32 cycles; 72 °C, 120 s; C1000 Touch Thermal Cycler, Bio-Rad) with fluorescence monitoring (CFX-96 Real-Time System, Bio-Rad). Samples were quantitated using single-threshold Cq determination mode (400 RFU). Supernatant background was subtracted from respective single-particle measurements. Background-subtracted replicates were averaged and %RSD calculated (Supporting Information S3).
Plate samples were centrifuged using a swing bucket rotor (2 min, 400 rcf). For each bead population replicate set (n = 3), one sample was transferred into a clean 96-well plate and diluted (1:10,000 in BTPWB). PCR mixture contained Taq DNA polymerase (0.05 U/μL), oligonucleotide primer 5′-CCTCTCTATGGGCAGTCGGTGATGCCGCCCAGTCCTGCTCGCTTCGCTAC-3′ (0.3 μM), SYBR Green (0.2×, Life Technologies), DMSO (6%), betaine (1 M), MgCl2 (1 mM), and PCR buffer (1×). Each amplicon dilution was added (2 μL, 1 μL, 0.5 μL) with a corresponding NGS barcode (XXXXXXXXXX) oligonucleotide primer (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAGXXXXXXXXXXGATGTGGCACAACAACTGGCGGGCAAAC-3′, 12 pmol) to separate amplification reactions (40 μL). The plate was thermally cycled ([95 °C, 8 s; 70 °C, 24 s; 72 °C, 16 s] x 24 cycles; 72 °C, 120 s). Aliquots of barcoded amplicons with similar quantitation (Cq = 12.8 ± 0.2) were pooled (0.1, 0.35, 1.0, 3.5, 10.5, 35, 16.8 μL; 3, 10, 30, 100, 300, 1000, 3000 bead samples, respectively) and purified by native PAGE (6%, 1 × TBE, 4 W, 30 min) with SYBR Gold staining (Life Technologies). Gel slices containing 248-bp DNA products were excised and placed in a tube (0.6 mL) punctured at the bottom using a syringe needle (18 gauge). The punctured tube was placed inside a larger tube (1.5 mL) and centrifuged (5 min, 10 000 rcf). The extruded gel slices were combined with DI H2O (150 μL), incubated (overnight, RT, 8 rpm), and centrifuged (5 min, 14 000 rcf). The supernatant was removed to a clean tube. An aliquot was used for standard NGS sample preparation and sequencing (Ion Proton, Life Technologies).
Confocal LIF Detection System
Droplet fluorescence was detected on-chip using a two-channel confocal LIF microscope that was built in-house using 30-mm cage system components (Thorlabs, Inc., Newton, NJ). A long-pass dichroic mirror (500 nm, 25.2 × 35.6 mm dichroic long-pass filter, Edmund Optics, Barrington, NJ) directs light from an optically pumped semiconductor laser (488 nm, 20 mW, OBIS-488 20LS, Coherent Inc., Santa Clara, CA) into a microscope objective (40×, 0.60 NA, 2.8 mm WD, Motic, Richmond, Canada), which focuses the excitation beam on the microfluidic channel and collects fluorescence emission. The dichroic transmits the fluorescence signal to a short-pass dichroic (550 nm, 25.2 × 35.6 mm, dichroic short-pass filter, Edmund Optics). Emitted light between 500 and 550 nm is spectrally filtered through a bandpass filter (520BP10, Omega Optical Inc., Brattleboro, VT) before being focused by a plano-convex lens (f/D = 30 mm/25.4 mm). A pinhole (8 μm) spatially filters the focused light prior to detection with a photon counting PMT (H7828, Hamamatsu, Middlesex, NJ). Emitted light longer than 550 nm is directed to a long-pass dichroic (600 nm, 25.2 × 35.6 mm, dichroic long-pass filter, Edmund Optics), which reflects light between 550 and 600 nm to a bandpass filter (570BP10, Omega Optical) and an otherwise identical plano-convex lens/pinhole/PMT optical train.
LabVIEW code written in-house controls signal processing and droplet sorting decisions. The PMT signals were digitized by a data acquisition board (DAQ, NI USB-6341, National Instruments, Austin, TX), and binned into packets of counts (Δt = 0.5 ms). Median-filter smoothing (window width = 7) is applied to the signal in real-time, droplet signal regions are identified by the 570 nm channel signal (countsn > 500) and 520 nm channel signal maxima (max = countsn–1 when countsn < countsn–1) returned as “droplet fluorescence.” When a hit droplet is detected (droplet fluorescence < 1000 counts), LabVIEW outputs a TTL pulse from the DAQ board to a waveform generator (Agilent 33210A, Agilent Technologies, Santa Clara, CA), triggering a defined square wave pulse output (0—5 V, 10 kHz, 80 cycles) that is amplified (gain = 100 V/V, TREK Model 2210 high-voltage power amplifier, TREK Inc., Lockport, NY) and conducted through needle-fitted tubing filled with salt water (4 M NaCl) into a microfabricated electrode channel (VAC).
Microfluidic Device Fabrication and Calibration
Channel structures were fabricated using soft lithography.64 Devices were fabricated using two-tone patterned PDMS as previously described.45 Briefly, avobenzone (34 mg) was dissolved in toluene (200 μL) and mixed into PDMS prepolymer (5.5 g, 10:1 elastomer base/curing agent). Degassed avobenzone-PDMS prepolymer was loaded into a disposable syringe (3 mL, BD Medical, Franklin Lakes, NJ) and applied over the incubation channel and bead-introduction reservoir regions of the master. After partial curing (80 °C, 12 min), native, degassed PDMS prepolymer (44 g, 10:1) was poured on top of the master and cured to completion (80 °C, 1 h). After peeling the PDMS mold from the wafer, fluidic access ports were punched with a biopsy punch (0.75 mm, World Precision Instruments, Inc., Sarasota, FL). Microfluidic devices were prepared and fitted with integrated waveguides.46 Waveguide performance was calibrated for each device to ensure reproducible compound liberation. To calibrate, a solution of Brilliant Violet 510 (BV510, 4 μg/mL in MES buffer) was pumped through the calibration channel (0.5 μL/min). The dye was excited with UV via integrated waveguide (LED current = 115 mA, 130 mA) and emission was measured in the 520 nm channel (100 Hz). Afterward, the calibration channel was rinsed with water, dried, and filled with trimethylsiloxy-terminated PDMS.
Integrated Circuit Operation
Prior to droplet generation, the incubation channel was backfilled with oil phase. OIL1 (1.5 μL/min) was pumped into the circuit through OIL1 inlet and aqueous phase was pumped into the circuit through AQ1 (0.3 μL/min) and AQ2 (0.2 μL/min). Fluorogenic CatD peptide probe (1.5 nmol, AnaSpec) and R-phycoerythrin (12.5 μg) in BHB (250 μL) were driven into the circuit via AQ1. CatD (610 ng, 0.36 U) in protease assay buffer (400 μL) was driven into the circuit via AQ2. A mixture of DNA-encoded photocleavable pepstatin A positive control inhibitor beads 1 and negative control N-acetyl-Glu beads 2 serves as a model bead library. The DNA-encoded model library was suspended in BHB (180 μL), filtered (CellTrics 20-μm mesh, Sysmex-Partec), and drawn into syringe (1 mL, slip-tip disposable tuberculin syringe, Becton Dickinson, Franklin Lakes, NJ) fitted with a needle (30 gauge, 0.5 in., Precisionglide, Becton Dickinson) while preserving head space. A PDMS plug-sealed pipet tip (200 μL, Rainin BioClean LTS tips, Mettler-Toledo, Columbus, OH) was filled with model library bead suspension by inserting the needle through the PDMS plug and expelling the bead suspension (∼170 μL) to fill the tip without air pockets. The puncture hole in the PDMS was sealed with hot glue, and the tip was inserted into the LIB port of the device.40 Water-in-oil droplets were generated (190—220 pL, 43—37 Hz) at the flow-focusing channel intersection, directed to the waveguide illumination region for UV irradiation, then driven through the incubation channel (18 min) before detection and sorting. Droplet spacing oil (3.5 μL/min) and droplet guiding oil (2.5 μL/min) enter the device at OIL2 and OIL3, respectively. Droplets were interrogated via confocal laser-induced fluorescence detection (2 kHz acquisition). Droplets were dielectrophoretically deflected in an electric field generated between one saltwater electrode connected to a high-voltage source (500 VAC, 10 kHz square wave, 8 ms pulse) and another electrode connected to ground. Deflected droplets flow to the HIT output for collection. After model library screening, droplets collected through the HIT output are visualized using an inverted epifluorescence microscope (2.5×, 0.075 NA, Axio Observer A1, Zeiss, Thornwood, NY) equipped with a CCD camera (AxioCam ICm1, Zeiss). Droplets were imaged in brightfield mode and fluorescence mode using Zeiss filter set 38HE (FAM channel, λex = 470/40 nm; λdc = 495 nm; λem = 525/50 nm) and Zeiss filter set 20 (TMR channel, λex = 546/12 nm; λdc = 560 nm; λem = 608/65 nm).
NGS Decoding and Bead Index Cutoff
Ion Proton FASTQ files for each screening sample set were imported into R, each sequence was matched to the degenerate 10-position reference sequence “ATGGNNNNNNNNTCANNNNNNNNGTTNNNNNNNNCTANNNNNNNNTTCNNNNNNNNCGCNNNNNNNNGTANNNNNNNNTGGNNNNNNNNTCTNNNNNNNNAAGNNNNNNNNGCCTCCCAAACnnnnnnnnGTT” (overhangs in bold, N = n = any DNA base) allowing for up to 3 mismatched bases, then excess sequence was trimmed. All NNNNNNNN encoding sequences were matched with the known encoding set (“AAGAGGCA”, “ACGGAGCA”, “ACGAGATT”, “AAGGAGGT”, “AGAAAGCA”, “ATAGAGCC”, “CAGAAGGA”, “GAGGAACA”, “TGAAGGAA”, “TTGAGGAT”, “CCTCCTAA”, “AACCTCAA”, “ATTCTCGG”, “AACCCTAC”, “GACTCCGC”, “CATTTCAA”, “CCCTCCGG”, “CGTTCCTG”, “TTCTTCAT”, “TCTCCTCC”). Hamming distances were calculated for all nonmatched NNNNNNNN encoding sequences. Those with Hamming distance = 1 from a member of the known encoding set were replaced with the correct sequence. Any read containing an encoding sequence with Hamming distance >1 was removed. Identical sequences including the UMI (nnnnnnnn) were aggregated as a single sequence, trimmed to terminate at “GCCTCCCAAC”, and both the UMI sequence (nnnnnnnn) and read counts for each UMI were preserved. Identical sequences were aggregated (now without UMI consideration) as a single sequence, and a random sample of up to 5000 UMI sequences for each sequence was selected. For each sequence, the set of UMIs was examined and for any given UMI pair with a Hamming distance = 1, the UMI represented by the fewest read counts was discarded. After this UMI “distance filter”, each sequence’s UMI set was counted. Sequences were rank-ordered by UMI counts, assigned a “Bead Index” number, then plotted. For each bead-lot sample (30, 100, 300, 1000, 3000, hit collection), a weighted mean UMI count (UMIw) was generated at each bead index, using a rolling window (width = 10), and a normal weighted distribution (e–kx; k = 0.01). The transition from bead-derived sequences to background sequences was approximated by determining the inflection point of each curve, equal to the bead index yielding a maximum %ΔUMIw/UMIw (% change in UMIw). The inflection’s x coordinate is called the bead index cutoff.
hvSABR Hit Bead Recovery and Preparation for NGS
The water-in-oil emulsion (∼0.6 mL) was centrifuged (5 min, 12000 rcf), 2/3 of the oil phase was removed, and fresh DMF-A-6CS silicone oil (Shin-Etsu) was added (400 μL). After oil exchange, the tube contents were agitated by flicking. This process was repeated thrice more and BTPWB (2 μL) was added. The tube was agitated and centrifuged (5 min, 12,000 rcf). BTPWB (100 μL) was added, and the tube was centrifuged (5 min, 12,000 rcf). Most oil was removed, BTPWB (300 μL) was added, and the tube was centrifuged (5 min, 12,000 rcf). The remaining oil and excess aqueous phase were removed with a clean polyurethane swab (Berkshire Corporation, Great Barrington, MA). Beads were transferred to PCR tube (0.2 mL) in BTPWB (4 × 50 μL), then the tube was centrifuged (30 s, 6000 rcf) and all supernatant except ∼10 μL was removed.
qPCR mixture contained Taq (0.05 U/μL), oligonucleotide primers 5′-GCCGCCCAGTCCTGCTCGCTTCGCTAC-3′ and 5′-GTGGCACAACAACTGGCGGGCAAAC-3′ (0.3 μM each), and SYBR Green (0.2×, Life Technologies) in GC-PCR buffer (1×). Template standards (100 amol, 10 amol, 1 amol, 100 zmol, 10 zmol, 1 zmol, 100 ymol, and 10 ymol in BTPWB) were added into BTPWB (10 μL) in separate reaction wells of a PCR tube strip (8 tubes), and qPCR mix (30 μL) was added. Hit beads in the PCR tube were lightly vortexed to free bead pellet, sonicated (30 s), then qPCR mix (30 μL) was added prior to thermal cycling. The hit bead PCR tube and standards were thermally cycled (96 °C, 10 s; [95 °C, 8 s; 72 °C, 24 s] × 27 cycles; 72 °C, 120 s; C1000 Touch Thermal Cycler, Bio-Rad) with fluorescence monitoring (CFX-96 Real-Time System, Bio-Rad). The hit bead PCR tube was centrifuged (1 min, 1000 rcf), and supernatant was diluted (1:10000 in BTPWB). The amplicons were barcoded and prepared for NGS as described previously (Ion Proton, Life Technologies).
Acknowledgments
B.M.P. gratefully acknowledges the support of a NIH Director’s New Innovator Award (OD008535) and the DARPA Fold F(X) Program (N66001-14-2-4057).
Glossary
Abbreviations
- NGS
next-generation sequencing
- HTS
high-throughput screening
- DEL
DNA-encoded library
- hνSABR
light-induced and -graduated high-throughput screening after bead release
- OBOC
one bead one compound
- FAM
fluorescein
- TMR
tetramethylrhodamine
- DESPS
DNA-encoded solid-phase synthesis
- HDNA
headpiece DNA
- catD
cathepsin D
- LIF
laser-induced fluorescence
- BHB
bead hopper buffer
- UMI
unique molecular identifier
- ER
encoding region
- BSB
bead-specific barcode
- OP
oligonucleotide paired stock
- FDR
false discovery rate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombsci.6b00192.
Experimental details and additional results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gartner Z. J.; Liu D. R. The Generality of DNA-Templated Synthesis as a Basis for Evolving Non-Natural Small Molecules. J. Am. Chem. Soc. 2001, 123, 6961–6963. 10.1021/ja015873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin D. R.; Harbury P. B. DNA Display I. Sequence-Encoded Routing of DNA Populations. PLoS Biol. 2004, 2, E173. 10.1371/journal.pbio.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkko S.; Scheuermann J.; Dumelin C.; Neri D. Encoded Self-Assembling Chemical Libraries. Nat. Biotechnol. 2004, 22, 568–574. 10.1038/nbt961. [DOI] [PubMed] [Google Scholar]
- Clark M. A.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; et al. Design, Synthesis and Selection of DNA-Encoded Small-Molecule Libraries. Nat. Chem. Biol. 2009, 5, 647–654. 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Brenner S.; Lerner R. A. Encoded Combinatorial Chemistry. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 5381–5383. 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. P. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Hanes J.; Pluckthun A. In Vitro Selection and Evolution of Functional Proteins by Using Ribosome Display. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 4937–4942. 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. W.; Szostak J. W. RNA-Peptide Fusions for the in Vitro Selection of Peptides and Proteins. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 12297–12302. 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y.; Clark M. A. Robust Suzuki-Miyaura Cross-Coupling on DNA-Linked Substrates. ACS Comb. Sci. 2015, 17, 1–4. 10.1021/co5001037. [DOI] [PubMed] [Google Scholar]
- Satz A. L.; Cai J.; Chen Y.; Goodnow R.; Gruber F.; Kowalczyk A.; Petersen A.; Naderi-Oboodi G.; Orzechowski L.; Strebel Q. DNA Compatible Multistep Synthesis and Applications to DNA Encoded Libraries. Bioconjugate Chem. 2015, 26, 1623–1632. 10.1021/acs.bioconjchem.5b00239. [DOI] [PubMed] [Google Scholar]
- Eidam O.; Satz A. L. Analysis of the Productivity of DNA Encoded Libraries. MedChemComm 2016, 7, 1323–1331. 10.1039/C6MD00221H. [DOI] [Google Scholar]
- Disch J. S.; Evindar G.; Chiu C. H.; Blum C. A.; Dai H.; Jin L.; Schuman E.; Lind K. E.; Belyanskaya S. L.; Deng J.; et al. Discovery of Thieno[3,2-D]Pyrimidine-6-Carboxamides as Potent Inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56, 3666–3679. 10.1021/jm400204k. [DOI] [PubMed] [Google Scholar]
- Encinas L.; O’Keefe H.; Neu M.; Remuiñán M. J.; Patel A. M.; Guardia A.; Davie C. P.; Pérez-Macías N.; Yang H.; Convery M. A.; et al. Encoded Library Technology as a Source of Hits for the Discovery and Lead Optimization of a Potent and Selective Class of Bactericidal Direct Inhibitors of Mycobacterium Tuberculosis InhA. J. Med. Chem. 2014, 57, 1276–1288. 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- Deng H.; Zhou J.; Sundersingh F. S.; Summerfield J.; Somers D.; Messer J. A.; Satz A. L.; Ancellin N.; Arico-Muendel C. C.; Sargent Bedard K. L.; et al. Discovery, SAR, and X-Ray Binding Mode Study of BCATm Inhibitors From a Novel DNA-Encoded Library. ACS Med. Chem. Lett. 2015, 6, 919–924. 10.1021/acsmedchemlett.5b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Graybill T. L.; Zeng X.; Platchek M.; Zhang J.; Bodmer V. Q.; Wisnoski D. D.; Deng J.; Coppo F. T.; Yao G.; et al. Cell-Based Selection Expands the Utility of DNA-Encoded Small-Molecule Library Technology to Cell Surface Drug Targets: Identification of Novel Antagonists of the NK3 Tachykinin Receptor. ACS Comb. Sci. 2015, 17, 722–731. 10.1021/acscombsci.5b00124. [DOI] [PubMed] [Google Scholar]
- Satz A. L. DNA Encoded Library Selections and Insights Provided by Computational Simulations. ACS Chem. Biol. 2015, 10, 2237–2245. 10.1021/acschembio.5b00378. [DOI] [PubMed] [Google Scholar]
- Beaudry A. A.; Joyce G. F. Science 1992, 257, 635–641. 10.1126/science.1496376. [DOI] [PubMed] [Google Scholar]
- Tawfik D. S.; Griffiths A. Man-Made Cell-Like Compartments for Molecular Evolution. Nat. Biotechnol. 1998, 16, 652–656. 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- Blakskjær P.; Heitner T.; Hansen N. J. V. Fidelity by Design: Yoctoreactor and Binder Trap Enrichment for Small-Molecule DNA-Encoded Libraries and Drug Discovery. Curr. Opin. Chem. Biol. 2015, 26, 62–71. 10.1016/j.cbpa.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Furka A.; Sebestyen F.; Asgedom M.; Dibo G. General-Method for Rapid Synthesis of Multicomponent Peptide Mixtures. Int. J. Pept. Protein Res. 1991, 37, 487–493. 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- Houghten R. A.; Pinilla C.; Blondelle S. E.; Appel J. R.; Dooley C. T.; Cuervo J. H. Generation and Use of Synthetic Peptide Combinatorial Libraries for Basic Research and Drug Discovery. Nature 1991, 354, 84–86. 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- Lam K. S.; Salmon S. E.; Hersh E. M.; Hruby V. J.; Kazmierski W. M.; Knapp R. J. A New Type of Synthetic Peptide Library for Identifying Ligand-Binding Activity. Nature 1991, 354, 82–84. 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- Silen J. L.; Lu A. T.; Solas D. W.; Gore M. A.; Maclean D.; Shah N. H.; Coffin J. M.; Bhinderwala N. S.; Wang Y. W.; Tsutsui K. T.; et al. Screening for Novel Antimicrobials From Encoded Combinatorial Libraries by Using a Two-Dimensional Agar Format. Antimicrob. Agents Chemother. 1998, 42, 1447–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell H. E.; Pérez L.; Stavenger R. A.; Tallarico J. A.; Cope Eatough E.; Foley M. A.; Schreiber S. L. A One-Bead, One-Stock Solution Approach to Chemical Genetics: Part 1. Chem. Biol. 2001, 8, 1167–1182. 10.1016/S1074-5521(01)00085-0. [DOI] [PubMed] [Google Scholar]
- You A. J.; Jackman R. J.; Whitesides G. M.; Schreiber S. L. A Miniaturized Arrayed Assay Format for Detecting Small Molecule-Protein Interactions in Cells. Chem. Biol. 1997, 4, 969–975. 10.1016/S1074-5521(97)90305-7. [DOI] [PubMed] [Google Scholar]
- Upert G.; Merten C. A.; Wennemers H. Nanoliter Plates-Versatile Tools for the Screening of Split-and-Mix Libraries on-Bead and Off-Bead. Chem. Commun. 2010, 46, 2209–2211. 10.1039/b927017e. [DOI] [PubMed] [Google Scholar]
- Borchardt A.; Liberles S.; Biggar S.; Crabtree G.; Schreiber S. Small Molecule-Dependent Genetic Selection in Stochastic Nanodroplets as a Means of Detecting Protein-Ligand Interactions on a Large Scale. Chem. Biol. 1997, 4, 961–968. 10.1016/S1074-5521(97)90304-5. [DOI] [PubMed] [Google Scholar]
- Thorsen T.; Roberts R.; Arnold F.; Quake S. Dynamic Pattern Formation in a Vesicle-Generating Microfluidic Device. Phys. Rev. Lett. 2001, 86, 4163–4166. 10.1103/PhysRevLett.86.4163. [DOI] [PubMed] [Google Scholar]
- Anna S.; Bontoux N.; Stone H. Formation of Dispersions Using “Flow Focusing” in Microchannels. Appl. Phys. Lett. 2003, 82, 364–366. 10.1063/1.1537519. [DOI] [Google Scholar]
- Song H.; Tice J.; Ismagilov R. A Microfluidic System for Controlling Reaction Networks in Time. Angew. Chem., Int. Ed. 2003, 42, 768–772. 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]
- Song H.; Chen D. L.; Ismagilov R. F. Reactions in Droplets in Microfluidic Channels. Angew. Chem., Int. Ed. 2006, 45, 7336–7356. 10.1002/anie.200601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baret J.-C.; Miller O. J.; Taly V.; Ryckelynck M.; El-Harrak A.; Frenz L.; Rick C.; Samuels M. L.; Hutchison J. B.; Agresti J. J.; et al. Fluorescence-Activated Droplet Sorting (FADS): Efficient Microfluidic Cell Sorting Based on Enzymatic Activity. Lab Chip 2009, 9, 1850–1858. 10.1039/b902504a. [DOI] [PubMed] [Google Scholar]
- Sciambi A.; Abate A. R. Accurate Microfluidic Sorting of Droplets at 30 kHz. Lab Chip 2015, 15, 47–51. 10.1039/C4LC01194E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher G. F.; Bergstein J.; End N. B.; Poon M.; Nguyen C.; Anna S. L. Coalescence and Splitting of Confined Droplets at Microfluidic Junctions. Lab Chip 2009, 9, 1102–1109. 10.1039/b813062k. [DOI] [PubMed] [Google Scholar]
- Frenz L.; Blank K.; Brouzes E.; Griffiths A. D. Reliable Microfluidic on-Chip Incubation of Droplets in Delay-Lines. Lab Chip 2009, 9, 1344–1348. 10.1039/B816049J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenz L.; Blouwolff J.; Griffiths A. D.; Baret J.-C. Microfluidic Production of Droplet Pairs. Langmuir 2008, 24, 12073–12076. 10.1021/la801954w. [DOI] [PubMed] [Google Scholar]
- Xu L.; Lee H.; Panchapakesan R.; Oh K. W. Fusion and Sorting of Two Parallel Trains of Droplets Using a Railroad-Like Channel Network and Guiding Tracks. Lab Chip 2012, 12, 3936–3942. 10.1039/c2lc40456g. [DOI] [PubMed] [Google Scholar]
- Chen D.; Liu Y.; Liu W.; Kuznetsov A.; Mendez F. E.; Philipson L. H.; Ismagilov R. F.; et al. The Chemistrode: a Droplet-Based Microfluidic Device for Stimulation and Recording with High Temporal, Spatial, and Chemical Resolution. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16843–16848. 10.1073/pnas.0807916105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Slaney T. R.; Kennedy R. T. Label Free Screening of Enzyme Inhibitors at Femtomole Scale Using Segmented Flow Electrospray Ionization Mass Spectrometry. Anal. Chem. 2012, 84, 5794–5800. 10.1021/ac3011389. [DOI] [PubMed] [Google Scholar]
- Price A. K.; MacConnell A. B.; Paegel B. M. Microfluidic Bead Suspension Hopper. Anal. Chem. 2014, 86, 5039–5044. 10.1021/ac500693r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois F.; Olguin L. F.; Whyte G.; Bratton D.; Huck W. T. S.; Abell C.; Hollfelder F. An Integrated Device for Monitoring Time-Dependent in Vitro Expression From Single Genes in Picolitre Droplets. ChemBioChem 2008, 9, 439–446. 10.1002/cbic.200700536. [DOI] [PubMed] [Google Scholar]
- Mazutis L.; Baret J.-C.; Treacy P.; Skhiri Y.; Araghi A. F.; Ryckelynck M.; Taly V.; Griffiths A. D. Multi-Step Microfluidic Droplet Processing: Kinetic Analysis of an in Vitro Translated Enzyme. Lab Chip 2009, 9, 2902–2908. 10.1039/b907753g. [DOI] [PubMed] [Google Scholar]
- Fallah-Araghi A.; Baret J.-C.; Ryckelynck M.; Griffiths A. D. A Completely in Vitro Ultrahigh-Throughput Droplet-Based Microfluidic Screening System for Protein Engineering and Directed Evolution. Lab Chip 2012, 12, 882–891. 10.1039/c2lc21035e. [DOI] [PubMed] [Google Scholar]
- Autour A.; Westhof E.; Ryckelynck M. iSpinach: a Fluorogenic RNA Aptamer Optimized for in Vitro Applications. Nucleic Acids Res. 2016, 44, 2491–2500. 10.1093/nar/gkw083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A. K.; Paegel B. M. Discovery in Droplets. Anal. Chem. 2016, 88, 339–353. 10.1021/acs.analchem.5b04139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A. K.; MacConnell A. B.; Paegel B. M. hνSABR: Photochemical Dose-Response Bead Screening in Droplets. Anal. Chem. 2016, 88, 2904–2911. 10.1021/acs.analchem.5b04811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciambi A.; Abate A. R. Generating Electric Fields in PDMS Microfluidic Devices with Salt Water Electrodes. Lab Chip 2014, 14, 2605–2609. 10.1039/c4lc00078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacConnell A. B.; McEnaney P. J.; Cavett V. J.; Paegel B. M. DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb. Sci. 2015, 17, 518–534. 10.1021/acscombsci.5b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes K. R.; Malone M. L.; Ndungu J. M.; Suponitsky-Kroyter I.; Cavett V. J.; McEnaney P. J.; MacConnell A. B.; Doran T. M.; Ronacher K.; Stanley K.; et al. High-Throughput Identification of DNA-Encoded IgG Ligands That Distinguish Active and Latent Mycobacterium Tuberculosis Infections. ACS Chem. Biol. 2017, 12, 234–243. 10.1021/acschembio.6b00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appell K. C.; Chung T. D. Y.; Ohlmeyer M. J. H.; Sigal N. H.; Baldwin J. J.; Chelsky D. Biological Screening of a Large Combinatorial Library. J. Biomol. Screening 1996, 1, 27–31. 10.1177/108705719600100111. [DOI] [Google Scholar]
- Doran T. M.; Gao Y.; Mendes K.; Dean S.; Simanski S.; Kodadek T. Utility of Redundant Combinatorial Libraries in Distinguishing High and Low Quality Screening Hits. ACS Comb. Sci. 2014, 16, 259–270. 10.1021/co500030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Chung T.; Oldenburg K. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 1999, 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Sun S.; Kennedy R. T. Droplet Electrospray Ionization Mass Spectrometry for High-Throughput Screening for Enzyme Inhibitors. Anal. Chem. 2014, 86, 9309–9314. 10.1021/ac502542z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight C. G.; Barrett A. J. Interaction of Human Cathepsin D with the Inhibitor Pepstatin. Biochem. J. 1976, 155, 117–125. 10.1042/bj1550117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E. Z.; Basu A.; Satija R.; Nemesh J.; Shekhar K.; Goldman M.; Tirosh I.; Bialas A. R.; Kamitaki N.; Martersteck E. M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A. M.; Mazutis L.; Akartuna I.; Tallapragada N.; Veres A.; Li V.; Peshkin L.; Weitz D. A.; Kirschner M. W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone M. L.; Paegel B. M. What Is a “DNA-Compatible” Reaction?. ACS Comb. Sci. 2016, 18, 182–187. 10.1021/acscombsci.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossett D. R.; Tse H. T. K.; Lee S. A.; Ying Y.; Lindgren A. G.; Yang O. O.; Rao J.; Clark A. T.; Di Carlo D. Hydrodynamic Stretching of Single Cells for Large Population Mechanical Phenotyping. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 7630–7635. 10.1073/pnas.1200107109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvortrup K.; Nielsen T. E. In-Bead Screening of Hydroxamic Acids for the Identification of HDAC Inhibitors. Angew. Chem., Int. Ed. 2016, 55, 4472–4475. 10.1002/anie.201511308. [DOI] [PubMed] [Google Scholar]
- Lian W.; Upadhyaya P.; Rhodes C. A.; Liu Y.; Pei D. Screening Bicyclic Peptide Libraries for Protein-Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-A Antagonist. J. Am. Chem. Soc. 2013, 135, 11990–11995. 10.1021/ja405106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh T. B.; Upadhyaya P.; Qian Z.; Pei D. Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci. 2016, 18, 75–85. 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-W.; Kim G.-J.; Lee S.; Kim J.; deMello A. J.; Chang S.-I. A Droplet-Based Fluorescence Polarization Immunoassay (dFPIA) Platform for Rapid and Quantitative Analysis of Biomarkers. Biosens. Bioelectron. 2015, 67, 497–502. 10.1016/j.bios.2014.09.013. [DOI] [PubMed] [Google Scholar]
- Chan T. R.; Hilgraf R.; Sharpless K. B.; Fokin V. V. Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis. Org. Lett. 2004, 6, 2853–2855. 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- Duffy D.; McDonald J.; Schueller O.; Whitesides G. Anal. Chem. (Washington, DC, U. S.) 1998, 70, 4974–4984. 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.