

Abstract
Toll-like receptors (TLRs) such as TLR2 and TLR4 have been implicated in host response to mycobacterial infection. Here, mice deficient in the TLR adaptor molecule myeloid differentiation factor 88 (MyD88) were infected with Mycobacterium tuberculosis (MTB). While primary MyD88–/– macrophages and DCs are defective in TNF, IL-12, and NO production in response to mycobacterial stimulation, the upregulation of costimulatory molecules CD40 and CD86 is unaffected. Aerogenic infection of MyD88–/– mice with MTB is lethal within 4 weeks with 2 log10 higher CFU in the lung; high pulmonary levels of cytokines and chemokines; and acute, necrotic pneumonia, despite a normal T cell response with IFN-γ production to mycobacterial antigens upon ex vivo restimulation. Vaccination with Mycobacterium bovis bacillus Calmette-Guérin conferred a substantial protection in MyD88–/– mice from acute MTB infection. These data demonstrate that MyD88 signaling is dispensable to raise an acquired immune response to MTB. Nonetheless, this acquired immune response is not sufficient to compensate for the profound innate immune defect and the inability of MyD88–/– mice to control MTB infection.
Introduction
Tuberculosis caused by Mycobacterium tuberculosis (MTB) is a highly infectious respiratory infection. One-third of the world’s population has been in contact with the pathogen, but approximately 90% of infected persons contain the infection with no overt clinical symptoms (1). A better understanding of the mechanisms of the host that lead to protective immunity is important to develop novel therapies and vaccines. The need of functional APCs and T cells in concert with several cytokines and chemokines to initiate an adaptive immunity has been established (2). The recent discovery of the Toll-like receptor (TLR) family has provided new insights on how pathogens recognize and activate innate immune cells and thereby link innate and adaptive immunity (3). MTB, its cell wall, and secreted components such as 19-kDa lipoprotein, lipomannan, and mannosylated phosphatidylinositol activate macrophages and DCs through TLR2 (4–9), but the contribution of other TLRs or pattern-recognition receptors (PRRs) in MTB recognition is not excluded.
Previous in vivo investigations demonstrated that TLR2, TLR4, or TLR6 play no role or only a minor role in the early in vivo host response to MTB infection (10–13), but TLR2 (14) and TLR4 (10) may be required to control the chronic stage of infection. These data suggest a partial redundancy of TLR2, TLR4, and TLR6 or other TLRs that may be involved in MTB-induced cell recognition.
Most TLRs, except TLR3, use myeloid differentiation factor 88 (MyD88), an intracellular adaptor protein of the IL-1 receptor/IRAK pathway, to link TLR recognition with activation of IRAK and TRAF, translocation of NF-κB, and gene transcription (3). Absence of MyD88 resulted in a dramatic reduction of host resistance to several infectious agents (15–21).
Here, we investigated the role of MyD88 in the control of MTB infection using MyD88–/– mice and demonstrate an essential role of MyD88 in the host response to an aerogenic infection with H37Rv MTB. MyD88–/– mice succumbed within 4 weeks to acute, necrotic pulmonary MTB infection with uncontrolled bacterial growth despite their ability to mount an adaptive immune response. Interestingly, vaccination with Mycobacterium bovis bacillus Calmette-Guérin (BCG) conferred initial protection to acute MTB infection but did not prevent chronic infection. Therefore, the MyD88 signaling pathway is crucial for the control of MTB infection.
Results
Reduced cytokine production, but normal upregulation of costimulatory molecules, in mycobacteria-activated macrophages from MyD88–/– mice.
TNF-α and IL-12 play important roles in the control of local immune responses to intracellular organisms such as MTB. We therefore investigated the ability of MyD88–/– BM-derived macrophages and DCs to secrete TNF-α and IL-12 p40 in response to mycobacteria in vitro. TNF and IL-12 p40 production after stimulation with MTB H37Rv, H37Ra, or M. bovis BCG was drastically reduced in MyD88-deficient macrophages and DCs compared with wild-type controls (Figure 1, A–D). Similar results were obtained after 24 or 48 hours of stimulation, except that the low cytokine levels were merely due to delayed kinetics (data not shown). We further tested the MyD88 dependence of mycobacteria-induced iNOS activation and demonstrate a dramatic reduction of nitrite production in MyD88-deficient macrophages and little effect in DCs (Figure 1, E and F). Furthermore, we investigated the cytokine production at the site of infection in the lung. TNF production by interstitial lung macrophages stimulated with mycobacteria was undetectable, and IL-6 production was substantially reduced in the absence of MyD88 (Figure 1, G and H).
Figure 1.

Impaired proinflammatory cytokine and NO production in MyD88–/– macrophages and DCs. BM-derived macrophages (A, C, and E) and DCs (B, D, and F) (5 × 105 cells/ml) prepared from MyD88–/– (white bars) and wild-type (black bars) mice were incubated with LPS (100 ng/ml), M. bovis BCG, MTB H37Ra, or MTB H37Rv (all at 2 bacteria per cell). After 24 hours, the production of TNF (A and B), IL-12 p40 (C and D), or nitrite (E and F) was determined in the supernatant by ELISA or Griess reaction. TNF and IL-6 production by pulmonary macrophages stimulated in the same conditions was also measured (G and H). Upregulation of CD40 (I) and CD86 (J) expression by DCs stimulated with LPS or M. bovis BCG were analyzed by FACS. Data are from 1 experiment, representative of 3 independent experiments with n = 2 mice per genotype; mean values ± SD are shown. MFI, mean fluorescence intensity.
In contrast, the expression of the costimulatory molecules CD40 and CD86 was upregulated in mycobacteria-infected MyD88-deficient DCs (Figure 1, I and J) and macrophages (not shown) as in wild-type control cells. Therefore, the production of cytokines, but not the expression of costimulatory molecules, in response to mycobacterial infection is MyD88 dependent. Based on these results, we hypothesized that MyD88 signaling might be critical for the innate immune response to MTB infection.
Lethal MTB infection with necrotic pneumonia in the absence of MyD88.
MyD88–/– mice infected with a low dose (200 CFU per mouse) of virulent MTB H37Rv started to lose body weight around 3 weeks (Figure 2A) and died within 4 weeks after infection (Figure 2B). MyD88–/– mice exhibited dramatically increased lung weight (Figure 2C) and a bacterial load in the lungs that was more than 2 log10 higher than that in wild-type controls at 4 weeks (Figure 2D). This was accompanied by bacterial dissemination into the spleen (Figure 2E) and the liver (data not shown). In view of the low proinflammatory response in MTB-stimulated macrophages from MyD88–/– mice, we compared mice deficient in MyD88 with mice deficient in TNF. TNF–/– mice lost weight rapidly and were unable to control MTB infection, dying slightly, but significantly, earlier than MyD88-deficient mice (P < 0.01; Figure 2, A and B). Acute pulmonary infection, illustrated by a dramatic increase in lung weight, was found in both TNF- and MyD88-deficient mice 4 weeks after infection (Figure 2C). Therefore, MyD88–/– mice are highly susceptible to MTB infection, similar to TNF-deficient mice (22).
Figure 2.

MyD88–/– mice are unable to control an MTB infection. MyD88–/– (open circles), TNF–/– (open squares), and wild-type (filled squares) mice were exposed to a low aerogenic dose of MTB H37Rv (200 CFU per mouse i.n.) and monitored for body weight (A; mean values of n = 6–8 mice per group from 1 representative experiment out of 7 independent experiments) and survival (B; n = 13–24 mice pooled from several experiments; P – 0.01 between MyD88–/– or TNF–/– mice and wild-type controls, and between TNF- and MyD88-deficient mice). (C) Lung wet weight of TNF–/– mice (gray bars), MyD88–/– mice (white bars), and wild-type controls (black bars) were measured 17 and 27 days after infection. The numbers of viable bacteria present in the lungs (D) and spleen (E) of MyD88–/– (white bars) and wild-type (black bars) mice were determined after 27 days of infection (mean ± SD of n = 3–4 lung or n = 5–7 spleen samples from 1 representative experiment out of 2 or 3 independent experiments, respectively; **P – 0.01).
Macroscopically, the lungs of MyD88–/– mice displayed pleural adhesions and effusions and large subpleural and confluent nodules (Figure 3A), as compared with wild-type controls (Figure 3B). Microscopic investigation of the lungs of MyD88–/– mice revealed massive mononuclear and neutrophil infiltrations with extensive confluent necrosis in the absence of proper granuloma formation at 27 days (Figure 3, C–F) and abundant mycobacteria in macrophages and also in the extracellular space (Figure 3, G–H). In the liver and spleen, only a few small granuloma were found as a sign of dissemination of infection in both MyD88-deficient and wild-type mice (data not shown). The observed lung lesions are similar to those induced by mycobacterial infection in TNF-deficient mice (22, 23). Thus, in the absence of MyD88, mice developed massive necrosis and infiltration of inflammatory cells in the lungs with uncontrolled MTB growth.
Figure 3.

MyD88–/– mice exhibit acute necrotic pneumonia with large nodules but defective granuloma formation in response to MTB infection. Lung tissue from MyD88–/– (A, C, E, and G) and wild-type (B, D, F, and H) mice was analyzed 27 days after MTB H37Rv infection (200 CFU i.n.). Lungs of MyD88–/– mice showed large and confluent nodules (A) in comparison with those of wild-type mice (B). Microscopic examination showed extensive inflammation and necrosis in infected MyD88–/– lungs (C–F; H&E) with abundant mycobacteria in the extracellular space (G and H; Ziehl-Neelsen). Low-power micrographs of representative lung sections are shown in C and D (magnification, ×50), and higher magnification shows details in E–H (magnification, ×400).
Increased recruitment of myeloid cells in MyD88–/– mice.
In view of the acute and uncontrolled pulmonary infection of MyD88–/– mice, we asked how the recruitment of immunoinflammatory cells in the lung parenchyma is modulated in the absence of MyD88. The total number of cells recovered from lung of both groups was comparable. Flow cytometric analysis revealed similar amounts of CD4+ and CD8+ T cells expressing the activation marker CD44 in MyD88-deficient and control lungs at 4 weeks of infection (Figure 4A). Consistent with the histology, we found increased numbers of Ly6G+ and CD11b+ cells in the lung homogenates of MyD88–/– mice (Figure 4B), with expression of class II on the CD11b+ cells (Figure 4C). CD11c+ cells were normally recruited and activated in the absence of MyD88 (Figure 4, B and C). Thus, the data demonstrate that the high infectious burden in MTB-infected MyD88-deficient mice is accompanied by a vigorous inflammatory response with increased neutrophils and macrophages, and apparently normal recruitment and activation of T lymphocytes in the lung, that occurs in the absence of MyD88 signaling.
Figure 4.

T lymphocyte and myeloid cell recruitment and activation in the lung of MyD88–/– infected mice. Infiltrating cells from the lungs of MyD88–/– and control mice were isolated 27 days after infection and analyzed by flow cytometry for the expression of CD4, CD8, CD44, CD11b, CD11c, Ly-6G, and MHC class II IA–IE. Typical dot plots of FACS analysis are shown for CD44 expression in CD4+ and CD8+ T cells (A; gated on T lymphocytes). Graphic representations of the different leukocyte populations (B) and of the expression of MHC class II on APCs (C) are shown. Results are expressed as absolute numbers of positive cells. Mean ± SD from 2 MyD88–/– mice are shown and are representative of 2 independent experiments.
Activation of T cells in MyD88–/– mice.
As MyD88 signaling has been shown to be required for an antigen-specific Th1 immune response (18), we asked whether the T cell responses in vivo and in vitro to mycobacterial antigens are altered in vaccinated MyD88–/– mice. First, we tested delayed type hypersensitivity (DTH) reaction in mice vaccinated with M. bovis BCG. DTH-induced swelling was absent in MyD88–/– mice, as in naive mice, although a clear response was measured in vaccinated wild-type mice (Figure 5A). The absence of DTH-induced swelling in MyD88–/– mice might be due to the impaired production of proinflammatory cytokines rather than to a reduced cell-mediated immune response. Therefore, we tested the antigen-specific response in vitro by restimulation of splenic T cells from wild-type and MyD88–/– animals vaccinated with M. bovis BCG at 4 weeks. Antigen-specific IFN-γ secretion was induced in both MyD88–/– and wild-type splenocytes restimulated with soluble BCG antigens with no significant statistical difference but was not induced in splenocytes restimulated with unrelated Listeria antigens (Figure 5B). Similar results were obtained with splenic CD3+ purified T cells upon stimulation with soluble BCG antigens, in the presence of either wild-type or MyD88-deficient macrophages (data not shown). In order to further demonstrate the T cell origin of the IFN-γ response, the intracellular expression of IFN-γ in pulmonary T cells was analyzed. Upon BCG infection, both MyD88–/– and wild-type mice exhibited IFN-γ–producing CD4 T cells and, to a lesser extent, CD8 T cells (Figure 5C and Table 1), although the percentages of IFN-γ–producing T cells seemed slightly lower in MyD88-deficient mice. Furthermore, after BCG vaccination, the percentage of CD4+ spleen T cells expressing IFN-γ intracellularly upon restimulation with BCG soluble antigens was similar in MyD88–/– and in wild-type mice (4.0% and 3.6%, respectively) and higher than in unvaccinated MyD88–/– and control mice (2.2% and 2.9%, respectively). The response was specific, because lower response was obtained after restimulation with heat-killed Listeria monocytogenes (HKLM) (1.3–2.4%). Therefore, antigen-specific activation of T cells and induction of a Th1 response to mycobacterial antigens do occur in the absence of MyD88.
Figure 5.

Reduced DTH response, but normal IFN-γ secretion, to mycobacterial antigens stimulation in MyD88–/– mice. (A) Cutaneous DTH response was performed 3 weeks after vaccination with M. bovis BCG (105 CFU s.c.; v) by assessing the footpad swelling in response to PPD injection as described. Paw swelling is defined as the difference in thickness between the left paw injected with PPD and the right paw injected with saline. DTH responses of nonvaccinated MyD88–/– and MyD88+/+ mice are shown as control (nv). Data are expressed as mean ± SD (n = 7–8) and are from 1 representative experiment out of 2. **P – 0.01. (B) For the T cell response, spleen cells from M. bovis BCG–vaccinated MyD88-deficient and control mice were harvested 4 weeks after inoculation (105 CFU s.c.) and were restimulated in vitro in the presence of soluble BCG antigens (SupBCG, 10 μg/ml) or unrelated antigen (HKLM, 100 bacteria per cell). Naive MyD88–/– mice and wild-type mice were used as negative control. IFN-γ production was quantified in the supernatants after 48 hours of incubation. Results are mean ± SD from n = 2 mice per genotype and are representative of 3 independent experiments. (C) Intracellular IFN-γ staining of CD4+ or CD8+ splenocytes from BCG-infected MyD88-deficient and control mice 4 weeks after infection, restimulated for 18 hours in the presence of soluble BCG antigens (SupBCG, 10 μg/ml) or unrelated antigen (HKLM, 100 bacteria per cell). Typical dot blots are shown for CD4+ T cells (numbers indicate percentage of IFN-γ–positive CD4+ cells).
Table 1.
Intracellular IFN-γ staining of CD4+ or CD8+ splenocytes from BCG-infected MyD88-deficient mice upon restimulation

BCG immunization protects MyD88–/– mice from acute MTB infection.
To confirm further that MyD88-dependent signaling is not required for antigen-specific T cell response, we tested whether BCG immunization would confer protection to a challenge with aerogenic MTB (200 CFU). We found that BCG immunization 5 weeks prior to the MTB challenge conferred protection against acute infection. At 4 weeks after MTB challenge, MyD88–/– mice behaved like wild-type controls, while nonimmunized MyD88–/– mice rapidly lost weight (Figure 6A). Vaccinated MyD88–/– mice started to succumb between 6 and 10 weeks after MTB challenge (Figure 6B). Lung weights at 4 weeks after MTB challenge were not increased in vaccinated MyD88–/– mice (Figure 6C), and mycobacterial burden in the lungs was comparable to that of wild-type controls (Figure 6D). We verified that BCG infection did not induce mortality in MyD88-deficient mice, which survived the 8-month observation period, although they developed chronic pneumonitis (2 × 106 CFU i.v.) (24).
Figure 6.

BCG vaccination confers an initial protection to MTB-challenged MyD88–/– mice. MyD88–/– mice and control mice were either immunized by s.c. injection of M. bovis BCG (105 CFU) or left naive and 5 weeks later challenged by MTB H37Rv aerogenic infection (200 CFU per mouse i.n.). The mice were monitored for body weight (A) or survival (B) or were sacrificed at 4 weeks after MTB challenge and analyzed for lung weight (C) and bacterial counts (D) as in Figure 2 (A: n = 7–8 mice per group from 1 representative experiment out of 2; B: n = 11–13 mice per group from 2 experiments; C and D: n = 4 per group from 1 representative experiment out of 3; *P – 0.05; **P – 0.01).
Macroscopically, the lungs of BCG-vaccinated MyD88-deficient mice appeared normal at 4 weeks after MTB challenge, with no large nodules on the pleura, unlike nonvaccinated MyD88-deficient mice (Figure 7, A and B). The lung micrographs of vaccinated MyD88-deficient mice revealed a robust mononuclear cell infiltration with abundant focal and perivascular lymphocytes, activated macrophages, and no signs of necrosis, suggesting that infection was controlled (Figure 7, C and E). Vaccinated wild-type mice displayed similar morphologic signs of an active immune response with lymphocyte recruitment (Figure 7, D and F) as the vaccinated MyD88-deficient mice. The flow cytometric analysis revealed some increase in the relative recruitment of T cells after BCG immunization in MyD88–/– and wild-type mice; however, this was not apparent in absolute numbers of lung-infiltrating lymphocytes (Figure 8, A and B) in vaccinated MyD88–/– and wild-type mice as compared with nonvaccinated mice (Figure 4). Furthermore, the relative recruitment and activation of myeloid cells was comparable in both vaccinated groups, although absolute numbers of myeloid cells were lower for vaccinated MyD88-deficient mice (Figure 8, B and C), likely due to the lower number of infiltrating cells obtained from the lungs of infected MyD88-deficient mice at 4 weeks after MTB challenge. Few mycobacteria were detected in the lungs of vaccinated MyD88-deficient mice at this stage (data not shown). At the time of death, however, vaccinated MyD88–/– mice presented large, confluent pulmonary nodules; reduced airspace with confluent, necrotic lesions; and abundant mycobacteria (data not shown), indicating that they succumbed to overwhelming infection. These data suggest that T cell immunity can confer an initial protective effect in the absence of the MyD88 pathway, but this is not sufficient to compensate the profound defect of the innate immune response in MyD88–/– mice.
Figure 7.
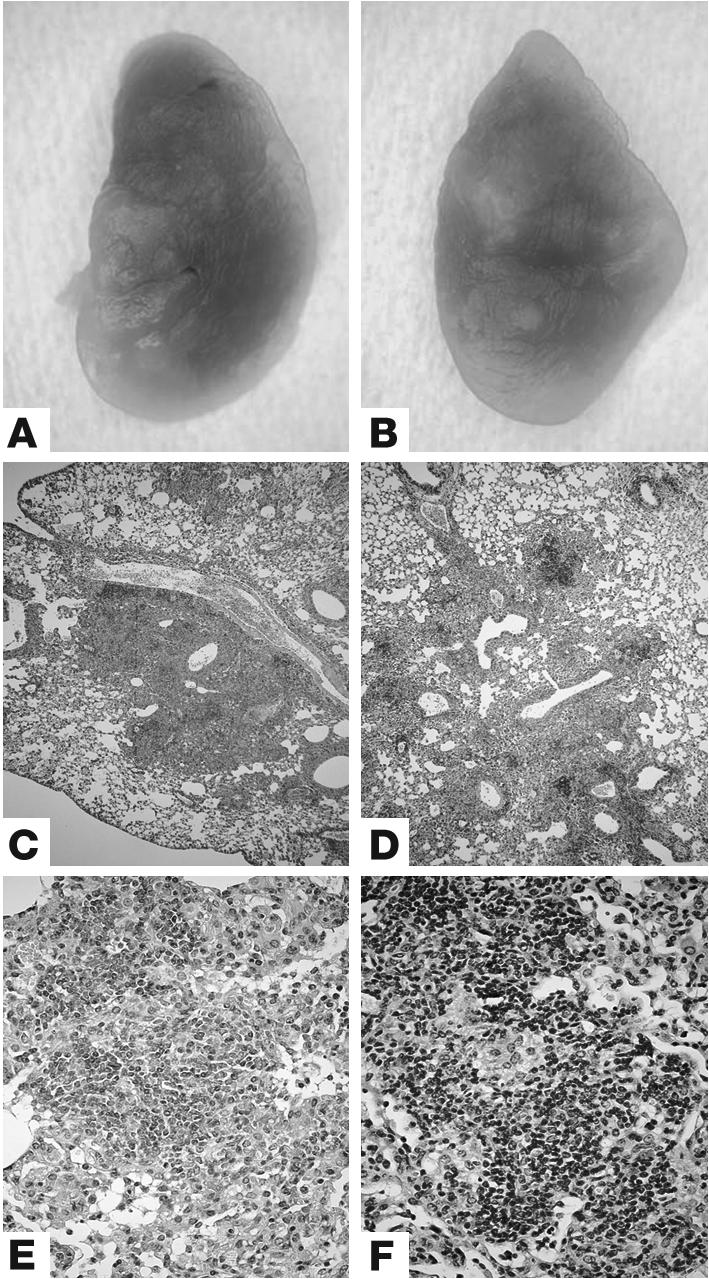
BCG vaccination prevents necrotic pneumonia by aerogenic MTB in MyD88–/– mice. Lung tissue from BCG-vaccinated MyD88–/– (A, C, and E) and wild-type (B, D, and F) mice was analyzed at 4 weeks after MTB H37Rv infection (200 CFU i.n.). Lungs of MyD88–/– mice display small nodules (A), similar to vaccinated wild-type mice (B). Microscopic examination reveals a striking increase of mononuclear cells including abundant lymphocytes in the lungs of vaccinated and infected MyD88–/– and wild-type mice (C–F; H&E). Low-power micrographs of representative lung sections are shown in C and D (magnification, ×50), and higher magnification shows details in E and F (magnification, ×400).
Figure 8.

Cell recruitment in the lungs of vaccinated and MTB-challenged MyD88–/– mice. MyD88–/– and control mice were immunized by s.c. injection of M. bovis BCG (105 CFU) and 5 weeks later challenged by MTB aerogenic infection (200 CFU) as in Figure 6. Leukocytes from infected lung were isolated 4 weeks after infection and analyzed by flow cytometry for CD44 expression in CD4- and CD8-positive T cells (A and B); for CD11b-, CD11c-, and Ly-6G–positive cells (B); and for MHC class II IA–IE expression (C) as in Figure 4. Results are expressed as absolute numbers of positive cells. Mean ± SD from 2 MyD88–/– mice are shown and are representative of 2 independent experiments.
Reduced cytokine and chemokine pulmonary levels in MTB-infected MyD88–/– mice upon vaccination.
MyD88-deficient cells show a defect in TNF and IL-12 p40 production in response to mycobacteria in vitro (Figure 1). Further, we showed impaired early in vivo TNF production in the airways of MyD88-deficient mice 6 hours after local exposure to killed mycobacteria (data not shown). However, massive leukocyte infiltration in the lungs of MTB-infected MyD88-deficient mice at 4 weeks after MTB challenge suggested an increase in the production of cytokines and/or chemokines. In order to assess the local cytokine and chemokine levels, we homogenized infected lungs at 25 days after MTB inoculation and assayed the supernatants. High levels of IL-1β, IFN-γ, and TNF-α were detected in the lungs of nonvaccinated MyD88–/– mice, whereas cytokine levels were lower upon vaccination in MyD88-deficient mice and in wild-type mice (Figure 9, A–C). Low levels of IL-10, IL-12, and TGF-β were detected in all 4 groups of mice (data not shown).
Figure 9.

The high pulmonary levels of cytokines and chemokines in MTB-infected MyD88–/– mice is reduced upon vaccination. Cytokine and chemokine concentrations were determined in lung homogenates from MyD88–/– and control mice immunized by s.c. injection of M. bovis BCG (105 CFU) and challenged 5 weeks later by MTB aerogenic infection (200 CFU) as in Figure 6. IL-1β (A), IFN-γ (B), TNF (C), MIP-1α (D), MCP-1 (E), and RANTES (F) were quantified by SearchLight protein array. Results are expressed as mean ± SD from 4 mice per group (*P – 0.05; **P – 0.01).
Levels of the chemokines MIP-1α and MCP-1 were significantly elevated in the lungs of nonvaccinated MyD88–/– mice as compared with vaccinated MyD88–/– mice and wild-type controls (Figure 9, D and E). RANTES levels were high in all 4 groups, but most elevated in lungs of nonvaccinated MyD88–/– mice (Figure 9F). Therefore, nonvaccinated MyD88-deficient mice showed highly increased levels of IL-1β, IFN-γ, TNF-α, MIP-1α, and MCP-1 as compared with control mice. Cytokine and chemokine levels were lower upon vaccination, concomitant with the reduced bacillary burden in these animals.
Discussion
We show here that the MyD88-mediated signaling pathway is critically involved in the development of innate, but not adaptive, immunity in response to MTB infection. Activation of APCs by mycobacteria with the production of cytokines and nitrite is MyD88 dependent, while the expression of costimulatory molecules on APCs is MyD88 independent. Most significantly, the T cell response to mycobacterial antigens, with IFN-γ production, is conserved, suggesting a MyD88-independent induction of adaptive immunity to live mycobacteria. This may be a peculiarity of responses to intracellular pathogens, as the induction of an antigen-specific Th1 response to Listeria monocytogenes (LM) has been reported to be MyD88 independent (25). These results contrast with a preferential Th2 response induced by parasites (17, 26, 27) and soluble antigens such as ovalbumin in the absence of MyD88 (18). In this last study, maturation of DCs by mycobacteria was reported to require MyD88 (18), whereas we show here a strong expression of costimulatory molecules in response to live M. bovis BCG in MyD88-deficient DCs. A possible explanation for the difference between the 2 studies may be the use of heat-killed H37Ra, as we show that heat-killed mycobacteria have strong TLR2 agonist signaling through MyD88, whereas live BCG is able to stimulate some cytokine and costimulatory expression independent of TLR2 (28, 29).
A further strong argument for the emergence of adaptive immunity in the absence of MyD88 is the fact that BCG vaccination conferred an initial protection to acute MTB infection. Vaccinated MyD88–/– mice displayed a rapid recruitment and activation of lymphocytes to the lung with little inflammation and controlled pulmonary infection, like wild-type mice, while nonvaccinated MyD88–/– mice developed fatal necrotic pneumonia within 4 weeks of MTB challenge.
Clearly, although MyD88-deficient mice presented a defect of immediate proinflammatory cytokine release in the airways after local administration of mycobacteria (our unpublished data), nonvaccinated MyD88-deficient mice showed highly increased levels of IL-1β, IFN-γ, and TNF as compared with nonvaccinated control mice 4 weeks after MTB infection. The amount of cytokines expressed in vivo seemed to correlate with the bacillary burden, which was highest in nonvaccinated MyD88-deficient mice at this time point. Vaccination was associated with lower cytokine secretion in MyD88-deficient mice, along with lower bacterial load in the lungs.
Therefore, the MyD88-deficient host is able to compensate for the absence of the MyD88 signaling pathway in response to high mycobacterial burden by triggering alternative pathways leading to increased cytokine secretion. Macrophages and DCs from MyD88-deficient mice show minute cytokine or nitrite production upon stimulation with mycobacterial antigens in vitro, but their response is not null. In fact, similarly low levels of TNF, IL-12 p40, or IL-6 are obtained after stimulation of MyD88-deficient APCs by mycobacteria or by LPS. LPS is a recognized TLR4 agonist using both MyD88-dependent and MyD88-independent pathways, the latter involving the adaptor TRIF (30). TRIF has been implicated in an IFN-β autocrine/paracrine loop leading to costimulatory molecule expression independent of MyD88. We show that live mycobacteria are also able to induce expression of costimulatory molecules such as CD40 or CD86 in MyD88-deficient macrophages and DCs, similar to LPS. In addition, since mycobacteria interact with other receptors including scavenger, mannose, and complement receptors as well as DC-specific ICAM-3 grabbing nonintegrin (DC-SIGN), TLR-independent signaling pathways might provide proinflammatory signals. Identifying which of these pathways contribute to the MyD88-independent cytokine response to high mycobacterial infection, or initiate the adaptive response to mycobacteria, will require the generation of double-deficient mice for these receptors.
To address the mechanism of the vigorous inflammatory response in the lung of MTB-infected MyD88-deficient mice, we measured chemokine levels in lung homogenates of both nonvaccinated and vaccinated mice. Levels of RANTES, MCP-1, and MIP-1α were strongly increased in the lungs of nonvaccinated MyD88-deficient mice, correlating with the high bacillary burden in these mice 4 weeks after MTB infection. The increased levels of chemokines are likely to account for the increased influx of neutrophils and macrophages into the lungs of MyD88-deficient mice. Therefore, these data may suggest a more severe pneumonitis and vigorous inflammatory response in conjunction with higher bacterial burden, and increased secretion of cytokines and chemokines, in MyD88-deficient mice.
The present results were not predicted by the recent report showing unimpaired killing of MTB in vitro by IFN-γ–stimulated MyD88–/– macrophages (31). However, the same study demonstrated that the IFN-γ inducibility of several genes was dramatically reduced in MyD88–/– macrophages, favoring the hypothesis that the innate immune system is hyporesponsive or nonresponsive (31). Further, we show reduced TNF and IL-6 production by IFN-γ–stimulated MyD88–/– lung macrophages (Figure 1, G and H), which may contribute to the increased susceptibility to MTB infection, as IL-6–deficient mice are highly susceptible to high-dose systemic MTB infection (32), although they could contain bacterial growth after low-dose aerosol infection (33).
In order to link innate and antigen-specific T cell responses, the present results suggest that MTB antigens recognized by TLRs or other PRRs might signal through MyD88-independent pathways, allowing antigen-specific immunity to develop in the absence of MyD88. Since the T cell response emerges late during MTB infection, the MyD88–/– host with a profound defect of innate immunity is confronted with a load of virulent bacteria that is too high and therefore uncontrollable.
We hypothesize that there may be an immediate component of the innate immune response in the absence of MyD88, sufficient to initiate an adaptive immune response, which is able to control the growth of nonvirulent bacteria. In a second phase of established cellular adaptive response, the long-term containment of infection requires a competent innate system. Along this line, systemic infection of MyD88-deficient mice with the nonvirulent, attenuated M. bovis BCG leads to a long-term control of the infection (24), whereas TNF-deficient mice succumb within 5 weeks under the same conditions (23). Thus, a nonvirulent infection may be controlled, even if the initial innate response is deficient. In the case of the BCG vaccination, the enhanced T cell response with release of IFN-γ activates MyD88-deficient APCs, which have a reduced but residual function, allowing the initial control of a virulent MTB infection (Figure 6). However, the defect of APCs is only temporarily compensated for, and the vaccinated mice start to succumb to infection after 2 months. The present data demonstrate that absence of MyD88 signaling precludes long-term control of chronic MTB infection, as shown previously in TLR2- or TLR4-deficient mice (10, 14).
The possibility that absence of IL-18 and/or IL-1 signaling were also responsible for the defect observed in MyD88–/– mice cannot be excluded, although previous reports on MTB infection of IL-18–/– (34), IL-1β–/–, or IL-R1–/– mice showed less-striking defects in host resistance (35, 36). Under our experimental conditions, caspase-1–deficient mice (37) did not show an enhanced susceptibility to MTB infection, as they controlled acute infection and survived for more than 6 months without loss of body weight (data not shown).
Recently, MyD88-deficient mice were shown to be sensitive to an intravenous Mycobacterium avium infection, an opportunistic pathogen found in immunosuppressed individuals. MyD88-deficient mice succumbed within 9–14 weeks to systemic M. avium infection with reduced early neutrophil recruitment in the liver and a profound defect in IFN-γ response (21). There are substantial differences in the host response to MTB versus M. avium infection, not the least being that the clearance of M. avium is enhanced in the absence of iNOS (38), while the same enzyme is critical to control MTB infection (39). A normal control and granuloma formation in response to MTB infection in the absence of MyD88 has been reported (40). The discrepancy with the results presented here may be related to differences in the virulence of the strain used, the Kiruno strain of MTB, or in the protocol and follow-up of infection. By contrast, increased susceptibility of MyD88-deficient mice to MTB infection has been shown in a low-dose aerosol infection (41). Here, we demonstrate T cell priming to mycobacterial antigens in the absence of MyD88, in contrast to results from M. avium (21) and MTB infection (41). However, T cell priming in the absence of MyD88 has been shown for another intracellular pathogen, LM. In fact, only a modest impairment of CD4 response and a normal CD8 response to LM was reported in immunized MyD88–/– mice, and transfer of primed CD8 T cells from MyD88–/– mice conferred protection to LM infection (25). Therefore, the data from our studies and those involving LM infection suggest that adaptive immunity can be generated and provide total or partial protective immunity in the absence of MyD88.
In summary, our data show that MyD88 signaling is essential for efficient mycobacteria-induced cytokine and NO production, while the expression of costimulatory molecules on APCs is not affected. This initial defect of innate response is associated with rapid death upon MTB infection, as the MyD88-deficient mice cannot control the infection although they exhibit an antigen-specific T cell response. Second, the antigen-specific cellular immunity is MyD88 independent, explaining the partial protection to MTB infection by BCG vaccination. Indeed, BCG vaccination primed a T cell response and allowed the initial control of virulent MTB infection. Third, as a result of the profound defect of the innate immune response, MyD88–/– mice are unable to durably control the infection despite the adaptive response. Therefore, the development of innate immunity to intracellular pathogen MTB is MyD88-dependent, while adaptive immunity develops in the absence of MyD88.
Methods
Mice.
MyD88–/– (15) and TNF–/– mice (42) backcrossed on the C57BL/6 background (N10 and N6, respectively) were bred in our specific pathogen-free animal facility at CNRS. For experiments, adult (8- to 15-week-old) animals were kept in sterile isolators in a biohazard animal unit. The infected mice were monitored regularly for clinical status and weighed weekly. Permission was obtained from the Regional Ethics Committee for Animal Experiments of Toulouse, France, for analysis of murine models of mycobacterial infection.
Bacteria and infection.
MTB H37Rv (Pasteur) and M. bovis BCG (Pasteur strain 1173P2) were grown to mid-log phase in Middlebrook 7H9 liquid medium (Difco Laboratories), supplemented with 10% oleic acid/albumin/dextrose/catalase (OADC; Difco Laboratories) at 37°C. Aliquots were prepared and frozen at –80°C. Before use, an aliquot was thawed, briefly vortexed, and diluted in sterile saline containing 0.04% Tween 20, and clumping was disrupted by 20 repeated aspirations through a 29-gauge needle (Omnican). Pulmonary infection with MTB H37Rv was performed by delivering approximately 200 bacteria into both nasal cavities (20 μl each) under xylazine-ketamine anesthesia, and the inoculum size was verified by sacrificing mice 48 hours after infection and determining bacterial load in the lungs of infected mice. Furthermore, we compared the intranasal (i.n.) route of infection with that induced by the aerosol generator using the Middlebrook chamber (Glas-Col) as described previously (10) and confirmed effective and comparable lung infection (data not shown).
Vaccination with M. bovis BCG.
MyD88–/– and C57BL/6 mice were injected s.c. with 105 CFU M. bovis BCG as described previously (43). Five weeks later, mice were infected i.n. with MTB H37Rv (200 CFU). Half of the mice were sacrificed 4 weeks later for histology and CFU determination, and the other half were used for a survival study. The experiments were performed twice.
Bacterial load in tissues.
Bacterial loads in the lung, liver, and spleen of infected mice were evaluated at different time points after infection with MTB H37Rv as described (44). Organs were weighed and defined aliquots were homogenized in 0.04% Tween 20 PBS. Tenfold serial dilutions of organ homogenates were plated in duplicates onto Middlebrook 7H10 agar plates containing 10% OADC and incubated at 37°C. Colonies were enumerated at 3 weeks and results are expressed as log10 CFU per organ.
Histopathological analysis.
For histological analysis, lungs were removed at different time points of infection from both wild-type and MyD88–/– mice (12 and 14 mice, respectively), fixed in 4% phosphate-buffered formalin and embedded in paraffin. Two- to 3-micron sections were stained with H&E and a modified Ziehl-Neelsen stain. The latter involved staining in a prewarmed (60°C) carbol-fuchsin solution for 10 minutes followed by destaining in 20% sulphuric acid and 90% ethanol before counterstaining with methylene blue.
Fluorescence-activated cell-sorting analysis of infiltrating cells from infected lung.
FACS analysis of inflammatory cells from infected lung was performed as described (45, 46). In brief, mice were deeply anesthetized with xylazine-ketamine and perfused with 0.02% EDTA-PBS until the tissue turned white. After removal, lung tissue was sliced into 1- to 2-mm3 pieces and was incubated in RPMI 1640 (Invitrogen Corp.) containing 5% FCS, antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin), 10 mM HEPES (Invitrogen Corp.), collagenase (150 U/ml), and DNase (50 U/ml; Sigma-Aldrich). After 1.5 hours of incubation at 37°C, single-cell suspension was obtained by vigorous pipetting. Cells were washed 3 times in PBS containing 0.01% NaN3 and 0.5% BSA and were then stained according to antibody manufacturer protocols. Rat anti-mouse CD4–peridinin-chlorophyll-protein complex (PerCP; clone RM4-5), CD8-FITC (clone 53-6.7), CD44-PE (clone IM7), Ly6G-PE (clone RD6-8C5), CD11b-PE (clone M1/70), I-A/I-E-FITC (clone 2G9), CD19-PerCP-Cy5-5 (clone 1D3), and hamster anti-mouse CD11c-APC (clone HL3) were purchased from BD Biosciences — Pharmingen. Stained cells were washed twice, fixed with 1% paraformaldehyde (FACS Lysing Solution; BD), and analyzed by flow cytometry on a LSR analyzer (BD). A large gate including myeloid and lymphoid cells and excluding only very low forward scatter (FSC) cells was used. Data were processed with CellQuest software (BD Biosciences — Immunocytometry Systems).
Interstitial lung macrophage isolation.
Lung cells obtained as described above (46) were washed twice in HBSS with 2% of FCS and antibiotics and resuspended in RPMI supplemented with 10% FCS, 2 mM L-glutamin, antibiotics, 1% nonessential amino acids, 1 mM pyruvate, and 10 mM HEPES. The cell suspension (20 × 106 cells in 10 ml) was incubated for 1.5 hours on 90-mm–diameter Petri dishes (Costar-Corning) at 37°C. Nonadherent cells were removed by triple vigorous washing with warm HBSS containing 2% of FCS. Adherent cells were detached from the plastic by incubating the monolayers in 0.02% EDTA-HBSS for 30 minutes at room temperature. Cell suspensions were obtained by pipetting, washed twice with HBSS/FCS, and resuspended in culture medium.
Primary macrophage and DC cultures.
Murine BM cells were isolated from femurs and differentiated into macrophages after culturing at 106 cells/ml for 7 days in DMEM (Sigma-Aldrich) supplemented with 20% horse serum and 30% L929 cell-conditioned medium as a source of M-CSF (47). Three days after washing and reculturing in fresh medium, the cell preparation contained a homogenous population of macrophages. Alternatively, murine BM cells were differentiated into myeloid DCs after culturing (change on days 3, 6, and 8) at 2 × 105 cells/ml for 10 days in RPMI supplemented with 10% FCS and 4% J558L cell-conditioned medium as a source of GM-CSF as described previously (48).
Stimulation of macrophages and DCs.
BM-derived macrophages, pulmonary macrophages, and DCs were plated in 96-well microculture plates (at 105 cells per well) and stimulated with LPS (Escherichia coli, serotype O111:B4 [Sigma-Aldrich] at 100 ng/ml) or MTB H37Rv (heat-killed for 40 minutes at 80°C; 2 bacteria per cell) or infected with M. bovis BCG or MTB H37Ra (both from Pasteur Institute; at a MOI of 2 bacteria per cell). Cell supernatants were harvested after 24 hours of stimulation in the presence of IFN-γ (100 U/ml) for TNF, IL-12 p40, and IL-6 quantification using commercial ELISA (Duoset; R&D Systems) and nitrite measurements by Griess reagents as described previously (49).
Antigen-specific IFN-γproduction.
T cell priming was assessed by the production of IFN-γ upon antigen restimulation ex vivo. Single-cell suspensions of splenocytes were prepared from wild-type and MyD88–/– mice 4 weeks after s.c. immunization with 105 live M. bovis BCG. Splenocytes were cultured at 5 × 105 cells/ml in RPMI 1640 with glutamine, 5% FCS, and antibiotics and stimulated with 2.5 μg/ml concanavalin A (Con A; Sigma-Aldrich), a lyophilized soluble fraction from BCG culture (10 μg/ml), or HKLM (100 bacteria per cell) for 2 days at 37°C. IFN-γ production in the supernatant was quantified by ELISA (Duoset; R&D Systems). Alternatively, splenocytes from BCG-infected mice restimulated as above were analyzed for intracellular IFN-γ staining of CD4+ or CD8+ T cells: after 12 hours of restimulation, addition of GolgiStop to block protein transport, and incubation for an additional 6 hours, cells were labeled with anti-CD4–PerCP (clone RM4-5) or CD8-APC (clone 53-6.7) antibodies, fixed, and permeabilized, and intracellular IFN-γ was stained with antibodies to IFN-γ–FITC (clone XMG1.2; according to BD Cytofix/Cytoperm kit manual; all reagents from BD Biosciences).
Cutaneous DTH reaction.
DTH reaction was performed as described previously (44). MyD88-deficient and control mice were vaccinated with 105 live M. bovis BCG (s.c.). Three weeks later, mice were injected with 20 IU tuberculin/purified protein derivative (PPD; Bovituber; Merial) in 50 μl in the left footpad. Swelling was measured 48 hours later with a micrometer (Mitotuyo) and compared with that in the saline-injected right footpad.
Preparation and analyses of lung homogenates for cytokine/chemokine determination.
At day 25 after infection, mice were deeply anesthetized with xylazine-ketamine and perfused with 0.02% EDTA-PBS until the tissue turned white. Whole lungs were harvested, weighed, and placed in 0.5 ml of 4°C PBS solution. The contents were transferred into sterile 50-ml plastic tubes and homogenized in Dispomix homogenizer (Medic Tools) for 20 seconds at 6,000 rpm. Homogenates were centrifuged at 20,200 g, sterilized by filtration through a 0.22-μm filter (Costar-Corning), and stored at –80°C until determination of IL-1β, IL-10, IL-12 p70, IFN-γ, TNF-α, MIP-1α, JE (MCP-1), RANTES, and TGF-β levels by SearchLight protein array technology (Perbio).
Statistical analysis.
Analysis was performed using Student’s t and ANOVA tests and P values less than or equal to 0.05 were considered significant.
Acknowledgments
The authors gratefully acknowledge grant support by Le Studium, Orléans, France (to V. Yeremeev); the Fondation de la Recherche Médicale, France; French and South African Research cooperation grant; the Wellcome Trust, Medical Research Council; and National Research Foundation, Cape Town, South Africa.
Footnotes
See the related Commentary beginning on page 1699.
Cecile M. Fremond and Vladimir Yeremeev contributed equally to this work.
Vladimir Yeremeev is on sabbatical leave from the Central Institute for Tuberculosis, Moscow, Russia.
Nonstandard abbreviations used: BCG, bacillus Calmette-Guérin; DC-SIGN, DC-specific ICAM-3 grabbing nonintegrin; DTH, delayed type hypersensitivity; FSC, forward scatter; HKLM, heat-killed Listeria monocytogenes; i.n., intranasal(ly); LM, Listeria monocytogenes; MTB, Mycobacterium tuberculosis; MyD88, myeloid differentiation factor 88; OADC, oleic acid/albumin/dextrose/catalase; PerCP, peridinin-chlorophyll-protein complex; PPD, purified protein derivative; PRR, pattern-recognition receptor; TLR, Toll-like receptor.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Dye C, Williams BG, Espinal MA, Raviglione MC. Erasing the world’s slow stain: strategies to beat multidrug-resistant tuberculosis. Science. 2002;295:2042–2046. doi: 10.1126/science.1063814. [DOI] [PubMed] [Google Scholar]
- 2.Flynn JL, Chan J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 3.Akira S. Mammalian Toll-like receptors. Curr. Opin. Immunol. 2003;15:5–11. doi: 10.1016/s0952-7915(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 4.Aliprantis AO, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 5.Thoma-Uszynski S, et al. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 6.Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. U. S. A. 1999;96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilleron M, Quesniaux VF, Puzo G. Acylation state of the phosphatidylinositol hexamannosides from mycobacterium bovis BCG and mycobacterium tuberculosis H37Rv and its implication in TLR response. J. Biol. Chem. 2003;278:29880–29889. doi: 10.1074/jbc.M303446200. [DOI] [PubMed] [Google Scholar]
- 8.Guerardel Y, et al. Lipomannan and lipoarabinomannan from a clinical isolate of Mycobacterium kansasii: novel structural features and apoptosis-inducing properties. J. Biol. Chem. 2003;278:36637–36651. doi: 10.1074/jbc.M305427200. [DOI] [PubMed] [Google Scholar]
- 9.Quesniaux VJ, et al. Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J. Immunol. 2004;172:4425–4434. doi: 10.4049/jimmunol.172.7.4425. [DOI] [PubMed] [Google Scholar]
- 10.Abel B, et al. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J. Immunol. 2002;169:3155–3162. doi: 10.4049/jimmunol.169.6.3155. [DOI] [PubMed] [Google Scholar]
- 11.Reiling N, et al. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J. Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 12.Sugawara I, et al. Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol. Immunol. 2003;47:327–336. doi: 10.1111/j.1348-0421.2003.tb03404.x. [DOI] [PubMed] [Google Scholar]
- 13.Heldwein KA, et al. TLR2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J. Leukoc. Biol. 2003;74:277–286. doi: 10.1189/jlb.0103026. [DOI] [PubMed] [Google Scholar]
- 14.Drennan MB, et al. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am. J. Pathol. 2004;164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 17.Muraille E, et al. Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. J. Immunol. 2003;170:4237–4241. doi: 10.4049/jimmunol.170.8.4237. [DOI] [PubMed] [Google Scholar]
- 18.Schnare M, et al. Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 19.Mun HS, et al. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int. Immunol. 2003;15:1081–1087. doi: 10.1093/intimm/dxg108. [DOI] [PubMed] [Google Scholar]
- 20.Scanga CA, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 21.Feng CG, et al. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J. Immunol. 2003;171:4758–4764. doi: 10.4049/jimmunol.171.9.4758. [DOI] [PubMed] [Google Scholar]
- 22.Flynn JL, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs M, et al. Correction of defective host response to Mycobacterium bovis BCG infection in TNF-deficient mice by bone marrow transplantation. Lab. Invest. 2000;80:901–914. doi: 10.1038/labinvest.3780094. [DOI] [PubMed] [Google Scholar]
- 24.Nicolle D, et al. Chronic pneumonia despite adaptive immune response to Mycobacterium bovis BCG in MyD88-deficient mice. Lab. Invest. 2004;84:1305–1321. doi: 10.1038/labinvest.3700149. [DOI] [PubMed] [Google Scholar]
- 25.Way SS, Kollmann TR, Hajjar AM, Wilson CB. Protective cell-mediated immunity to Listeria monocytogenes in the absence of myeloid differentiation factor 88. J. Immunol. 2003;171:533–537. doi: 10.4049/jimmunol.171.2.533. [DOI] [PubMed] [Google Scholar]
- 26.Netea MG, et al. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J. Infect. Dis. 2002;185:1483–1489. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- 27.Chen M, et al. Involvement of MyD88 in host defense and the down-regulation of anti-heat shock protein 70 autoantibody formation by MyD88 in Toxoplasma gondii-infected mice. J. Parasitol. 2002;88:1017–1019. doi: 10.1645/0022-3395(2002)088[1017:IOMIHD]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 28.Fremond CM, Nicolle DM, Torres DS, Quesniaux VF. Control of Mycobacterium bovis BCG infection with increased inflammation in TLR4-deficient mice. Microbes Infect. 2003;5:1070–1081. doi: 10.1016/j.micinf.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Nicolle, D., et al. 2005. Long-term control of Mycobacterium bovis BCG infection in the absence of Toll-like receptors: investigation on TLR2, TLR6 or TLR2-TLR4 deficient mice. Infect. Immun. In press. [DOI] [PMC free article] [PubMed]
- 30.Hoebe K, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 31.Shi S, et al. MyD88 primes macrophages for full-scale activation by interferon-{gamma} yet mediates few responses to Mycobacterium tuberculosis. J. Exp. Med. 2003;198:987–997. doi: 10.1084/jem.20030603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ladel CH, et al. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 1997;65:4843–4849. doi: 10.1128/iai.65.11.4843-4849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saunders BM, Frank AA, Orme IM, Cooper AM. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect. Immun. 2000;68:3322–3326. doi: 10.1128/iai.68.6.3322-3326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugawara I, et al. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 1999;67:2585–2589. doi: 10.1128/iai.67.5.2585-2589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juffermans NP, et al. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J. Infect. Dis. 2000;182:902–908. doi: 10.1086/315771. [DOI] [PubMed] [Google Scholar]
- 36.Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab. Invest. 2000;80:759–767. doi: 10.1038/labinvest.3780079. [DOI] [PubMed] [Google Scholar]
- 37.Glaccum MB, et al. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol. 1997;159:3364–3371. [PubMed] [Google Scholar]
- 38.Gomes MS, Florido M, Pais TF, Appelberg R. Improved clearance of Mycobacterium avium upon disruption of the inducible nitric oxide synthase gene. J. Immunol. 1999;162:6734–6739. [PubMed] [Google Scholar]
- 39.MacMicking JD, et al. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugawara I, Yamada H, Mizuno S, Takeda K, Akira S. Mycobacterial infection in MyD88-deficient mice. Microbiol. Immunol. 2003;47:841–847. doi: 10.1111/j.1348-0421.2003.tb03450.x. [DOI] [PubMed] [Google Scholar]
- 41.Scanga CA, et al. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect. Immun. 2004;72:2400–2404. doi: 10.1128/IAI.72.4.2400-2404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marino MW, et al. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeremeev VV, et al. Proteins of the Rpf family: immune cell reactivity and vaccination efficacy against tuberculosis in mice. Infect. Immun. 2003;71:4789–4794. doi: 10.1128/IAI.71.8.4789-4794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobs M, Brown N, Allie N, Ryffel B. Fatal Mycobacterium bovis BCG infection in TNF-LT-alpha-deficient mice. Clin. Immunol. 2000;94:192–199. doi: 10.1006/clim.2000.4835. [DOI] [PubMed] [Google Scholar]
- 45.Botha T, Ryffel B. Reactivation of latent tuberculosis infection in TNF-deficient mice. J. Immunol. 2003;171:3110–3118. doi: 10.4049/jimmunol.171.6.3110. [DOI] [PubMed] [Google Scholar]
- 46.Lyadova IV, et al. Comparative analysis of T lymphocytes recovered from the lungs of mice genetically susceptible, resistant, and hyperresistant to Mycobacterium tuberculosis-triggered disease. J. Immunol. 2000;165:5921–5931. doi: 10.4049/jimmunol.165.10.5921. [DOI] [PubMed] [Google Scholar]
- 47.Muller M, et al. Correction or transfer of immunodeficiency due to TNF-LT alpha deletion by bone marrow transplantation. Mol. Med. 1996;2:247–255. [PMC free article] [PubMed] [Google Scholar]
- 48.Lutz MB, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 49.Green LC, et al. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]