

Abstract
Treatments for allergic disease block the effects of mediators released from activated mast cells and blood basophils. A panel of fullerene derivatives was synthesized and tested for their ability to preempt the release of allergic mediators in vitro and in vivo. The fullerene C70‐tetraglycolic acid significantly inhibited degranulation and cytokine production from mast cells and basophils, while C70‐tetrainositol blocked only cytokine production in mast cells and degranulation and cytokine production in basophils. The early phase of FcɛRI inhibition was dependent on the blunted release of intracellular calcium stores, elevations in reactive oxygen species, and several signaling molecules. Gene microarray studies further showed the two fullerene derivatives inhibited late phase responses in very different ways. C70‐tetraglycolic acid was able to block mast cell‐driven anaphylaxis in vivo, while C70‐tetrainositol did not. No toxicity was observed with either compound. These findings demonstrate the biological effects of fullerenes critically depends on the moieties added to the carbon cage and suggest they act on different FcɛRI‐specific molecules in mast cells and basophils. These next generation fullerene derivatives represent a new class of compounds that interfere with FcɛRI signaling pathways to stabilize mast cells and basophils. Thus, fullerene‐based therapies may be a new approach for treating allergic diseases. Clin Trans Sci 2010; Volume 3: 158–169
Keywords: drug action, drug design, inflammation, mast cell, basophil, nanomedicine
Introduction
Mast cells (MC) are granule‐rich tissue cells that significantly contribute to a wide range of diseases through the release of noxious mediators. Peripheral blood basophils (PBB) are similar to MC in that they are the only leukocytes that contain prestored histamine within their granules. The high‐affinity IgE receptor, FcɛRI, is one of many ways in which MC/PBB can be activated for mediator release. Cross‐linking Immunoglobulin E (IgE)‐primed FcɛRI leads to the release of various preformed and newly generated mediators that can cause allergic disease. 1 Moreover, MC are suspected to play a role in other inflammatory disorders such as arthritis and cardiovascular disease through non‐FcɛRI mediated mechanisms. 2 , 3 ) Basophils, also established effector cells in allergic disease, have recently been implicated as playing a major role in adaptive and innate immunity. 4 , 5 Thus, finding new ways of preventing MC/PBB activation and subsequent mediator release is of clinical importance.
Nanotechnology encompasses a multidisciplinary scientific field that is rapidly increasing and has the potential to play an important role in many branches of science. 6 One type of nanomaterial is buckminsterfullerene (fullerene), which are carbon spheres. 7 Fullerenes have a unique cage structure with delocalized π molecular orbital electrons. This structure confers unusual activity in electron transfer systems due to their low reorganization energy, low‐lying excited states (singlet and triplet), and extended triplet lifetimes. Further, the spherical configuration of the planar benzene rings imposes an unusual constraint on these π electron orbitals. As the core carbon cage is insoluble in aqueous solutions, fullerene derivatives (FD) have been developed with a wide variety of side chain moieties that make them more compatible with biological systems.
These unique properties of FD have led to their potential as a new way for treating a wide range of diseases and pathologies. 8 These include multiple sclerosis, 9 neurodegenerative disease, 10 HIV infection, 11 cancer, 12 radiation exposure, 13 ischemia, 14 and general inflammation. 15 , 16 Moreover, mice chronically treated with FD have significantly extended lifespans compared to littermate controls. 17 While it is clear that these molecules possess traits that may potentially be beneficial for therapies, they have yet to be tested in humans given the lack of a clearly efficacious and characterized candidate for a specific disease. Safety is a concern as it is for any new class of compounds. 18 Studies investigating one class of structurally related FD cannot be extrapolated to represent all FD; each new molecular entity must be investigated separately. Proof‐of‐principle studies to identify a specific candidate need to be established before relevant toxicity and pharmacokinetic studies of that candidate can be performed. We showed previously that water‐soluble FD could blunt MC and PBB responses. 19 However, the FD used previously were mixtures of many different isomers limiting their therapeutic potential and made dissecting their mechanism of action difficult. The goal of this study was to develop and test next generation FD (highly purified and characterized) to establish proof‐of‐principle as candidates for diseases strongly influenced by MC and PBB activation through FcɛRI.
To achieve this goal, a panel of water‐soluble FD was synthesized and tested in different concentrations for their ability to inhibit FcɛRI‐mediated MC/PBB activation. It is shown for the first time that the biological function of FD on MC/PBB critically depends on the class of moieties added to the carbon cage; some constructs had no effect, others were effective at blocking degranulation and cytokine release, and another group blocked cytokine production only. Two representative FD, C70 tetragycolate (TGA) and C70 tetrainositol (Inos) were further examined for differences in their mechanisms of action. We examined early activation events (reactive oxygen species [ROS], Ca2+, and phosphorylation of signaling molecules) and later events (gene expression by microarray and quantification of those FcɛRI‐activated signaling molecules most affected by FD pretreatment). For select FD (TGA), it is further shown that this behavior translates to in vivo inhibition of MC‐dependent anaphylaxis. These results show that appropriate FD may be effective treatments for diseases that are influenced by MC activation. More importantly, we demonstrate for the first time that FD can be engineered at the nano‐scale level to control specific signal transduction pathways affecting cell function.
Methods
Fullerene derivatives
All FD were synthesized at Luna Innovations Incorporated. A representative synthesis protocol is given in Supplementary Figure S1. Each derivative was characterized using matrix assisted laser desorption ionization mass spectrometry, nuclear magnetic resonance, high‐performance liquid chromatography, and dynamic light scatter. All FD were tested for cell toxicity by incubation with increasing concentrations up to 100 μg/mL and viability counts taken on days 3, 6, and 9. No toxicity was observed with any of the FD compared to control cells (not shown).
MC/PBB FD culture and FcɛRI‐mediated activation
Human skin tissue was received from the Cooperative Human Tissue Network and MC purified and cultured as described. 20 The MC were cultured in media containing stem cell factor that is removed from the culture 24 hours prior to experimentation. PBB were obtained from two sources: donors recruited under an Internal Review Board (IRB)‐approved protocol after informed consent or from leukopheresis packs obtained from the Johns Hopkins Hemapheresis Center. PBB were purified to ≥99% purity as previously described. 21 , 22 , 23 Purified PBB were incubated overnight (20 hours) with FD (5 μg/mL) or vehicle control and a minimal (nonstimulatory) concentration of IL‐3 (2 pg/mL) to prevent apoptosis. 22 The next day, cells were washed and aliquoted for the histamine release assay by treatment with 0.1 mg/mL of goat polyclonal anti‐IgE, buffer alone (spontaneous release) or perchloric acid (total histamine determination). Histamine was quantified in cell free supernatants using automated fluorimetry in duplicate. In a second set of experiments, the two FD (at 5 μg/mL) were incubated with PBB for 20 hours, washed cells were stimulated with 15 ng/mL anti‐IgE for 18 hours in duplicate, and supernatants were collected for quantification of IL‐13 by in‐house enzyme‐linked immunosorbent assay (ELISA). The doses of anti‐IgE are optimal for activation of PBB based on previous studies.
For activation, MC were suspended in fresh medium (without cytokines) and incubated for 16 hours with or without FD at 37°C in a 6% CO2 incubator. The 16‐hour time point was chosen based on preliminary experiments demonstrating this was optimal for inhibition of mediator release (not shown) and uptake within FcɛRI cells. 24 The next morning, cells were washed and stimulated with anti‐FcɛRI antibodies (Abs) (3B4; 1 μg/mL) for 30 minutes (β‐hexosaminidase) or overnight (granulocyte macrophage colony‐stimulating factor [GMC‐SF]) at 37°C in a 6% CO2 incubator and mediator release measured as described previously. 25 All MC mediator release studies were performed in triplicate.
Western blotting and phospho‐signaling quantification
Cell lysate preparation and Western blotting were performed using a protocol optimized for extracting phosphoproteins from human MC. 26 Following activation, cells were lysed directly in boiling denaturing sample buffer consisting of tris‐buffered saline with triton‐X‐100 (0.5%) and protease and phosphatase inhibitors. The cell suspension was then passed through a 20‐gauge needle, boiled, and centrifuged to remove cell debris. Proteins were separated on 8% or 10% NuPage Tris‐Bis gels using Licor running buffer. Western blotting was performed using Licor blocking buffer and IR800 and IR700 antirabbit F(ab)2 secondary Abs (1:1,000). Primary Abs were from Cell Signaling Technologies (Danvers, MA, USA) or Santa Cruz Biotechnology (Santa Cruz, CA, USA) unless otherwise noted. Band intensities were captured using the Odyssey Imaging System (Li‐Cor Biosciences, Lincoln, NE, USA) and bands quantified by measuring the number of pixels in each band using a box drawn for the same area of measurement for each separate blot. The band intensity was then normalized for loading by dividing the number of pixels in each band with the housekeeping band intensity (β‐actin) performed on the same blot.
Calcium and ROS measurement
MC were pretreated with or without FD as above, washed with Tyrodes buffer supplemented with bovine serum albumin (BSA), and incubated with Fura‐2 AM (2 μm) for 30 minutes at 37°C. Cells were washed, stimulated with anti‐FcɛRI, and calcium flux measured in real time on a Perkin Elmer LS55 Spectrofluorometer. For ROS production, cells were exposed to FD as above. After washing, cells were resuspended in X‐Vivo medium containing 5 μm dichlorodihydrofluorescein (DCF) at 37°C for 30 minutes, washed, and activation‐induced changes in mean fluorescence were measured with excitation at 502 nm and emission at 523 nm for 15 minutes. 19 The data are presented as fluorescence intensity of the 523 nm emission over time. All experiments were performed in triplicate and degranulation was measured in parallel. Separate experiments were performed to ensure that the FD do not interfere with indicator dye binding (not shown).
Gene microarray studies and validation using Western blotting or flow cytometry
MC (1 × 107 cell/condition; each condition performed in triplicate) were incubated with or without FD as above and incubated with or without anti‐FcɛRI antibodies for 10 minutes, supernatants were removed (to remove preformed mediators), and fresh warm medium containing anti‐FcɛRI antibodies (1 μg/mL) added for 2 hours. Cells were centrifuged, the supernatant and the pellet immediately frozen, and microarray performed using the Human Whole Genome OneArrayTM gene expression profiling service (Phalanx Biotech Group, Palo Alto, CA, USA). Separate samples, assayed in parallel, were lysed and protein expression was analyzed by Western blotting or flow cytometry (CD45) as described. 27
For microarray, RNA was isolated using the Ambion MessageAmp aRNA kit (Applied Biosystems/Ambion, Austin, TX, USA); all samples passed the internal quality control checks. For hybridization, each sample was run in triplicate. Optical density was measured by NanoDrop ND‐1000. The ratio of absorbance at 260 nm and 280 nm provides an estimate of RNA purity. Samples were found to have ratios between 1.8 and 2.2 indicating highly pure samples. The reactive amino group of 5‐(3‐aminoallyl)‐UTP/5‐(3‐aminoallyl)‐dUTP was used to conjugate the purified aRNA/cDNA with the NHS‐CyDye. Labeling efficiency was calculated by the concentration of CyDye and aRNA/cDNA that was above 10. For hybridization, 10 μg Cy5‐labeled aRNA was utilized by the Phalanx Hybridization Protocol Array Version HOA 4.3.
Pearson correlation tables (R values) for each technical repeat were calculated from raw log2 intensity (R) and normalized log2 intensity (N) values and compared to each other. Only probes with p value (detected) less than 0.05 were included in the calculation. This analysis showed good correlation between platforms when filtered stringently for fold change and loosely for significance (p value). Greater than 95% of the samples had R values of 0.8 or greater indicating strong correlation between the two parameters.
In order to examine the genes most affected by FD preincubation, the resulting values were sorted and filtered the following way. First, the mean (±standard deviation [SD]) of the six values from each condition for each of the 30,970 genes was calculated. Second, only normalized value intensities of >100 were included so that follow‐up detection of protein levels would be more likely to be successful. Third, these data were further truncated to include only normalized data in which the average FcɛRI activation (without pretreatment) significantly (p < 0.05) increased at least 10% compared to non‐FcɛRI resting cells. Fourth, the genes with >10% upregulation by FcɛRI stimulation alone were compared to those samples pretreated with each FD and only those showing at least >20% inhibition or >20% upregulation (nontreated +anti‐FcɛRI compared to treated + anti‐FcɛRI) were examined. Data are presented as the average percentage downregulated with FD treatment derived from the following equation.
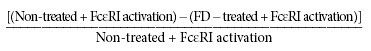 |
Downregulation observed at the gene level was verified at the protein level for several representative molecules using Western blotting or flow cytometry. A complete list of those genes downregulated or upregulated by each FD (>20%) is shown in Supplemental File S1.
Mouse models of anaphylaxis and assessment of toxicity
All studies were approved by the Virginia Commonwealth University IACUC. MC‐dependent anaphylaxis and treatments are described. 19 Briefly, female C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME, USA) aged 10–12 weeks were injected i.p. with 50 μg IgE‐dinitrophenol (DNP). Two hours later mice were injected i.p. with TGA, Inos (100 ng/200 μL in phosphate buffered saline [PBS]), or 200 μL PBS alone as a vehicle control. After 16 hours, mice were challenged i.p. with 100 μg DNP‐BSA (Sigma‐Aldrich, St. Louis, MO, USA) in 100 μL PBS. Body temperature measurements were recorded with a digital rectal thermometer every 10 minutes for a total of 50 minutes. Peripheral blood was harvested by cardiac puncture 50 minutes after antigen challenge and serum histamine measurements determined by ELISA.
To examine potential toxicity, alanine aminotranferease (ALT) and aspartate aminotransferase (AST) were measured in serum. These enzymes leak out into the general circulation when the liver is injured. 28 Mice were treated with or without 1,000 μg/100 μL (50 times more than used for the in vivo study) of TGA or Inos by tail vein injection and i.p. After 2 and 14 days, mice were sacrificed and blood obtained by cardiac puncture. ALT and AST activity were measured as described. 28 Data are presented as an average of four (untreated) or four (treated) mice ± SD.
Results
FD differentially affect FcɛRI‐mediated degranulation
A panel of FD with various moieties attached to the carbon cage was developed using cyclopropanation reactions to determine the effect various group additions have on the ability to inhibit FcɛRI stimulation. For MC, 35 different FD developed to date were tested and 12 of these significantly (p < 0.05) inhibited degranulation and cytokine production by >10%. For PBB, 15 different FD were screened and three inhibited PBB degranulation >10%, while 11 inhibited cytokine production >10% (Supplementary Figure S2). Two representative FD are shown in 1 , 2 , 3 , 4 ; TGA was observed to be one of the most efficient inhibitors and significantly reduced both degranulation and cytokine production in MC and PBB. However, Inos significantly inhibited degranulation and cytokine production in PBB ( Tables 3 and 4 ) but significantly inhibited only cytokine production in MC; degranulation was not affected in MC. IC50 values were calculated by setting the FD dose that resulted in maximum inhibition to 100% and then extrapolating the dose at which 50% inhibition was seen. The majority of the 35 compounds tested to date had no significant effect on FcɛRI mediator release. These data demonstrate selective inhibition of FcɛRI mediator release from primary human MC/PBB using nano‐engineered FD that depends on the moieties added to the carbon cage.
Table 1.
Degranulation (±SD) and GMCSF‐cytokine production (±SD) in TGA treated and untreated mice.
| Mean percent degranulation (±SD) in TGA treated and untreated MC | |||||
|---|---|---|---|---|---|
| Mast cells | |||||
| no XL | XL | TGA+XL*† | %inh | IC50 (μg/mL) | |
| Exp 1 | 3.4 | 76.7 | 39.1 | 48.9 | |
| Exp 2 | 3.2 | 81.1 | 50.1 | 38.2 | |
| Exp 3 | 5.5 | 74.5 | 53.4 | 28.3 | |
| Exp 4 | 4 | 77.9 | 40.1 | 48.5 | |
| Exp 5 | 8.1 | 78.5 | 52.7 | 32.8 | |
| Average | 4.8 (±2.0) | 77.7 (±2.4) | 47.1 (±6.9) | 39.3 (±9.2) | 10.9 (±3.5) |
| Mean percent GMCSF‐cytokine production (±SD) in TGA treated and untreated MC | |||||
|---|---|---|---|---|---|
| Mast cells | |||||
| no XL | XL | TGA+XL*† | %inh | IC50 (μg/mL) | |
| Exp 1 | 50.4 | 211.4 | 69.5 | 67.8 | |
| Exp 2 | 135.4 | 893.8 | 121.9 | 84.9 | |
| Exp 3 | 91.6 | 309.5 | 104.5 | 66.2 | |
| Exp 4 | 65.4 | 271.4 | 74.1 | 72.7 | |
| Exp 5 | 105.4 | 741 | 43.4 | 95.2 | |
| Average | 89.6 (±33.4) | 485.4 (±309.8) | 82.7 (±30.8) | 77.4 (±12.4) | 3.3 (±0.6) |
* TGA Treatment @ 10 μg/mL.
† p value = 0.0001.
* TGA Treatment @ 10 μg/mL.
†p value = 0.02.
Table 2.
Degranulation (±SD) and GMCSF‐cytokine production (±SD) in Inos treated and untreated mast cells.
| Mean percent degranulation (±SD) in Inos treated and untreated MC | |||||
|---|---|---|---|---|---|
| Mast cells | |||||
| no XL | XL | Inos+XL*† | %inh | IC50 (μg/mL) | |
| Exp 1 | 3.4 | 76.7 | 78.1 | 1.9 | |
| Exp 2 | 3.2 | 81.1 | 80.3 | 1 | |
| Exp 3 | 5.5 | 74.5 | 70.2 | 5.8 | |
| Exp 4 | 4 | 77.9 | 75.5 | 3.1 | |
| Exp 5 | 8.1 | 78.5 | 91.6 | 16.8 | |
| Average | 4.8 (±2.0) | 77.7 (±2.4) | 79.1 (±7.9) | 3.3 (±2.4) | |
| Mean percent GMCSF‐cytokine production (±SD) in Inos treated and untreated MC | |||||
|---|---|---|---|---|---|
| Mast cells | |||||
| no XL | XL | Inos+XL*† | %inh | IC50 (μg/mL) | |
| Exp 1 | 50.4 | 211.4 | 11.9 | 94.4 | |
| Exp 2 | 135.4 | 893.8 | 20.4 | 97.7 | |
| Exp 3 | 91.6 | 309.5 | 23.5 | 92.4 | |
| Exp 4 | 65.4 | 271.4 | 63.1 | 76.8 | |
| Exp 5 | 105.4 | 741 | 43.4 | 95.2 | |
| Average | 89.6 (±33.4) | 485.4 (±309.8) | 32.5 (±20.6) | 91.3 (±8.3) | 1.9 (±1.1) |
*Inos Treatment @ 10 μg/mL.
†p value = 0.358.
* Inos Treatment @ 10 μg/mL.
†p value = 0.006.
Table 3.
Degranulation (±SD) and IL‐13 cytokine production (±SD) in TGA treated and untreated peripheral blood basophils.
| Mean percent degranulation (±SD) in TGA treated and untreated PBB | |||||
|---|---|---|---|---|---|
| Peripheral Blood Basophils | |||||
| no XL | XL | TGA+XL*† | %inh | IC50 (μg/mL) | |
| Exp 1 | 7.3 | 35 | 31 | 11.4 | |
| Exp 2 | 22.2 | 37 | 34 | 8.1 | |
| Exp 3 | 16.8 | 36 | 24 | 33.3 | |
| Exp 4 | 8.1 | 43 | 40 | 6.9 | |
| Exp 5 | 9.5 | 21 | 17 | 19 | |
| Exp 6 | 17.8 | 62.5 | 51 | 18.4 | |
| Exp 7 | 9.2 | 64 | 63 | 1.56 | |
| Exp 8 | 11 | 83 | 78 | 6 | |
| Exp 9 | 19.2 | 27.5 | 17 | 38.1 | |
| Average | 13.5 (±5.5) | 45.4 (±20.1) | 39.4 (±7.0) | 15.8 (±4.2) | 9.9 (±2.9) |
| Mean percent IL‐13‐cytokine production (±SD) in TGA treated and untreated PBB | |||||
|---|---|---|---|---|---|
| Peripheral Blood Basophils | |||||
| no XL | XL | TGA+XL*† | %inh | IC50 (μg/mL) | |
| Exp 1 | 42.8 | 252 | 119.5 | 52.6 | |
| Exp 2 | 12.6 | 297.5 | 35 | 88.2 | |
| Exp 3 | 16.4 | 102.5 | 52.5 | 48.8 | |
| Exp 4 | 39.4 | 137.5 | 110 | 20 | |
| Average | 27.8 (±92.4) | 197.4 (±92.4) | 79.3 (±41.8) | 52.4 (±28.0) | 10.6 (±3.4) |
* TGA Treatment @ 5 μg/mL.
†p value = 0.001.
* TGA Treatment @ 5 μg/mL.
† p value = 0.029.
Table 4.
Degranulation (±SD) and IL‐13 cytokine production (±SD) in Inos treated and untreated peripheral blood basophils.
| Mean percent degranulation (±SD) in Inos treated and untreated PBB | |||||
|---|---|---|---|---|---|
| Peripheral Blood Basophils | |||||
| no XL | XL | Inos+XL*† | %inh | IC50 (μg/mL) | |
| Exp 1 | 7.3 | 35 | 23 | 34.2 | |
| Exp 2 | 16.8 | 36 | 21 | 41.6 | |
| Exp 3 | 12.6 | 41 | 36 | 12.1 | |
| Exp 4 | 10.4 | 19 | 11 | 42.1 | |
| Exp 5 | 12.9 | 62.5 | 60 | 4 | |
| Exp 6 | 12.5 | 86 | 68 | 20.9 | |
| Exp 7 | 11.4 | 76 | 76 | 0 | |
| Average | 12 (±2.9) | 50.8 (±24.5) | 42.1 (±25.7) | 22.1 (±17.5) | 17.8 (±3.2) |
| Mean percent IL‐13‐cytokine production (±SD) in Inos treated and untreated PBB | |||||
|---|---|---|---|---|---|
| Peripheral Blood Basophils | |||||
| no XL | XL | Inos+XL *† | %inh | IC50 (μg/mL) | |
| Exp 1 | 42.8 | 252 | 7 | 97.2 | |
| Exp 2 | 16.4 | 102.5 | 5 | 95.1 | |
| Exp 3 | 1.2 | 51.8 | 10.1 | 80.5 | |
| Average | 29.6 (±18.7) | 177.3 (±105.7) | 6.0 (±1.4) | 90.9 (±9.0) | 2.6 (±1.2) |
* Inos Treatment @ 5 μg/mL.
†p value = 0.007.
* Inos Treatment @ 5 μg/mL.
†p value = 0.05.
Mechanisms of MC‐FcɛRI inhibition
In order to understand how TGA and Inos differentially influence FcɛRI‐dependent mediator release, we first examined those events that occur immediately (within 30 minutes) upon FcɛRI activation. The activation of MC and PBB FcɛRI leading to degranulation is calcium dependent and induces elevated cellular levels of ROS. 29 , 30 We hypothesized that the underlying mechanism of inhibition involved the blocking of FcɛRI‐mediated calcium and ROS responses based on previous studies with mixed isomer FDs. 19 We compared the responses of MC treated with TGA or Inos. The increase in FcɛRI‐induced intracellular calcium stores release ( Figure 1A ) and ROS ( Figure 1B ) upon FcɛRI cross‐linking was inhibited with TGA. However, Inos preincubation did not affect calcium ( Figure 1C ) or ROS ( Figure 1D ) levels. Gene microarray data further suggests that TGA and Inos differentially influence FcɛRI‐associated signaling molecules involved in calcium stores release and oxidative stress (manuscript in preparation). Thus, TGA and Inos differentially affect ROS and calcium responses induced by FcɛRI activation.
Figure 1.
Fullerene derivatives inhibit anti‐FcɛRI‐dependent increases in intracellular calcium and ROS levels. Cells were incubated with or without FD (10 μg/mL) overnight. The next day cells were challenged with anti‐FcɛRI (3B4; 1 μg/mL) and calcium stores release was determined by the 340/380 nm ratio (A and B) and ROS measured by DCF detection at 523 nm (C and D). A and C contain cells incubated with the TGA FD and B and D contain cells incubated with the Inos FD (10 μg/mL). The dark gray squares are the unchallenged negative control, dark thick solid black line is the anti‐FcɛRI challenged positive control cells, and the two light gray lines (hashed and double line) are the anti‐FcɛRI challenged cells pretreated with respective FD. Results are representative of at least three separate experiments.
FD block early FcɛRI‐activated signaling molecules
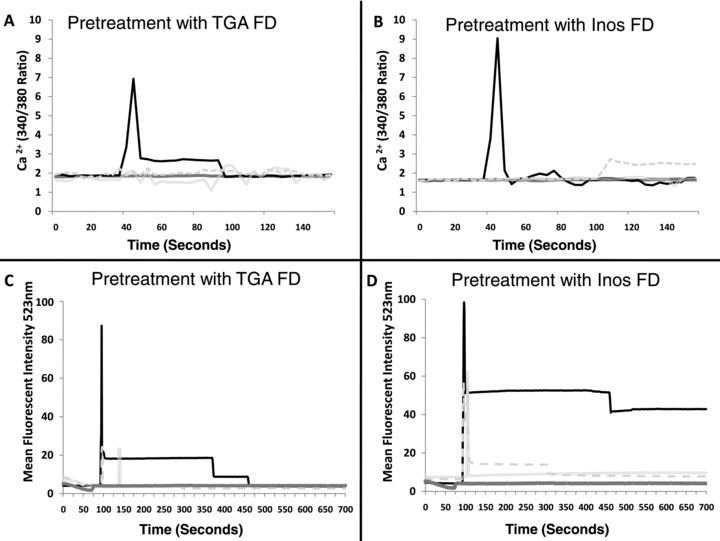
To further investigate the early signaling events in FcɛRI‐dependent mediator release that are affected by TGA or Inos, we performed Western blotting analysis using phospho‐specific antibodies. The phosphorylation of signaling intermediates is an important early step in FcɛRI‐induced mediator release. 31 As seen in Figure 2 , several signaling intermediates that were phosphorylated by FcɛRI activation were inhibited by preincubation with both FD including phosphorylation of extracellular signal regulated kinases 1/2 (ERK1/2), p38‐mitogen‐activated protein kinase (p38 MAPK), linker of activated T cells (LAT), Rho family GTPase (RAC) serine threonine protein kinase (AKT), phosphoinositide 3‐kinase (PI3‐K), and v‐src sarcoma viral oncogene homolog (SRC). For example, FD inhibition of MC signaling was time dependent as preincubation with TGA inhibited AKT phosphorylation 10%, 36%, and 43% at 3, 5 and, 10 minutes, while Inos inhibited AKT phosphorylation 18%, 48%, and 42% at 3, 5 and, 10 minutes. Both FD dramatically reduced LAT phosphorylation at later time points with 36%, 72%, and 69% for TGA and 33%, 79%, and 70% for Inos at 3, 5 and, 10 minutes, respectively. PI3‐kinase, a kinase strongly associated with calcium flux, was dramatically inhibited by both compounds with TGA reducing phosphorylation by 15%, 35%, and 50%, while Inos reduced phosphorylation by 8%, 40%, and 56% at 3, 5, and 10 minutes, respectively. There were also variations in the phosphorylation of other signaling intermediates examined at several time points. Little change was observed with a phospho‐Lyn Ab to the negative regulatory tyrosine, a kinase with positive and negative signaling roles in MC. 32 Thus, molecules previously implicated in the release of calcium stores and ROS production in response to FcɛRI aggregation were inhibited by certain FD. However, differences in how TGA and Inos inhibit FcɛRI mediator release were not due to differential phosphorylation of the signaling molecules examined.
Figure 2.
FcɛRI‐mediated activation of early signaling molecules is inhibited by FD. MC were pretreated with Inos or TGA (10 μg/mL) overnight at 37°C/6% CO2. The next day washed cells were activated with or without anti‐FcɛRI (1 μg/mL) for the indicated times and lysed using protocols described above. Two separate 10% Tris‐Glycine gels were used for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blotting performed with the indicated phosphorylation‐specific (i.e., phosph‐Lyn) antibodies. To ensure equal loading, an antiactin antibody was used in parallel. The intensity of each band was detected and quantified using an Odyssey imaging system. Band intensities are presented as a ratio compared to antiactin band intensities probed in the same lane and presented for each time point in the colored graphs on the right side. Results are representative of three separate experiments. The two gels were run side‐by‐side, so that each time point could be examined, and are shown next to each other.
Gene microarray analysis of TGA and Inos‐treated MC
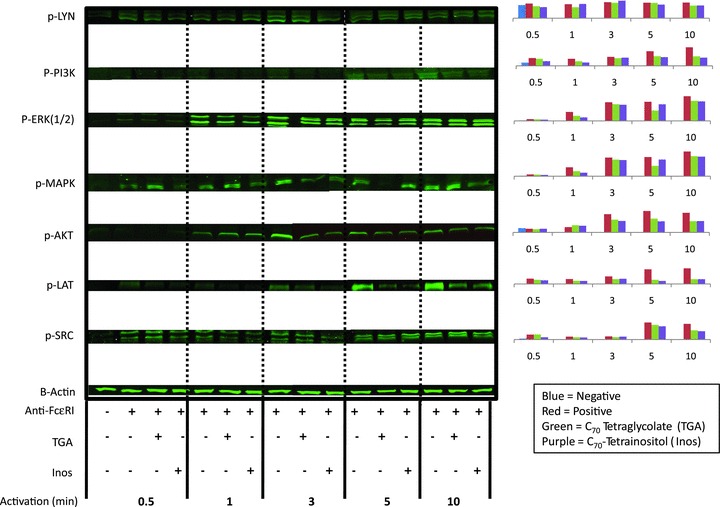
Given that optimal inhibition of MC or PBB secretion was observed after overnight preincubation with FD, we hypothesized that a transcriptional mechanism of action was involved. Microarray analysis was used to obtain a broad overview of those FcɛRI‐associated signaling molecules influenced by TGA and Inos preincubation following FcɛRI activation at later (2 hours) times. Of the approximately 31,000 genes examined, the level of expression of 1,771 increased at least 10% in MCs following cross‐linking of FcɛRI. In cells pretreated with TGA 2,603 of those were decreased at least 20% compared to non‐TGA treated cells and 515 demonstrated greater than 50% inhibition. Figure 3A represents several of those genes that were upregulated after 2 hours of FcɛRI activation and significantly inhibited by TGA preincubation. The gene microarray data was verified at the protein level using Western blotting. For example, the gene for the tyrosine kinase Lyn was upregulated 293% upon FcɛRI stimulation. However, gene expression levels were reduced by 51% at 2 hours that resulted in a 92% inhibition at the protein level observed at 6 hours ( Figure 3 ). E74‐like factor 2 (ELF‐2), which transcriptionally regulates Lyn, was also downregulated 47% suggesting Lyn is controlled at the transcription/translation level and protein level after two hours of FcɛRI aggregation but not after 10 minutes of activation. Conversely, Fyn, which was not inhibited at the genetic level (not shown) had no change in protein levels when preincubated with TGA ( Figure 3 ‐insert). Several other molecules, including ADAM10, MAP2K, BTK, and Syk, were inhibited at the gene level that resulted in lower protein levels. The ability to downregulate multiple components of a signaling pathway is novel and may provide a platform for engineering potent anti‐inflammatory compounds. Our results indicate the ability to block both the activation as well as the expression of distinct signaling molecules within the FcɛRI pathway.
Figure 3.
Microarray analysis of FcɛRI activated genes affected by TGA. MC were pretreated with or without 10 μg/mL TGA overnight at 37°C/6% CO2. The next day cells were washed and challenged with (+) or without (−) anti‐FcɛRI (1 μg/mL) for 2 hours. Cell pellets were used for RNA isolation and gene microarray as described above or used for Western blotting. Each condition was performed in triplicate. What is shown is the mean of the relative intensities of each gene (±SD of six observed values). The red bar/numbers is the percent increase in resting cells compared to FcɛRI‐activated cells and the black bar/numbers is the percent inhibition comparing FD pretreated and activated cells compared to non‐FD‐treated FcɛRI‐treated cells. For Western blotting, approximately 200,000 cell equivalents/lane were loaded onto a 10% tris‐glycine gel and Western blotted with the indicated Abs as described previously. 20
There was a dramatic difference in the FcɛRI genes affected by Inos compared to TGA. In cells preincubated with this FD, only 741 genes were decreased at least 20% compared to non‐FD treated cells and 92 demonstrated greater than 50% inhibition. Instead, there was a dramatic increase in expression of genes associated with inhibitory signaling. The Inos FD had 6 times more (1,847) genes upregulated compare to TGA (322) (Supplemental File 2). Unlike with TGA, Inos induced the upregulation of over 20 protein tyrosine phosphatases (PTP) and dual‐specificity phosphatases (DUSP). The upregulation of several of these inhibitory molecules is shown in Figure 4 . For example, DUSP1 was upregulated 126% at the gene level at 2 hours and 99% at 6 hours at the protein level ( Figure 4 ‐insert). Therefore, the inhibition of cytokine production by Inos (opposed to TGA) was due in part to increasing protein expression of PTP. Since TGA and Inos may be differentially affecting two groups of enzymes (kinases vs. PTP), the FD may be recognizing a common regulatory mechanism or motif in the signaling molecule.
Figure 4.
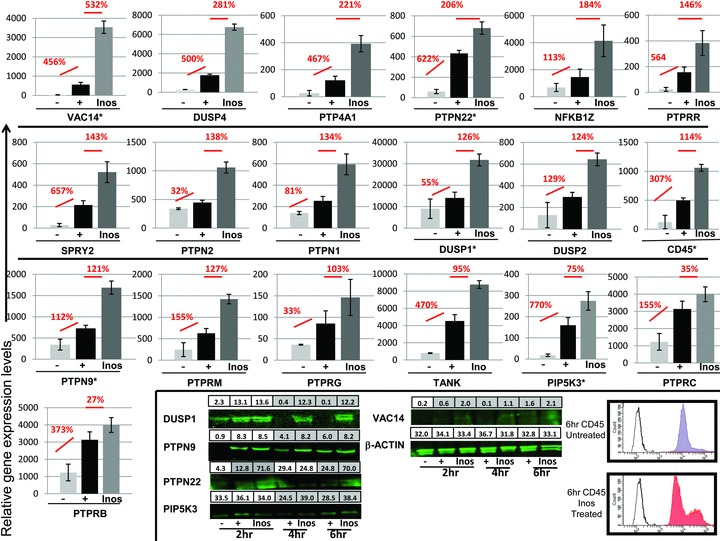
Microarray analysis of FcɛRI activated genes affected by Inos. MC were treated and analyzed as in Figure 3. Given the large size of CD45 (220 kDa) and difficulty in gel‐to‐membrane transfer FACS analysis was used to monitor upregulation: mouse IgG Isotype control (black line), untreated FITC labeled CD45 (gray filled), and Inos treated Fluorescein isothiocyanate (FITC) labeled CD45 (red filled).
Effects of FD on MC FcɛRI‐induced anaphylaxis and toxicological assessment
To explore the in vivo effects of FD on MC responses, we tested the degranulation and cytokine‐blocking TGA and the cytokine blocking Inos on MC‐induced anaphylaxis. DNP‐IgE‐sensitized animals injected i.p. with DNP‐BSA demonstrated a characteristic drop in core body temperature resulting from MC‐driven anaphylactic shock. When mice were injected with TGA before FcɛRI challenge, there was a significant reduction in the anaphylactic‐induced drop in core body temperature and behavioral responses that accompany anaphylactic shock ( Figure 5A ). However, Inos had no effect on MC‐induced anaphylaxis ( Figure 5B ). As expected, serum histamine levels were significantly lower in animals treated with the degranulation and cytokine inhibitor compared to controls ( Figure 5C ). There was no significant increase in serum activity of ALT and AST between the untreated and fullerene‐treated mice injected with FD concentrations 50‐fold higher than that needed for in vivo efficacy ( Table 5 ; tail vein or i.p. routes gave the same result). The injections were well tolerated and no change in behavior or body weights was noted. These experiments demonstrate that the efficacy of FD in vivo depends on how the carbon cage is derivatized and suggest they can be engineered at the nano‐scale level to perform specific cellular functions.
Figure 5.
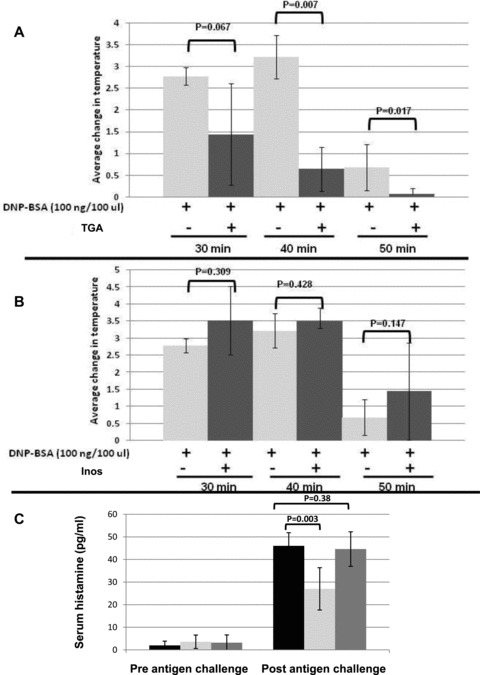
FD blunt MC‐driven anaphylaxis in vivo. Mice (5 per group) were sensitized with 50 μg DNP‐IgE in 100 μl PBS. Two hours later the mice were injected i.p. with PBS alone or 100 ng of FD that inhibit degranulation and cytokine production (A) or cytokine production only (B) in 100 μL PBS. The following day rectal temperatures were recorded before mice were challenged i.p. with 100 μg of DNP‐BSA in 100 μL PBS. Temperatures were recorded every 10 minutes (up to 50 minutes) following challenge with DNP‐BSA. (C) Increases in antigen‐induced serum histamine release is blunted by FD. Blood was collected from control (black bars), degranulation inhibiting FD (light gray bars) or cytokine inhibiting FD (dark gray bars) at 50 minutes and histamine content determined by ELISA.
Table 5.
No liver toxicity is detected following FD injection
| Day 2 | Day 14 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inositol Treated | TGA Treated | Untreated | Inositol Treated | TGA Treated | Untreated | |||||||
| n | Activity | n | Activity | n | Activity | n | Activity | n | Activity | n | Activity | |
| Aspartate amino‐transferase (AST) | 3 | 27.6 (±2.1) | 3 | 8.5 (±2.3) | 3 | 29.5 (±9.2) | 3 | 33.5 (±4.2) | 3 | 29.8 (±9.9) | 3 | 51.1 (±4.1) |
| Alanine amino‐transferase (ALT) | 3 | 26.4 (±2.0) | 3 | 29.2 (±2.5) | 3 | 64.3 (±11.5) | 3 | 44.2 (±1.5) | 3 | 36.7 (±6.6) | 3 | 47.0 (±2.9) |
n = Number of Mice evaulated in Duplicates.
Untreated = Normal Mice with PBS injection.
Treated = Tail Vein injection of 100 μl of C70‐inositol or C70‐tetragylcolate.
Discussion
This study examines how FD impact MC and PBB responses following FcɛRI challenge. The impetus for these studies was based on the observation that fullerenes are potent ROS scavengers, and there is evidence that ROS is involved in FcɛRI signaling. In addition, we had shown previously that water‐soluble FD could affect MC/PBB responses, although the compounds used in those studies are not viable therapeutic candidates. Thus, next generation FD were developed and investigated to determine which side chains most affected FcɛRI signaling pathways. Several new FD were identified that inhibited MC/PBB FcɛRI responses and two were selected for further in vitro and in vivo studies. Several observations were noted. First, there were striking differences in the biological activity of FD that was dependent on the side chain moieties added to the carbon cage. Second, the differences in FD structures revealed selectivity in FcɛRI pathway inhibition between MC and PBB; Inos inhibited cytokine production only in MC but inhibited both degranulation and the cytokine release in PBB. Third, it is shown for the first time that FD affect both phosphorylation of signaling molecules as well as gene expression. Fourth, while previous studies suggest FD induce oxidative stress33, 34, 35, 36 and apoptosis, 35 , 37 our studies contradict these findings and suggest FD block oxidative stress and apoptosis‐related genes that were upregulated by FcɛRI cross‐linking. Lastly, this study expands the wide range of biological pathways that FD affect and may represent a new way to control MC responses before they occur.
Complete inhibition of FcɛRI MC/PBB mediator release was not observed with TGA, yet it was sufficient to improve disease outcomes. This result is consistent with those reported on MC‐blunting pharmaceuticals currently prescribed to patients. Cromolyn, an MC‐stabilizer and current treatment for asthmatic patients (Nasalcrom, Intal, Opticrom), has been shown to inhibit MC activation in vitro in the same range that FD inhibit MC mediator release (35%–80% inhibition). 38 , 39 , 40 , 41 , 42 Cromolyn was first discovered as an MC stabilizer (such as FD) and is currently prescribed by physicians to prevent diseases associated with MC activation. Similarly, the anti‐IgE therapy Xolair (which indirectly blocks MC degranulation by depleting IgE levels) does not completely inhibit FcɛRI mediator release. 43 , 44 , 45 , 46 Therefore, MC‐targeting and complete inhibition of FcɛRI mediator release in vitro is not necessary for in vivo efficacy.
Our results are consistent with studies demonstrating a lack of toxicity of FD using in vitro cell culture assays. 18 No toxicity was observed using up to 100 μg/mL, well above concentrations in which efficacy was observed in vitro and in vivo. In contrast to most studies conducted previously on FD toxicity, our current study employed thoroughly purified and characterized FD, limiting the likelihood of confounding results due to sample impurities. In separate experiments, the in vivo administration (i.v. daily; 200 ng/3 weeks) of TGA to mice showed no notable differences in behavior, and there were no abnormalities observed upon gross pathological examination. No mutagenic potential (using the Ames test) was observed (not shown). No adverse reactions were observed in the anaphylaxis model, when FD were injected and no liver damage was noted at the concentrations sufficient for in vivo efficacy. A full toxicity profile is currently being performed for each candidate based on their MC/PBB inhibitory capabilities. Taken together, the derivatives described herein are not cytotoxic to several cell lines tested and appear to have no acute in vivo cytotoxic effects.
Empty cage fullerenes—without appropriate side chain moieties—are generally not biologically suitable for therapeutic development. In order to harness the biological potential of the core carbon fullerene cage, chemical structures are added directly to the cage to make them compatible with biological systems. A similar strategy has previously been employed to target the antioxidant properties of FD for neuronal protection. 9 We used a similar strategy to add distinct side chain moieties with various chemical properties that were hypothesized to interfere with FcɛRI mediator release. We chose to focus on the use of carbon cages with C70 structures, instead of C60, as bis additions to C70 occurs exclusively on each pole of the oblate spheroid cage. This strategy offers enhanced control over the topography of the addition groups thus reducing the potential number of isomers, a key issue in the FDA approval process. We have also observed that the C70 is a more efficient scavenger of ROS compared to C60 (submitted for publication, Bioconjugate Chemistry), which is understandable given the increased number of C = C bonds in the C70 cage.
The sugar inositol is involved in several biological pathways in inflammation including FcɛRI signaling. 47 Interestingly, unlike TGA, Inos does not interfere with degranulation in MC but is highly effective at blocking cytokine production in MC, while it does inhibit degranulation in PBB. This suggests that the Inos interacts and/or indirectly inhibits a signaling molecule found in human MC FcɛRI signaling and not in PBB. Mechanistically, the variations in how Inos and TGA differentially inhibit intercellular FcɛRI responses between MC and PBB may be explained in how they affect Lyn. In mouse Lyn ‐/‐ basophils, FcɛRI‐mediated degranulation is inhibited. 48 However, Lyn ‐/‐ BMMCs can have the opposite phenotype with degranulation being upregulated 49 or inhibited depending on the Lyn/Fyn ratio. 50 In rat basophil leukaemia (RBL) cells, degranulation can be inhibited, while TNF‐α secretion is unaffected by overexpressing Lyn. 51 There is also precedence demonstrating that intracellular FcɛRI‐signaling pathways diverge subsequent to activation in MC and PBB. It is well known that concentrations of FcɛRI cross‐linking agents leading to optimal cytokine production are consistently lower than concentrations needed for optimal degranulation. 52 The release of preformed mediators through FcɛRI‐mediated degranulation follows the activation of protein kinase C (PKC) and calcium mobilization; cytokine and chemokine production requires activation of mitogen‐activated protein kinases p38 and c‐Jun N‐terminal kinases (JNK), whereas lipid mediator production follows the activation of ERK1/2 pathway. 53 A central control point that possibly mediates FcɛRI‐signals leading to cytokine production and mediator release occurs at LAT; 54 the phospho‐activation of this signaling molecule was affected significantly (see below). In mice, tumor necrosis factor‐associated factor 6 (TRAF6) is specifically required for cytokine generating FcɛRI‐signals of NF‐κB, p38 MAP kinase, and JNK yet is not required for proximal signaling and subsequent degranulation. 55 Our plan is to identify what signaling molecule(s) bind to TGA and Inos using cell lysates from differentially activated MC and PBB.
To begin to explore at what point the FD affect early FcɛRI‐signaling pathways postactivation and why certain FD differentially affected mediator release, we performed Western blotting with several phospho‐specific antibodies for FcɛRI‐associated signaling molecules. It is shown for the first time that FD can inhibit the phosphorylation of signaling intermediates involved with calcium and ROS generation. The Inos and TGA both reduced the phosphorylation of several intermediates, especially LAT and PI3‐K. Our results are consistent with other findings showing that LAT is critical for calcium mobilization 53 and MC from LAT‐deficient mice have inhibited FcɛRI‐mediated degranulation. Similarly, PI3‐K is critical for functional responses in MC as the PI3‐K inhibitors wortmannin and LY294002 inhibit antigen‐induced calcium mobilization, degranulation and cytokine production by murine and human MC. 31 However, the activation of these signaling molecules is dependent on prior activation of Lyn. In our current study, the early (10 minutes) phosphorylation of Lyn was not influenced by either FD.
To better understand the differences in Inos and TGA effects on MC FcɛRI‐signaling, gene microarray experiments were used. These studies revealed that TGA profoundly reduced the FcɛRI‐induced activation of over 1,000 genes that was selectively verified at the protein level. Several important discoveries were made from these experiments. First, many upregulated genes were identified that were previously not associated with MC/PBB FcɛRI signaling. These include tank‐binding kinase 1 (TBK‐1) involved in mediating NFκB activation, 56 Poly (ADP‐ribose) polymerase 1 (PARP1) previously shown to be a target for the development of new therapeutic strategies in the treatment of lung disorders such as asthma, 57 calumenin (CALU) that is a calcium‐binding protein localized in the endoplasmic reticulum (ER) involved in protein folding and sorting, a disintegrin and metalloproteinase (ADAM10), SOX9, tumor necrosis factor alpha‐induced protein 1 (TNFAIP1) an immediate‐early response gene of endothelium induced by TNF‐α, 58 and SAMD9 involved in the regulation of TNF‐α signaling. 59 , 60 Second, these are the first data to demonstrate that FD can influence gene expression. Given that current dogma suggest their biological activity depends solely on their antioxidant properties, these results suggest FD effects are not entirely due to ROS scavenging capabilities. Third, it proves that fullerenes as a class cannot be considered to behave the same in vitro, in situ, and in vivo. Two C70‐based derivatives with very similar molecular weights had practically no common effects in the microarray studies. This further reiterates the key message from these studies that the biological effects of FD critically depend on the side chains added to the core carbon cage.
TGA inhibits MC FcɛRI‐mediated activation in vivo as shown in the anaphylaxis model used in Figure 5 . Interestingly, Inos, the FD that have no effect on MC FcɛRI degranulation but are a potent cytokine inhibitor, did not significantly inhibit anaphylaxis using the same model ( Figure 5B ) suggesting FD are inhibiting preformed MC mediators. We provide initial mechanistic evidence for the ability of FD to suppress MC and PBB secretion by affecting transcription of a select group of FcɛRI‐signaling molecules. While the FD are likely to be influencing other cell types, our goal herein was to first identify MC inhibitors and then begin to explore their effects on other cell types. To this end, preliminary experiments using TGA suggest it may interfere with human B cell IgE production (not shown).
Conclusion
These studies describe a platform for engineering MC/PBB inhibitors at the nano‐scale based on FD and establish proof‐of‐principle for their use as a new way to treat MC/PBB‐influenced diseases. Currently, MC/PBB‐targeting FD are being developed that will be able to selectively deliver FD that inhibit activation responses alone or in combination with other therapeutics. Future studies are also aimed at developing next generation FD that are even more potent inhibitors of MC/PBB mediator release and investigating their use as new therapeutics for patients.
Supporting information
Supplementary Figure S1: Representative schematic demonstrating the synthesis of FD compound TGA.
Supplementary File S1: Microarray analysis of genes up‐ or downregulated by TGA and Inos. The details of FD preincubation, cell activation, and transcript analysis are described methods.
Supplementary Figure S2: Fullerene derivatives reduce degranulation and cytokine production from MC after anti‐FcRI activation. Cells were cultured with fixed concentrations of FD (10 μg/mL; A and C) or different concentration (B and D) for 16 hours, washed and stimulated for 30 minutes (A and B) or 24 hours (C and D) with optimal concentrations of αanti‐FcRI Abs (3B4; 1 μg/mL). Cells were centrifuged and β‐hexosaminidase release and GMCSF levels determined by ELISA. In A and C data shown are means ± SE of triplicate samples that is representative of at least four separate experiments with separate MC cultures. All values shown demonstrated a significant (p < 0.05) inhibition of at least >10% inhibition compared to nontreated samples. In B, C70‐(OH)12 (black square), C70‐tetraphosphate (gray diamond), C70‐tetrapyridine (black X), C70‐niacin (gray circle), C70‐(PC)4 (gray triangle), and CCC (black +). In D, C70‐tetrapyridine (black diamond), C70‐tetraphosphate (black *), C70‐tetrasulfonate (gray triangle), C60‐ethanolamine (black X), and CCC (gray circle). FD with no effect (approximately 76% of those tested) are not shown.
Please note: Wiley‐Blackwell Publishing is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
This material is available as part of the online article from http://www.ctsjournal.com.
Supporting info item
Supporting info item
Supporting info item
Acknowledgment
CLK acknowledges NIH Grants 1R01GM083274, 1 R21 ES015696, and 1R43HL087578. We thank Andrew Saxon for helpful discussion. We thank Dr. Lois S. Weisman for VAC antibodies.
References
- 1. Ryan JJ, Kashyap M, Bailey D, Kennedy S, Speiran K, Brenzovich J, Barnstein B, Oskeritzian C, Gomez G. Mast cell homeostasis: a fundamental aspect of allergic disease. Crit Rev Immunol. 2007; 27: 15–32. [DOI] [PubMed] [Google Scholar]
- 2. Nigrovic PA, Lee DM. Mast cells in inflammatory arthritis. Arthritis Res Ther. 2005; 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kovanen PT. Mast cells: multipotent local effector cells in atherothrombosis. Immunol Rev. 2007; 217: 105–122. [DOI] [PubMed] [Google Scholar]
- 4. Alter SC, Schwartz LB. Effect of histamine and divalent cations on the activity and stability of tryptase from human mast cells. Biochim Biophys Acta. 1989; 991: 426–430. [DOI] [PubMed] [Google Scholar]
- 5. Bergmann U, Scheffer J, Köller M, Schönfeld W, Erbs G, Müller FE, König W. Induction of inflammatory mediators (histamine and leukotrienes) from rat peritoneal mast cells and human granulocytes by Pseudomonas aeruginosa strains from burn patients. Infect Immun. 1989; 57: 2187–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. FASEB J. 2005; 19: 311–330. [DOI] [PubMed] [Google Scholar]
- 7. Kroto HW, Allef AW, Balm SP. Buckminsterfullerene . Chem Revs. 1991; 91: 1213–1235. [Google Scholar]
- 8. Bakry R, Vallant RM, Najam‐ul‐Haq M, Rainer M, Szabo Z, Huck CW, Bonn GK. Medicinal applications of fullerenes. Int J Nanomedicine. 2007; 2: 639–649. [PMC free article] [PubMed] [Google Scholar]
- 9. Basso AS, Frenkel D, Quintana FJ, Costa‐Pinto FA, Petrovic‐Stojkovic S, Puckett L, Monsonego A, Bar‐Shir A, Engel Y, Gozin M, Weiner HL. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008; 118: 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dugan LL, Turetsky DM, Du C, Lobner D, Wheeler M, Almli CR, Shen CK, Luh TY, Choi DW, Lin TS. Carboxyfullerences as neuroprotective agents. Proc Natl Acad Sci U S A. 1997; 19;94: 9434–9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bosi S, Da RT, Spalluto G, Balzarini J, Prato M. Synthesis and Anti‐HIV properties of new water‐soluble bis‐functionalized[60]fullerene derivatives. Bioorg Med Chem Lett. 2003; 13: 4437–4440. [DOI] [PubMed] [Google Scholar]
- 12. Mroz P, Tegos GP, Gali H, Wharton T, Sarna T, Hamblin MR. Photodynamic therapy with fullerenes. Photochem Photobiol Sci. 2007; 6: 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin HS, Lin TS, Lai RS, D’Rosario T, Luh TY. Fullerenes as a new class of radioprotectors. Int J Radiat Biol. 2001; 77: 235–239. [DOI] [PubMed] [Google Scholar]
- 14. Lai YL, Murugan P, Hwang KC. Fullerene derivative attenuates ischemia‐reperfusion‐induced lung injury. Life Sci. 2003; 72: 1271–1278. [DOI] [PubMed] [Google Scholar]
- 15. Huang ST, Ho CS, Lin CM, Fang HW, Peng YX. Development and biological evaluation of C60 fulleropyrrolidine‐thalidomide dyad as a new anti‐inflammation agent. Bioorg Med Chem. 2008; 16: 8619–8626. [DOI] [PubMed] [Google Scholar]
- 16. Dellinger A, Zhou Z, Lenk R, Macfarland D, Kepley CL. Fullerene nanomaterials inhibit phorbol myristate acetate‐induced inflammation. Exp Dermatol. 2009; 18: 1079–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quick KL, Ali SS, Arch R, Xiong C, Wozniak D, Dugan LL. A carboxyfullerene SOD mimetic improves cognition and extends the lifespan of mice. Neurobiol Aging. 2008; 29: 117–128. [DOI] [PubMed] [Google Scholar]
- 18. Kolosnjaj J, Szwarc H, Moussa F. Toxicity studies of fullerenes and derivatives. Adv Exp Med Biol. 2007; 620: 168–180. [DOI] [PubMed] [Google Scholar]
- 19. Ryan JJ, Bateman HR, Stover A, Gomez G, Norton SK, Zhao W, Schwartz LB, Lenk R, Kepley CL. Fullerene nanomaterials inhibit the allergic response. J Immunol. 2007; 179: 665–672. [DOI] [PubMed] [Google Scholar]
- 20. Kepley CL. Antigen‐induced reduction in mast cell and basophil functional responses due to reduced Syk protein levels. Int Arch Allergy Immunol. 2005; 138: 29–39. [DOI] [PubMed] [Google Scholar]
- 21. Vonakis B M, Gibbons S Jr, Sora R, Langdon JM, MacDonald SM. Src homology 2 domain‐containing inositol 5‘ phosphatase is negatively associated with histamine release to human recombinant histamine‐releasing factor in human basophils. J Allergy Clin Immunol. 2001; 108: 822–831. [DOI] [PubMed] [Google Scholar]
- 22. Miura K, Saini SS, Gauvreau G, MacGlashan DW Jr. Differences in functional consequences and signal transduction induced by IL‐3, IL‐5, and nerve growth factor in human basophils. J Immunol. 2001; 167: 2282–2291. [DOI] [PubMed] [Google Scholar]
- 23. Saini SS, Paterniti M, Vasagar K, Gibbons SP Jr., Sterba PM, Vonakis BM. Cultured mast cells from chronic idiopathic urticaria patients spontaneously degranulate upon IgE sensitization: relationship to expression of Syk and SHIP‐2. Clin Immunol. 2009; 132: 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dellinger A, Zhou Z, Norton SK, Lenk R, Conrad D, Kepley CL. Uptake and distribution of fullerenes in human mast cells. Nanomedicine. 2010; doi:10.1016/j.nano.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao W, Kepley CL, Morel PA, Okumoto LM, Fukuoka Y, Schwartz LB. Fc gamma RIIa, not Fc gamma RIIb, is constitutively and functionally expressed on skin‐derived human mast cells. J Immunol. 2006; 177: 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tkaczyk C, Metcalfe DD, Gilfillan AM. Determination of protein phosphorylation in FceRI‐activated human mast cells by immunoblot analysis requires protein extraction under denaturing conditions. J Immunol Methods. 2002; 268: 239–243. [DOI] [PubMed] [Google Scholar]
- 27. Kepley CL, Pfeiffer J, Wilson BW, Schwartz LB, Oliver JM. Identification and characterization of umbilical cord blood‐derived human basophils. J Leukocyte Biol. 1998; 64: 474–483. [DOI] [PubMed] [Google Scholar]
- 28. Booth G, Newham P, Barlow R, Raines S, Zheng B, Han S. Gene expression profiles at different stages of collagen‐induced arthritis. Autoimmunity. 2008; 41: 512–521. [DOI] [PubMed] [Google Scholar]
- 29. Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol. 2008; 9: 1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Swindle EJ, Metcalfe DD. The role of reactive oxygen species and nitric oxide in mast cell‐dependent inflammatory processes. Immunol Rev. 2007; 217: 186–205. [DOI] [PubMed] [Google Scholar]
- 31. Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009; 228: 149–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rivera J, Fierro NA, Olivera A, Suzuki R. New insights on mast cell activation via the high affinity receptor for IgE. Adv Immunol. 2008; 98: 85–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sayes CM, Gobin AM, Ausman KD, Mendez J, West JL, Colvin VL. Nano‐C‐60 cytotoxicity is due to lipid peroxidation. Biomaterials. 2005; 26: 7587–7595. [DOI] [PubMed] [Google Scholar]
- 34. Moller P, Jacobsen NR, Folkmann JK, Danielsen PH, Mikkelsen L, Hemmingsen JG, Vesterdal LK, Forchhammer L, Wallin H, Loft S. Role of oxidative damage in toxicity of particulates. Free Radic Res. 2010; 44: 1–46. [DOI] [PubMed] [Google Scholar]
- 35. Nishizawa C, Hashimoto N, Yokoo S, Funakoshi‐Tago M, Kasahara T, Takahashi K, Nakamura S, Mashino T. Pyrrolidinium‐type fullerene derivative‐induced apoptosis by the generation of reactive oxygen species in HL‐60 cells. Free Radic Res. 2009; 43: 1240–1247. [DOI] [PubMed] [Google Scholar]
- 36. Zhu X, Zhu L, Lang Y, Chen Y. Oxidative stress and growth inhibition in the freshwater fish Carassius auratus induced by chronic exposure to sublethal fullerene aggregates. Environ Toxicol Chem. 2008; 27: 1979–1985. [DOI] [PubMed] [Google Scholar]
- 37. Yamawaki H, Iwai N. Cytotoxicity of water‐soluble fullerene in vascular endothelial cells. Am J Physiol Cell Physiol. 2006; 290: C1495–C1502. [DOI] [PubMed] [Google Scholar]
- 38. Free CA, Hall LE. Antiallergic properties of SQ 13,847, an orally effective agent. II. Activity in vitro. J Pharmacol Exp Ther. 1980; 213: 437–440. [PubMed] [Google Scholar]
- 39. Green WF, Konnaris K, andWoolcock AJ. Effect of salbutamol, fenoterol, and sodium cromoglycate on the release of heparin from sensitized human lung fragments challenged with dermatophagoides pteronyssinus allergen. Am J Respir Cell Mol Biol. 1993; 8: 518–521. [DOI] [PubMed] [Google Scholar]
- 40. Kano S, Tyler E, Salazar‐Rodriguez M, Estephan R, Mackins CJ, Veerappan A, Reid AC, Silver RB, Levi R. Immediate hypersensitivity elicits renin release from cardiac mast cells. Int Arch Allergy Immunol. 2008; 146: 71–75. [DOI] [PubMed] [Google Scholar]
- 41. Gonzalez R, Garcia M, De L. V, Arruzazabala ML, Perez H. Allergic contact dermatitis due to Centella asiatica: a new case. Allergol Immunopathol (Madr.) 1979; 7: 125–134. 88897 [Google Scholar]
- 42. Ma H, Kovanen PT. Inhibition of mast‐cell‐dependent conversion of cultured macrophages into foam cells with antiallergic drugs. Arterioscler Thromb Vasc Biol. 2000; 20: E134–E142. [DOI] [PubMed] [Google Scholar]
- 43. Mirkina I, Schweighoffer T, Kricek F. Inhibition of human cord blood‐derived mast cell responses by anti‐Fc epsilon RI mAb 15/1 versus anti‐IgE Omalizumab. Immunol Lett. 2007; 109: 120–128. [DOI] [PubMed] [Google Scholar]
- 44. Beck LA, Marcotte GV, MacGlashan D, Togias A, Saini S. Omalizumab‐induced reductions in mast cell Fc{epsilon}RI expression and function. J Allergy Clin Immunol. 2004; 114: 527–530. [DOI] [PubMed] [Google Scholar]
- 45. MacGlashan DW Jr, Bochner BS, Adelman DC, Jardieu PM, Togias A, McKenzie‐White J, Sterbinsky SA, Hamilton RG, Lichtenstein LM. Down‐regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti‐IgE antibody. J Immunol. 1997; 158: 1438–1445. [PubMed] [Google Scholar]
- 46. Saini SS, MacGlashan DWJ, Sterbinsky SA, Togias A, Adelman DC, Lichtenstein LM, Bochner BS. Down‐regulation of human basophil IgE and FC epsilon RI alpha surface densities and mediator release by anti‐IgE‐infusions is reversible in vitro and in vivo. J Immunol. 1999; 162: 5624–5630. [PubMed] [Google Scholar]
- 47. Ma HT, Beaven MA. Regulation of Ca2+ signaling with particular focus on mast cells. Crit Rev Immunol. 2009; 29: 155–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Charles N, Watford WT, Ramos HL, Hellman L, Oettgen HC, Gomez G, Ryan JJ, O'Shea JJ, Rivera J. Lyn kinase controls basophil GATA‐3 transcription factor expression and induction of Th2 cell differentiation. Immunity. 2009; 30: 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parravicini V, Gadina M, Kovarova M, Odom S, Gonzalez‐Espinosa C, Furumoto Y, Saitoh S, Samelson LE, O'Shea JJ, Rivera J. Fyn kinase initiates complementary signals required for IgE‐dependent mast cell degranulation. Nat Immunol. 2002; 3: 741–748. [DOI] [PubMed] [Google Scholar]
- 50. Yamashita Y, Charles N, Furumoto Y, Odom S, Yamashita T, Gilfillan AM, Constant S, Bower MA, Ryan JJ, Rivera J. Cutting edge: genetic variation influences Fc epsilonRI‐induced mast cell activation and allergic responses. J Immunol. 2007; 179: 740–743. [DOI] [PubMed] [Google Scholar]
- 51. Vonakis BM, Gibbons SP Jr, Rotte MJ, Brothers EA, Kim SC, Chichester K, MacDonald SM. Regulation of rat basophilic leukemia‐2H3 mast cell secretion by a constitutive lyn kinase interaction with the high affinity IgE Receptor (Fc{epsilon}RI). J Immunol. 2005; 175: 4543–4554. [DOI] [PubMed] [Google Scholar]
- 52. MacGlashan D Jr. Signal mechanisms in the activation of basophils and mast cells. Immunology Series. 1992; 57: 273–299. [PubMed] [Google Scholar]
- 53. Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast‐cell activation. Nat Rev Immunol. 2006; 6: 218–230. [DOI] [PubMed] [Google Scholar]
- 54. Rivera J, Gilfillan AM. Molecular regulation of mast cell activation. J Allergy Clin Immunol. 2006; 117: 1214–1225. [DOI] [PubMed] [Google Scholar]
- 55. Yang YJ, Chen W, Carrigan SO, Chen WM, Roth K, Akiyama T, Inoue J, Marshall JS, Berman JN, Lin TJ. TRAF6 specifically contributes to FcεRI‐mediated cytokine production but not mast cell degranulation. J Biol Chem. 2008; 283: 32110–32118. [DOI] [PubMed] [Google Scholar]
- 56. Pomerantz JL, Baltimore D. NF‐kB activation by a signaling complex containing TRAF2, TANK, and TBK1, a novel IKK‐related kinase. EMBO J. 1999; 18: 6694–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boulares AH, Zoltoski AJ, Sherif ZA, Jolly P, Massaro D, Smulson ME. Gene knockout or pharmacological inhibition of poly(ADP‐ribose) polymerase‐1 prevents lung inflammation in a murine model of asthma. Am J Respir Cell Mol Biol. 2003; 28: 322–329. [DOI] [PubMed] [Google Scholar]
- 58. Sarma V, Wolf FW, Marks RM, Shows TB, Dixit VM. Cloning of a novel tumor necrosis factor‐alpha‐inducible primary response gene that is differentially expressed in development and capillary tube‐like formation in vitro. J Immunol. 1992; 148: 3302–3312. [PubMed] [Google Scholar]
- 59. Topaz O, Indelman M, Chefetz I, Geiger D, Metzker A, Altschuler Y, Choder M, Bercovich D, Uitto J, Bergman R, Richard G, Sprecher E. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet. 2006; 79: 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chefetz I, Ben AD, Browning S, Skorecki K, Adir N, Thomas MG, Kogleck L, Topaz O, Indelman M, Uitto J, Richard G, Bradman N, Sprecher E. Normophosphatemic familial tumoral calcinosis is caused by deleterious mutations in SAMD9, encoding a TNF‐alpha responsive protein. J Invest Dermatol. 2008; 128: 1423–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: Representative schematic demonstrating the synthesis of FD compound TGA.
Supplementary File S1: Microarray analysis of genes up‐ or downregulated by TGA and Inos. The details of FD preincubation, cell activation, and transcript analysis are described methods.
Supplementary Figure S2: Fullerene derivatives reduce degranulation and cytokine production from MC after anti‐FcRI activation. Cells were cultured with fixed concentrations of FD (10 μg/mL; A and C) or different concentration (B and D) for 16 hours, washed and stimulated for 30 minutes (A and B) or 24 hours (C and D) with optimal concentrations of αanti‐FcRI Abs (3B4; 1 μg/mL). Cells were centrifuged and β‐hexosaminidase release and GMCSF levels determined by ELISA. In A and C data shown are means ± SE of triplicate samples that is representative of at least four separate experiments with separate MC cultures. All values shown demonstrated a significant (p < 0.05) inhibition of at least >10% inhibition compared to nontreated samples. In B, C70‐(OH)12 (black square), C70‐tetraphosphate (gray diamond), C70‐tetrapyridine (black X), C70‐niacin (gray circle), C70‐(PC)4 (gray triangle), and CCC (black +). In D, C70‐tetrapyridine (black diamond), C70‐tetraphosphate (black *), C70‐tetrasulfonate (gray triangle), C60‐ethanolamine (black X), and CCC (gray circle). FD with no effect (approximately 76% of those tested) are not shown.
Please note: Wiley‐Blackwell Publishing is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
This material is available as part of the online article from http://www.ctsjournal.com.
Supporting info item
Supporting info item
Supporting info item